

Abstract
Objective Identifying disease-mutation relationships is a significant challenge in the advancement of precision medicine. The aim of this work is to design a tool that automates the extraction of disease-related mutations from biomedical text to advance database curation for the support of precision medicine.
Materials and Methods We developed a machine-learning (ML) based method to automatically identify the mutations mentioned in the biomedical literature related to a particular disease. In order to predict a relationship between the mutation and the target disease, several features, such as statistical features, distance features, and sentiment features, were constructed. Our ML model was trained with a pre-labeled dataset consisting of manually curated information about mutation-disease associations. The model was subsequently used to extract disease-related mutations from larger biomedical literature corpora.
Results The performance of the proposed approach was assessed using a benchmarking dataset. Results show that our proposed approach gains significant improvement over the previous state of the art and obtains F-measures of 0.880 and 0.845 for prostate and breast cancer mutations, respectively.
Discussion To demonstrate its utility, we applied our approach to all abstracts in PubMed for 3 diseases (including a non-cancer disease). The mutations extracted were then manually validated against human-curated databases. The validation results show that the proposed approach is useful in a real-world setting to extract uncurated disease mutations from the biomedical literature.
Conclusions The proposed approach improves the state of the art for mutation-disease extraction from text. It is scalable and generalizable to identify mutations for any disease at a PubMed scale.
Keywords: precision medicine, disease-mutation relationship, automated extraction, machine learning, text mining, breast cancer, prostate cancer
INTRODUCTION
Finding relationships between genomic mutations and disease risk is one of the main challenges in developing supportive databases for personalized medicine,1 treatment or therapies that are guided by the differences in an individual’s genome. Such findings will lead to a better understanding of new pathways and disease mechanisms, which can then be translated to clinical practice. While some of these findings are discovered through clinical trials,2 most of them are buried in unstructured text3 within the biomedical literature. Manual curation of the current exponentially growing body of biomedical literature is practically an impossible task. Robust automated or semiautomated curation tools are a solution to this problem.4,5
Some well-known databases, such as ClinVar,2 Online Mendelian Inheritance in Man,6 Swiss-Prot,7 and SNPedia,8 contain human-curated information about disease-mutation relationships. Most of these databases contain categorized information about proteins and DNA mutations for specific disease phenotypes. Some of these databases, such as ClinVar, contain information from clinical trials and therefore are not synchronized with other documented research findings. SNPedia contains information about single-nucleotide polymorphism-associated diseases. Other resources focus on specific diseases, such as cancer,9 or specific chromosomal locations.10 All these databases are currently constructed and curated manually, which is a slow process that limits the number of cancer mutations available to the biomedical community and the potential use of these databases in clinical practice, even though most patients were found to be receptive to molecular testing for personalized cancer therapy.11
Recent efforts in the direction of automated or semiautomated approaches include extraction of mutational information from biomedical text. Overall, most methods have focused only on mutation extraction without connecting mutations to their associated diseases. Examples include MutationFinder,12 tmVar,13 EMU,3 and others.14 Among these tools, EMU is one of the most recent efforts. It provides a semiautomated approach to extracting disease-related mutations from PubMed abstracts and full text. While this approach automatically extracts mutations along with genes from the text, establishing mutation-disease relationships involves human curation and validation. Approaches prior to EMU include MuGeX,15 EnzyMiner,16 and OSIRIS.17 Each of these has been reported to have limitations and overspecialization.3 One notable method recently developed by Kuipers et al.18 introduces an automatic method for extracting and validating mutations for a single disease, Fabry disease. In all the above approaches, disease-to-mutation relationships in the text are not explicitly investigated or utilized for extraction. Therefore, developing an efficient, robust, and fully automated approach to extracting disease-related mutations is still a challenge.
In this article, we propose a novel, fully automated machine-learning approach for identifying disease-related point mutations from biomedical literature repositories. In this study, we primarily focus on 2 specific diseases, breast cancer and prostate cancer, because it has been noted that “oncology is the clear choice for … precision medicine,”19 and these are 2 diseases for which we have human-annotated ground truth for system development and evaluation. In order to identify relationships between mutations and diseases, we constructed several features, such as statistical features, distance features, and sentiment features. We trained our model with a pre-labeled dataset consisting of manually curated information about mutation-disease relationships. We subsequently used our model to extract mutation-disease relationships referenced in larger biomedical literature corpora.
To assess the validity of our approach, we compared it with several baseline systems on benchmarking datasets. We then applied our method to all literature in PubMed related to 3 diseases and validated the results against 2 human-curated databases.
