

Abstract
Recycling endosomes recently have emerged as major regulators of cytokinesis and abscission steps of cell division. Rab11-endosomes in particular were shown to transport proteins to the mitotic ingression furrow and play a key role in establishing the abscission site. Rab11 GTPase function by binding and activations various effector proteins, such as Rab11 family interacting proteins (FIPs). FIPs appear to be at the core of many Rab11 functions, with FIP3 playing a role in targeting of the Rab11-endosomes during mitosis. Here we summarize the newest finding regarding the roles and regulation of FIP3 and Rab11 complex, as well as describe the methods developed to analyze membrane and cytoskeleton dynamics during abscission step of cytokinesis.
Keywords: cytokinesis, abscission, Rab11, FIP3, recycling endosomes, midbody, mitosis
INTRODUCTION
The last step of cell division is a physical separation of two daughter cells via a process known as cytokinesis (Barr and Gruneberg 2007; Pollard 2010). After replication of the genetic material, the mother cell divides by the formation of the cleavage furrow that constricts cytoplasm leaving two daughter cells connected by a thin intracellular bridge (ICB). The resolution of this bridge (abscission) results in separation of two daughter cells. Although the mechanisms that govern abscission are not fully understood, recent evidence suggests that actin cytoskeleton, endosomes and the ESCRT-III protein complex play a critical role in this process.
ESCRT complexes (complexes 0, I, II and III) were originally described as regulators of multivesicular body formation (Babst, Katzmann et al. 2002). Since then several ESCRT proteins, namely Tsg101, Alix, and ESCRT-III complex proteins were demonstrated to be required for cytokinesis (Carlton and Martin-Serrano 2007; Carlton, Agromayor et al. 2008). The model of ESCRT recruitment to the ICB is as follows: Alix and/or Tsg101 are recruited to the midbody by binding to the midbody protein CEP55. These components then recruit various ESCRT-III complex members to the midbody. The ESCRT-III complex has the ability to form ~5 nm filaments that are proposed to mediate abscission (Elia, Sougrat et al. 2011; Guizetti, Schermelleh et al. 2011). How ESCRT-III complex proteins move from the midbody to the abscission site remains unclear, but we have shown that localized actin depolymerization and narrowing of the ICB (secondary ingression) are required to establish the abscission site and recruit the ESCRT-III complex (Figure 1).
Figure 1. Mechanisms mediating abscission.
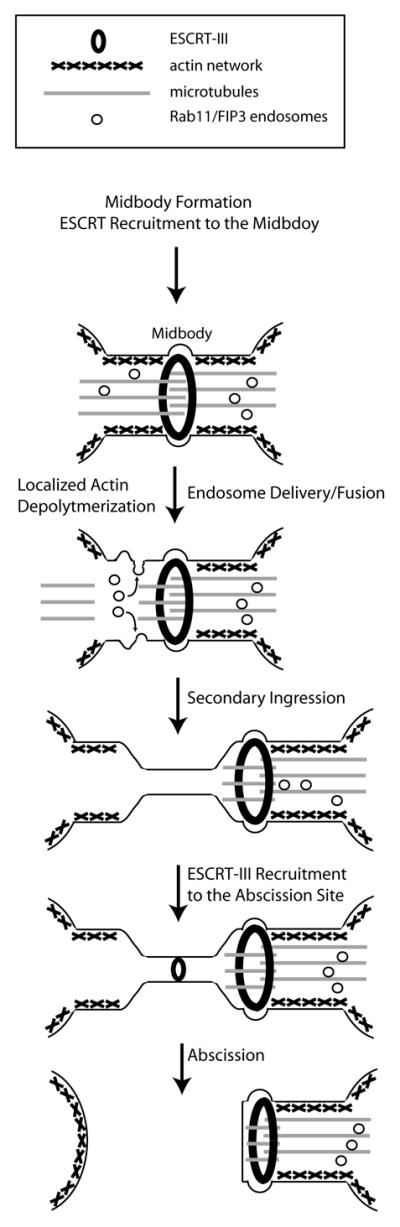
In early telophase, as midbody is formed from central spindle microtubules, ESCRT-III complex is recruited to the midbdoy. In late telophase, Rab11/FIP3 and Rab35 endosomes (likely different endosomal populations) are delivered and fuse with cleavage furrow plasma membrane. Among other factors, these organelles deliver OCRL and p50RhoGAP leading to the localized disassembly of actin cytoskeleton and severing of central spindle microtubules. Actin depolymerization induces formation of the secondary ingression and ESCRT-III translocation from the midbody to the abscission site. Delivery of the ESCRT-III to the secondary ingression leads to a final scission event and separation of daughter cells.
