

Abstract
The cannabinoids are members of a deceptively simple class of terpenophenolic secondary metabolites isolated from Cannabis sativa highlighted by (−)-Δ9-tetrahydrocannabinol (THC), eliciting distinct pharmacological effects mediated largely by cannabinoid receptor (CB1 or CB2) signaling. Since the initial discovery of THC and related cannabinoids, synthetic and semisynthetic classical cannabinoid analogs have been evaluated to help define receptor binding modes and structure–CB1/CB2 functional activity relationships. This perspective will examine the classical cannabinoids, with particular emphasis on the structure–activity relationship of five regions: C3 side chain, phenolic hydroxyl, aromatic A-ring, pyran B-ring, and cyclohexenyl C-ring. Cumulative structure–activity relationship studies to date have helped define the critical structural elements required for potency and selectivity toward CB1 and CB2 and, more importantly, ushered the discovery and development of contemporary nonclassical cannabinoid modulators with enhanced physicochemical and pharmacological profiles.
Keywords: classical cannabinoids, phytocannabinoids, cannabidiol, tetrahydrocannabinol, Cannabis sativa
History of Cannabis Sativa
Cannabis sativa has one of the longest histories of plants used for medicinal and recreational uses by humans. Cannabis, commonly known as marijuana, has been used throughout human history to treat a wide variety of ailments, with some of the earliest known references dating back to 2600 BC in ancient Chinese texts prescribing its use for relieving pain and cramping. While the medicinal and psychoactive properties of cannabis were well known for thousands of years, it was not until the late 19th century that cannabis fell under scientific scrutiny to understand the underlying mechanisms of these actions. The first scientific report in the Western world on the medicinal use of cannabis came from an Irish physician, Sir William B. O’Shaughnessy, who noted in 1843 that hemp “possesses, in small doses, an extraordinary power of stimulating the digestive organs, exciting the cerebral system, of acting also on the generative apparatus.”1 This report also noted the ability of hemp oil to alleviate pain, both rheumatic and otherwise in origin, and perhaps most remarkably noted the effects of hemp oil in reducing seizures in infants, a use now being heavily explored for medical marijuana and therapeutic use of cannabinoids.
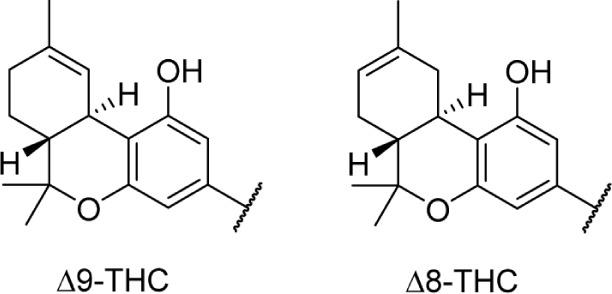
With the rise of research on natural products and the isolation of alkaloids such as morphine and cocaine, from the opium poppy and coca plant, respectively, cannabis was thought to possess similar chemical constituents. Much of the early research conducted on cannabis and hemp oil cantered on the search for alkaloids and other amine natural products and attempts to develop colorimetric tests for cannabinoids.2 The search for psychoactive compounds in cannabis, however, led not to a mixture of alkaloids but to the discovery of new terpenes. Most isolation experiments followed a similar procedure for nearly 100 years; hemp oil would be extracted with organic solvents, filtered, followed by removal of the solvent and fractional distillation of the resulting residue.3 This residue, referred to as red oil, possessed biological activity similar to that of the plant material, and when further fractionally distilled would yield an active fraction at 180 °C–190 °C (1 mm), called purified red oil. It was this purified red oil that was used in most chemical studies from the 1840s until the 1940s, when the adoption of chromatographic methods became more prevalent. The first secondary metabolite of cannabis isolated was cannabinol, in 1896, which was from purified red oil derived from hemp by Wood et al.4 Their work was unable to be repeated until 1933 when Cahn reported a partial structure of cannabinol; the structure was fully elucidated by two groups in 1940 (Fig. 1).5–7
Figure 1.

Structures of cannabinol, Δ9-THC, and cannabidiol.
It was initially thought that cannabinol was the active constituent of cannabis, but these early reports, likely obtained using impure cannabinol extracts, were proved erroneous in the 1930s.3,8 From 1940 until the 1960s, several other noncannabinoid natural products in cannabis were isolated, including cannabidiol (CBD).9 The active component of cannabis was finally discovered in 1964 by Gaoni and Mechoulam, with the report of the structure and partial synthesis of (–)-Δ9-tetrahydrocannabinol (THC).10 The discovery of new compounds in cannabis has continued, with over 100 phytocannabinoids reported to date.
Phytocannabinoids
Phytocannabinoids are found throughout all major morphologies of cannabis. Cannabinoids are mixed polyketides derived from malonyl-CoA, hexanoyl-CoA units prenylated with geranyl phosphate.11–13 This biosynthetic pathway, shown in Figure 2 with the synthesis of CBD, THC, and CBN, produces several classes of phytocannabinoids.
Figure 2.

Phytocannabinoid biosynthesis.
Of the more than hundred phytocannabinoids isolated and characterized, THC and CBD, depicted in Figure 1, have received the most attention in both basic science and clinical research. THC is marketed as dronabinol (Marinol®) and is currently approved for the treatment of anorexia in Acquired Immune Deficiency Syndrome (AIDS) patients and chemotherapy-induced nausea and vomiting.14 CBD has not been approved by the US Food and Drug Administration (FDA), but clinical trials are underway exploring the use of CBD, branded as Epidiolex®, in the treatment of epilepsy and Dravet syndrome, a severe seizure disorder in children.15,16 Despite the long-standing traditional medicinal uses of cannabis, and the culmination of scientific evidence leading to FDA approval of THC, the mechanism of action of cannabinoids in humans remained a conundrum until recently. The cannabinoid (CB) receptors remained elusive for 30 years after the discovery of THC. Both CBD and THC exert their therapeutic effects through the CB receptors, in addition to G-protein-coupled receptor 55 (GPR55), 5-hydroxytryptamine (5-HT)-3A ligand-gated ion channel, transient receptor potential cation channel Ankyrin type 1 (TRPA1), and transient receptor potential cation channel vanilloid type 1 (TRPV2).17–20 The pharmacology of CBD is not entirely understood with respect to the treatment of seizures; CBD has been shown to block both CB receptors, activate several TRP cation channels, and activate the 5-HT1A receptor.18,19,21
CB Receptors and the Endocannabinoid System
The G-protein-coupled receptor (GPCR) superfamily of genes encodes for 800 GPCRs. This superfamily of genes is further divided into five major families: glutamate, rhodopsin, adhesion, frizzled/taste2, and secretin.22 The largest of these is the rhodopsin family, containing 672 GPCR genes, approximately 300 of which encode known GPCRs, with the remainder classified as orphan receptors whose structure, endogenous ligand(s), and function remain unknown.23–25 GPCRs are characterized by having a transmembrane domain unit with seven alpha-helices coupled to a G-protein consisting of three subunit proteins: Gα, β, and γ. Upon binding of an agonist ligand to the transmembrane domain, the G-protein subunits catalyze downstream functions by coupling to another cellular protein (eg, adenylyl cyclase, protein kinases, etc.).26 Because of their significant role in human cellular functions, drugs targeting GPCRs make up 30%–40% of all drugs on the market.27
The CB receptors are members of the rhodopsin-like family of GPCRs. The first evidence of a CB receptor surfaced in 1984, with Howlett et al demonstrating that select cannabinoids decreased cyclic adenosine monophosphate (cAMP) concentrations in neuroblastoma cells.28,29 Further work in 1986 showed that cannabinoids induce a decrease in cAMP production, and this effect was eliminated by exposing cells to pertussis toxin, a known Gαi (commonly referred to simply as the Gi protein) protein inhibitor, which strongly suggested the presence of a CB binding GPCR.30 In 1988, the same group characterized a CB-specific receptor in rat brain, and in 1990, the CB1 receptor was finally cloned from a cDNA library from rat cerebral cortex tissue.31 In the same year that CB1 was discovered, tissue distribution studies showed that CB1 was one of the most abundantly expressed receptors in the brain, nearly equivalent to the expression of glutamate and GABA receptors.32,33 The correlation between CB1 localization in the brain and the known pharmacological effects of CB agonists was made immediately clear; CB1 expression in the basal ganglia and cerebellum was associated with the effects on gait, and expression in the cerebral cortex and hippocampus was associated with the effects on cognition and memory. More recent studies have found CB1 expression in the spleen, tonsils, gastrointestinal tract, uterus, prostate, vascular smooth muscle cells, and adrenal glands.34
While the CB1 receptor was commonly referred to as the CB receptor, this receptor did not account for the well-documented immunomodulatory effects of cannabis. The search for an explanation led to the discovery of the CB2 receptor in a human promyelocytic leukemia cell line in 1993.33,35 The human CB1 and CB2 receptors share 48% sequence identity and are both coupled to G proteins; neither CB1 nor CB2 has been crystallized to date.36 While the CB receptors are primarily coupled to Gi proteins, there is mounting evidence that much of the pharmacological effects of CB1 are mediated through Gs and Go proteins as well; this is in contrast with CB2, which thus far has only been found coupled to Gi.36–38 The pharmacology of the CB2 receptor has a long, complicated history since its discovery, with numerous contradicting reports and flawed methodologies, leading some to call CB2 “a receptor with an identity crisis.”39 The CB2 receptor, unlike CB1, is not highly expressed in the CNS. For several years after its discovery, CB2 was known as the peripheral CB receptor, owing to its high expression levels in the spleen and immune cells and relative absence from the brain.40 This was proven incorrect, however, with CB2 protein expression in microglial cells in the brain and reports of CB2 expression in neurons. CB2 expression in the brain correlated with neuroinflammation, with one study in 2005 showing a 200-fold upregulation of CB2 receptors in microglial cells in an in vitro autoimmune encephalomyelitis model.41
These and many other results, however, have been called into question, as anti-CB2 antibodies used in these immunohistochemical methods have been demonstrated to have nonspecific binding with other proteins.42,43 The immunomodulatory role of CB2 has remained unchallenged, and CB2 has been heavily implicated in neurodegenerative diseases such as Huntington’s and Alzheimer’s diseases.44,45 Increased expression of CB2 in the brain was confirmed with CB2-selective positron emission tomography (PET) tracers in Alzheimer’s mice models; this increased expression was concomitant with the formation of amyloid-beta plaques, suggesting a potential utility for CB2 PET tracers as diagnostic for the onset of neuroinflammation.
Activation of either CB1 or CB2 produces a dose-dependent decrease in cellular cAMP levels and modulation of intracellular Ca2+ and K+ levels.46 Stimulation of CB receptors results in activation of the p42/44 mitogen-activated protein kinases (MAPK), otherwise known as the extracellular signal-regulated kinases 1 and 2 (ERK1 and ERK2), respectively, as well as p38 MAPK and c-Jun N-terminal kinases.47,48 Signal transduction studies have linked this CB1/2 mediation of ERK1/2 to downstream regulation of genes, controlling cytokine synthesis, transcription regulation, and cell differentiation (Fig. 3).49,50
Figure 3.

Neuronal CB signaling. Activation of a CB receptor with an agonist causes several downstream effects: inhibition of adenylcyclase and inwardly rectifying calcium channels, and activation of potassium channels as well as the mitogen-activated protein kinase pathway. Activation of MAPK modulates gene expression, depending on downstream signaling, cell types, etc. Gene expression can also be modulate as a downstream effect of adenylyl cyclase inhibition through the activation of protein kinase A.
Abbreviations: MAPK, mitogen-activated protein kinases; AC, adenylyl cyclase; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A.
Note: Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Drug Discovery, copyright 2004.150
Endocannabinoid System
While the discovery (and the naming) of the CB receptors was driven by a desire to understand the pharmacological effects of cannabis, both receptors are involved in extensive signaling pathways known as the endocannabinoid system. The presence of CB GPCRs suggested the existence of endogenous ligands, and as most phytocannabinoids are highly lipophilic, it was assumed that these ligands would likely be lipids.
The identification of anandamide (AEA) by the Mechoulam group in 1992 confirmed its role as an endogenous ligand for the CB receptors, with a Ki of 61.0 nM at CB1 and 1,930 nM at CB2.51,52 AEA produces similar effects to that of the exogenous phytocannabinoids, with administration to rodents of AEA inducing hypothermia, analgesia, catalepsy, and appetite stimulation.53,54 Furthermore, its tissue distribution is highly similar to that of CB1: the highest levels of AEA are found in the hippocampus and cerebellum and to a lesser degree in the spleen and heart tissue.55 Soon after the discovery of AEA followed the identification of several more endocannabinoids, namely, 2-arachidonoylglycerol (2-AG), with Ki values of 472 ± 55 nM at CB1 and 1,400 ± 172 nM for CB2, O-arachidonoyl ethanolamine, with Ki values of 1,900 nM at CB1 and 1,400 nM at CB2, and 2-arachidonoyl glycerol ether, with Ki values of 21.2 nM at CB1 and 480 nM at CB2 (Fig. 4).56–59 Although initially considered an insignificant component of the endocannabinoid system, the role of 2-AG has evolved to that of one of the more important signaling molecules in the brain. 2-AG has been linked to the modulation of feeding, hypotension, neuroprotection, cell proliferation, and other interesting physiological processes.60–63
Figure 4.