MATERIALS AND METHODS
We developed our method for identifying mutation-disease relationships for multiple diseases using 2 text sources: (1) manually annotated corpora for breast and prostate cancer, and (2) comprehensive test sets built from all literature in PubMed related to the 3 diseases. The manually annotated corpora used in our approach were created by Doughty et al. and were the same sets used for developing the EMU tool. The prostate cancer corpus (EMU_PCa) contains 141 PubMed IDs (PMIDs), and the breast cancer corpus (EMU_BCa) contains 203 PMIDs. We refer to these corpora collectively as the EMU_dataset. In the Experimental Results section of this paper, we compare the performance of our approach to the EMU tool using this EMU_dataset.
In addition to the manually annotated EMU_dataset, we also generated a PubMed_dataset for the purpose of evaluating the utility of our method. The PubMed_dataset consists of 3 test sets, each of which contains all articles with abstracts in PubMed related to 1 of the 3 given diseases: prostate cancer, breast cancer, and age-related macular degeneration (AMD). We used the following query to collect the abstracts: “disease_name [tiab] AND English [lang] AND has_abstract [filter].” For the prostate cancer corpus (PubMed_PCa), we obtained 66 320 PMIDs with this query. For breast cancer (PubMed_BCa) and macular degeneration (PubMed_AMD), we obtained 155 512 PMIDs and 11 383 PMIDs, respectively. We obtained the title and abstract texts for these PMIDs using the PubTator tool4 and subsequently identified the disease mentions and mutations in these sets using the DNorm20 and tmVar13 tools. Based on the results of the DNorm tool, we found 66 104 (99.67%) PMIDs for prostate cancer, 154 815 (99.55%) PMIDs for breast cancer, and 11 331 (99.54%) PMIDs for AMD that contained mentions of these diseases in the text. tmVar processing of these PMIDs identified 1998 (3.02%) prostate cancer, 6490 (4.19%) breast cancer, and 803 (7.05%) AMD PMIDs that contained a mutation mention. Thus, a total of 1998 prostate cancer, 6490 breast cancer, and 803 AMD PMIDS contained both a mutation and a disease mention. Unlike the EMU_dataset, there is no ground truth available for the PubMed_dataset. We use it to show the utility and broad applicability of our method to extract mutations related to a given disease.
Our approach for identifying disease-mutation relationships is portrayed schematically in Figure 1 and can be summarized as follows. (The various text mining outputs contained in this work are available upon request. Our algorithm uses 2 publicly available tools, tmVar and DNorm. We also used the Weka tool for building our ML classifiers, which is open source as well. The source code required to generate features is readily available upon request.) First, the proposed approach uses the tmVar tool to identify mutation names in the text of the corpus. In parallel, the DNorm tool is used to identify all the disease names mentioned in the text of the articles in the corpus. In the second step, for each of the mutations identified by tmVar, the nearest disease mention is obtained. In the third step, we construct a set of features for each mutation-disease pair. Given a target disease, a training dataset is constructed to train a binary machine-learning (ML) classifier model to predict whether a mutation is related to the target disease or not. Finally, our approach consists of a trained model to automatically predict mutations related to a target disease in any given corpus.
Figure 1.
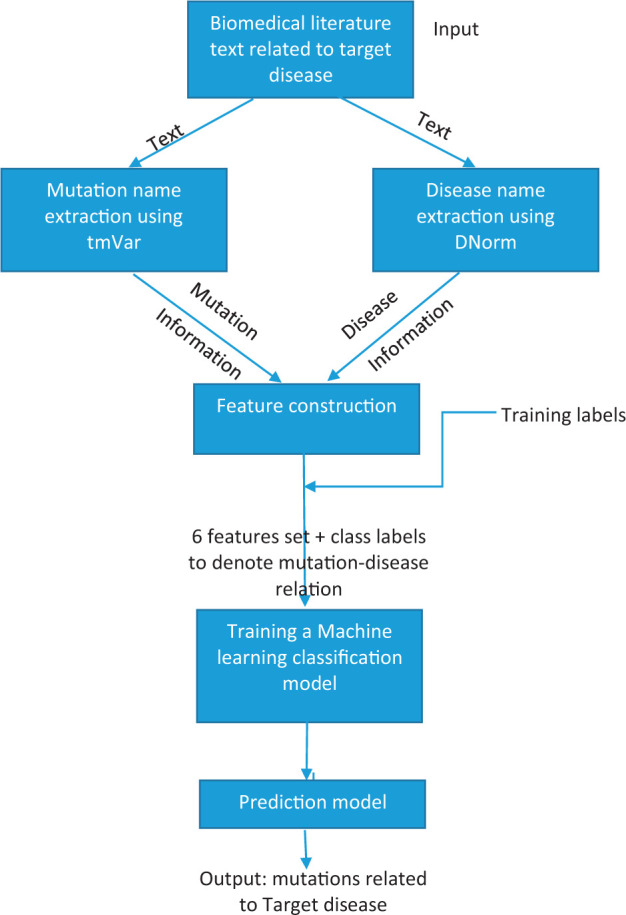
Schematic of the proposed approach for disease-related mutation prediction
Input text corpus
The input for our approach consists of the text from biomedical literature repositories. In this work, we use the text content in the title and abstract of the biomedical research articles.
