

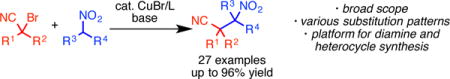
Abstract
Copper catalysis now enables the efficient C-alkylation of nitroalkanes with α-bromonitriles. Using a simple and inexpensive catalyst, this process provides access to β-cyanonitroalkanes. The method is highly tolerant of various functional groups and substitution patterns. These functionally dense products serve as orthogonally masked 1,3-diamines, which can be revealed selectively for access to differentially substituted diamines. These products can also be exploited for the formation of complex cyanoalkenes and 5-aminoisoxazoles.
Graphical abstract
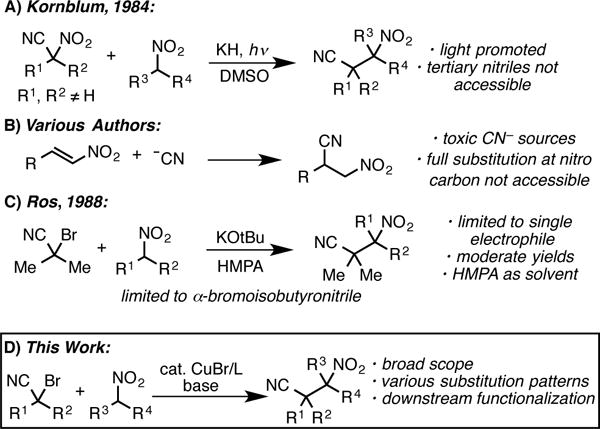
β-Cyanonitroalkanes are highly appealing and synthetically valuable building blocks, particularly because of their potential to serve as orthogonally masked diamines.1 Several prior entries into β-cyanonitroalkanes have been reported, but all are limited with respect to generality (Scheme 1A–C). In 1984, Kornblum reported that nitronates could be alkylated using α-nitronitriles under photolytic conditions.2 However, only fully substituted α-nitronitriles could be utilized as electrophiles, which significantly limits the accessible substitution patterns. Moreover, only simple alkyl substrates were examined, providing little evidence of functional group compatibility. β-cyanonitroalkanes have also been accessed via the conjugate addition of cyanide to simple nitroalkenes.3 However, these reactions require the use of toxic cyanide reagents (or equivalents) and β-cyanonitroalkanes bearing full substitution at the nitro center are not accessible via this method.4
Scheme 1.
Synthesis of β-Cyanonitroalkanes.
An alternative route for the preparation of β-cyanonitroalkanes would be alkylation of nitroalkanes with α-bromonitriles. This is attractive as α-bromonitriles are stable, readily available compounds that can be prepared on multi-gram scale from aldehydes or alkyl nitriles.5 Although these reagents have recently found wide use in catalytic reactions,6,7 their use to alkylate nitroalkanes has not been well developed. The non-catalyzed addition of nitronate anions to these reagents was studied by Ros in 1988.8 Although α-bromoisobutyronitrile could be used to alkylate simple, unfunctionalized nitronate anions in modest yields (36–70%), only a single example of another α-bromonitrile was reported. In this latter case, the product was formed in only 16% yield, along with significant amounts of elimination products (56%). No examples of α-bromonitriles bearing alpha hydrogens were reported, drawing into question the generality of the procedure.
In general, nitroalkanes are highly versatile synthetic intermediates, particularly as precursors to nitrogen containing molecules. The nitro group serves as a diverse functional handle that can undergo myriad reactions including oxidation, reduction to amines, arylation, allylation, additions to aldehydes, and conjugate additions.9 However, simple C-alkylation of nitroalkanes using alkyl electrophiles has been challenging, as non-catalyzed alkylation reactions are dominated by O-alkylation.10 This challenge might inform on the difficulties observed in the Ros study.
Recently, however, our group has developed inexpensive copper catalysts that allow the C-alkylation of nitroalkanes using simple alkyl halides.11 In previous studies, we have shown that benzylic, heterobenzylic, and α-bromocarbonyls can serve as the electrophiles in these reactions.11 Preliminary evidence suggests that these reactions proceed via stabilized radical intermediates.
As nitrile groups can also serve as potential radical stabilizing groups, we reasoned that copper catalysis of the alkylation of nitroalkanes with α-bromonitriles might be possible. If so, this system would provide straightforward access to β-cyanonitroalkanes. In this paper, we now report that a wide range of β-cyanonitroalkanes can be accessed using this strategy.