Recycling endosomes (RE) have emerged as important players in mediating abscission (Wilson, Fielding et al. 2004; Fielding, Schonteich et al. 2005; Schiel, Childs et al. 2013). Several reports demonstrated that pronounced changes occur in endocytic recycling during mitosis, and that these changes are required for successful completion of cytokinesis. Originally, it was proposed that REs initiate abscission by fusing with each other and the plasma membrane, thus building a separating membrane in a manner similar to a formation of the phragmoplasts in plant cells. However, recent data from our laboratory (Schiel, Park et al. 2011; Schiel, Simon et al. 2012) have shown that fusion of REs mediates the formation of a “secondary ingression”, thus initiating ESCRT-III recruitment to the abscission site (Figure 1).
Rab11, a small GTPase that functions in RE-mediated trafficking of plasma membrane receptors has recently emerged as a key regulator of RE transport to the ICB during abscission (Wilson, Fielding et al. 2004; Fielding, Schonteich et al. 2005). All Rab GTPases function by binding and recruiting various effector proteins. While several Rab11 effector proteins have been identified, Rab11 regulates RE delivery to the ICB predominantly via binding to its FIP3 effector protein (Wilson, Fielding et al. 2004; Fielding, Schonteich et al. 2005). The FIP3/Rab11 complex accumulates at the ICB during mitosis, and depletion of FIP3 by siRNA arrests cells in late telophase, while having no effect on anaphase/early telophase (Wilson, Fielding et al. 2004; Simon, Schonteich et al. 2008). Interestingly, recent data show that FIP3-endosomes deliver p50RhoGAP (known RhoA GAP) to the ingression furrow during late telophase. Upon delivery of p50RhoGAp to the ICB, it mediates nested depolymerization of the actin cytoskeleton, leading to the formation of the “secondary ingression” and abscission (Schiel, Simon et al. 2012) (Figure 1).
The past few years have seen a dramatic increase in our understanding of abscission. It is clear that highly dynamic regulation of endosomes, cytoskeleton and ESCRT complexes is the key to the successful completion of cytokinesis (Schiel, Childs et al. 2013). Here, we describe a set of novel techniques/approaches that will allow the further dissection of the machinery governing cytokinesis and abscission.
METHODS FOR ANALYZING PROTEINS REGULATING CYTOKINESIS
Based on the studies from many research groups led to development of several abscission models. These studies are based on the use of various techniques. The temporary nature of the abscission event makes it difficult to study. Furthermore, the abscission site is very small and difficult to image using standard light microscopy techniques. Finally, there are currently no good markers to clearly identify the abscission site. Consequently, most of the techniques used to detect the abscission defects usually measure the failure of cells to separate rather then the actual abscission event. Unfortunately, the separation of daughter cells is a very complex event that can be affected by many factors. Accordingly, each of the techniques (described bellow) has its shortcomings and a combination of multiple approaches should be used to test the involvement of the candidate protein in the abscission event.
(A) Multi-nucleation analysis
One of the most commonly used and easiest techniques for investigating proteins that regulate cytokinesis is to measure the multi-nucleation of cells. The idea behind this approach is that the inhibition of abscission results in the regression of the cleavage furrow, thus leading to bi- or multi-nucleated cells. This technique has been commonly used to detect defects in the formation and ingression of the cleavage furrow. One drawback of using this approach to analyze the abscission event is the failure of cleavage furrow to regress in cells arrested at late telophase. Instead, these cells stay connected with the ICB, often long enough to start a second round of division. Therefore, to better analyze the effect on cell abscission, cells are usually scored for the number of bi- and multi-nucleated cells along with the number of cells connected by ICBs.
Procedure
Coat 22 mm glass coverslips with collagen (placed in 6-well dish). Make sure to clean the glass coverslips with 3% acetic acid, followed by wash with water, beforehand to ensure a even coating of collagen. Alternatively, poly-L-lysine can also be used as a substrate.
Under a cell culture hood, plate HeLa cells on collagen-coated glass coverslips. Make sure that cells are no more than 30% confluent. Plating cells too densely will affect their division rates, thus directly affecting the levels of multi-nucleation. It is paramount to always plate equal number of cells, since the cell density will directly affect cell division rates.
Incubate cells in serum-supplemented media for 24 hours.
Rinse cells with phosphate-buffered saline (PBS) and fix for 15 minutes with 4% paraformaldehyde (in PBS).
Permeabilize cells with 2 mls of blocking solution (PBS, 2% Fetal Bovine Serum, 1% Albumin, 2% Saponin) by incubating for 20 minutes.
Incubate cells with primary anti-acetylated tubulin antibody (Sigma-Aldrich, cat#T7451; dilution 1:100) for 1 hour by overlaying 100 μl of blocking solution with primary antibody. Make sure to place a small piece of parafilm on top of the coverslip and wrap 6-well dish in moist paper towel to minimize evaporation.