Mutation extraction using tmVar
We employ tmVar to identify all the mutations in the input text. Several occurrences of a mutation are identified, along with their position within the text. To determine whether a mutation-disease relationship exists, it is useful to obtain information about all the occurrences of a mutation in the text, as explained in the feature construction step. Details of the tmVar method and performance are provided in the Appendix.
Disease name extraction using DNorm
In our approach, DNorm is used to extract all the disease mentions in the input text. While we are interested in knowing the relationship of the mutations with a given target disease, for the proposed approach it is important to obtain information about the presence of each disease along with the position of its mentions in the text to construct features used to develop the ML model. Details of the DNorm method and its performance are given in the Appendix.
Feature construction
This step is one of the main contributions of this work. Here we design a novel feature set to determine relationships between a mutation mention and a target disease for an input text. We designed 6 features, and they are described as follows:
- 1. Nearness to Target Disease Score (NTDS): For a mutation identified in the text, its NTDS is an integer denoting a cumulative score of all the times this mutation has the target disease as the closest disease mentioned in the text. A high positive NTDS signifies that the target disease was often the nearest disease mentioned in the text for this mutation. A negative NTDS denotes that diseases other than the target disease were often mentioned nearest to the mutation in the text. The nearness of a disease to a mutation is derived from the character count between the mutation position and the disease position in the text. It is mathematically described in the following manner:
where mi is the ith mutation, mij is the jth occurrence of ith mutation in the text. 2. Target Disease Frequency Score (TDFS): This score is computed as the frequency count for the target disease mentioned in the input text. This feature adds information about the dominance of target disease mentions in the text.
3. Other Disease Frequency Score (ODFS): Unlike the TDFS, which captures information about the target disease in the text, the ODFS denotes the frequency of the next most frequent disease mention in the text other than the target disease. In some cases, it is possible that the other disease is more frequent than the target disease. In that case, the ODFS is greater than the TDFS. Both scores, in combination, describe the predominance of the target disease or other diseases in the text. Note that these scores are calculated independent of the mutation name in the text.
4. Same Sentence Disease-Mutation Co-occurrence Score (DMCS): For a mutation name and its nearest disease mentioned in the text, the DMCS is a binary score denoting the co-occurrence of the mutation and its nearest disease in the same sentence. The DMCS is 1 if both the mutation and its nearest disease are mentioned in the same English sentence. The DMCS is 0 if they are not in the same sentence.
5. Within Text Sentiment Score (WTSS): For a mutation name in the text and its corresponding nearest disease mentioned in the text, we extracted the “within text,” which refers to the text between the mutation and the nearest disease mentioned. This text is then analyzed for its sentiment using the TextBlob library.21 The sentiment score is based on the polarity of different words that appear in the within text. A negative polarity denotes that the within text contains negative terms based on the NLTK corpora used by the TextBlob library. The polarity score is a float within the range [−1.0, 1.0]. For several instances of the same mutation in the text, the WTSS is computed as the minimum of all the polarity scores for that mutation.
6. Text Sentiment Subjectivity Score (TSSS): The TSSS corresponds to the subjectivity of the sentiment score computed in the previous feature. It provides an estimate of the reliability of the sentiment score. The subjectivity is a float within the range [0.0, 1.0] where 0.0 is extremely objective and 1.0 is subjective. A value of 0.0 for this score says that the estimated text sentiment score is more reliable than if its subjectivity score was 1.0.
Training labels
We built a training set using the labels provided in the EMU_dataset. We assumed that the labels provided in this dataset denote whether a particular disease-mutation relationship exists or not. These are binary labels. If the mutation has some relationship with the target disease, the label is 1, otherwise is it 0. Using these labels, we were able to train the classification model discussed in the next step.
Training a machine learning classification model
Using the feature set constructed for the mutations identified in the EMU_dataset and the training labels associated with the mutations, we used the Weka3.6 tool22 to define several ML classifiers for our approach. We tested a few ML classifiers, such as the C4.5 decision tree, multilayer perceptron, and Bayesian logistic regression. In this work, we only report the results for the C4.5 decision tree23 because of its superior performance. The following parameters were used: confidenceFactor = 0.25, minNumObj = 2, andnumFold = 3; the other parameters were default settings in the Weka J48 (C4.5). Results of other models are briefly stated in the Appendix.
Prediction Model
As stated earlier, we used the EMU_dataset for the purposes of development and testing of our tool via cross-validation experiments. Once the entire system was validated against human-annotated data, we used the entire EMU_dataset consisting of 236 and 344 training samples, for prostate cancer and breast cancer, respectively, to develop ML models to extract disease-mutation associations from the larger PubMed_dataset.