We set out to develop a general, robust, and functional group tolerant route to β-cyanonitroalkanes. Based upon our mechanistic hypothesis for copper-catalyzed nitroalkane alkylation, we began by examining the alkylation of 1-nitropropane with 1-bromocyclohexanecarbonitrile (1). Gratifyingly, using our previously identified catalytic system for the alkylation of nitronates with benzyl halides (20 mol % CuBr, 25 mol % ligand 2, 1.2 equiv NaOtBu),11a the desired β -cyanonitroalkane was formed smoothly in excellent yield (Scheme 2). The reaction proceeds at room temperature and uses an inexpensive, easily accessed catalyst. No product is formed in the absence of catalyst.
Scheme 2.
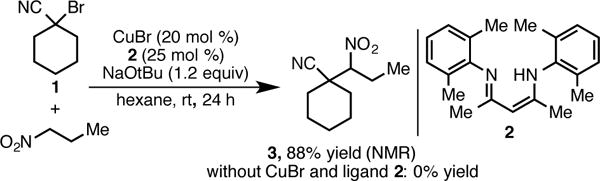
Identification of Catalyst Conditions.
Further investigations revealed that the scope of this transformation is broad (Schemes 3 and 4). A variety of primary nitroalkanes were subjected to the reaction using 1-bromocyclohexanecarbonitrile as an alkylating reagent. Nitroalkanes bearing alkenes, amides, esters, acetates, and aryl ethers were all alkylated in good to excellent yields (4–8). Nitromethane was alkylated smoothly, although an excess of the nucleophile was required (9). 2-Nitropropane participated in the alkylation reaction to provide product 10 bearing two contiguous quaternary centers in moderate yield. This reaction demonstrates the ability of the alkylation protocol to form highly congested carbon-carbon bonds and potentially access sterically congested diamines. It is notable that formation of product 8 (as well as products 16, 24 and 28 discussed below) utilizes a nitroalkane prepared from benzylation of simple nitroalkanes using our previously developed procedure.11a This serves to demonstrate how copper-catalyzed nitroalkane alkylation can be exploited to generate complexity from simple, inexpensive starting materials.
Scheme 3.
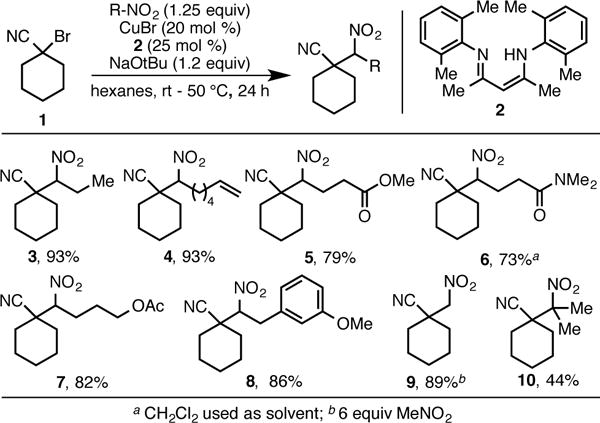
Scope of the Alkylation Reaction with Respect to Nitroalkane.
Scheme 4.
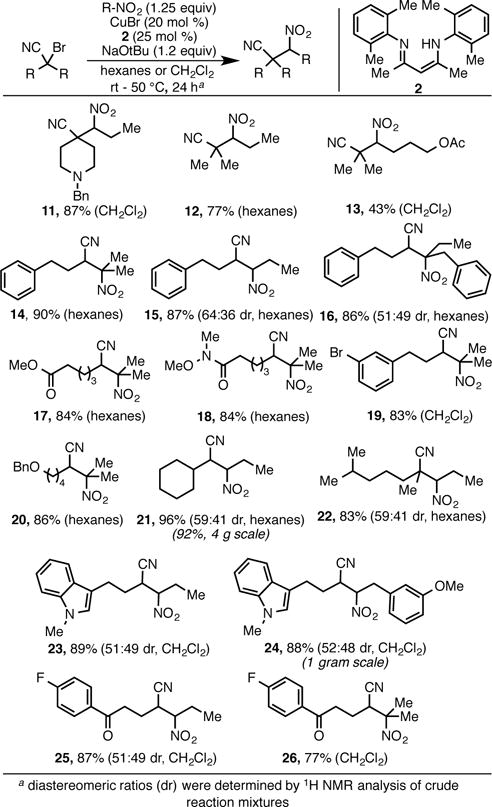
Scope of the Alkylation Reaction with Respect to α–Bromonitriles.