Wash cells three times (5 minutes each) with 2 mls of blocking solution. Overlay cells with 100 μl of blocking solution with secondary antibody, DAPI and rhodamine-phalloidin (Invitrogen, cat#R415; dilution 1:50). We typically use anti-mice IgG antibody conjugated to Alexa488. Incubate for 30 minutes.
Wash cells three times (5 minutes each) with 2 mls of blocking solution.
Mount cells in Vectashield (Vector Laboratories, cat#H-1000). Seal edges with nail polish.
Image cells using florescent micropscopy. Rhodamine-phalloidin will allow you to visualize the edges of the cells to count the number of nuclei within each cell. Similarly, anti-acetylated tubulin antibodies will stain the central spindle, allowing visualization of the ICBs.
Data Interpretation
Typically, untreated HeLa cells have around 2–5% of multi-nucleated cells. Similarly, 3–6% of cells are still connected with the ICB and therefore in the late telophase stage. Knock-down of known key regulators of abscission, such as FIP3 or CHMP4B (ESCRT-III subunit) usually results in about 20% multi-nucleation and about 25% cells that remain connected with ICBs (Carlton, Agromayor et al. 2008; Schiel, Childs et al. 2013). This is presumably due to the fact that some cells that are arrested in late telophase will eventually undergo apoptosis. Indeed, FIP3 knock-down leads to a significant decrease in cell survival (Schiel, Simon et al. 2012). Interestingly, a large portion of the telophase-arrested cells will resolve their ICBs using traction-dependent cytokinesis, generating forceful breakage of the extended ICBs and the eventual separation of daughter cells. Thus, if the candidate protein is involved in abscission, one should not expect to see more than 20–25% multi-nucleated cells.
(B) Measuring delay in abscission
Due to the redundancy in cytokinesis machinery knock-down of any single protein involved in mediating abscission may not completely block cell division. A more direct method of evaluating the involvement of candidate proteins in mediating the abscission event is to measure the time required for daughter cells to complete cytokinesis. Indeed, many well-established abscission regulators dramatically increase division time while having a very moderate effect on multi-nucleation (Dambournet, Machicoane et al. 2011; Schiel, Simon et al. 2012).
Procedure
Plate Hela cells on collagen-coated glass bottom Grid-50 dishes (Ibidi, cat#81148). Make sure that cells are plated at 20–30% confluency since cells will be imaged for 24 hours, during which the time majority of cells will divide at least once. Plating cells to densely will make it difficult to visualize the timing of abscission.
Let cells attach for 3–4 hours in serum-supplemented media. Replace media to remove floating cells.
Set up cells for imaging on an inverted microscope equipped with a X-Y-Z motorized stage and an environmental control system.
Image cells with a 20X objective. Pick at least 5–6 imaging fields (with 10–15 cells each) and image by field-contrast at 30 minute time-lapses for 24 hours.
During cell division the ICB and midbody are quite dynamic and often can leave the field of focus. To ensure visualization of each abscission event at every time-point a mini-Z-stack should be imaged. Typically taking 10 images separated by 1 μm Z-step is sufficient to ensure that ICB and midbody can be evaluated at every time point.
Data Interpretation
Once imaging is complete, the time required for cytokinesis is measured for every cell in each field. Typically, the metaphase is considered to be time-point 0, while the resolution of the ICB is marked as the last step in daughter cell separation. The time required for cells to complete mitosis is quite variable and can range from 60 minutes to 3–4 hours. As a result, a large sample of cells is needed (80–100 cells for every condition) to derive a meaningful and statistically sound data about the timing of cytokinesis.
(C) Analyzing the establishment of the abscission site
Measuring the formation of abscission site is the most direct way of testing the effect of candidate proteins in regulating abscission. Endosomes initiate the abscission site by regulating localized depolymerization of actin cytoskeleton by either delivering p50RhoGAP (Rab11/FIP3-endosomes) or by modulating PI(4,5)P2 levels (Rab35-endosomes) (Dambournet, Machicoane et al. 2011; Schiel, Simon et al. 2012). Actin depolymerization then leads to the formation of the secondary ingression, leading to the formation of the abscission site (Schiel, Park et al. 2011). The abscission site can be identified by several methods. First, central spindle microtubules are heavily acetylated and are clearly identifiable following staining with anti-acetylated tubulin antibody. Since abscission involves localized spastin-dependent cutting of microtubules (Connell, Lindon et al. 2009) the abscission site can be identified as a gap in the central spindle (Elia, Sougrat et al. 2011; Schiel, Park et al. 2011). Note that central spindle stained with anti-acetylated tubulin antibodies always has a gap in the middle that is caused by limited access of antibodies to a dense midbody structure. The abscission site usually forms on one or both sides of this midbody-gap and can be identified as secondary gaps in anti-acetylated tubulin staining (Figure 2). Alternatively, the ESCRT-III complex can also be used as an abscission site marker. Typically, CHMP4B-GFP is used in these types of studies, since good anti-CHMP4B antibodies are not available. Along with other ESCRT complex members, CHMP4B-GFP is first recruited to the midbody (Figure 1) (Elia, Sougrat et al. 2011; Schiel, Simon et al. 2012). After formation of the secondary ingression part of CHMP4B-GFP translocates to the abscission site and can be identified as a puncta on one or both sides of the midbody. If cells are co-stained with anti-acetylated tubulin antibodies, these CHMP4B puncta will be present in the secondary gaps within central spindle microtubules (Elia, Sougrat et al. 2011; Schiel, Simon et al. 2012).