EXPERIMENTAL RESULTS
In this section, we present the results of 2 experiments we conducted to evaluate the performance of the proposed approach. The first experiment is a comparison against the state-of-the-art EMU tool, which extracts mutations for a given target disease. In the second experiment, we utilized the PubMed_PCa, PubMed_BCa, and PubMed_AMD test sets to generate mutations related to prostate cancer, breast cancer, and AMD.
Experimental results for the EMU_dataset
In this section our experiments and results with the EMU_datasets are described. For clarification, the EMU_dataset is used here as a benchmark dataset, so we only used the PMIDs mentioned in this set. Training for the proposed approach was done using the labeled data (10-fold cross-validation); to ensure separation of training and testing sets, we obtained classification results on the entire dataset. The classification results from cross-validation were used for comparison with the baselines.
Baselines
A. EMU only: We used the results stated in Doughty et al.3 for the EMU tool’s performance on the breast cancer and prostate cancer datasets. These results correspond to the mutations extracted by the EMU tool in PubMed abstracts that were identified to be related to breast cancer or prostate cancer. The EMU mutations were related to the disease if the abstract containing the mutation also contained a disease term (identified using MetaMap24).
B. tmVar only: Similar to (A), tmVar was used to extract mutations in the PubMed articles instead.
C. EMU+ Nearest Disease Mention (EMU+NDM): We enhanced the EMU-only baseline by identifying the NDM (using the character count between the mutation and the disease name) as the disease related to that mutation. The mutations were identified using the EMU tool, and the disease names were identified using DNorm (not MetaMap). This differs from the EMU-only baseline in that it does not assume all identified mutations to be related to the target disease; it uses the nearest disease name as the related disease for a given mutation.
Tables 1 and 2 show the summary comparison of the proposed approach (tmVar + ML) with the baselines discussed above for disease-mutation associations. The tmVar + ML results were obtained using the 10-fold cross-validation technique described earlier for the C4.5 decision tree classifier. We used 3 accuracy metrics (precision, recall, and F-measure) to compare the performance of various approaches.
Table 1.
Performance comparison of the proposed approach (tmVar + ML) with baselines (EMU, tmVar, and EMU + NDM) on the prostate cancer dataset
| EMU_PCa | EMU | tmVar | EMU + NDM | tmVar + ML |
|---|---|---|---|---|
| Precision | 0.729 | 0.720 | 0.845 | 0.904 |
| Recall | 0.803 | 0.903 | 0.681 | 0.856 |
| F-measure | 0.764 | 0.801 | 0.754 | 0.880 |
Table 2.
Performance comparison of the proposed approach (tmVar + ML) with baselines (EMU, tmVar, and EMU + NDM) on the breast cancer dataset
| EMU_BCa | EMU | tmVar | EMU + NDM | tmVar + ML |
|---|---|---|---|---|
| Precision | 0.806 | 0.757 | 0.924 | 0.878 |
| Recall | 0.852 | 0.923 | 0.600 | 0.813 |
| F-measure | 0.828 | 0.832 | 0.730 | 0.845 |
As shown in Table 1, the proposed approach (tmVar + ML) shows significant improvement in all 3 metrics against the EMU-only baseline. In precision, it gives a 24% improvement over the EMU-only baseline. It also results in a 15% improvement in the F-measure when compared to the EMU-only baseline. With the EMU + NDM baseline, the performance of tmVar + ML is better in all 3 metrics. In comparison with the tmVar baseline, tmVar + ML significantly improves in precision and F-measure (0.904 versus 0.720 and 0.880 versus 0.801, respectively) at the cost of recall (0.856 versus 0.903).
For the breast cancer dataset (in Table 2), the tmVar + ML improves over the EMU-only baseline in terms of precision and F-measure (9% and 2%, respectively) at the cost of recall (0.813 versus 0.852). In comparison to the EMU + NDM baseline, there is significant improvement in terms of recall and F-measure (35% and 16%, respectively), while the precision is lowered (0.878 versus 0.924). tmVar + ML performs better in both precision and F-measure compared to tmVar only.
In addition to these benchmarking experiments, we also compared our results directly with the EMU results for gene-mutation-disease associations. Here we added gene association to the text-mined mutations by the nearest gene mention to the mutation. The gene mentions were extracted from the abstract texts using PubTator4 gene annotations. For comparison purposes, we evaluated the results only on the prostate cancer dataset (EMU_PCa), because manual annotation was required to normalize gene information from the EMU_dataset.
Table 3 summarizes a comparison of our results for the full mutation (including gene information) and disease association with EMU results for full mutation and disease association. As shown in the table, tmVar + ML’s performance (precision) is significantly higher than all the other baselines.
Table 3.