A number of functionalized α–bromonitriles were also examined in the alkylation reaction. While fully substituted α–bromonitriles often underwent smooth coupling at room temperature, secondary bromides required slightly elevated temperature (50 °C) to avoid significant production of protodebrominated starting materials. While hexanes is the solvent of choice for nonpolar substrates, dichloromethane is often superior to improve solubility in the case of more polar substrates. Basic amines (11), esters (17), Weinreb amides (18), ethers (20) and ketones (25, 26) proved compatible with the reaction. Product 11 is particularly notable due to its potential use as a latent triamine. Aryl bromides were also tolerated without incident (19), providing a convenient handle for further functionalization. Acyclic, fully substituted bromonitriles (22), as well as heterocycles (23, 24) were tolerated under the reaction conditions. The scalability of the reaction was also demonstrated, as product 21 could be synthesized on a 4 g scale without incident.
Although the diastereoselectivity of the reaction is moderate, it should be noted that in almost all cases diastereomers were easily separated by standard column chromatography. It appears that the observed diastereomeric ratio is kinetic in origin. For example, the alkylation reaction produces compound 21 as a 59:41 mixture of diastereomers. Analysis of aliquots (NMR) shows that this ratio of products is constant over the course of the reaction. However, subjecting isolated samples of each diastereomer to mild base (sodium propylnitronate) results in equilibration to a ~40:60 diastereomeric mixture with the opposite sense of diastereomeric enrichment.12
Bromoacetonitrile proved to be a particularly recalcitrant electrophile in the alkylation reaction, providing only trace amounts of product under our optimized conditions. We assume this is due to the difficulty in formation of the putative primary radical intermediate. However, a survey of reaction conditions revealed that a weaker base, sodium trimethylsilanolate, in conjunction with the use of the nitroalkane as the limiting reagent provided cyanomethylated products (27, 28) in moderate yield (Scheme 5). Notably, these products, as well as many outlined in Schemes 3 and 4 are not accessible via the aforementioned photolytic or conjugate addition methods, highlighting the complementarity of this work to existing methods.
Scheme 5.
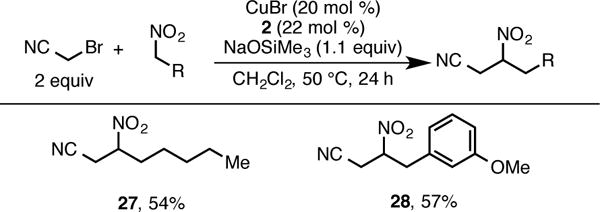
Alkylation of Nitroalkanes with Bromoacetonitrile.
With convenient access to β-cyanonitroalkanes in hand, we sought to investigate the reactivity of these functionally rich compounds. Exposure of the β-cyanonitroalkanes to DBU resulted in rapid formation of the corresponding cyanoalkenes in excellent yield (Scheme 6). In the case of compound 31, high selectivity for the Z product was observed (as determined by nOe analysis).
Scheme 6.
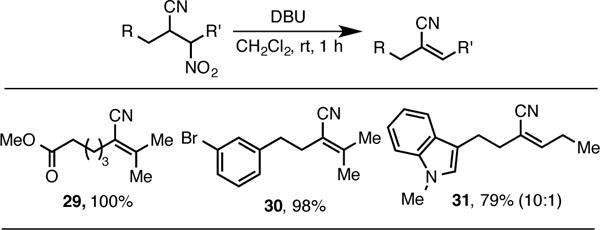
Synthesis of Cyanoalkenes.
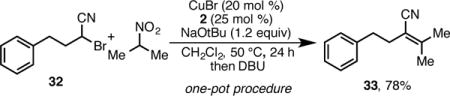
Isolation of the β-cyanonitroalkane was not required to achieve this elimination reaction. After simply filtering the salts formed in the alkylation reaction, DBU could be added to the crude reaction mixture, providing convenient access to the cyanoalkene (eq 1).
 |
(1) |
β-Cyanonitroalkanes can also be utilized in the synthesis of nitrogen-rich heterocycles. 5-Aminoisoxazoles have been accessed via the spontaneous cyclization of α-cyanooximes, but access to the oxime intermediates often requires harsh or highly toxic conditions.13,14 We envisioned that controlled reduction of the nitro group of our products to the oxime would allow interception of this pathway in a convergent and mild fashion. Carreira has reported a streamlined process for the conversion of nitroalkanes to oximes using benzyl bromide in the presence of tetrabutylammonium iodide.10,15 After optimization, we found that treatment of the β-cyanonitroalkanes under similar conditions resulted in formation of α-cyanooximes, which undergo spontaneous cyclization to form 5-aminoisoxazoles.