Figure 2. Models of asymmetric and symmetric abscission.

In many dividing cells abscission sites are established bi-laterally on both sides of the midbody. That leads to a shedding of the midbody to the extracellular space (left panel). However, abscission site can also be formed only on one side of the midbody, leading to asymmetric abscission (right panel). This type of asymmetric abscission results in midbody inheritance by one of the daughter cells.
Procedure
-
1
Under a cell culture hood, plate HeLa cells on collagen or polycoated glass coverslips. Make sure that cells are no more then 30% confluent. Plating cells too densely will make it difficult to visualize central spindle and the midbody.
-
3
Incubate cells in serum-supplemented media for 24 hours.
-
4
If using CHMP4B-GFP as an abscission marker, transfect cells and incubate another 28 hours.
-
4
Rinse cells with phosphate-buffered saline (PBS) and fix for 15 minutes with 4% paraformaldehyde (in PBS).
-
5
Permeabilize cells with 2 mls of blocking solution (PBS, 2% Fetal Bovine Serum, 1% Albumin, 2% Saponin) by incubating for 20 minutes.
-
6
Incubate cells with primary anti-acetylated tubulin antibody (Sigma-Aldrich, cat#T7451; dilution 1:100) for 1 hour by overlaying 100 μl of blocking solution with primary antibody. Make sure to place a small peace of parafilm on top of the coverslip and wrap 6-well dish in a moist paper towel to minimize evaporation.
-
7
Wash cells three times (5 minutes each) with 2 mls of blocking solution. Overlay cells with 100 μl of blocking solution with secondary antibody and DAPI. We typically use anti-mice IgG antibody conjugated to Alexa594 to allow co-imaging of acetylated tubulin with CHMP4B-GFP. Incubate for 30 minutes.
-
8
Wash cells three times (5 minutes each) with 2 mls of blocking solution.
-
9
Mount cells in Vectashield (Vector Laboratories, cat#H-1000). Seal edges with nail polish.
-
10
Image cells by fluorescent microscopy. Anti-acetylated tubulin antibodies will stain the central spindle. If cells were transfected with CHMP4B-GFP, it can also be used to identify abscission sites.
Data Interpretation
The abscission rates can be evaluated by counting the percentage of telophase cells that have clearly established an abscission site, as determined by the presence of a gap in the central spindle. Care must be taken not to count a midbody-associated gap. Similarly, abscission sites can be counted by determining the percentage of the cells that contain CHMP4B-GFP puncta within the ICB, but outside the midbody. In addition to counting cells that have an established abscission site, one can also analyze the effect of experimental treatment on the rates of asymmetric abscission. In about 40% of HeLa cells abscission usually occurs on both sides of the midbody leading to the release of the midbody into the media (Figure 2). Interestingly, in a majority of the cell divisions the abscission site is only established on one side of the midbody (Kuo, Chen et al. 2011). As a result of this asymmetric abscission, one daughter cell will inherit the post-mitotic midbody (Figure 2). Importantly, midbody inheritance has been proposed as a mechanism for regulating cell fate and differentiation in daughter cells (Ettinger, Wilsch-Brauninger et al. 2011; Kuo, Chen et al. 2011; Chen, Ettinger et al. 2012). Thus, it will be important to identify the factors that mediate the establishment of these asymmetric abscissions.
One of the potential draw-backs of using CHMP4B-GFP over-expression as an abscission site marker is the fact that high levels of CHMP-GFP can inhibit abscission presumably by interfering with ESCRT-III function. Thus, care must be taken to analyze only cells expressing low to moderate levels of CHMP4B-GFP.