Results of comparison of EMU, tmVar, and tmVar + ML for complete mutation and disease association results
| Precision (TP, FP) | EMU | EMU + seq_filtera | tmVar | tmVar + ML |
|---|---|---|---|---|
| Disease-Gene-Mutation | 0.39 (151, 237) | 0.59 (127, 89) | 0.57 (134, 102) | 0.72 (128, 50) |
a The number of PMIDs in the EMU + seq_filter dataset is less than the number used in the EMU dataset, which is the benchmark dataset for evaluating tmVar and tmVar + ML performance.
In the next section, we demonstrate the performance of the proposed approach on the PubMed_dataset, which contains all the PubMed literature corresponding to the 3 diseases.
Experimental results for the PubMed_dataset
The PubMed_dataset serves as a testing platform to demonstrate the practical utility of the tool developed in this work. The ML model based on the EMU_dataset was used to distinguish between the relevant and irrelevant mutations for the target diseases.
The mutations identified by our approach are compared against 3 manually curated databases containing disease-mutation relationship information: (1) the ClinVar curated database,2 (2) the manually curated ground truth dataset for EMU,3 and (3) the SNPedia database.8 We used the following ClinVar query to collect the results: “(prostate [Disease/Phenotype]) AND cancer [Disease/Phenotype].” SNPedia contains information about the SNPs related to diseases. We used the ground truth labels of the EMU dataset (EMU_PCa and EMU_BCa) to validate the results of mining the PubMed_dataset. (All the PMIDs in the training set were excluded from the PubMed_dataset.) While we used these for validation of our findings, they still may not be an exhaustive resource for this validation. Our extractions were ranked on the basis of their frequency of appearance in the literature: mutations with mentions in several articles were ranked higher than mutations with mentions in only a few articles. A mutation was considered irrelevant to the target disease if the majority of times (of all its appearances in the input corpus) it was classified as unrelated to the target disease. In our results, we ranked the extracted mutations by their frequency of occurrence in the literature. The results of this comparison are displayed below.
Tables 4 and 5 contain a sample of the top 10 results from our prediction compared with the 3 validation sources. The “is mutation” column denotes whether the returned entity is a mutation or not. The next 3 columns provide information about each entity’s match with the 3 databases. The top 5th, 7th, 9th, and 10th mutations shown in Table 4 were found in at least 1 database and are therefore important. The 6th and 8th mutations were not found in any database, but they received high ranks from our approach. P504S (6th rank) is not a mutation but a cytoplasmic protein commonly related to prostate cancer. This is an error of mutation extraction using tmVar. For the p.G84E mutation (8th rank), we find the evidence of its relationship with prostate cancer in PMID: 23393222, “The G84E mutation of HOXB13 is associated with increased risk for prostate cancer: results from the REDUCE trial.” Interestingly, none of the 3 databases has curated this mutation, which was discovered with our automated approach.
Table 4.
Top 10 validation results of tmVar + ML for prostate cancer The prediction results are validated against 3 sources containing manually curated information about mutation to disease relationship.
| Rank | Mutation | Cumulative frequency | Is mutation? | In ClinVar? | In EMU GS? | In SNPedia? |
|---|---|---|---|---|---|---|
| 1 | rs1447295 | 41 | Yes | 0 | 0 | 1 |
| 2 | p.T877A | 40 | Yes | 0 | 1 | 0 |
| 3 | p.V89L | 36 | Yes | 0 | 1 | 1 |
| 4 | rs10993994 | 33 | Yes | 0 | 0 | 1 |
| 5 | rs6983267 | 33 | Yes | 0 | 0 | 1 |
| 6 | P504S | 29 | No | 0 | 0 | 0 |
| 7 | rs4430796 | 29 | Yes | 0 | 0 | 1 |
| 8 | p.G84E | 26 | Yes | 0 | 0 | 0 |
| 9 | p.R462Q | 25 | Yes | 1 | 1 | 1 |
| 10 | p.A49T | 21 | Yes | 0 | 1 | 1 |
Table 5.
Top 10 validation results of tmVar + ML for breast cancer The prediction results are validated against 3 sources containing manually curated information about mutation to disease relationship.