Using this method, several such heterocycles were prepared in good yield. β-Cyanonitroalkanes bearing ketones (36), aryl ethers (38), arenes (35) and heteroarenes (37, 38) all cyclized smoothly, allowing a highly convergent entry into these interesting products. Importantly, β-cyanonitroalkanes may be used as a mixture of diastereomers, rendering the stereoselectivity of the nitroalkane alkylation reaction inconsequential for the preparation of 5-aminoisoxazoles.
β-Cyanonitroalkanes are exceptionally versatile in their ability to be used as latent diamines. The presence of the orthogonal nitrile and nitro groups allows simple chemoselective functional group interconversions, allowing easy access to differentially functionalized 1,3-diamines. To illustrate this, we targeted the synthesis of bispiperidine 42 (Scheme 8). This motif serves the core for a series of amides 43 that have recently been shown to be nanomolar inhibitors of glycine transporter 1 (GlyT1).16 This enzyme regulates glycine concentrations in the brain and has been implicated in the treatment of schizophrenia and other cognitive disorders.17
Scheme 8.
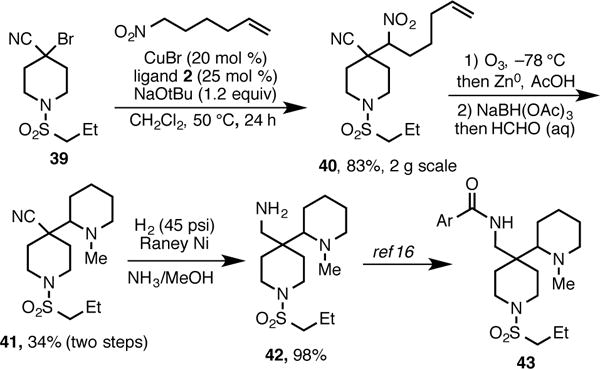
Synthesis of Bispiperidine Core of GlyT1 Inhibitor 43.
Nitroalkane 40 was accessed smoothly on a 2 g scale via our alkylation strategy. Ozonolysis using a reductive workup provided an intermediate aminoaldehyde. Sequential reductive amination and reduction of the nitrile provided amine 42,18 which is the reported intermediate for the synthesis of amides 43.16
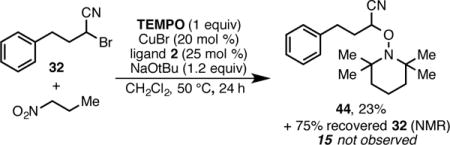
To investigate the mechanism of the alkylation reaction, several experiments were performed. First, when the reaction was run in the presence of one equivalent of TEMPO, a known radical scavenger, no alkylation product (15) was formed (eq 2). Instead, only remaining starting material and 44 (the adduct of TEMPO with the starting material) were observed. This adduct likely results from the radical recombination of TEMPO with a transient α-cyano radical.19 Additionally, no alkylation was observed when the reaction
 |
(2) |
was run in the presence of galvinoxyl free radical, also a known radical scavenger. These results are consistent with a mechanism involving transient radicals. Second, the reaction of substrate 45 results exclusively in ring-opened product 46 (Scheme 9). Fragmentation of the cyclopropane ring also suggests a radical intermediate.20,21 These results are consistent with the thermal redox catalysis pathway that we have previously proposed.11a,22
Scheme 9.
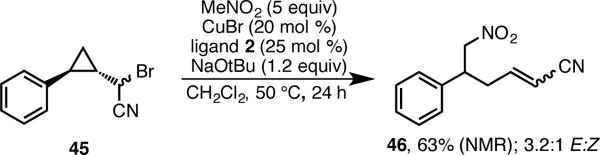
Fragmentation/Alkylation of the Cyclopropylcarbinyl Radical Resulting from Substrate 45.
In conclusion, we have demonstrated a facile and convergent synthesis of β-cyanonitroalkanes from nitroalkanes and α-bromonitriles. The synthesis is mild and tolerant of a variety of functional groups and substitution patterns, and may be performed on the benchtop utilizing standard anaerobic technique. The densely functionalized products obtained therein may be utilized in the synthesis of various synthetically valuable targets including cyanoalkenes, 1,3-diamines and 5- aminoisoxazoles.