Structures and CB receptor affinities of major endocannabinoids.
For a very broad overview, endocannabinoid signaling typically occurs in retrograde fashion, from post- to presynaptic neurons (Fig. 5). Due to the highly hydrophobic nature of endocannabinoids like 2-AG, it was initially thought that endocannabinoids were synthesized in the same cell in which receptor binding occurs. This was supported by the observation that endocannabinoids could approach a receptor by moving laterally through the cell membrane, through postsynaptic nonretrograde signaling.64 However, the identification of AEA in interstitial fluid and cell incubation media suggested additionally that AEA, and likely other endocannabinoids, can travel across a synapse by either passive diffusion or active transport mechanisms, although a specific mechanism has yet to be resolved.65–68
Figure 5.

Synaptic endocannabinoid signaling. Dashed arrows indicate inactivation of endocannabinoid.
Abbreviations: AA, arachidonic acid; DAGs, diacylglycerols; ER, endoplasmic reticulum; MAPK, mitogen-activated protein kinases; PIP2, phosphoinositide bisphosphate; PKA, protein kinase A; PLCβ, phospholipase Cβ; PPARs, peroxisome proliferator-activated receptors; TRPs, transient receptor potential channels; VGCCs, voltage-gated calcium channels.
Note: Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Neuroscience, copyright 2015.44
In retrograde signaling, endocannabinoids cause a variety of downstream effects. Presynaptic CB1 activation causes two major neurotransmitter inhibition mechanisms: short-term and long-term plasticity.69 In short-term plasticity, elevation of the intracellular Ca2+ levels by postsynaptic depolarization stimulates the production of 2-AG, which diffuses across the neuronal synapse to CB1 receptors on the presynaptic neuron.70–72 Activation of CB1 causes inhibition of Ca2+ influx via voltage-gated Ca2+ channels and subsequent downregulation of neurotransmitter release. Long-term plasticity, while initiating in a similar fashion, involves the suppression of neurotransmitter release via the downregulation of cAMP production and protein kinase A inhibition.
Once released, endocannabinoids are rapidly deactivated by two enzymes: fatty acid amide hydrolase 1 and monoacylglycerol lipase.44 The distribution of these enzymes provides some evidence as to the signaling mechanisms carried out by the endocannabinoid system, as fatty acid amide hydrolase 1 is located postsynaptically and monoacylglycerol lipase presynaptically.
The role of CB2 is less well defined than CB1 in the endocannabinoid system. The involvement of CB2 in the endocannabinoid signaling system has been relegated to that of an immunomodulatory mediator. Like CB1, CB2 also decreases the production of cAMP, although to a lesser degree, and unlike CB1, it has not been found to be coupled to G proteins other than Gi, somewhat limiting its inhibitory effect on Ca2+ and K+ channels.36 Also unique to the activation of CB2 receptors is an initial decrease in cAMP production, followed by a sustained increase up to 10-fold in T-cell cAMP levels, which can lead to suppression of T-cell signaling, manifesting phenotypically as an immunosuppressant effect.73 Immunohistochemical and mRNA analyses show CB2 localization to occur primarily in microglial cells in the brain, neutrophils, macrophages, monocytes, and lymphocytes peripherally, with significantly increased expression under inflammatory conditions.41,74,75 Increased endocannabinoid production in immune cells has been linked to pro-inflammatory stimuli and hematopoietic stem cell differentiation.76,77 For a more in-depth discussion of endocannabinoid signaling, several timely reviews have been published.68,78–82
Synthetic Classical Cannabinoids
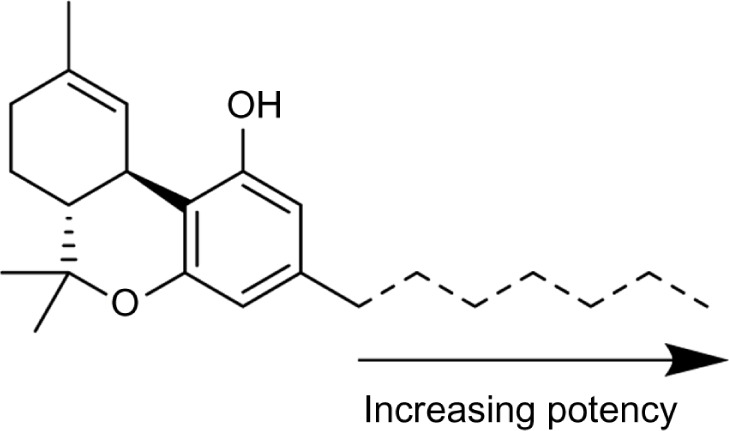
Since the initial discovery of THC and other related cannabinoids, numerous modifications and analogs were synthesized in an attempt to define the structure–activity relationship (SAR) of THC at both CB1 and CB2. Δ9-THC contains five major structural features, depicted in Figure 6: the C3 side chain, phenolic hydroxyl, and three rings: the aromatic A-ring, pyran B-ring, and cyclohexenyl C-ring. While not present in any natural cannabinoids, some important synthetic analogs replace the pyran B-ring with a substituted aliphatic chain, and as such, the southern aliphatic region is included as a major structural feature of classical cannabinoids.
Figure 6.

SAR of classical cannabinoids (left) major pharmacophores of classical cannabinoids, and common regions of functionalization and analog synthesis; (right) dibenzopyran numbering of Δ9-THC.
Many of the earliest SAR studies on classical cannabinoids involved modifications to the C3 side chain. While most analogs contain saturated straight or branched alkyl chains, a number of C3 side chains incorporating unsaturated alkyl chains, heteroatoms, and functional groups such as esters, carboxylic acids, ethers, nitriles, and heterocycles were reported, with varying effects on CB1 and CB2 potency and selectivity.
C3 alkyl analogs
The length of the C3 side chain of THC directly correlates with CB1 and CB2 binding affinity; an increase in chain length leads to an increase in binding affinity at both receptors (Fig. 7). CB binding affinity data for methyl- or ethyl-substituted THC analogs have not been published; however, a study conducted in 2011 examined the functional activity of these THC analogs, demonstrating decrease in the receptor affinity in a linear fashion with decreasing chain length.83 Interestingly, this study found an inverse relationship between chain length and TRPA1 channel activity; a one-carbon chain was found to be a potent TRPA1 agonist, this effect decreasing with additional carbons on the chain. This may be a contributing factor to the biological significance of cannabinoids like propyl-substituted Δ9-tetrahydrocannabivarin, which does not activate either CB receptor yet retains numerous biological effects.
Figure 7.
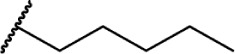
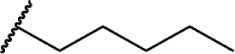
Relationship between alkyl chain length and CB receptor binding affinity.
Utilizing the C3 alkyl chain as a point of diversification in classical cannabinoids has remained a staple of CB research since the discovery of Δ9-THC. Numerous analogs, containing a variety of carbon chains and rings with and without heteroatom incorporation, provided a well-defined and predictable SAR profile for this portion of the THC scaffold.
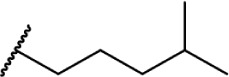
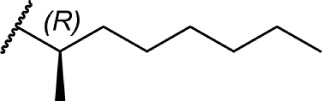
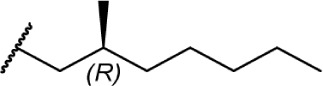
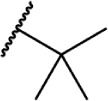
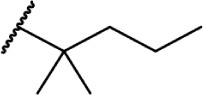
Of the major pharmacophores defined for classical CB SAR, the C-3 side chain seems to have the largest influence on binding affinity for the CB receptors. Table 1 illustrates a homolog series of Δ8-THC and Δ9-THC analogs with various saturated aliphatic substitutions. Entries 1 and 2, Δ9-THC and Δ8-THC, respectively, are nearly equipotent at both CB receptors, displaying partial agonist functional activity. As such, Δ9-THC and Δ8-THC are interchangeable in most SAR studies. Examining various alkyl chain lengths (entries 2–9), there is a definite requirement for at least 3 carbons, with 5–8 carbon length chains being optimal.84,85 Binding affinity at CB1 receptors is further enhanced by the addition of methyl groups on the alkyl chain, preferably at the 1′ and 2′ positions. A systematic study of methylated Δ8-THC analogs (entries 10–16) revealed 1′ and 2′ substitutions to be optimal, with a slight decrease in affinity in 3′-methyl analogs 14–15 and a sharp decrease in 4′-methyl analog 16, with little difference in affinity observed between the R and S isomers.86 Given the near doubling of receptor affinity by the 7-carbon chain analog 8, a similar study of compounds with a 7-carbon alkyl chain, entries 17–27, showed a similar trend, although less pronounced than the 5-carbon chain analogs.87 Again, there were minimal differences between R and S isomers, with binding affinity optimal at the 1′ and 2′ positions in 17–20, slowly decreasing as the methyl substitutions is moved down the alkyl chain (entries 21–27). Expanding on this trend, 1′,2′-dimethylheptyl analogs 28–31 possessed subnanomolar binding affinity, with essentially no difference in activity observed between the four diastereomers.88 A series of 1′,1′-dimethylalkyl analogs (entries 32–42) with chains lengths of 2–12 carbons displayed a similar trend to that of the unsubstituted homolog series (entries 2–9), with optimal binding affinity for CB1 achieved between 5 and 9 carbons.89 The 1′,1′-dimethyl substitution clearly has a significant effect on the affinity however, as none of the entries in this series, with the exception of the 12-carbon chain analog 42, had binding affinities greater than 100 nM. The 1′,1′-dimethylalkyl-substituted analogs have typically been preferred over the 1′,2′-dimethylakyl congeners, since the latter possesses two stereogenic centers, which add unnecessary complexity in synthesis with the potential for formation of diastereomeric mixtures.
Table 1.
C3 alkyl analogs of Δ8-THC and Δ9-THC (human CB1 and CB2).
| # | CB | NAME | CB1 KI (nM) | CB2 KI (nM) | FUNCTIONAL | REF | |
|---|---|---|---|---|---|---|---|
|
|||||||
| 1 |
|
A9-THC | Δ9-THC | 40.7 ± 1.7 | 36 ± 10 | Partial Agonist | 132,133 |
| 2 |
|
Δ8-THC | Δ8-THC | 44 ± 12 | 44 ± 17 | 134 | |
| 3 |

|
Δ9-THC | |||||
| 4 |

|
Δ9-THC | |||||
| 5 |

|
Δ9-THC | Δ9-THCV | 75.4 | 62.8 | Mixed | 84 |
| 6 |

|
Δ8-THC | JWH-130 | 65 ± 13 | 85 | ||
| 7 |
|
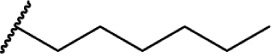
Δ8-THC | JWH-124 | 41 ± 3.8 | 85 | ||
| 8 |

|
Δ8-THC | JWH-091 | 22 ± 3.9 | 85 | ||
| 9 |

|
Δ8-THC | JWH-138 | 8.5 ± 1.4 | 85 | ||
| 10 |
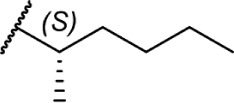
|
Δ8-THC | 20 ± 4 | 86 | |||
| 11 |

|
Δ8-THC | 7.6 ± 0.6 | 86 | |||
| 12 |

|
Δ8-THC | 11 ± 1 | 86 | |||
| 13 |
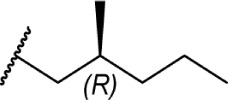
|
Δ8-THC | 19 ± 5 | 86 | |||
| 14 |
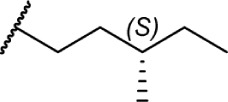
|
Δ8-THC | 53 ± 1 | 86 | |||
| 15 |
|
Δ8-THC | JWH-359 | 38 ± 3 | 86 | ||
| 16 |
|
Δ8-THC | 141 ± 52 | 86 | |||
| 17 |
|
Δ8-THC | 0.51 ± 0.02 | 87 | |||
| 18 |

|
Δ8-THC | 2.0 ± 0.3 | 87 | |||
| 19 |
|
Δ8-THC | 1.4 ± 0.2 | 87 | |||
| 20 |

|
Δ8-THC | 2.0 ± 0.8 | 87 | |||
| 21 |
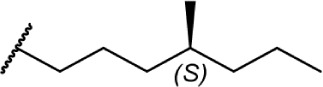
|
Δ8-THC | 9.5 ± 2.9 | 87 | |||
| 22 |
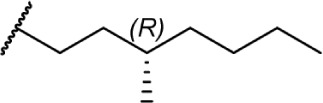
|
∆8-THC | 1.3 ± 0.2 | 87 | |||
| 23 |
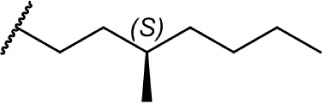
|
Δ8-THC | 18 ± 2 | 87 | |||
| 24 |

|
Δ8-THC | 32 ± 5 | 87 | |||
| 25 |
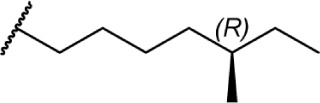
|
Δ8-THC | 75 ± 9 | 87 | |||
| 26 |

|
Δ8-THC | 38 ± 5 | 87 | |||
| 27 |

|
Δ8-THC | 19 ± 1 | 87 | |||
| 28 |

|
Δ8-THC | 0.46 ± 0.04 | 88 | |||
| 29 |
|
Δ8-THC | 0.81 ± 0.08 | 88 | |||
| 30 |
|
Δ8-THC | 0.84 ± 0.21 | 88 | |||
| 31 |