TIME-LAPSE ANALYSIS OF ENDOSOME AND ACTIN DYNAMICS DURING CYTOKINESIS
Microscopy analysis of fixed cells remains a powerful tool for the initial characterization of candidate proteins that regulate abscission. However, static image analysis will not uncover the properties and regulation of dynamic changes in action cytoskeleton and membrane dynamics during cell division. Thus, time-lapse analysis of live dividing cells is needed to investigate rapid and localized changes in cytoskeleton and endosomes during abscission. The approaches listed below will allow researchers to investigate the changes in Rab11/FIP3-endosomes (A) or actin and microtubules (B).
(A) Analyzing Rab11/FIP3-endosome transport during cytokinesis
Rab11/FIP3-endosomes have recently emerged as key mediators of abscission (Wilson, Fielding et al. 2004; Schiel, Simon et al. 2012). Importantly, the dynamic movement and localization of Rab11/FIP3-endosomes change dramatically as cells proceed from metaphase to telophase (Figure 3). During metaphase Rab11/FIP3-endosomes accumulate around centrosomes, where they stay until telophase (Figure 3) (Schiel, Park et al. 2011). After cells form the midbody and start progressing to late telophase, Rab11/FIP3-endosomes move along the central spindle microtubules to the cleavage furrow (Figure 3) (Simon, Schonteich et al. 2008; Schiel, Park et al. 2011). This leads to the translocation of all Rab11/FIP3-endosomes from centrosomes to the endocytic pools situated on each side of the midbody (Figure 3). The translocation of the Rab11/FIP3-endosomes is usually non-symmetric, with endosomes from one centrosome arriving at the midbody slightly ahead of the endosomes from the other side (Schiel, Park et al. 2011). Since Rab11/FIP3-endosomes initiate the formation of the secondary ingression and the establishment of the abscission site, potentially this asynchronous arrival at the cleavage furrow determines asymmetric abscission and midbody inheritance by one daughter cell. Despite the potential impact on cell fate and differentiation, the machinery mediating asynchronous translocation of Rab11/FIP3-endosomes to the cleavage furrow remains essentially unknown. Thus, time-lapse analysis of FIP3-GFP dynamics during cell division is perfectly suited to decipher the mechanisms governing Rab11/FIP3-endosome dynamics and midbody inheritance.
Figure 3. Rab11/FIP3-endosome dynamics during cytokinesis.

During metaphase and anaphase Rab11/FIP3-endosomes associate with centrosomes. As dividing cell progresses to telophase and midbody is formed, Rab11/FIP3-endosomes start translocated along central spindle microtubules to the close proximity of the midbody. My late telophase almost all Rab11/FIP3-endosmes are localize at and around the midbody. This translocation precedes and is required for the final abscission event.
Procedure
Transfect HeLa cells with FIP3-GFP and plate cells on collagen-coated glass bottom Grid-50 dishes (Ibidi, cat#81148). Make sure that cells are plated at 30–50% confluency to allow clear visualization of FIP3-GFP dynamics. Plating cells too dense will result in the formation of short intercellular bridges, making visualization more difficult.
Set up cells for imaging on an inverted microscope equipped with an environmental control system. Cells need to be imaged at 37°C to ensure proper endosome dynamics and cytokinesis. Imaging cells at room temperature often leads to either mitotic arrest or dramatically increase the time required for abscission.
Image cells with a 63X oil-objective. Pick cells at metaphase and image it every 10–15 minutes. Since during metaphase cells round up, small Z-stack with 500 nm Z-step. About 30 stack images will be needed to images the entire cell.
Imaging cells every 10–15 minutes will uncover the steady-state changes in FIP3-GFP localization but will not allow tracking of individual cells. To visualize and analyze the speed and dynamics of individual FIP3-GFP-endosomes 300 msec time-lapses will be needed. Unfortunately, imaging dividing metaphase cells using such short time-lapses will lead to phototoxicity and can dramatically affect the cell’s ability to divide. Therefore, to analyze the dynamics of individual FIP3-endosomes we usually pick cells at early telophase that contain a formed midbody which substantially reduces the number of time points needed to visualize FIP3-GFP dynamics through the abscission event. Generally, no more then 50–80 time points should be taken for each cell. Furthermore, picking cells at telophase results in much flatter cells compared to metaphase cells, thus eliminating the need to do z-stacks and further reducing imaging-induced photodamage.
Data Interpretation
Time-lapse analysis is a powerful tool for investigating many different aspects of Rab11/FIP3-endosome dynamics. Using 10–15 minute time-lapse windows any changes in the steady-state Rab11/FIP3-endosome distribution at different mitotic stages can be captured. Using 300 msec time-lapse windows will allow for measurement of the directionality, processivity and speed of individual Rab11/FIP3-endosomes.