| Rank | Mutation | Cumulative frequency | Is mutation? | In ClinVar? | In EMU GS? | In SNPedia? |
|---|---|---|---|---|---|---|
| 1 | T47D | 1675 | No | 0 | 0 | 0 |
| 2 | c.C677T | 79 | Yes | 0 | 1 | 0 |
| 3 | p.R72P | 55 | Yes | 0 | 1 | 0 |
| 4 | rs3803662 | 53 | Yes | 0 | 0 | 1 |
| 5 | p.R399Q | 52 | Yes | 0 | 0 | 0 |
| 6 | rs2981582 | 51 | Yes | 0 | 0 | 1 |
| 7 | p.H1047R | 45 | Yes | 1 | 1 | 0 |
| 8 | c.A1298C | 45 | Yes | 0 | 1 | 0 |
| 9 | c.C3435T | 37 | Yes | 0 | 0 | 0 |
| 10 | rs1219648 | 36 | Yes | 0 | 0 | 1 |
In Table 5, we find 3 predictions in the top 10 that were not found in either database. The first prediction corresponds to a cell line that was falsely identified as a mutation in tmVar. The 5th prediction is a p.R399Q mutation that corresponds to the rs25487 SNP, but the only evidence supporting a relationship between this mutation and breast cancer is PMID: 18669164, which states that the mutation has a positive association in North Indian women. This mutation may require a deeper analysis for its relationship with breast cancer. However, the 9th prediction, c.C3435T mutation (rs1045642), is clearly related to breast cancer in PMID: 24070710, as indicated by the phrase “may contribute to individual susceptibility to breast cancer.”
DISCUSSION
This section is organized into 2 parts. In the first part we discuss the comparative performance. In the second part, we analyze the overall results of our prediction on the PubMed_dataset and highlight some interesting findings.
Discussion of results for the EMU_dataset
The comparison of the results of our approach with baselines is summarized in Tables 1–3. For the prostate cancer dataset, the performance of tmVar + ML was better than all the baselines in both precision and F-measure. The tmVar-only baseline had the highest recall (highest coverage) among all the approaches. But its significantly low precision (high redundancy) results in a lower F-measure. tmVar + ML balances coverage and redundancy using a classification scheme to distinguish between the disease-related and unrelated mutations. For the breast cancer dataset, the EMU + NDM baseline achieves the highest precision, while the tmVar-only baseline achieves the highest recall. However, in either case the trade-off between the precision and recall increases.
In summary, the proposed approach is better than the state-of-the-art approach (EMU) and its enhancement (EMU + NDM) in terms of mutation-disease-only extraction and gene-mutation-disease association extraction from the biomedical literature. The tmVar + ML learns to distinguish redundant predictions using the feature set constructed for each mutation, and hence improves the precision. In comparison, the baseline approaches have large trade-offs between precision and recall.
Discussion of results for the PubMed_dataset
The results presented in the previous section (with reference to Tables 4 and 5) show the effectiveness of the proposed approach in automatically extracting correct disease-related mutations. We found 2 mutations in our top 10 results that are not currently mentioned in any of the 3 databases. Also, an interesting observation from the results in Tables 4 and 5 is that the 3 disease-mutation relationship resources have few overlapping mutation curations among them. Since the results of the proposed approach largely overlap with the aggregation of the 3 databases (see the last column in Tables 4 and 5), our results can serve as a single database from which to curate important disease-related mutations.
We also analyze our mutation predictions for known errors and false negatives. Known errors are mutations that our approach classified as unrelated to the disease but were found to be related in any of the 3 resources. The aggregate of the 3 resources contained 172 and 450 mutations related to prostate and breast cancer, respectively. Interestingly, we found that our approach missed only 6 and 29 mutations related to prostate cancer and breast cancer, respectively. These errors can be attributed to the following:
Indirect or infrequent disease references: In such cases, the text referred to the target disease by general terms such as “tumor” and “affected tissue.” DNorm fails to filter disease references such as these. Such cases could be avoided by increasing the weight of target disease frequency in comparison to other disease mentions.
Unrelated documents: In a few cases, the documents were not directly related to the target disease. This can be improved by more comprehensive extraction of documents related to the target disease.
Disease name ambiguity: In some cases, the DNorm tool identifies a non-disease mention (sometimes a reference to a mutation in parentheses) as a disease, and the feature set is disturbed due to close proximity of the mutation with the false disease identification. These errors occur due to ambiguous abbreviations that resemble disease names. For example, in PMID: 17487399, we find a mutant cell line “DDS (R384W)” mentioned many times in the abstract, although the disease studied is prostate cancer. Here DDS is mistaken for Denys-Drash syndrome, a disease.
Mutation errors: Some prediction errors may occur due to mutation identification errors with the tmVar tool. An example is shown in Table 4, row 1: T47D, a cell, is wrongly identified as a mutation. Its unusually high frequency also distinguishes it from other mutations. Similarly, in Table 5, row 8, the entity P504S is a protein, not a mutation. Such errors could be avoided by incorporating a post-processing filter.