|
Δ8-THC | 0.60 ± 0.15 | 88 | |||
| 32 |
|
Δ8-THC | 14 ± 1.8 | 89 | |||
| 33 |

|
Δ8-THC | 14 ± 0.9 | 89 | |||
| 34 |
|
Δ8-THC | 10.9 ± 1.7 | 89 | |||
| 35 |
|
Δ8-THC | JWH-133 | 3.9 ± 0.9 | 89 | ||
| 36 |

|
Δ8-THC | 2.7 ± 1.2 | 89 | |||
| 37 |
|
Δ8-THC | 0.83 | 0.49 | 89 | ||
| 38 |

|
Δ8-THC | 0.09 ± 0.1 | 89 | |||
| 39 |

|
Δ8-THC | 1.6 ± 0.4 | 89 | |||
| 40 |
|
Δ8-THC | 6.1 ± 1.8 | 89 | |||
| 41 |

|
Δ8-THC | 25.8 ± 5.8 | 89 | |||
| 42 |
|
∆8-THC | 126 ± 18 | 89 | |||
| 43 |
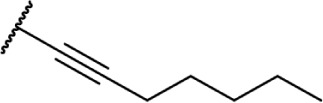
|
∆8-THC | O-964 | 0.65 ±.012 | 3.1 ± 0.13 | Agonist | 135 |
| 44 |
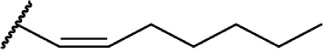
|
∆8-THC | O-1317 | 0.86 ± 0.09 | Agonist | 85 | |
| 45 |
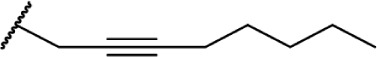
|
∆8-THC | O-584 | 4.9 ± 2.0 | Partial Agonist | 85, 136 | |
| 46 |

|
∆8-THC | O-1020 | 9.0 ± 1.3 | Agonist | 85 | |
| 47 |
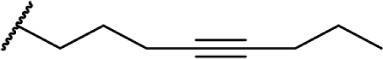
|
∆8-THC | O-1052 | 19 ± 1.3 | Agonist | 85 | |
| 48 |
|
∆8-THC | O-1004 | 367 ± 23 | 85 | ||
|
|||||||
| 49 |
|
∆8-THC | 703 ± 98 | 90 | |||
| 50 |

|
∆8-THC | 402.4 | 161.5 | 91 | ||
| 51 |
|
∆8-THC | AM855 | 22.3 | 58.6 | 91 | |
| 52 |
|
∆8-THC | 542.1 | 455.6 | 91 | ||
| 53 |
|
∆8-THC | 126.0 ± 22 | 137 | |||
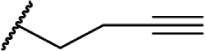
Introducing unsaturation on the alkyl chain does not significantly modulate CB1 receptor binding, with activity retained between homologs of the same chain length [eg, compare the unsaturated 8-carbon analogs 45–47 (Ki = 4.9 ± 2.0 nM, 9.0 ± 1.3 nM, and 19 ± 1.3 nM, respectively) to the saturated 8-carbon analog 9 (Ki = 8.5 ± 1.4 nM)].85 Similar to the unsaturated series, chain length has a significant influence on affinity, with the terminal acetylene 4-carbon analog 48 showing modest affinity at CB1 (Ki = 367 ± 23 nM).
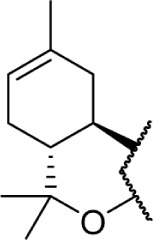
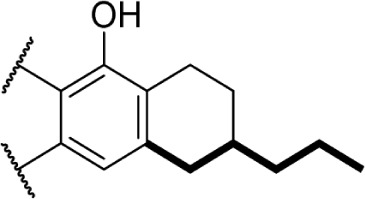
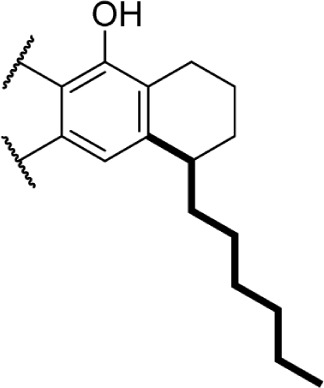
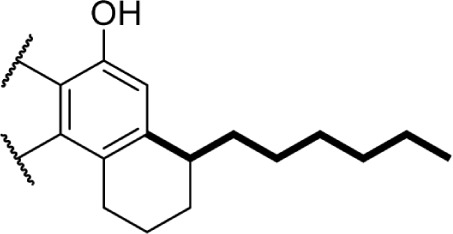
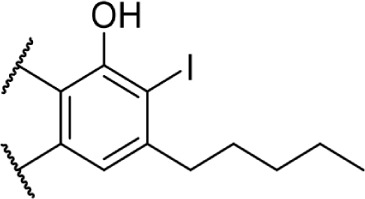
A series of rotationally restricted Δ8-THC analogs, entries 49–52, narrow the possible conformers the alkyl chain can adopt. Entry 49, reported by Huffman and Yu incorporates the typical 5-carbon chain with a six-membered ring bridging the aryl C-2 and alkyl C2′, exhibited low affinity for the CB1 receptor.90 This is reinforced by a series of compounds (entries 50–52), reported by Khanolkar et al, in which the 7-carbon homolog 50 displayed equally poor affinity for both CB1 and CB2.91 Shifting the alkyl chain to the adjacent carbon on the alkyl chain restores the affinity to 22.3 and 58.6 nM for CB1 and CB2, respectively. The significant loss in affinity for 49, 50, and 52 suggests that the lateral orientation of the alkyl chain is not the relevant conformer for receptor binding, rather the conformer 51 with the chain orienting downward, away from phenol. It is unlikely that the ring in 50 and 51 is occupying space required by either receptor, as the 2-iodo-substituted 53 retains modest affinity at CB1; the C4–C2′ ring of 54, however, follows a similar pattern of other C4-substituted analogs with significant loss of affinity, suggesting that this space is required in the CB1 receptor for binding.92 Substitution with an adamantyl group in place of the alkyl chain also retained potency at both CB1 and CB2, suggesting that favorable hydrophobic interactions can be made with the receptor in the C1′ and C2′ positions on the chain.93
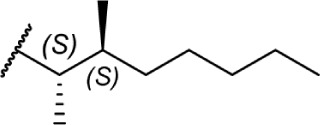
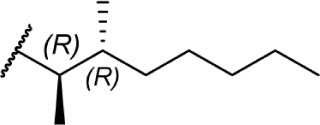
These aliphatic side chain analogs offer several conclusions that can be inferred with regard to CB receptor binding requirements. A 5–8 carbon length alkyl chain is optimal, with binding affinity decreasing when shorter and longer alkyl chains are incorporated. Restricting the flexibility of the alkyl chain lends some insight into the optimal conformation for receptor binding. First, introducing methyl substitutions in various positions on the chain adds steric congestion that restricts the number of possible rotamers, in addition to providing hydrophobic bulk that may be interacting with the receptors. Branching close to the aromatic ring provides a minimum 10-fold increase in affinity for CB1 (17 and 18 vs. 8), with affinity diminishing as the methyl group is moved down the chain further from the aryl ring (19–27), and the best affinity is achieved with 1′,2′- and 1′,1′-dimethyl substitutions. Second, restricting rotation by introducing double and triple bonds on the chain follows a similar pattern: cis-double bonds and triple bonds in the C1′ position (43 Ki = 0.65 nM, 44 Ki = 0.86 nM) have no change in receptor binding, with affinity decreasing as the triple bond is moved down the chain to the C2′ position (45 Ki = 4.9 nM), C3′ (46 Ki = 9.0 nM), and C4′ positions (47 Ki = 19 nM). Third, restricting the C1′ and C2′ carbons in a ring results in the chain orienting in a linear (50 Ki = 402.4 nM) or downward fashion (51 Ki = 22.3 nM), illustrated in Figure 8.
Figure 8.

Rotational conformers of 3-heptyl-Δ8-THC. Studies of rotationally restricted alkyl chains determined that the optimal conformation of the chain is oriented downward away from the phenol, rather than linearly away from the phenyl ring.
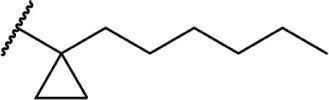
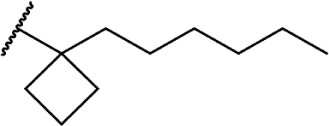
To investigate the SAR requirements of C1′ substitutions on the side chain, several series of compounds containing aliphatic rings and heterocycles were synthesized, as illustrated in Table 2. Transforming the 1′,1′-geminal dimethyl substitution into a compact cyclopropyl ring further enhances activity at both receptors.94 Expanding the ring size to 4, 5, and 6 carbons modulates the activity as a function of the lowest energy conformation of the alkyl chain, as well as the presence of hydrophobic bulk, potentially creating steric clashes with the putative binding site in the receptor. Quantitative structure–activity relationship studies confirm that the cyclopropyl and cyclopentyl rings force the chain into similar favorable orientations, with the cyclobutyl- and cyclohexyl-substituted chains adopting less favorable conformations for optimal receptor binding.95 This orientation, with the alkyl chain oriented perpendicular to the aromatic ring of the THC scaffold, appears to be the optimal conformer for receptor binding.
Table 2.
C3 analogs of ∆8-THC and ∆9-THC.
| # | CB | NAME | CB1 KI (nM) | CB2 KI (nM) | FUNCTIONAL | REF | |
|---|---|---|---|---|---|---|---|
| 54 |
|
Δ8-THC | AMG-41 | 0.44 ± 0.07 | 0.86 ± 0.16 | 94 | |
| 55 |
|
Δ8-THC | 1.5 ± 0.2 | 11.5 ± 3.4 | 138 | ||
| 56 |
|
Δ8-THC | AMG-36 | 0.45 ± 0.07 | 1.92 ± 0.4 | 139 | |
| 57 |
|
Δ8-THC | 18.4 ± 2.1 | 23.5 ± 4.1 | 138 | ||
| 58 |
|
Δ8-THC | 13.6 ± 2.4 | 143 ± 31.5 | 138 | ||
| 59 |
|
Δ8-THC | 3.9 ± 0.5 | 4.9 ± 0.7 | 138 | ||
| 60 |

|
Δ8-THC | 0.32 ± 0.08 | 0.52 ± 0.17 | 135 | ||
| 61 |
|
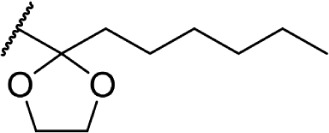
Δ8-THC | 0.52 ± 0.11 | 0.22 ± 0.06 | 139 | ||
| 62 |
|
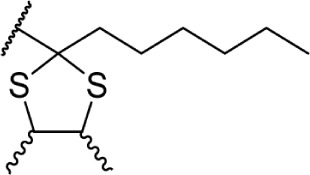
Δ8-THC | 32.3 ± 4.0 | 19.7 ± 2.7 | 139 | ||
| 63 |
|
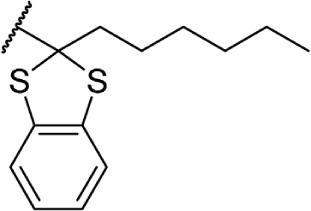
Δ8-THC | 56.9 ± 6.8 | 257 ± 41 | 139 |
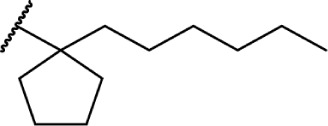
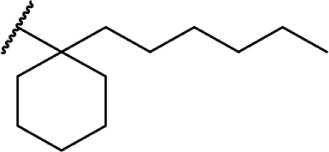
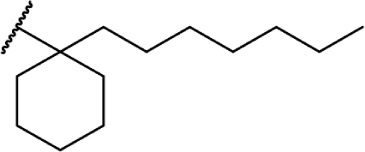
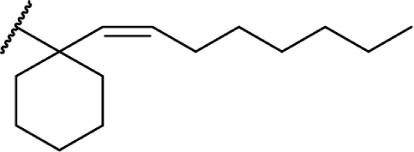
In addition to the alkyl chain conformation, the increased affinity of these compounds also suggests the presence of a hydrophobic binding subsite near the phenyl ring in both CB1 and CB2. Both receptors appear to be indifferent to heteroatoms at this position, illustrated by the high affinity of analogs 60–62; however, CB2 has a preference for smaller rings, as larger substitutions such as the cyclohexyl analog 58 and bulky benzodithiolane 63 show decreased affinity for CB2 but not for CB1.
The replacement of the alkyl chain with various ring structures results in a variety of effects on receptor binding (Table 3). To explore the necessity of hydrophobic bulk near the phenolic ring, a series of saturated alkyl ring analogs were synthesized and evaluated. Bulky bornyl and adamantyl derivatives 64–67 display interesting profiles, depending on the substitution pattern of the rings. An epimer change from bornyl-substituted 64 and isobornyl 65 results in 10-fold selectivity for CB2, and the location of the link between the adamantyl and phenolic rings results in either CB1 preference for 66 or CB2 for 67. The differences in binding affinity of these compounds can be explained, at least in part, by conformational analysis of each compound. The allowable conformational space of 65 and 67 both occupy a larger volume than that of their respective isomers 64 and 66, suggesting that such compounds are better accommodated by the CB2 receptor. This hypothesis is further reinforced by the loss of selectivity by the more flexible analog 68, which is capable of accessing both conformational spaces.93,96,97
Table 3.
C3 analogs of ∆8-THC and ∆9-THC.
| # | CB | NAME | CB1 KI (nM) | CB2 KI (nM) | FUNCTIONAL | REF | |
|---|---|---|---|---|---|---|---|
| 64 |
|
Δ8-THC | AM735 | 8.9 ± 1.0 | 7.4 ± 0.8 | 97 | |
| 65 |
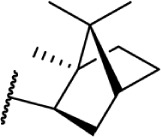
|
Δ8-THC | AM731 | 60.2 ± 0.8 | 6.1 ± 0.6 | 97 | |
| 66 |
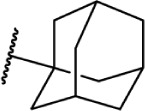
|
Δ8-THC | AM411 | 6.80 | 52.0 | Agonist | 93 |
| 67 |