(B) Analyzing actin or microtubule dynamics during cytokinesis
In addition to changes in Rab11/FIP3-endosome dynamics, the actin and microtubule cytoskeleton also undergo dramatic and very dynamic changes. As described earlier, during telophase central spindle microtubules are compacted together to form a very dense cellular structure known as the midbody. The spastin-dependent localized severing of these central spindle microtubules is a key step in determining the location and timing of the abscission. Although the machinery determining the site of spastin-dependent microtubule severing remains unclear the location of “cut” appears to depend on central spindle bending during late telohase (Simon, Schonteich et al. 2008).
Actin filaments also dynamically change during cytokinesis. During early anaphase actin forms a filamentous acto-myosin contractile “ring” at the midzone of the dividing cell. The contraction of this acto-myosin ring leads to the formation and ingression of the cleavage furrow. As ingression progresses, the acto-myosin ring gets compacted leading to the dramatic increase in filamentous actin amount at the plasma membrane of the cleavage furrow. However, once the ingression is complete and the midbody has formed, the acto-myosin network undergoes a very rapid and localized disassembly, the step that is controlled by endocytic transport and is required for the abscission (Dambournet, Machicoane et al. 2011; Schiel, Simon et al. 2012).
These findings clearly demonstrate the dynamic nature of cytoskeleton and support the ides that to fully understand the regulation of cytokinesis it is imperative to perform time-lapse microscopy to analyze filamentous actin and microtubule dynamics. Microtubules are typically analyzed using GFP or mCherry-tagged tubulin. This type of analysis usually works better in cells stably expressing tagged-tubulin. Similarly, actin dynamics can be analyzed in cells transfected with GFP-actin. However, some published reports have suggested that GFP-tagging can affect filamentous actin dynamics. Therefore, alternatives for imaging actin have recently been developed, including LifeAct and utrophin. LifeAct is a recombinant reporter utilizing a yeast-derived peptide that binds specifically to filamentous actin (Riedl, Crevenna et al. 2008). Tagging LifeAct with either GFP or mCherry enables visualization of the formation and dynamics of actin filaments. Alternatively, actin-binding peptide derived from utrophin can also be tagged with GFP and used to visualize actin filaments. Although LifeAct is more commonly used, we found that both these actin biosensors work very similarly (Schiel, Simon et al. 2012). Once transfected with tubulin-GFP, actin-GFP, LifeAct-GFP or utrophin-GFP cells are then analyzed by time-lapse microscopy as described in part (A).
(C) Cytokinesis and apical lumen formation in 3D cultures
While the main function of cytokinesis is to physically separate a mitotic cell into two daughter cells, several non-canonical functions for cytokinesis have been recently proposed. For example, asymmetric abscission leads to midbody inheritance by one of the daughter cells and has been implicated in regulating cell fate (Chen, Ettinger et al. 2012). Additionally, we recently proposed that cytokinesis and midbody formation is the first symmetry-breaking event that determines the timing and location of apical lumen during epithelial tissue morphogenesis (Li, Mangan et al. 2014) (Figure 4). Based on these findings, several approaches have been developed to image cytokinesis during the process of epithelia polarization and apical lumen formation. One of them is time-lapse analysis of mitosis and epithelia polarization in 3D Matrigel cultures (Li, Mangan et al. 2014).
Figure 4. Model depicting the role of midbody-associated AMIS formation and endosome transport during lumenogenesis in epithelial cells.
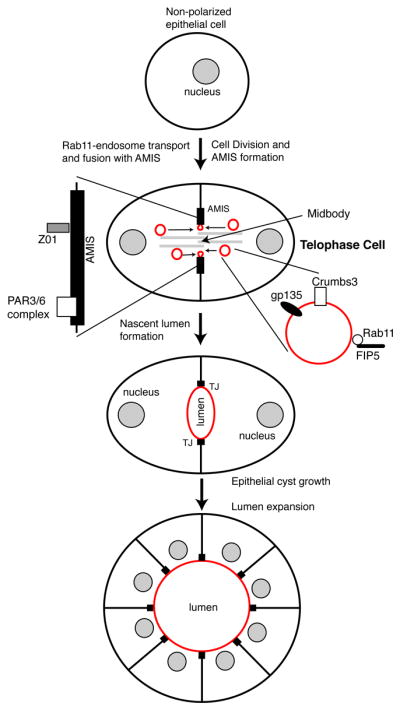
Upon division of non-polarized epithelial cell, apical membrane initiation site (AMIS) forms abound midbody in late telophase. Rab11/FIP3-endosomes containing apical cargo, such as Crumbs3 and gp135, are then delivered along centrals spindle microtubules to the AMIS, where they fuse and form nascent apical lumen. As cells polarized epithelial cells divide these nascent lumens mature to become a function apical lumens.