Supplementary Material
Scheme 7.
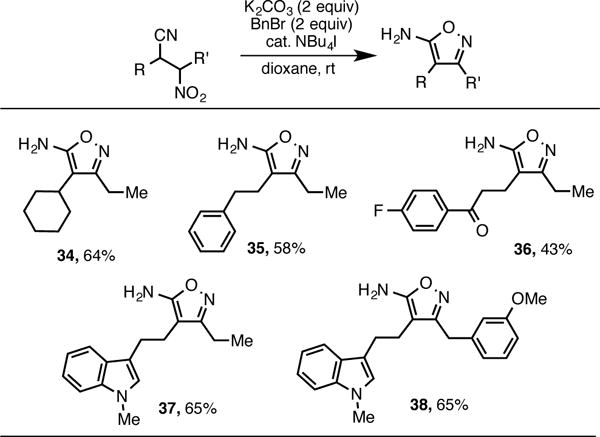
Synthesis of 5-Aminoisoxazoles.
Acknowledgments
The University of Delaware, the University of Delaware Research Foundation, the Research Corp. Cottrell Scholars Program, and the NIH NIGMS (R01 GM102358) are gratefully acknowledged for support. KWS thanks the University of Delaware for a graduate fellowship. Benjamin L. Prather (University of Delaware) is acknowledged for help with starting material synthesis. Data was acquired at UD on instruments obtained with the assistance of NSF and NIS funding (NSF CHE0421224, CHE1229234, and CHE0840401; NIH P20 GM104316, P30 GM110758, S10 RR02692, and S10 OD016267).
Footnotes
Experimental procedures and spectral data (PDF).
Notes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
References
- 1.Kizirian J-C. Chemical Reviews. 2008;108:140–205. doi: 10.1021/cr040107v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kornblum N, Boyd SD, Stuchal FW. J Am Chem Soc. 1970;92:5783–5784. [Google Scholar]; (b) Kornblum N, Singh HK, Boyd SD. J Org Chem. 1984;49:358–362. [Google Scholar]
- 3.Anderson JC, Blake AJ, Mills M, Ratcliffe PD. Org Lett. 2008;10:4141–4143. doi: 10.1021/ol801691c. [DOI] [PubMed] [Google Scholar]
- 4.(a) Bernardi L, Fini F, Fochi M, Ricci A. Synlett. 2008;2008:1857–1861. [Google Scholar]; (b) Bernal P, Fernández R, Lassaletta JM. Chem Eur J. 2010;16:7714–7718. doi: 10.1002/chem.201001107. [DOI] [PubMed] [Google Scholar]; (c) North M, Watson JM. ChemCatChem. 2013;5:2405–2409. doi: 10.1002/cctc.201300215. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Lin L, Yin W, Fu X, Zhang J, Ma X, Wang R. Org Biomol Chem. 2012;10:83–89. doi: 10.1039/c1ob05899a. [DOI] [PubMed] [Google Scholar]; (e) Jakhar A, Sadhukhan A, Khan N-uH, Saravanan S, Kureshy RI, Abdi SHR, Bajaj HC. Chem Cat Chem. 2014;6:2656–2661. [Google Scholar]
- 5.(a) Stevens CL. J Am Chem Soc. 1948;70:165–167. [Google Scholar]; (b) Stevens CL, Coffield TH. J Am Chem Soc. 1951;73:103–105. [Google Scholar]; (c) Fleming FF, Zhang Z, Liu W, Knochel P. J Org Chem. 2005;70:2200–2205. doi: 10.1021/jo047877r. [DOI] [PubMed] [Google Scholar]; (d) Watahiki T, Ohba S, Oriyama T. Org Lett. 2003;5:2679–2681. doi: 10.1021/ol0348295. [DOI] [PubMed] [Google Scholar]
- 6.(a) Liu Q, Yi H, Liu J, Yang Y, Zhang X, Zeng Z, Lei A. Chem Eur J. 2013;19:5120–5126. doi: 10.1002/chem.201203694. [DOI] [PubMed] [Google Scholar]; (b) Nakatani A, Hirano K, Satoh T, Miura M. Chem Eur J. 2013;19:7691–7695. doi: 10.1002/chem.201301350. [DOI] [PubMed] [Google Scholar]; (c) Tang S, Liu C, Lei A. Chem Commun. 2013;49:2442–2444. doi: 10.1039/c3cc00029j. [DOI] [PubMed] [Google Scholar]; (d) Zhang F, Du P, Chen J, Wang H, Luo Q, Wan X. Org Lett. 2014;16:1932–1935. doi: 10.1021/ol5004687. [DOI] [PubMed] [Google Scholar]; (e) Zhang X, Yi H, Liao Z, Zhang G, Fan C, Qin C, Liu J, Lei A. Org Biomol Chem. 2014;12:6790–6793. doi: 10.1039/c4ob00813h. [DOI] [PubMed] [Google Scholar]; (f) Liao Z, Yi H, Li Z, Fan C, Zhang X, Liu J, Deng Z, Lei A. Chem Asian J. 2015;10:96–99. doi: 10.1002/asia.201403097. [DOI] [PubMed] [Google Scholar]