|
Δ8-THC | AM744 | 34.9 | 14.0 | Agonist | 93 |
| 68 |
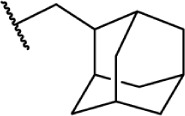
|
Δ8-THC | AM757 | 79.7 | 76.0 | Agonist | 93 |
| 69 |

|
Δ8-THC | 95.5 ± 16.7 | 71.8 ± 12.2 | 140 | ||
| 70 |

|
Δ8-THC | 11.7 ± 1.6 | 9.4 ± 1.5 | 140 | ||
| 71 |
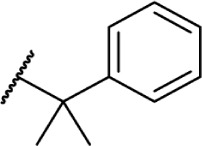
|
Δ8-THC | KM-233 | 12.3 ± 0.6 | 0.9 ± 0.1 | 141 | |
| 72 |
|
Δ8-THC | 76.1 ± 1.5 | 12.4 ± 0.2 | 141 | ||
| 73 |

|
Δ8-THC | 18.8 ± 1.4 | 1.7 ± 0.2 | 141 | ||
| 74 |
|
Δ8-THC | 5.03 ± 0.4 | 1.5 ± 0.2 | 141 | ||
| 75 |
|
Δ8-THC | 3.1 ± 0.4 | 0.9 ± 0.1 | 141 | ||
| 76 |
|
Δ8-THC | 5.3 ± 0.9 | 0.9 ± 0.02 | 141 |
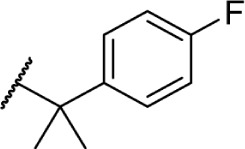
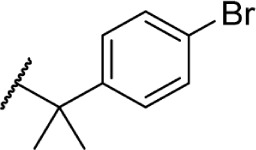
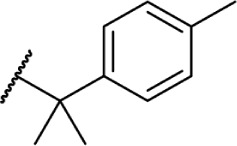
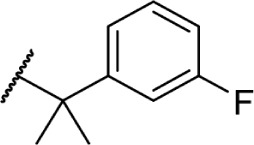
Using more planar phenyl rings, analogs 69–75 display modest activity at both receptors. Replacing the alkyl chain in Δ8-THC with an aromatic ring (69) leads to a reduction in affinity, and expansion of the ring to a naphthyl analog 70 enhanced affinity. Reincorporating the gem-dimethyl linker in 71–75 restores activity to low nanomolar affinity; substituting the phenyl ring in the para-position requires a bulky, non-polar group, with affinity increasing from para-fluoro analog 72 to p-methyl 75. Moving these groups to the meta-position negates any loss in receptor activity by substitution, as illustrated by meta-fluoro analog 76.
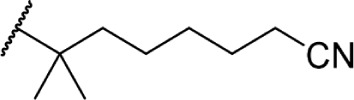
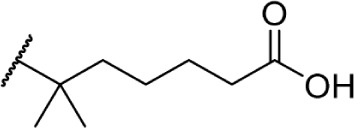
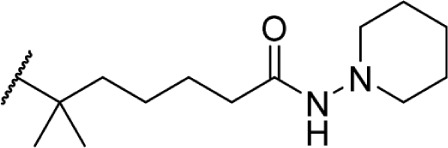
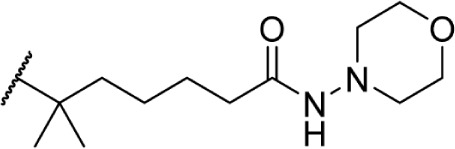
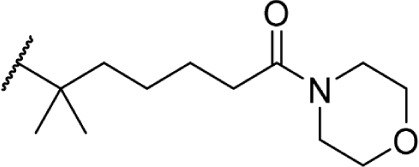
The addition of heteroatom substitutions on the terminal position of the alkyl chain provides analogs that display increases in binding affinity as well as enhanced polarity (Table 4). The use of halogens and nitrile pseudohalogens on the terminal position of the alkyl chain for both the pentyl (77) and dimethylheptyl (79) analogs provides an increase in binding affinity over their hydrocarbon homologs, while substitution with smaller halogens such as fluorine (78) cause a loss of affinity at CB1. One postulation on this change between fluorine and bromine is that the 5′-bromide chain (77) is closer to length of a 7-carbon chain, with the bromine essentially serving as a bioisostere for two carbons.98 The addition of a more polar carboxyl group (81) causes a significant loss in CB1 affinity to 222 ± 63 nM, while retaining CB2 affinity at 4.00 ± 1.35 nM. Using substituted carboxamide groups (82–86), with the exception of 85, shows similarly high affinities for both receptors. Removing the carboxamide linker and directly coupling a heterocycle to the alkyl chain is not detrimental to binding affinity, as seen in morpholine 87 and imidazole 88. These analogs provide a welcomed decrease in lipophilicity, as the vast majority of classical CB analogs are insoluble in water, severely hampering their potential utility as therapeutic candidates.
Table 4.
C3 analogs of ∆8-THC and ∆9-THC.
| # | CB | NAME | CB1 KI (nM) | CB2 KI (nM) | FUNCTIONAL | REF | |
|---|---|---|---|---|---|---|---|
| 77 |
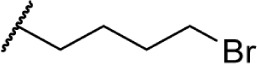
|
Δ8-THC | 10.8 ± 1.8 | 137 | |||
| 78 |
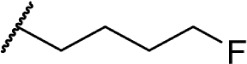
|
Δ8-THC | 81 ± 3.5 | 137 | |||
| 79 |
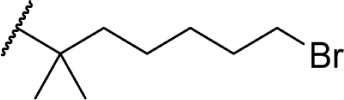
|
Δ8-THC | 1.27 ± 0.13 | 142 | |||
| 80 |
|
Δ8-THC | O-774 | 0.6 ± 0.05 | 2.94 ± 1.40 | Agonist | 143, 144 |
| 81 |
|
Δ8-THC | O-607 | 222 ± 66.3 | 4.00 ± 1.35 | Agonist | 144 |
| 82 |

|
Δ8-THC | O-1125 | 0.86 ± 0.06 | 1.98 ± 0.26 | Agonist | 143, 145 |
| 83 |
|
Δ8-THC | 1.2 ± 0.2 | 3.23 ± 0.29 | 143, 144 | ||
| 84 |
|
Δ8-THC | 6.0 ± 0.65 | 11 ± 0.91 | 146 | ||
| 85 |

|
Δ8-THC | 112 ± 14 | 389 ± 46 | 146 | ||
| 86 |
|
Δ8-THC | 1.3 ± 0.12 | 0.57 ± 0.04 | 146 | ||
| 87 |

|
Δ8-THC | O-704 | 3.0 ± 2.0 | 1.14 ± 0.54 | Agonist | 143, 144 |
| 88 |
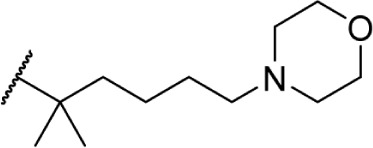
|
Δ8-THC | 2.8 ± 0.35 | 1.0 ± 0.16 | 146 | ||
| 89 |

|
Δ8-THC | O-2545 | 1.3 ± 0.17 | 0.12 ± 0.01 | 146 |
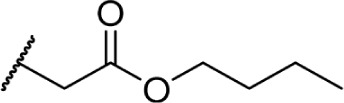
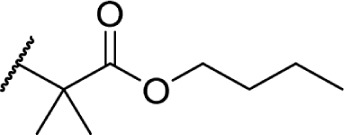
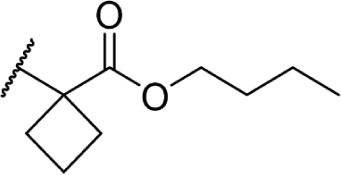
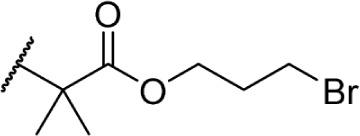
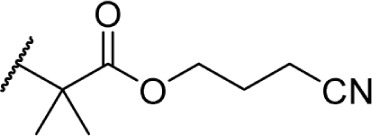
In another effort to improve the pharmacokinetics and bioavailability of classical type cannabinoids, Nikas et al.99 and Sharma et al.100 synthesized a series of side chain analogs containing a labile ester, in order to increase polarity and solubility and to extend the half-life in vivo (Table 5). Installing an ester in the 2′ position on the side chain afforded analogs with enhanced binding affinities at both CB1 and CB2. Carboxyester 90, while retaining nanomolar activity at both receptors suffered from an extremely short half-life of 0.7 minutes in mouse plasma.100 Adding the usual 1′,1′-dimethyl substitution in 91 increased the half-life substantially to 120 minutes, with a 90-fold and 30-fold increase in affinity at CB1 and CB2, respectively. Analogs containing a cyclobutyl ring exhibited improved half-lives to 263 minutes with no binding affinity cost at either receptor. Introducing steric hindrance near the ester carbonyl demonstrated a clear strategy for increasing the half-life for analogs 93–95 through inhibition of ester hydrolysis.
Table 5.
C3 analogs of ∆8-THC and ∆9-THC.

C1 phenol analogs
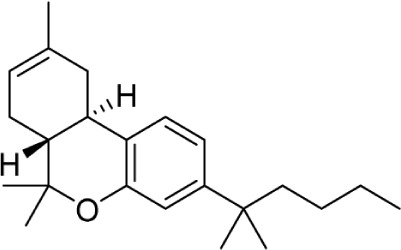
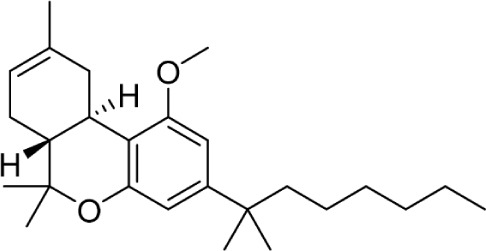
Another major point of structure modification on the THC scaffold is the C1 phenol (Table 6). Analogs lacking the phenolic hydroxy group or those that have been modified with minor changes to the phenolic group can result in drastic changes in the pharmacological activity of these compounds. It was quickly realized that etherification or removal of the phenol generated compounds that displayed significant selectivity for CB2. For example, the deoxy-Δ8-THC analog 96 is >300-fold selective for CB2; addition of the 1′,1′-demethyl group in analog 97 results in a slight decrease in selectivity to 200-fold. Conversion of the phenol to a methyl ester in analog 98 results in approximately 800-fold selectivity for CB2 over CB1.
Table 6.
C1 analogs of ∆8-THC and ∆9-THC.



Analogs 99 and 100 represent two compounds that demand additional comment. It was discovered that forcing the orientation of the phenol oxygen lone pair toward the cyclohexene ring resulted in analogs with optimal CB receptor activity.101 Both of these compounds were tested before the discovery of the CB receptors, making in vitro analysis unavailable (and not been determined since); however, intraperitoneal injection of these compounds in animal assays showed similar potency to Δ8-THC for 100, and almost no activity for 99. The presumption for some time was that the free phenol served as an important hydrogen bond donor, but the reanalysis of these compounds showed that while this may be true for CB1, there is no necessity for hydrogen bonding in this position for CB2. Computational modeling studies suggested that the diminished selectivity of the deoxy analogs compared to the methoxy analogs could be a result of inverted binding to the receptor, where the pyran oxygen can serve as a hydrogen bond acceptor in the CB1 receptor.102
C9/C11 analogs
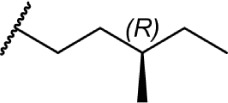
The C11 methyl group is another major pharmacophore at which minor structural changes can significantly modulate receptor binding (Table 7). Substitutions at this position do not confer selectivity when compared to analogs modified at the C1 phenol; however, binding affinity can be greatly enhanced. Methylene analogs 101 and 102 retain similar affinities to their respective homologs 37 and 98, with slightly higher CB2 selectivity for 102 (1,000-fold) compared to 98. Conversion of the methylene group to a carbonyl almost completely eliminates this selectivity in 104. Nabilone, 103, is one of two marketed CB therapeutics, approved by the US FDA in 1985 as an antiemetic. Hydroxylation of the C11 methyl group is a major metabolite of THC, which retains activity at both receptors at subnanomolar affinities. This analog, 105, better known as HU-210, has been widely used in CB pharmacology studies. Removal of the phenol in 106 confers a slight selectivity for CB2, but this is mitigated by the presence of the C11 hydroxy group. Aldehyde 107 and carboxylic acid 108 are also major metabolites of THC, which maintain modest affinity at the CB receptors. Compound 108, ajulemic acid, is currently undergoing clinical trials for cystic fibrosis, diffuse cutaneous systemic sclerosis, and dermatomysitis under the name Resunab.103–105 The decreased selectivity of the oxygen-substituted analogs 103–108 compared to their more selective homologs was the subject of a computational study, which suggested that the phenolic hydroxyl group was necessary for CB1 binding by hydrogen bonding with lysine 192, and the introduction of polar group on the C-11 position may satisfy this requirement.106 The removal of the unsaturation at C9 introduces another stereogenic center, which can dramatically affect receptor binding. Hydroxy analogs 109 and 110 show remarkably diverse activities; the R-diastereomer 109 is 6-fold and 17-fold more potent at CB1 and CB2, respectively, compared to S-diastereomer 110. This effect does not appear to be guided by hydrogen bonding interactions with either receptor, as the (R)-methyl analog 111 binds with higher affinity at both receptors. Compound 111 is also the only reported Δ6-THC analog.
Table 7.
C9/C11 analogs of ∆8-THC and ∆9-THC.
| # | CB | CB | NAME | CB1 KI (nM) | CB2 KI (nM) | FUNCTIONAL | REF |
|---|---|---|---|---|---|---|---|
| 101 |