Case study of age-related macular degeneration (AMD)
We tested the generalizability of the proposed approach. Since obtaining manually annotated datasets for mutation-disease association is challenging, we tested to see whether the ML model trained using the annotated EMU cancer dataset could be used for extracting mutations for a different disease from the literature. We chose AMD to test the generalizability of the trained model because it is a non-cancer disease. We used the prostate cancer EMU_PCa for training. The model was tested on 861 unique mutations extracted from 11 383 PubMed abstracts. However, in the absence of any known gold standard for this disease, we present the top 10 most frequently referenced mutations classified as disease-related in the text-mined results in Table 6. Since the EMU gold standard is not available for AMD, we recruited a domain expert to manually annotate these top 10 results as well as a randomly drawn sample of 68 from the 739 disease-mutation associations. These random samples were drawn by portioning the 739 output pairs by frequency count into low (fewer than 4), medium (4–10), and high (greater than 10) frequency categories (ie, the number of associated publications in PubMed). From each category, a maximum of 25 disease-mutation associations were mined. Since the high frequency category had fewer than 25 and a few pairs lacked adequate information in PubMed abstracts for the human annotator to label their accuracy, we ended up with 68 disease-mutation associations. We obtained an average precision of 0.882 for these 68 disease-mutation associations and observed that precision was higher for frequent mutations than infrequent ones.
Table 6.
Evaluation of top 10 text-mined mutations for AMD disease using 3 resources
| Rank | Mutation | Cumulative frequency | Is mutation? | In ClinVar? | In manual curation? | In SNPedia? |
|---|---|---|---|---|---|---|
| 1 | p.Y402H | 209 | Yes | 0 | 1 | 0 |
| 2 | rs10490924 | 103 | Yes | 0 | 1 | 1 |
| 3 | rs1061170 | 95 | Yes | 0 | 1 | 1 |
| 4 | p.A69S | 85 | Yes | 1 | 1 | 0 |
| 5 | rs11200638 | 73 | Yes | 0 | 1 | 1 |
| 6 | rs800292 | 43 | Yes | 0 | 1 | 1 |
| 7 | p.I62V | 26 | Yes | 0 | 1 | 0 |
| 8 | rs1410996 | 25 | Yes | 0 | 1 | 1 |
| 9 | rs2230199 | 23 | Yes | 0 | 1 | 1 |
| 10 | p.R102G | 17 | Yes | 1 | 1 | 0 |
LIMITATIONS OF THE CURRENT APPROACH AND FUTURE WORK
We identified a few areas of improvement for the proposed method of extracting disease-related mutations. First, our approach extracts only point mutations (protein mutations, DNA mutations, and SNPs) from the text. However, some of these may be redundant (may map to a common concept), and we have not addressed this problem in the current work. In future work we plan to address this challenge by providing concept-level information about mutations.
Second, the current approach uses a simple proximity metric to address the problem of gene association. A more robust approach will be needed to reliably extract full gene-variant-disease triplets. Our previous work25 demonstrated that gene-mutation relationships can be mined with high accuracy using crowdsourcing. Crowdsourcing may prove to be a useful avenue for improvements.
Third, in this study, we focused on mining the biomedical literature for supporting precision medicine, whereas other studies have shown value in using additional text sources such as electronic health records26 and clinical trial data.27 Systematically integrating data and results from multiple textual sources might be worth exploring in future research. Also, we will extend the present approach to full-text articles, since many literature references contain mutation mentions within the main text and not the abstract.
CONCLUSIONS
Identifying correct disease-related mutations remains a crucial challenge in developing comprehensive disease-mutation databases, which are useful in precision medicine. In this work, we developed an automated approach to find relevant disease-related mutations from the biomedical literature. The proposed approach utilizes information from biomedical literature repositories to identify disease-related mutations. The comparative evaluation shows the effectiveness of the proposed approach in comparison to the state-of-the-art EMU tool’s performance. We also analyzed the performance of our approach on the entire body of literature in PubMed for 3 diseases and validated disease mutations identified in this manner against 3 manually curated resources. The results indicate that this approach will greatly benefit curation of mutation-disease databases, even on a mass scale.
ACKNOWLEDGEMENTS
We are grateful to Dr Chih-Hsuan Wei for helping with data access through PubTator and other support. We are also thankful to Emily Doughty and Dr Maricel Kann for providing the manually curated dataset for EMU. We also extend our special thanks to Dr Ioannis Xenarios for his helpful discussions on the paper’s topic.
CONTRIBUTORS
ZL conceived the project. AS implemented methods and performed the experiments. All authors participated in its design, analyzed the results, and wrote the manuscript. All authors read and approved the final manuscript.
FUNDING
This research was supported by the NIH Intramural Research Program, National Library of Medicine, and the NIH Medical Research Scholars Program, a public-private partnership supported jointly by the NIH and generous contributions to the Foundation for the NIH from the Doris Duke Charitable Foundation, the Howard Hughes Medical Institute, the American Association for Dental Research, the Colgate-Palmolive Company, and other private donors. No funds from the Doris Duke Charitable Foundation were used to support research that used animals.
COMPETING INTERESTS
The authors have no competing interests to declare.