- 7.(a) Choi J, Fu GC. J Am Chem Soc. 2012;134:9102–9105. doi: 10.1021/ja303442q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kadunce NT, Reisman SE. J Am Chem Soc. 2015;137:10480–10483. doi: 10.1021/jacs.5b06466. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Welin ER, Warkentin AA, Conrad JC, MacMillan DWC. Angew Chem Int Ed. 2015;54:9668–9672. doi: 10.1002/anie.201503789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ros F, De la Rosa J. J Org Chem. 1988;53:2868–2870. [Google Scholar]
- 9.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. [Google Scholar]
- 10.Hass HB, Bender ML. J Am Chem Soc. 1949;71:1767–1769. [Google Scholar]
- 11.(a) Gildner PG, Gietter AAS, Cui D, Watson DA. J Am Chem Soc. 2012;134:9942–9945. doi: 10.1021/ja304561c. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gietter AAS, Gildner PG, Cinderella AP, Watson DA. Org Lett. 2014;16:3166–3169. doi: 10.1021/ol5014153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.See the Supporting Information for further details of this analysis.
- 13.Bourbeau MP, Rider JT. Org Lett. 2006;8:3679–3680. doi: 10.1021/ol061260+. [DOI] [PubMed] [Google Scholar]
- 14.(a) Kong YC, Kim K, Park YJ. Heterocycles. 2001;55:75–89. [Google Scholar]; (b) Nishiwaki T, Saito T. J Chem Soc C. 1971:3021–3026. [Google Scholar]; (c) Alberola A, Gonzalez AM, Laguna MA, Pulido FJ. J Org Chem. 1984;49:3423–3424. [Google Scholar]; (d) Beccalli EM, Manfredi A, Marchesini A. J Org Chem. 1985;50:2372–2375. [Google Scholar]
- 15.Czekelius C, Carreira EM. Angew Chem Int Ed. 2005;44:612–615. doi: 10.1002/anie.200461879. [DOI] [PubMed] [Google Scholar]
- 16.Zhao Z, Leister WH, O’Brien JA, Lemaire W, Williams DL, Jr, Jacobson MA, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Hartman GD, Lindsley CW, Wolkenberg SE. Bioorg Med Chem Lett. 2009;19:1488–1491. doi: 10.1016/j.bmcl.2008.12.115. [DOI] [PubMed] [Google Scholar]
- 17.(a) Harvey RJ, Yee BK. Nat Rev Drug Discov. 2013;12:866–885. doi: 10.1038/nrd3893. [DOI] [PubMed] [Google Scholar]; (b) Möhler H, Boison D, Singer P, Feldon J, Pauly-Evers M, Yee BK. Biochem Pharmacol. 2011;81:1065–1077. doi: 10.1016/j.bcp.2011.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 19.(a) Beckwith ALJ, Bowry VW, Ingold KU. J Am Chem Soc. 1992;114:4983–4992. [Google Scholar]; (b) Bowry VW, Ingold KU. J Am Chem Soc. 1992;114:4992–4996. [Google Scholar]
- 20.(a) Griller D, Ingold KU. Acc Chem Res. 1980;13:317–323. [Google Scholar]; (b) Newcomb M. Tetrahedron. 1993;49:1151–1176. [Google Scholar]
- 21.(a) Newcomb M, Johnson CC, Manek MB, Varick TR. J Am Chem Soc. 1992;114:10915–10921. [Google Scholar]; (b) Martin-Esker AA, Johnson CC, Horner JH, Newcomb M. J Am Chem Soc. 1994;116:9174–9181. [Google Scholar]
- 22.Ros and co-workers also suggest a radical mechanism in thier study. See reference 8.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.