|
1.82 ± 0.11 | 0.58 ± 0.30 | 147 | |||
| 102 |
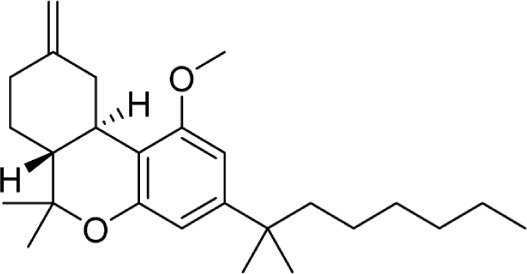
|
L-759,656 | >20,000 | 19.4 ± 3.8 | 147 | ||
| 103 |

|
Nabilone (Cesamet) | 2.19 ± 0.89 | 1.84 ± 0.42 | Agonist | 147 | |
| 104 |
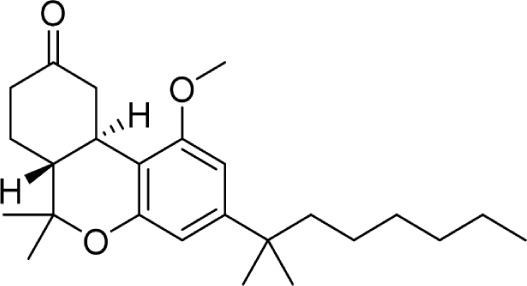
|
621 ± 215 | 132 ± 44 | 147 | |||
| 105 |
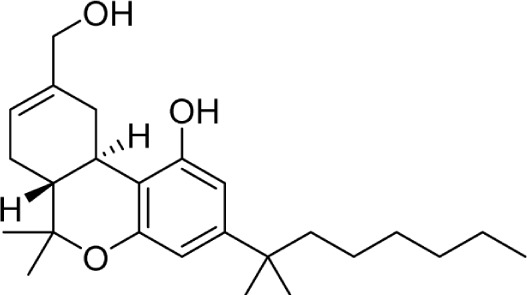
|
HU-210 | 0.73 ± 0.11 | 0.22 ± 0.18 | 133 | ||
| 106 |
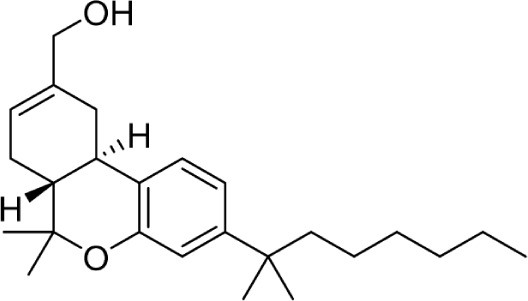
|
JWH-051 | 1.2 ± 0.1 | 0.032 ± 9.0 | 102 | ||
| 107 |
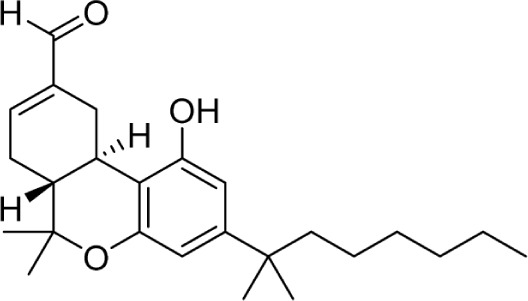
|
2.24 ± 0.05 | 2.61 ± 0.36 | 147 | |||
| 108 |
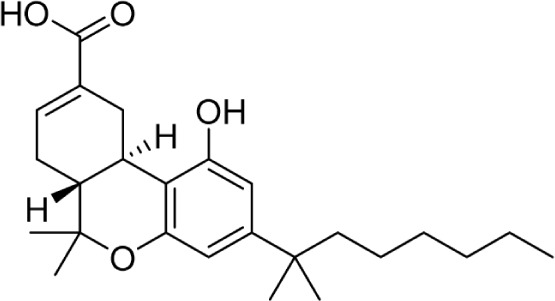
|
CT-3, JBT-101, Ajulemic Acid, Resunab | 628 | 51 | 103 | ||
| 109 |
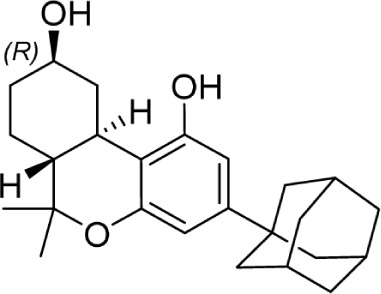
|
23.9 | 40.5 | 148 | |||
| 110 |

|
146.3 | 671.8 | 148 | |||
| 111 |

|
Δ6a-THC | 29 ± 12 | 18 ± 12 | 149 |
Miscellaneous analogs
Early SAR studies revealed that the pyran ring was not a requirement for cannabinergic activity in animal assays. Subsequent work on the core structure of 112 identified what is arguably one of the most important synthetic cannabinoids ever discovered, CP 55,940 (113); its radiolabeled [3H]-CP-55,940 isotope has been one of the most widely employed reference ligands used in pharmacology studies (Table 8). The use of radiolabeled 113 revealed the presence of CB1 receptor in rat brain, and it has been used as a standard in nearly every CB receptor assay developed.
Table 8.
Miscellaneous analogs of Δ8-THC and Δ9-THC.
| # | CB | CB | NAME | CB1 KI (nM) | CB2 KI (nM) | FUNCTIONAL | REF |
|---|---|---|---|---|---|---|---|
| 112 |

|
(−)-CP 47,497 | 21 ± 0.56 | ||||
| 113 |

|
(−)-CP-55,940 | 0.58 ± 0.07 | 0.69 ± 0.02 | Full Agonist | 133 |
Nonclassical Cannabinoids
CB1 selective
While classical CB development has remained a staple of compound synthesis, compounds adopting alternative scaffolds have become prominent. The lipophilicity of most classical cannabinoids has hampered their development into viable drugs, so efforts to increase the polarity and water solubility of new CB ligands has been an area of major focus.
Pravadoline (WIN 48,098) was a cyclooxygenase (COX) inhibitor developed by Bell et al. at Sterling-Winthrop in the 1980s, but its antinociceptive activity was significantly higher than other known COX inhibitors, despite its lower effect on prostaglandin synthesis compared to known nonsteroidal anti-inflammatory drugs.107 With animal assay results similar to known cannabinoids, the possibility was raised that the antinociceptive activity was derived not from COX binding, but from CB receptor activity. Structural optimization of pravadoline yielded one of the first nonclassical cannabinoids, WIN 55,212-2.108 Like CP-55,940, WIN 55,212-2 is a CB receptor agonist widely used in CB receptor assays as a reference compound. Since the discovery of WIN 55,212-2, numerous other potent, nonclassical cannabinoids were synthesized; two other notable compounds in this class are depicted in Figure 9. The (aminoalkyl)indole JWH-018 is nearly equipotent at both CB receptors, with slight selective toward CB2.109,110 In recent years, JWH-018 has gained notoriety for its illicit use in herbal blends known as “Spice.”111,112 Diarylpyrazole derivative SR141716 was first described as a CB1 receptor antagonist, but further studies found it to be a CB1 inverse agonist. SR141716, also known as rimonabant, was briefly approved and marketed as Acomplia by Sanofi in Europe as an antiobesity treatment from July 2006 to October 2008, when significant side effects such as suicidal thoughts and depression forced its withdrawal from the market.113
Figure 9.

Representative nonclassical CB1 ligands. The first nonclassical CB scaffold discoveries were the n-alkyl indoles, exemplified by WIN 55,212-2 and JWH-018, both non-selective CB1 and CB2 agonists. SR141716, a member of the diarylpyrazole class, was the first CB1 inverse agonist discovered.
CB2 selective
CB2-selective agonists have been explored for a number of therapeutic indications, most commonly as analgesic and anti-inflammatory compounds (Fig. 10). Cannabinor, formerly known as PRS-211,375, was tested in humans for the treatment of several modes of pain; it ultimately failed due to a lack of efficacy in a Phase IIb study in the treatment of pain following third molar tooth extraction.114 Cannabinor may have also suffered from modulation of bladder activity, as animal studies have demonstrated a CB receptor-linked effect increasing urination frequency.115,116 GW-842,166X was also explored for the treatment of third molar tooth extraction and osteoarthritis, but failed in Phase II trials due to lack of efficacy.117,118 S-777,469 completed Phase II trials for atopic dermatitis in 2011, but no clinical data or future plans for clinical testing of this compound have been released, indicating that its development may have been halted.119–121
Figure 10.

CB2-selective agonists. PRS-211,375 and GW-842,166X were both investigated in clinical trials for the treatment of pain; both failed for lack of efficacy and presence of adverse side effects. S-777,469 completed Phase II trials, but no results have been reported and development has presumably halted.
After several failed clinical trials, the possibility that years of immunostaining results were inaccurate and halted CB2 research programs, the progress of CB2-targeted therapeutics was seemingly at an impasse. Research efforts into therapeutic potential of CB2 selective agonists have recently focused on improving selectivity by several orders of magnitude. Although many of the conventional CB2 agonists were as much as 500-fold selective, this may not be effective in vivo, causing CB1 activation at higher doses.122 While comparison of their respective binding affinities may characterize these compounds as highly selective, the overall expression levels of the CB receptors in humans are not equivalent. In many disease states, overall CB1 expression can be significantly higher than CB2, causing activation of both receptors.123 High dosages used in human studies likely precipitated effects of CB1 and CB2 activation. Many preclinical animal models may also not be effective in studying CB2-selective activation because of interspecies differences in CB2 receptor expression and signaling.124
Since late 2014, several landmark improvements have been reported in the pharmacological tools used to study the CB2 receptor. Transgenic mice expressing a CB2-GFP reporter have allowed for highly specific and accurate studies of CB2 expression, especially in light of the specificity problems with most anti-CB2 antibodies.125 Many advances have been made in the development of CB2-selective compounds. New scaffolds have been employed in the synthesis of compounds that display selectivity as high as 35,000-fold, significantly greater than previously reported compounds (Fig. 11).
Figure 11.