Procedure
-
1
Plate MDCK cells on a 100 mm tissue culture plate in 10 mls MDCK media and let it grow for 24 hours at 37°C.
-
2
One to two hours before plating cells take an aliquot of Matrigel and set it on ice to thaw out. It is very important to keep Matrigel cold even while thawing, since it solidifies rapidly at room temperature.
-
3
Aspirate media and rinse cells with 10 mls PBS. This step is essential. Leaving some of the serum-supplemented media will inhibit trypsin and will make it very difficult to lift individual MDCK cells.
-
3
Add 2 mls 0.25% Trypsin-EDTA and let sit at 37°C for 10–15 minutes. MDCK cells are usually difficult to dislodge. Thus, if needed, they can be incubated for 20–25 minutes.
-
4
Dislodge cells by gently tapping at the side of the 100 mm dish. Harvest cells by adding 8 mls MDCK media to plate and transferring cells to a 15 ml tube. Sediment cells by centrifugation at 1000xrpm for 3 minutes.
-
5
Aspirate media and resuspend cells in 1 ml of MDCK media by pipetting up and down using 1 mL blue pipette tip. Pipetting up and down many times helps to separate cells. It is crucial for this assay to embed individual cells in Matrigel. Embedding cell clumps will lead to formation of cell aggregates with multiple lumens. We found that pipetting cells 50 times with a 1 mL pipette usually gives a maximum number of individual cells; however, this number may vary. Thus, it is advisable to initially pipette cells varying number of times, while monitoring the efficiency of cell separation.
-
6
Count cells to determine number of cells/mL and add 20,000 cells to 25 μl MDCK media in 1.5 ml Eppendorf tube. 7. Make a 75% Matrigel solution by adding 75 μl Matrigel to the 25 μL cell solution from step
-
7
Gently mix by pipetting up and down. Plate the cell/Matrigel mix immediately, by placing a drop onto the center of a 5 cm gridded glass-bottom dish. This step needs to be done quickly, since the Matrigel-cell mixture solidifies readily at room temperature. Place cells in the incubator and let it solidify in the tissue culture incubator at 37°C for 30 minutes. The percentage of Matrigel may also vary between 25–80%. The volume of media to suspend cells in and volume of Matrigel added can be adjusted to determine ideal conditions. In lower concentrations of Matrigel, cells may sink through the Matrigel matrix (during process of solidifying the Matrigel) and stick to the bottom of the plate, counteracting the function of the Matrigel to observe the cells suspended in the matrix. If the Matrigel concentration is too high, the cells might not be able to suspend in the matrix and instead would just sit on top, which is again not ideal for 3D imaging.
-
8
Add 5 mls of MDCK media to the dish and let cells acclimate for 6–12 hours at 37°C. Typically cells will start dividing within 12 hours after embedding. Imaging them within 6–12 hour time period will ensure that at least some cells will be undergoing first cell division.
-
9
Mount dish on fluorescent microscope and use a gridded chart to label the location of several individual cells, especially those that may be in metaphase (starting division). Adjust focus to set top and bottom of each cell and take initial 0.2 μm-step Z-stack images.
-
10
Repeat the imaging according to the time frame required for desired observation. This will result in taking a mini-Z-stack for every time point. Generally, avoid taking more than 50–80 time-lapse Z-stacks. To visualize apical lumen formation during cell division, we typically use a 10 minute time-lapse. To visualize lumen formation and expansion, we use 30 minute time-lapses. Finally, if the motility of individual Rab11/FIP3-endosomes needs to be observed, we use 200–500 msec time-lapse. Use grid etched in glass to locate various cells for imaging at different time points.
-
11
Taking mini-Z-stacks at every time point allows the use of post-acquisition image analysis to generate three-dimensional images of MDCK cells at each time point during lumen formation. Alternatively, individual images that best represent the lumen formation dynamics can be selected and displayed/analyzed for every time point.
SUMMARY
While the importance of polarized endosomal transport during cytokinesis is clearly established, the function and regulation of this transport is only beginning to emerge. Similarly, we still do not fully understand the machinery mediating localized reorganization of actin and microtubule cytoskeleton. This is in part due to the technical limitations in studying abscission, the very transient cellular event that is hard to image and quantify. Here, we described the newest techniques currently available to study membrane and cytoskeleton dynamics during abscission. It is important to note that while the emergence of live imaging techniques provided significant insights in understanding abscission, many of these techniques still have significance limitations in spatial and temporal resolution to directly analyze the cytokinesis and abscission events. Thus, the use of multiple approaches rather then reliance on a singe technique will be the key in understanding Rab11-endosome and cytoskeleton dynamics during cytokinesis.