REFERENCES
- 1.Overby CL, Tarczy-Hornoch P. Personalized medicine: challenges and opportunities for translational bioinformatics. Personalized Med. 2013;10(5):453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42(1):D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doughty E, Kertesz-Farkas A, Bodenreider O, et al. Toward an automatic method for extracting cancer- and other disease-related point mutations from the biomedical literature. Bioinformatics. 2011;27(3):408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wei C-H, Kao H-Y, Lu Z. PubTator: a web-based text mining tool for assisting Bio curation. Nucleic Acids Res. 2015;41(Web server issue 2013):W518–W522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeng J, Wu Y, Bailey A, et al. Adapting a natural language processing tool to facilitate clinical trial curation for personalized cancer therapy. AMIA Summits on Translational Sci Proceed. 2014;2014:126-131. [PMC free article] [PubMed] [Google Scholar]
- 6.Amberger J, Bocchini CA, Scott AF, Hamosh A. McKusick’s Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 2009;37 (Database issue):D793–D796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boeckmann B, Bairoch A, Apweiler R, et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31(1):365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cariaso M, Lennon G. SNPedia: a wiki supporting personal genome annotation, interpretation and analysis. Nucleic Acids Res. 2012; 40(Database issue): D1308–D1312. http://www.snpedia.com/index.php/SNPedia. Accessed September 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuhn K. The Cancer Biomedical Informatics Grid (caBIG™): Infrastructure and Applications for a Worldwide Research Community. Medinfo. 2007;1:330–334 [PubMed] [Google Scholar]
- 10.Claustres M, Horaitis O, Vanevski M, Cotton RGH. Time for a unified system of mutation description and reporting: a review of locus-specific mutation databases. Genome Res. 2002;12(5):680–688. [DOI] [PubMed] [Google Scholar]
- 11.Yusuf RA, Rogith D, Hovick SRA, et al. Attitudes toward molecular testing for personalized cancer therapy. Cancer. 2015;121:243–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Caporaso JG, Baumgartner WA, Randolph DA, Cohen KB, Hunter L. MutationFinder: a high-performance system for extracting point mutation mentions from text. Valencia A, ed. Bioinformatics (Oxford, England: ). 2007;23(14):1862–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wei C-H, Harris BR, Kao H-Y, Lu Z. tmVar: a text mining approach for extracting sequence variants in biomedical literature, Bioinformatics. 2013;29(11):1433–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimeno Yepes A, Verspoor K. Mutation extraction tools can be combined for robust recognition of genetic variants in the literature [version 1; referees: 3 approved with reservations] F1000Research 2014;3:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erdogmus M, Sezerman OU. Application of automatic mutation-gene pair extraction to diseases. J. Bioinform Comput Biol. 2007;5:1261–1275. [DOI] [PubMed] [Google Scholar]
- 16.Yeniterzi S, Sezerman U. EnzyMiner: automatic identification of protein level mutations and their impact on target enzymes from PubMed abstracts. BMC Bioinformatics. 2009;10(Suppl 8):S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonis J, Furlong LI, Sanz F. OSIRIS: a tool for retrieving literature about sequence variants. Bioinformatics. 2006; 22(20):2567–2569. [DOI] [PubMed] [Google Scholar]
- 18.Kuipers R, van den Bergh T, Joosten H-J, et al. Novel tools for extraction and validation of disease-related mutations applied to fabry disease. Hum Mutat. 2010; 31(9):1026–1032. [DOI] [PubMed] [Google Scholar]
- 19.Collins FS, Varmus H. A new initiative on precision medicine. New Engl J Med. 2015;372(9):793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leaman R, Doğan RI, Lu Z. DNorm: disease name normalization with pairwise learning to rank. Bioinformatics. 2013;29(22):2909–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. http://textblob.readthedocs.org/en/dev/#. Accessed August 2015.
- 22.Hall M, Eibe F, Holmes G, et al. The WEKA Data Mining Software: An Update. SIGKDD Explorations. 2009;11(1):10–18. [Google Scholar]
- 23.Salzberg S. C4.5: Programs for Machine Learning by J Ross Quinlan. Morgan Kaufmann Publishers, Inc., 1993. Machine Learning. 1994;16(3):235–240. [Google Scholar]
- 24.Aronson AR. Effective mapping of biomedical text to the UMLS Metathesaurus: the MetaMap program. Proc AMIA Symp. 2001:17–21. http://metamap.nlm.nih.gov/ [PMC free article] [PubMed]
- 25.Burger JD, Doughty E, Khare R, et al. Hybrid curation of gene-mutation relations combining automated extraction and crowdsourcing. Database. 2014:bau094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wei W-Q, Denny JC. Extracting research-quality phenotypes from electronic health records to support precision medicine. Genome Med. 2015;7(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng J, Wu Y, Bailey A, et al. Adapting a natural language processing tool to facilitate clinical trial curation for personalized cancer therapy. AMIA Summits Translational Sci Proceed. 2014:126–131. [PMC free article] [PubMed] [Google Scholar]