CB2-selective ligands reported in 2014 and 2015.
2015 has been a landmark year for drug discovery programs focusing on the development of selective CB ligands. Triazolopyrimidine 114 is a CB2 agonist developed by Roche, which displayed a 1,250-fold selectivity over CB1 and demonstrated a protective effect in inflammatory kidney damage models.126 Benzimidazole-containing scaffolds have also been reported, with RQ-00202730 showing over 4,000-fold selectivity for CB2 and a dose-dependent analgesic effect in rats.127 Both of these compounds displayed favorable pharmacokinetic parameters and were completely devoid of in vivo activity, traditionally associated with CB1 activity. Naphthyridin-2(1H)-one 115 possesses CB2 affinity in the picomolar range, with over 15,000-fold selectivity.128 This compound was one of many in a series employing this scaffold that allowed for the control of functional activity, highly dependent on substitution patterns. Pyridine analog RSR-056 and 4-oxo-quinoline analog RS-028 are >4,000-fold and >12,500-fold selective for CB2 over CB1, respectively, and have been used as 11C PET tracers in rat and mouse models.129,130 Proline analogs 116 and 117, developed by Boehringer Ingelheim Pharmaceuticals, are two of the most selective compounds reported to date with greater than 35,700-fold selectivity and 29,300-fold selectivity, respectively.131 Both of these demonstrated a dose-dependent reversal of hyperalgesia in a rat diabetic neuropathy model.
Outlook
Drug discovery programs focusing on CB receptor-targeted therapeutics will continue expanding with the identification and characterization of novel CB receptor-selective ligands. The development of receptor-selective modulators coupled to an understanding of their functional pharmacological activity (agonist; inverse agonist/antagonist) and mode of binding (orthosteric, allosteric) will prove critical for moving candidates to preclinical disease model studies. Classical cannabinoids, including both natural product and semisynthetic derivatives that have been described in this review, have ushered the development of nonclassical synthetic cannabinoids comprising novel scaffolds exhibiting receptor-selective profiles. Compounds selectively modulating the CB2 receptor may have utility in the treatment of inflammation, diabetes, cancer, pain, and other diseases. However, CB2-selective drug candidates thus far have failed in clinical trials due to a lack of efficacy and/or their propensity to mediate CB1 effects, even while displaying several hundred-fold selectivity profiles for CB2 over CB1.
Footnotes
ACADEMIC EDITOR: Yitzhak Tor, Editor in Chief
PEER REVIEW: Four peer reviewers contributed to the peer review report. Reviewers’ reports totaled 1,809 words, excluding any confidential comments to the academic editor.
FUNDING: This study was supported by an Institutional Development Award (IDeA) Grant Number P20GM104932 from the National Institute of General Medical Sciences (NIGMS) and the Research Core(s) of the COBRE, a component of the National Institutes of Health (NIH) under the grant number P20GM104932. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of NIGMS or NIH. The authors confirm that the funder had no influence over the study design, content of the article, or selection of this journal.
COMPETING INTERESTS: Authors disclose no potential conflicts of interest.
Paper subject to independent expert blind peer review. All editorial decisions made by independent academic editor. Upon submission manuscript was subject to anti-plagiarism scanning. Prior to publication all authors have given signed confirmation of agreement to article publication and compliance with all applicable ethical and legal requirements, including the accuracy of author and contributor information, disclosure of competing interests and funding sources, compliance with ethical requirements relating to human and animal study participants, and compliance with any copyright requirements of third parties. This journal is a member of the Committee on Publication Ethics (COPE). Provenance: the authors were invited to submit this paper.
Author Contributions
Wrote the first draft of the manuscript: EWB. Contributed to the writing of the manuscript: EWB, JMR. Agreed with manuscript results and conclusions: EWB, JMR. Jointly developed the structure and arguments for the paper: EWB, JMR. Made critical revisions and approved the final version: EWB, JMR. Both the authors reviewed and approved the final manuscript.
REFERENCES
- 1.O’Shaughnessy WB. On the preparations of the Indian Hemp, or Gunjah: Cannabis indica their effects on the animal system in health, and their utility in the treatment of tetanus and other convulsive diseases. Prov Med J Retrosp Med Sci. 1843;5(123):363–9. [PMC free article] [PubMed] [Google Scholar]
- 2.Blatt AH. A critical survey of the literature dealing with chemical constituents of Cannabis sativa. J Wash Acad Sci. 1938;28(11):465–77. [Google Scholar]
- 3.Adams R. Marihuana: Harvey Lecture, February 19, 1942. Bull N Y Acad Med. 1942;18(11):705–30. [PMC free article] [PubMed] [Google Scholar]
- 4.Wood TB, Spivey WTN, Easterfield TH. XL.-Charas. The resin of Indian hemp. J Chem Soc Trans. 1896;69(0):539–46. [Google Scholar]
- 5.Cahn RS. 326. Cannabis indica resin. Part IV. The synthesis of some 2: 2-dimethyldibenzopyrans, and confirmation of the structure of cannabinol. J Chem Soc. 1933;(0):1400–5. doi: 10.1039/JR9330001400. [DOI] [Google Scholar]
- 6.Jacob A, Todd AR. 119. Cannabis indica. Part II. Isolation of cannabidiol from Egyptian hashish. Observations on the structure of cannabinol. J Chem Soc. 1940;(0):649–53. doi: 10.1039/JR9400000649. [DOI] [Google Scholar]
- 7.Adams R, Baker BR, Wearn RB. Structure of cannabinol. III. Synthesis of cannabinol, 1-Hydroxy-3-n-amyl-6,6,9-trimethyl-6-dibenzopyran1. J Am Chem Soc. 1940;62(8):2204–7. [Google Scholar]
- 8.Marshall CR. The active principle of Cannabis indica. Br Med J. 1938;1(4039):1233–3. [Google Scholar]
- 9.Adams R, Hunt M, Clark JH. Structure of cannabidiol, a product isolated from the marihuana extract of Minnesota wild hemp. I. J Am Chem Soc. 1940;62(1):196–200. [Google Scholar]
- 10.Gaoni Y, Mechoulam R. Isolation, structure, and partial synthesis of an active constituent of hashish. J Am Chem Soc. 1964;86(8):1646–7. [Google Scholar]
- 11.Stout JM, Boubakir Z, Ambrose SJ, Purves RW, Page JE. The hexanoyl-CoA precursor for cannabinoid biosynthesis is formed by an acyl-activating enzyme in Cannabis sativa trichomes. Plant J. 2012;71(3):353–65. doi: 10.1111/j.1365-313X.2012.04949.x. [DOI] [PubMed] [Google Scholar]
- 12.Fellermeier M, Zenk MH. Prenylation of olivetolate by a hemp transferase yields cannabigerolic acid, the precursor of tetrahydrocannabinol. FEBS Lett. 1998;427(2):283–5. doi: 10.1016/s0014-5793(98)00450-5. [DOI] [PubMed] [Google Scholar]
- 13.Fellermeier M, Eisenreich W, Bacher A, Zenk MH. Biosynthesis of cannabinoids. Eur J Biochem. 2001;268(6):1596–604. doi: 10.1046/j.1432-1033.2001.02030.x. [DOI] [PubMed] [Google Scholar]
- 14.Administration FaD Electronic Orange Book. 2015. Available at: http://www.fda.gov/cder/ob/default.html.
- 15.Maa E, Figi P. The case for medical marijuana in epilepsy. Epilepsia. 2014;55(6):783–6. doi: 10.1111/epi.12610. [DOI] [PubMed] [Google Scholar]
- 16.Friedman D, Devinsky O. Cannabinoids in the treatment of epilepsy. N Engl J Med. 2015;373(11):1048–58. doi: 10.1056/NEJMra1407304. [DOI] [PubMed] [Google Scholar]
- 17.De Petrocellis L Di Marzo V. Non-CB1, Non-CB2 receptors for endocannabinoids, plant cannabinoids, and synthetic cannabimimetics: focus on G-protein-coupled receptors and transient receptor potential channels. J Neuroimmune Pharmacol. 2010;5(1):103–21. doi: 10.1007/s11481-009-9177-z. [DOI] [PubMed] [Google Scholar]
- 18.De Petrocellis L, Ligresti A, Moriello AS, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163(7):1479–94. doi: 10.1111/j.1476-5381.2010.01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Petrocellis L, Orlando P, Moriello AS, et al. Cannabinoid actions at TRPV channels: effects on TRPV3 and TRPV4 and their potential relevance to gastrointestinal inflammation. Acta Physiol. 2012;204(2):255–66. doi: 10.1111/j.1748-1716.2011.02338.x. [DOI] [PubMed] [Google Scholar]
- 20.Pertwee RG. Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr Med Chem. 2010;17(14):1360–81. doi: 10.2174/092986710790980050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150(5):613–23. doi: 10.1038/sj.bjp.0707133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fredriksson R, Lagerström MC, Lundin LG, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63(6):1256–72. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- 23.Lagerstrom MC, Schioth HB. Structural diversity of G protein-coupled receptors and significance for drug discovery. Nat Rev Drug Discov. 2008;7(4):339–57. doi: 10.1038/nrd2518. [DOI] [PubMed] [Google Scholar]
- 24.Bjarnadóttir TK, Gloriam DE, Hellstrand SH, Kristiansson H, Fredriksson R, Schiöth HB. Comprehensive repertoire and phylogenetic analysis of the G protein-coupled receptors in human and mouse. Genomics. 2006;88(3):263–73. doi: 10.1016/j.ygeno.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Tang XL, Wang Y, Li DL, Luo J, Liu MY. Orphan G protein-coupled receptors (GPCRs): biological functions and potential drug targets. Acta Pharmacol Sin. 2012;33(3):363–71. doi: 10.1038/aps.2011.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh E, Kumari P, Jaiman D, Shukla AK. Methodological advances: the unsung heroes of the GPCR structural revolution. Nat Rev Mol Cell Biol. 2015;16:69–81. doi: 10.1038/nrm3933. [DOI] [PubMed] [Google Scholar]
- 27.Stevens RC, Cherezov V, Katritch V, et al. The GPCR network: a large-scale collaboration to determine human GPCR structure and function. Nat Rev Drug Discov. 2013;12(1):25–34. doi: 10.1038/nrd3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Howlett AC, Fleming RM. Cannabinoid inhibition of adenylate cyclase. Pharmacology of the response in neuroblastoma cell membranes. Mol Pharmacol. 1984;26(3):532–8. [PubMed] [Google Scholar]
- 29.Howlett AC. Cannabinoid inhibition of adenylate cyclase. Biochemistry of the response in neuroblastoma cell membranes. Mol Pharmacol. 1985;27(4):429–36. [PubMed] [Google Scholar]
- 30.Howlett AC, Qualy JM, Khachatrian LL. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol Pharmacol. 1986;29(3):307–13. [PubMed] [Google Scholar]
- 31.Devane WA, Dysarz FA, III, Johnson MR, Melvin LS, Howlett AC. Determination and characterization of a cannabinoid receptor in rat brain. Mol Pharmacol. 1988;34(5):605–13. [PubMed] [Google Scholar]
- 32.Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC. Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci. 1991;11(2):563–83. doi: 10.1523/JNEUROSCI.11-02-00563.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Structure of a cannabinoid receptor and functional expression of the cloned cDNA. Nature. 1990;346(6284):561–4. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 34.Demuth DG, Molleman A. Cannabinoid signalling. Life Sci. 2006;78(6):549–63. doi: 10.1016/j.lfs.2005.05.055. [DOI] [PubMed] [Google Scholar]
- 35.Munro S, Thomas KL, Abu-Shaar M. Molecular characterization of a peripheral receptor for cannabinoids. Nature. 1993;365(6441):61–5. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 36.Howlett AC, Barth F, Bonner TI, et al. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol Rev. 2002;54(2):161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- 37.Bash R, Rubovitch V, Gafni M, Sarne Y. The stimulatory effect of cannabinoids on calcium uptake is mediated by Gs GTP-binding proteins and cAMP formation. Neurosignals. 2003;12(1):39–44. doi: 10.1159/000068915. [DOI] [PubMed] [Google Scholar]
- 38.Allyn CH, Lawrence CB, George DD. CB1 cannabinoid receptors and their associated proteins. Curr Med Chem. 2010;17(14):1382–93. doi: 10.2174/092986710790980023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Atwood BK, Mackie K. CB2: a cannabinoid receptor with an identity crisis. Br J Pharmacol. 2010;160(3):467–79. doi: 10.1111/j.1476-5381.2010.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galiègue S, Mary S, Marchand J, et al. Expression of central and peripheral cannabinoid receptors in human immune tissues and leukocyte subpopulations. Eur J Biochem. 1995;232(1):54–61. doi: 10.1111/j.1432-1033.1995.tb20780.x. [DOI] [PubMed] [Google Scholar]
- 41.Maresz K, Carrier EJ, Ponomarev ED, Hillard CJ, Dittel BN. Modulation of the cannabinoid CB2 receptor in microglial cells in response to inflammatory stimuli. J Neurochem. 2005;95(2):437–45. doi: 10.1111/j.1471-4159.2005.03380.x. [DOI] [PubMed] [Google Scholar]