Acknowledgments
I am grateful to Abitha Jacob and Dr. Alexander Blasky for critical reading of the manuscript. I apologize to all colleagues whose work could not be cited due to space limitations. I also acknowledge the financial support by NIH (DK064380), Cancer League of Colorado and Susan G. Komen for the Cure foundation (BCTR0706749).
References
- Babst M, Katzmann DJ, et al. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3(2):271–282. doi: 10.1016/s1534-5807(02)00220-4. [DOI] [PubMed] [Google Scholar]
- Barr FA, Gruneberg U. Cytokinesis: placing and making the final cut. Cell. 2007;131(5):847–860. doi: 10.1016/j.cell.2007.11.011. [DOI] [PubMed] [Google Scholar]
- Carlton JG, Agromayor M, et al. Differential requirements for Alix and ESCRT-III in cytokinesis and HIV-1 release. Proc Natl Acad Sci U S A. 2008;105(30):10541–10546. doi: 10.1073/pnas.0802008105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlton JG, Martin-Serrano J. Parallels between cytokinesis and retroviral budding: a role for the ESCRT machinery. Science. 2007;316(5833):1908–1912. doi: 10.1126/science.1143422. [DOI] [PubMed] [Google Scholar]
- Chen CT, Ettinger AW, et al. Resurrecting remnants: the lives of post-mitotic midbodies. Trends Cell Biol. 2012 doi: 10.1016/j.tcb.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JW, Lindon C, et al. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic. 2009;10(1):42–56. doi: 10.1111/j.1600-0854.2008.00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambournet D, Machicoane M, et al. Rab35 GTPase and OCRL phosphatase remodel lipids and F-actin for successful cytokinesis. Nat Cell Biol. 2011;13(8):981–988. doi: 10.1038/ncb2279. [DOI] [PubMed] [Google Scholar]
- Elia N, Sougrat R, et al. Dynamics of endosomal sorting complex required for transport (ESCRT) machinery during cytokinesis and its role in abscission. Proc Natl Acad Sci U S A. 2011;108(12):4846–4851. doi: 10.1073/pnas.1102714108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger AW, Wilsch-Brauninger M, et al. Proliferating versus differentiating stem and cancer cells exhibit distinct midbody-release behaviour. Nat Commun. 2011;2:503. doi: 10.1038/ncomms1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielding AB, Schonteich E, et al. Rab11-FIP3 and FIP4 interact with Arf6 and the exocyst to control membrane traffic in cytokinesis. Embo J. 2005;24(19):3389–3399. doi: 10.1038/sj.emboj.7600803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizetti J, Schermelleh L, et al. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science. 2011;331(6024):1616–1620. doi: 10.1126/science.1201847. [DOI] [PubMed] [Google Scholar]
- Kuo TC, Chen CT, et al. Midbody accumulation through evasion of autophagy contributes to cellular reprogramming and tumorigenicity. Nat Cell Biol. 2011;13(10):1214–1223. doi: 10.1038/ncb2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Mangan A, et al. FIP5 phosphorylation during mitosis regulates apical trafficking and lumenogenesis. EMBO Rep. 2014;15(4):428–437. doi: 10.1002/embr.201338128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Mechanics of cytokinesis in eukaryotes. Curr Opin Cell Biol. 2010;22(1):50–56. doi: 10.1016/j.ceb.2009.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl J, Crevenna AH, et al. Lifeact: a versatile marker to visualize F-actin. Nat Methods. 2008;5(7):605–607. doi: 10.1038/nmeth.1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiel JA, Childs C, et al. Endocytic transport and cytokinesis: from regulation of the cytoskeleton to midbody inheritance. Trends Cell Biol. 2013;23(7):319–327. doi: 10.1016/j.tcb.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiel JA, Park K, et al. Endocytic membrane fusion and buckling-induced microtubule severing mediate cell abscission. J Cell Sci. 2011;124(Pt 9):1411–1424. doi: 10.1242/jcs.081448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiel JA, Simon GC, et al. FIP3-endosome-dependent formation of the secondary ingression mediates ESCRT-III recruitment during cytokinesis. Nat Cell Biol. 2012;14(10):1068–1078. doi: 10.1038/ncb2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon GC, Schonteich E, et al. Sequential Cyk-4 binding to ECT2 and FIP3 regulates cleavage furrow ingression and abscission during cytokinesis. Embo J. 2008 doi: 10.1038/emboj.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GM, Fielding AB, et al. The FIP3-Rab11 Protein Complex Regulates Recycling Endosome Targeting to the Cleavage Furrow during Late Cytokinesis. Mol Biol Cell. 2004 doi: 10.1091/mbc.E04-10-0927. [DOI] [PMC free article] [PubMed] [Google Scholar]