- 42.Baek JH, Darlington CL, Smith PF, Ashton JC. Antibody testing for brain immunohistochemistry: brain immunolabeling for the cannabinoid CB2 receptor. J Neurosci Methods. 2013;216(2):87–95. doi: 10.1016/j.jneumeth.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 43.Marchalant Y, Brownjohn PW, Bonnet A, Kleffmann T, Ashton JC. Validating antibodies to the cannabinoid CB2 receptor: antibody sensitivity is not evidence of antibody specificity. J Histochem Cytochem. 2014;62(6):395–404. doi: 10.1369/0022155414530995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Marzo V, Stella N, Zimmer A. Endocannabinoid signalling and the deteriorating brain. Nat Rev Neurosci. 2015;16(1):30–42. doi: 10.1038/nrn3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Savonenko AV, Melnikova T, Wang Y, et al. Cannabinoid CB2 receptors in a mouse model of Aβ amyloidosis: immunohistochemical analysis and suitability as a PET biomarker of neuroinflammation. PLoS One. 2015;10(6):e0129618. doi: 10.1371/journal.pone.0129618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Derkinderen P, Valjent E, Toutant M, et al. Regulation of extracellular signal-regulated kinase by cannabinoids in hippocampus. J Neurosci. 2003;23(6):2371–82. doi: 10.1523/JNEUROSCI.23-06-02371.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turu G, Hunyady L. Signal transduction of the CB1 cannabinoid receptor. J Mol Endocrinol. 2010;44(2):75–85. doi: 10.1677/JME-08-0190. [DOI] [PubMed] [Google Scholar]
- 49.Derocq JM, Jbilo O, Bouaboula M, Ségui M, Clère C, Casellas P. Genomic and functional changes induced by the activation of the peripheral cannabinoid receptor CB2 in the promyelocytic cells HL-60: possible involvement of the CB2 receptor in cell differentiation. J Biol Chem. 2000;275(21):15621–8. doi: 10.1074/jbc.275.21.15621. [DOI] [PubMed] [Google Scholar]
- 50.Bouaboula M, Poinot-Chazel C, Bourrié B, et al. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem J. 1995;312(pt 2):637–41. doi: 10.1042/bj3120637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Devane WA, Hanus L, Breuer A, et al. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258(5090):1946–9. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 52.Lin S, Khanolkar AD, Fan P, et al. Novel analogues of arachidonylethanolamide (anandamide): affinities for the CB1 and CB2 cannabinoid receptors and metabolic stability. J Med Chem. 1998;41(27):5353–61. doi: 10.1021/jm970257g. [DOI] [PubMed] [Google Scholar]
- 53.Fride E, Mechoulam R. Pharmacological activity of the cannabinoid receptor agonist, anandamide, a brain constituent. Eur J Pharmacol. 1993;231(2):313–4. doi: 10.1016/0014-2999(93)90468-w. [DOI] [PubMed] [Google Scholar]
- 54.Jamshidi N, Taylor DA. Anandamide administration into the ventromedial hypothalamus stimulates appetite in rats. Br J Pharmacol. 2001;134(6):1151–4. doi: 10.1038/sj.bjp.0704379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Felder CC, Nielsen A, Briley EM, et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996;393(2–3):231–5. doi: 10.1016/0014-5793(96)00891-5. [DOI] [PubMed] [Google Scholar]
- 56.Mechoulam R, Ben-Shabat S, Hanus L, et al. Identification of an endogenous 2-monoglyceride, present in canine gut, that binds to cannabinoid receptors. Biochem Pharmacol. 1995;50(1):83–90. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 57.Hanuš L, Abu-Lafi S, Fride E, et al. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc Natl Acad Sci U S A. 2001;98(7):3662–5. doi: 10.1073/pnas.061029898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Porter AC, Sauer JM, Knierman MD, et al. Characterization of a novel endocannabinoid, virodhamine, with antagonist activity at the CB1 receptor. J Pharmacol Exp Ther. 2002;301(3):1020–4. doi: 10.1124/jpet.301.3.1020. [DOI] [PubMed] [Google Scholar]
- 59.Shoemaker JL, Joseph BK, Ruckle MB, Mayeux PR, Prather PL. The endocannabinoid noladin ether acts as a full agonist at human CB2 cannabinoid receptors. J Pharmacol Exp Ther. 2005;314(2):868–75. doi: 10.1124/jpet.105.085282. [DOI] [PubMed] [Google Scholar]
- 60.Sugiura T, Kodaka T, Kondo S, et al. 2-Arachidonoylglycerol, a putative endogenous cannabinoid receptor ligand, induces rapid, transient elevation of intracellular free ca2+in neuroblastoma × glioma hybrid NG108-15 cells. Biochem Biophys Res Commun. 1996;229(1):58–64. doi: 10.1006/bbrc.1996.1757. [DOI] [PubMed] [Google Scholar]
- 61.Sugiura T, Kondo S, Kishimoto S, et al. Evidence that 2-arachidonoylglycerol but not N-palmitoylethanolamine or anandamide is the physiological ligand for the cannabinoid CB2 receptor: comparison of the agonistic activities of various cannabinoid receptor ligands in HL-60 cells. J Biol Chem. 2000;275(1):605–12. doi: 10.1074/jbc.275.1.605. [DOI] [PubMed] [Google Scholar]
- 62.Panikashvili D, Shein Na A, Mechoulam R, et al. The endocannabinoid 2-AG protects the blood–brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol Dis. 2006;22(2):257–64. doi: 10.1016/j.nbd.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 63.Kirkham TC, Williams CM, Fezza F, Marzo VD. Endocannabinoid levels in rat limbic forebrain and hypothalamus in relation to fasting, feeding and satiation: stimulation of eating by 2-arachidonoyl glycerol. Br J Pharmacol. 2002;136(4):550–7. doi: 10.1038/sj.bjp.0704767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Xie XQ, Melvin LS, Makriyannis A. The conformational properties of the highly selective cannabinoid receptor ligand CP-55,940. J Biol Chem. 1996;271(18):10640–7. doi: 10.1074/jbc.271.18.10640. [DOI] [PubMed] [Google Scholar]
- 65.Fowler CJ. Transport of endocannabinoids across the plasma membrane and within the cell. FEBS J. 2013;280(9):1895–904. doi: 10.1111/febs.12212. [DOI] [PubMed] [Google Scholar]
- 66.Chicca A, Marazzi J, Nicolussi S, Gertsch J. Evidence for bidirectional endocannabinoid transport across cell membranes. J Biol Chem. 2012;287(41):34660–82. doi: 10.1074/jbc.M112.373241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fu J, Bottegoni G, Sasso O, et al. A catalytically silent FAAH-1 variant drives anandamide transport in neurons. Nat Neurosci. 2012;15(1):64–9. doi: 10.1038/nn.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gatta-Cherifi B, Cota D. Endocannabinoids and metabolic disorders. In: Pertwee GR, editor. Endocannabinoids. Cham: Springer International Publishing; 2015. pp. 367–91. [DOI] [PubMed] [Google Scholar]
- 69.Castillo Pablo E, Younts Thomas J, Chávez Andrés E, Hashimotodani Y. Endocannabinoid signaling and synaptic function. Neuron. 2012;76(1):70–81. doi: 10.1016/j.neuron.2012.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kreitzer AC, Regehr WG. Retrograde inhibition of presynaptic calcium influx by endogenous cannabinoids at excitatory synapses onto purkinje cells. Neuron. 2001;29(3):717–27. doi: 10.1016/s0896-6273(01)00246-x. [DOI] [PubMed] [Google Scholar]
- 71.Brown SP, Brenowitz SD, Regehr WG. Brief presynaptic bursts evoke synapse-specific retrograde inhibition mediated by endogenous cannabinoids. Nat Neurosci. 2003;6(10):1048–57. doi: 10.1038/nn1126. [DOI] [PubMed] [Google Scholar]
- 72.Wilson RI, Kunos G, Nicoll RA. Presynaptic specificity of endocannabinoid signaling in the hippocampus. Neuron. 2001;31(3):453–62. doi: 10.1016/s0896-6273(01)00372-5. [DOI] [PubMed] [Google Scholar]
- 73.Börner C, Smida M, Höllt V, Schraven B, Kraus J. Cannabinoid receptor type 1- and 2-mediated increase in cyclic AMP inhibits T cell receptor-triggered signaling. J Biol Chem. 2009;284(51):35450–60. doi: 10.1074/jbc.M109.006338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gong JP, Onaivi ES, Ishiguro H, et al. Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006;1071(1):10–23. doi: 10.1016/j.brainres.2005.11.035. [DOI] [PubMed] [Google Scholar]
- 75.Pacher P, Mechoulam R. Is lipid signaling through cannabinoid 2 receptors part of a protective system? Prog Lipid Res. 2011;50(2):193–211. doi: 10.1016/j.plipres.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Klein TW. Cannabinoid-based drugs as anti-inflammatory therapeutics. Nat Rev Immunol. 2005;5(5):400–11. doi: 10.1038/nri1602. [DOI] [PubMed] [Google Scholar]
- 77.Di Marzo V, Petrosino S. Endocannabinoids and the regulation of their levels in health and disease. Curr Opin Lipidol. 2007;18(2):129–40. doi: 10.1097/MOL.0b013e32803dbdec. [DOI] [PubMed] [Google Scholar]
- 78.Marzo VD, Piscitelli F. The endocannabinoid system and its modulation by phytocannabinoids. Neurotherapeutics. 2015;12(4):692–8. doi: 10.1007/s13311-015-0374-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Parsons LH, Hurd YL. Endocannabinoid signalling in reward and addiction. Nat Rev Neurosci. 2015;16(10):579–94. doi: 10.1038/nrn4004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Maccarrone M, Bab I, Bíró T, et al. Endocannabinoid signaling at the periphery: 50 years after THC. Trends Pharmacol Sci. 2015;36(5):277–96. doi: 10.1016/j.tips.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lutz B, Marsicano G, Maldonado R, Hillard CJ. The endocannabinoid system in guarding against fear, anxiety and stress. Nat Rev Neurosci. 2015;16(12):705–18. doi: 10.1038/nrn4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fonseca BM, Costa MA, Almada M, Correia-da-Silva G, Teixeira NA. Endogenous cannabinoids revisited: a biochemistry perspective. Prostaglandins Other Lipid Mediat. 2013;10(2–103):13–30. doi: 10.1016/j.prostaglandins.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 83.Andersson DA, Gentry C, Alenmyr L, et al. TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ9-tetrahydrocannabiorcol. Nat Commun. 2011;2:551. doi: 10.1038/ncomms1559. [DOI] [PubMed] [Google Scholar]
- 84.Thomas A, Stevenson LA, Wease KN, et al. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146(7):917–26. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Martin BR, Jefferson R, Winckler R, et al. Manipulation of the tetrahydrocannabinol side chain delineates agonists, partial agonists, and antagonists. J Pharmacol Exp Ther. 1999;290(3):1065–79. [PubMed] [Google Scholar]
- 86.Huffman JW, Lainton JAH, Kenneth Banner W, et al. Side chain methyl analogues of Δ8-THC. Tetrahedron. 1997;53(5):1557–76. [Google Scholar]
- 87.Huffman JW, Liddle J, Duncan SG, Jr, Yu S, Martin BR, Wiley JL. Synthesis and pharmacology of the isomeric methylheptyl-Δ8-tetrahydrocannabinols. Bioorg Med Chem. 1998;6(12):2383–96. doi: 10.1016/s0968-0896(98)80014-x. [DOI] [PubMed] [Google Scholar]
- 88.Huffman JW, Duncan SG, Jr, Wiley JL, Martin BR. Synthesis and pharmacology of the 1′,2′-dimethylheptyl-Δ8-THC isomers: exceptionally potent cannabinoids. Bioorg Med Chem Lett. 1997;7(21):2799–804. [Google Scholar]
- 89.Huffman JW, Miller JRA, Liddle J, et al. Structure–activity relationships for 1′,1′-dimethylalkyl-Δ8-tetrahydrocannabinols. Bioorg Med Chem. 2003;11(7):1397–410. doi: 10.1016/s0968-0896(02)00649-1. [DOI] [PubMed] [Google Scholar]
- 90.Huffman JW, Yu S. Synthesis of a tetracyclic, conformationally constrained analogue of Δ8-THC. Bioorg Med Chem. 1998;6(12):2281–8. doi: 10.1016/s0968-0896(98)80008-4. [DOI] [PubMed] [Google Scholar]
- 91.Khanolkar AD, Lu D, Fan P, Tian X, Makriyannis A. Novel conformationally restricted tetracyclic analogs of Δ8-tetrahydrocannabinol. Bioorg Med Chem Lett. 1999;9(15):2119–24. doi: 10.1016/s0960-894x(99)00355-8. [DOI] [PubMed] [Google Scholar]
- 92.Edery H, Grunfeld Y, Porath G, Ben-Zvi Z, Shani A, Mechoulam R. Structure-activity relationships in the tetrahydrocannabinol series. Modifications on the aromatic ring and it the side-chain. Arzneimittelforschung. 1972;22(11):1995–2003. [PubMed] [Google Scholar]
- 93.Lu D, Meng Z, Thakur GA, et al. Adamantyl cannabinoids: a novel class of cannabinergic ligands. J Med Chem. 2005;48(14):4576–85. doi: 10.1021/jm058175c. [DOI] [PubMed] [Google Scholar]
- 94.Papahatjis DP, Nikas SP, Andreou T, Makriyannis A. Novel 1′,1′-chain substituted Δ8-tetrahydrocannabinols. Bioorg Med Chem Lett. 2002;12(24):3583–6. doi: 10.1016/s0960-894x(02)00785-0. [DOI] [PubMed] [Google Scholar]
- 95.Durdagi S, Kapou A, Kourouli T, et al. The application of 3D-QSAR studies for novel cannabinoid ligands substituted at the C1′ position of the alkyl side chain on the structural requirements for binding to cannabinoid receptors CB1 and CB2. J Med Chem. 2007;50(12):2875–85. doi: 10.1021/jm0610705. [DOI] [PubMed] [Google Scholar]
- 96.Thakur GA, Duclos RI, Jr, Makriyannis A. Natural cannabinoids: templates for drug discovery. Life Sci. 2005;78(5):454–66. doi: 10.1016/j.lfs.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 97.Lu D, Guo J, Duclos RI, Bowman AL, Makriyannis A. Bornyl- and isobornyl-Δ8-tetrahydrocannabinols: a novel class of cannabinergic ligands. J Med Chem. 2008;51(20):6393–9. doi: 10.1021/jm8005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Seltzman HH. Structure and receptor activity for classical cannabinoids. Curr Med Chem. 1999;6(8):685–704. [PubMed] [Google Scholar]
- 99.Nikas SP, Sharma R, Paronis CA, et al. Probing the carboxyester side chain in controlled deactivation (−)-Δ8-tetrahydrocannabinols. J Med Chem. 2014;58(2):665–81. doi: 10.1021/jm501165d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sharma R, Nikas SP, Paronis CA, et al. Controlled-deactivation cannabinergic ligands. J Med Chem. 2013;56(24):10142–57. doi: 10.1021/jm4016075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Reggio PH, Seltzman HH, Compton DR, Prescott WR, Martin BR. Investigation of the role of the phenolic hydroxyl in cannabinoid activity. Mol Pharmacol. 1990;38(6):854–62. [PubMed] [Google Scholar]
- 102.Huffman JW, Yu S, Showalter V, et al. Synthesis and pharmacology of a very potent cannabinoid lacking a phenolic hydroxyl with high affinity for the CB2 receptor. J Med Chem. 1996;39(20):3875–7. doi: 10.1021/jm960394y. [DOI] [PubMed] [Google Scholar]
- 103.Tepper MA, Zurier RB, Burstein SH. Ultrapure ajulemic acid has improved CB2 selectivity with reduced CB1 activity. Bioorg Med Chem. 2014;22(13):3245–51. doi: 10.1016/j.bmc.2014.04.062. [DOI] [PubMed] [Google Scholar]
- 104.De Boeck K Amaral MD. Progress in therapies for cystic fibrosis. The Lancet Respiratory Medicine. 2016 doi: 10.1016/S2213-2600(16)00023-0. [DOI] [PubMed] [Google Scholar]
- 105.Pharmaceuticals C. 2016. Available at: http://www.corbuspharma.com/product-pipeline/resunab.
- 106.Song ZH, Bonner TI. A lysine residue of the cannabinoid receptor is critical for receptor recognition by several agonists but not WIN55212-2. Mol Pharmacol. 1996;49(5):891–6. [PubMed] [Google Scholar]
- 107.Bell MR, D’Ambra TE, Kumar V, et al. Antinociceptive (aminoalkyl)indoles. J Med Chem. 1991;34(3):1099–110. doi: 10.1021/jm00107a034. [DOI] [PubMed] [Google Scholar]
- 108.Compton DR, Gold LH, Ward SJ, Balster RL, Martin BR. Aminoalkylindole analogs: cannabimimetic activity of a class of compounds structurally distinct from delta 9-tetrahydrocannabinol. J Pharmacol Exp Ther. 1992;263(3):1118–26. [PubMed] [Google Scholar]
- 109.Huffman JW, Dai D, Martin BR, Compton DR. Design, synthesis and pharmacology of cannabimimetic indoles. Bioorg Med Chem Lett. 1994;4(4):563–6. [Google Scholar]
- 110.Pertwee RG, Griffin G, Lainton JAH, Huffman JW. Pharmacological characterization of three novel cannabinoid receptor agonists in the mouse isolated vas deferens. Eur J Pharmacol. 1995;284(3):241–7. doi: 10.1016/0014-2999(95)00318-f. [DOI] [PubMed] [Google Scholar]
- 111.Atwood BK, Huffman J, Straiker A, Mackie K. JWH018, a common constituent of ‘Spice’ herbal blends, is a potent and efficacious cannabinoid CB1 receptor agonist. Br J Pharmacol. 2010;160(3):585–93. doi: 10.1111/j.1476-5381.2009.00582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seely KA, Lapoint J, Moran JH, Fattore L. Spice drugs are more than harmless herbal blends: a review of the pharmacology and toxicology of synthetic cannabinoids. Prog Neuropsychopharmacol Biol Psychiatry. 2012;39(2):234–43. doi: 10.1016/j.pnpbp.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Di Marzo V, Despres JP. CB1 antagonists for obesity – what lessons have we learned from rimonabant? Nat Rev Endocrinol. 2009;5(11):633–8. doi: 10.1038/nrendo.2009.197. [DOI] [PubMed] [Google Scholar]
- 114.Nevalainen T. Recent development of CB2 selective and peripheral CB1/CB2 cannabinoid receptor ligands. Current Med Chem. 2014;21(2):187–203. doi: 10.2174/09298673113206660296. [DOI] [PubMed] [Google Scholar]
- 115.Gratzke C, Streng T, Stief CG, et al. Effects of cannabinor, a novel selective cannabinoid 2 receptor agonist, on bladder function in normal rats. Eur Urol. 2010;57(6):1093–100. doi: 10.1016/j.eururo.2010.02.027. [DOI] [PubMed] [Google Scholar]
- 116.Gratzke C, Streng T, Stief CG, et al. Cannabinor, a selective cannabinoid-2 receptor agonist, improves bladder emptying in rats with partial urethral obstruction. J Urol. 2010;185(2):731–6. doi: 10.1016/j.juro.2010.09.080. [DOI] [PubMed] [Google Scholar]
- 117.Ostenfeld T, Price J, Albanese M, et al. A randomized, controlled study to investigate the analgesic efficacy of single doses of the cannabinoid receptor-2 agonist GW842166, ibuprofen or placebo in patients with acute pain following third molar tooth extraction. Clin J Pain. 2011;27(8):668–76. doi: 10.1097/AJP.0b013e318219799a. [DOI] [PubMed] [Google Scholar]
- 118.Han S, Zhang FF, Qian HY, et al. Development of quinoline-2,4(1H,3H)-diones as potent and selective ligands of the cannabinoid type 2 receptor. J Med Chem. 2015;58(15):5751–69. doi: 10.1021/acs.jmedchem.5b00227. [DOI] [PubMed] [Google Scholar]
- 119.Odan M, Ishizuka N, Hiramatsu Y, et al. Discovery of S-777469: an orally available CB2 agonist as an antipruritic agent. Bioorg Med Chem Lett. 2012;22(8):2803–6. doi: 10.1016/j.bmcl.2012.02.072. [DOI] [PubMed] [Google Scholar]
- 120.Sekiguchi K, Fukumura K, Hasegawa H, Kanazu T. The metabolism and pharmacokinetics of [14C]-S-777469, a new cannabinoid receptor 2 selective agonist, in healthy human subjects. Xenobiotica. 2014;45(2):150–7. doi: 10.3109/00498254.2014.956158. [DOI] [PubMed] [Google Scholar]
- 121.Haruna T, Soga M, Morioka Y, et al. S-777469, a novel cannabinoid type 2 receptor agonist, suppresses itch-associated scratching behavior in rodents through inhibition of itch signal transmission. Pharmacology. 2015;95(1–2):95–103. doi: 10.1159/000371890. [DOI] [PubMed] [Google Scholar]
- 122.Rogers N. Cannabinoid receptor with an ‘identity crisis’ gets a second look. Nat Med. 2015;21(9):966–7. doi: 10.1038/nm0915-966. [DOI] [PubMed] [Google Scholar]
- 123.Pacher P, Kunos G. Modulating the endocannabinoid system in human health and disease – successes and failures. FEBS J. 2013;280(9):1918–43. doi: 10.1111/febs.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ndong C, O’Donnell D, Ahmad S, Groblewski T. Cloning and pharmacological characterization of the dog cannabinoid CB2 receptor. Eur J Pharmacol. 2011;669(1–3):24–31. doi: 10.1016/j.ejphar.2011.08.002. [DOI] [PubMed] [Google Scholar]
- 125.Schmöle AC, Lundt R, Gennequin B, et al. Expression analysis of CB2-GFP BAC transgenic mice. PLoS One. 2015;10(9):e0138986. doi: 10.1371/journal.pone.0138986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nettekoven M, Adam JM, Bendels S, et al. Novel triazolopyrimidine-derived cannabinoid receptor 2 agonists as potential treatment for inflammatory kidney diseases. ChemMedChem. 2015;11(2):179–89. doi: 10.1002/cmdc.201500218. [DOI] [PubMed] [Google Scholar]
- 127.Iwata Y, Ando K, Taniguchi K, Koba N, Sugiura A, Sudo M. Identification of a highly potent and selective CB2 agonist, RQ-00202730, for the treatment of irritable bowel syndrome. Bioorg Med Chem Lett. 2015;25(2):236–40. doi: 10.1016/j.bmcl.2014.11.062. [DOI] [PubMed] [Google Scholar]
- 128.Lucchesi V, Hurst DP, Shore DM, et al. CB2-selective cannabinoid receptor ligands: synthesis, pharmacological evaluation, and molecular modeling investigation of 1,8-naphthyridin-2(1H)-one-3-carboxamides. J Med Chem. 2014;57(21):8777–91. doi: 10.1021/jm500807e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Slavik R, Herde AM, Bieri D, et al. Synthesis, radiolabeling and evaluation of novel 4-oxo-quinoline derivatives as PET tracers for imaging cannabinoid type 2 receptor. Eur J Med Chem. 2015;92(0):554–64. doi: 10.1016/j.ejmech.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 130.Slavik R, Grether U, Müller Herde A, et al. Discovery of a high affinity and selective pyridine analog as a potential positron emission tomography imaging agent for cannabinoid type 2 receptor. J Med Chem. 2015;58(10):4266–77. doi: 10.1021/acs.jmedchem.5b00283. [DOI] [PubMed] [Google Scholar]
- 131.Riether D, Zindell R, Wu L, et al. Selective CB2 receptor agonists. Part 2: structure–activity relationship studies and optimization of proline-based compounds. Bioorg Med Chem Lett. 2015;25(3):581–6. doi: 10.1016/j.bmcl.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 132.Compton DR, Rice KC, De Costa BR, et al. Cannabinoid structure-activity relationships: correlation of receptor binding and in vivo activities. J Pharmacol Exp Ther. 1993;265(1):218–26. [PubMed] [Google Scholar]
- 133.Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278(3):989–99. [PubMed] [Google Scholar]
- 134.Huffman JW, Liddle J, Yu S, et al. 3-(1′,1′-Dimethylbutyl)-1-deoxy-Δ8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7(12):2905–14. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- 135.Papahatjis DP, Kourouli T, Abadji V, Goutopoulos A, Makriyannis A. Pharmacophoric requirements for cannabinoid side chains: multiple bond and C1′-substituted Δ8-tetrahydrocannabinols. J Med Chem. 1998;41(7):1195–200. doi: 10.1021/jm970277i. [DOI] [PubMed] [Google Scholar]
- 136.Coutts AA, Brewster N, Ingram T, Razdan RK, Pertwee RG. Comparison of novel cannabinoid partial agonists and SR141716 A in the guinea-pig small intestine. Br J Pharmacol. 2000;129(4):645–52. doi: 10.1038/sj.bjp.0703094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Charalambous A, Lin S, Marciniak G, et al. Pharmacological evaluation of halogenated Δ8-THC analogs. Pharmacol Biochem Behav. 1991;40(3):509–12. doi: 10.1016/0091-3057(91)90355-6. [DOI] [PubMed] [Google Scholar]
- 138.Papahatjis DP, Nahmias VR, Nikas SP, et al. C1′-cycloalkyl side chain pharmacophore in tetrahydrocannabinols. J Med Chem. 2007;50(17):4048–60. doi: 10.1021/jm070121a. [DOI] [PubMed] [Google Scholar]
- 139.Papahatjis DP, Nikas SP, Kourouli T, et al. Pharmacophoric requirements for the cannabinoid side chain. Probing the cannabinoid receptor subsite at C1‘. J Med Chem. 2003;46(15):3221–9. doi: 10.1021/jm020558c. [DOI] [PubMed] [Google Scholar]
- 140.Papahatjis DP, Nahmias VR, Andreou T, Fan P, Makriyannis A. Structural modifications of the cannabinoid side chain towards C3-aryl and 1′,1′-cycloalkyl-1′-cyano cannabinoids. Bioorg Med Chem Lett. 2006;16(6):1616–20. doi: 10.1016/j.bmcl.2005.12.026. [DOI] [PubMed] [Google Scholar]
- 141.Krishnamurthy M, Gurley S, Moore Ii BM. Exploring the substituent effects on a novel series of C1′-dimethyl-aryl Δ8-tetrahydrocannabinol analogs. Bioorg Med Chem. 2008;16(13):6489–500. doi: 10.1016/j.bmc.2008.05.034. [DOI] [PubMed] [Google Scholar]
- 142.Nikas S, Grzybowska J, Papahatjis D, et al. The role of halogen substitution in classical cannabinoids: a CB1 pharmacophore model. AAPS J. 2004;6(4):23–35. doi: 10.1208/aapsj060430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Singer M, Ryan WJ, Saha B, Martin BR, Razdan RK. Potent cyano and carboxamido side-chain analogues of 1‘,1‘-dimethyl-δ8-tetrahydrocannabinol. J Med Chem. 1998;41(22):4400–7. doi: 10.1021/jm9803875. [DOI] [PubMed] [Google Scholar]
- 144.Griffin G, Williams S, Aung MM, Razdan RK, Martin BR, Abood ME. Separation of cannabinoid receptor affinity and efficacy in delta-8-tetrahydrocannabinol side-chain analogues. Br J Pharmacol. 2001;132(2):525–35. doi: 10.1038/sj.bjp.0703827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Griffin G, Wray EJ, Martin BR, Abood ME. Cannabinoid agonists and antagonists discriminated by receptor binding in rat cerebellum. Br J Pharmacol. 1999;128(3):684–8. doi: 10.1038/sj.bjp.0702806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Martin BR, Wiley JL, Beletskaya I, et al. Pharmacological characterization of novel water-soluble cannabinoids. J Pharmacol Exp Ther. 2006;318(3):1230–9. doi: 10.1124/jpet.106.104109. [DOI] [PubMed] [Google Scholar]
- 147.Gareau Y, Dufresne C, Gallant M, et al. Structure activity relationships of tetrahydrocannabinol analogues on human cannabinoid receptors. Bioorg Med Chem Lett. 1996;6(2):189–94. [Google Scholar]
- 148.Thakur GA, Bajaj S, Paronis C, et al. Novel adamantyl cannabinoids as CB1 receptor probes. J Med Chem. 2013;56(10):3904–21. doi: 10.1021/jm4000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Rosati O, Messina F, Pelosi A, et al. One-pot heterogeneous synthesis of Δ3-tetrahydrocannabinol analogues and xanthenes showing differential binding to CB1 and CB2 receptors. Eur J Med Chem. 2014;85(0):77–86. doi: 10.1016/j.ejmech.2014.07.062. [DOI] [PubMed] [Google Scholar]
- 150.Marzo VD, Bifulco M, Petrocellis LD. The endocannabinoid system and its therapeutic exploitation. Nat Rev Drug Discov. 2004;3(9):771–84. doi: 10.1038/nrd1495. [DOI] [PubMed] [Google Scholar]