

Expression of a truncated allele of the Apc tumor suppressor causes intestinal tumors with a low rate of chromosomal instability (CIN). Increasing the rate of CIN suppresses tumor growth without inhibiting tumor initiation in both the small intestine and colon, suggesting that increasing CIN is a useful chemotherapeutic strategy.
Abstract
Aneuploidy, an abnormal chromosome number that deviates from a multiple of the haploid, has been recognized as a common feature of cancers for >100 yr. Previously, we showed that the rate of chromosome missegregation/chromosomal instability (CIN) determines the effect of aneuploidy on tumors; whereas low rates of CIN are weakly tumor promoting, higher rates of CIN cause cell death and tumor suppression. However, whether high CIN inhibits tumor initiation or suppresses the growth and progression of already initiated tumors remained unclear. We tested this using the ApcMin/+ mouse intestinal tumor model, in which effects on tumor initiation versus progression can be discriminated. ApcMin/+ cells exhibit low CIN, and we generated high CIN by reducing expression of the kinesin-like mitotic motor protein CENP-E. CENP-E+/−;ApcMin/+ doubly heterozygous cells had higher rates of chromosome missegregation than singly heterozygous cells, resulting in increased cell death and a substantial reduction in tumor progression compared with ApcMin/+ animals. Intestinal organoid studies confirmed that high CIN does not inhibit tumor cell initiation but does inhibit subsequent cell growth. These findings support the conclusion that increasing the rate of chromosome missegregation could serve as a successful chemotherapeutic strategy.
INTRODUCTION
Mitotic errors predicted to produce aneuploidy have been recognized as a characteristic of human cancer cells since the late 1800s (von Hansemann, 1890). Because of this correlation, aneuploidy was proposed to cause tumors in the early 1900s (Boveri, 1902, 1914). Aneuploidy is often accompanied by chromosomal instability (CIN), in which chromosomes are perpetually gained and lost during multiple divisions. Both aneuploidy and CIN serve as markers of poor prognosis in multiple tumor types (McGranahan et al., 2012; Zasadil et al., 2013).
Despite their high prevalence in human cancers, aneuploidy and CIN do not uniformly increase tumor incidence in mice. Numerous mouse models have been generated that develop aneuploidy and CIN as a consequence of chromosome missegregation (reviewed in Ricke et al., 2008; Holland and Cleveland, 2009; Zasadil et al., 2013). A portion of mutations that induce aneuploidy and CIN do result in an elevated level of spontaneous and/or carcinogen-induced tumors, as initially predicted (Iwanaga et al., 2007; Sotillo et al., 2007, 2010; Li et al., 2009; Ricke et al., 2011; Schvartzman et al., 2011). However, other models of aneuploidy and CIN exhibit tumor outcomes that do not differ from those in control animals (Cowley et al., 2005; Jeganathan et al., 2007; Baker et al., 2009; Li et al., 2010; Malureanu et al., 2010; Ricke et al., 2012). Most intriguingly, aneuploidy and CIN can promote tumors in certain contexts but inhibit them in others (Jallepalli et al., 2001; Wang et al., 2001; Yu et al., 2003; Chesnokova et al., 2005; Rao et al., 2005; Donangelo et al., 2006; Weaver et al., 2007; Fong et al., 2012; Silk et al., 2013). Thus the effects of aneuploidy and CIN on tumors depend on their method of induction, as well as on the cellular context.
One of the causes of aneuploidy and CIN that can both promote and suppress tumors is heterozygous loss of the mitotic kinesin-like motor protein centromere-associated protein E (CENP-E; Weaver et al., 2007; Silk et al., 2013). CENP-E is a large (∼312 kDa) protein that accumulates specifically during mitosis (Brown et al., 1994) and is required for accurate chromosome segregation. CENP-E localizes to kinetochores—the sites of chromosome attachment to spindle microtubules—through its C-terminal domain (Yen et al., 1991; Yao et al., 1997) and uses its 230-nm central stalk to allow its N-terminal motor to locate and bind microtubules (Kim et al., 2008). CENP-E functions in the mitotic checkpoint (also known as the spindle assembly checkpoint)—the major cell cycle checkpoint acting during mitosis to prevent chromosome missegregation—by facilitating recruitment of the mitotic checkpoint components BubR1, Mad1, and Mad2 to unattached kinetochores (Abrieu et al., 2000; Weaver et al., 2003). Reduction or inhibition of CENP-E results in the chronic misalignment of one or a few chromosomes at or near spindle poles (Schaar et al., 1997; McEwen et al., 2001; Putkey et al., 2002; Wood et al., 2010). Because the mitotic checkpoint is weakened, cells with reduced levels of CENP-E aberrantly enter anaphase in the presence of the misaligned, polar chromosomes in approximately one-fourth of divisions. As a result of these abnormal divisions, both sister chromatids of the polar chromosome are inherited by the same daughter cell, producing aneuploidy and a low rate of CIN. Cells with reduced levels of CENP-E have not shown discernible defects outside of mitosis (Weaver et al., 2003, 2007).
We recently demonstrated that it is the rate of chromosome missegregation, rather than the overall level of aneuploidy, that predicts the effect on tumors. Heterozygous loss of CENP-E did not cause tumor suppression in two contexts that did not exhibit prior CIN. In four contexts with existing low rates of CIN that developed tumors, reduction of CENP-E caused higher rates of CIN and cell death and reduced tumor incidence (Silk et al., 2013). However, it was unclear from these experiments whether high CIN inhibited tumor initiation or suppressed the growth and progression of already initiated tumors. This is a critical distinction because, in order to be a successful treatment strategy, increasing the rate of CIN must be sufficient to inhibit tumors that have already been initiated.
To test this question, we used tumor models expressing a mutant form of the adenomatous polyposis coli (APC) tumor suppressor. Mutations in APC, which predominantly encode truncated protein products, occur in ∼60% of spontaneous human colon cancers and cause the hereditary cancer syndrome familial adenomatous polyposis when present in the germline (Powell et al., 1992; Huang et al., 1996; Kerr et al., 2013). Expression of truncation mutants of APC in mouse and human cells results in missegregation of small numbers of chromosomes, producing a large population of aneuploid cells (Fodde et al., 2001; Kaplan et al., 2001; Tighe et al., 2004). Mice that express the multiple intestinal neoplasia (Min) allele of Apc, which produces a truncated product of 850 amino acids, develop tens to hundreds of early-onset adenomatous intestinal polyps (Moser et al., 1990; Su et al., 1992). Inhibition of tumor initiation versus tumor progression can be distinguished using intestinal tumor size and multiplicity. Here we show that combining low CIN from expression of the Min allele of Apc with low CIN due to reduction of CENP-E results in high CIN, elevated levels of cell death, and suppression of tumor growth, but not initiation, in both the small intestine and colon.
RESULTS AND DISCUSSION
CENP-E+/−;ApcMin/+ cells and tissues exhibit high CIN
Because expression of APC truncation mutants and reduction of CENP-E both cause low CIN, we predicted that combination of both insults would produce high CIN in doubly heterozygous cells. To test this, we crossed CENP-E+/− mice with ApcMin/+ animals to produce wild-type, ApcMin/+, CENP-E+/−, and CENP-E+/−;ApcMin/+ littermates. CENP-E+/−;ApcMin/+ animals were born at expected frequencies and were overtly normal. To measure CIN, we scored abnormal mitotic figures consistent with chromosome missegregation in primary murine embryonic fibroblasts (MEFs) generated from embryonic day 14.5 (E14.5) embryos. These included polar chromosomes, which become persistently associated with the spindle pole and are characteristic of CENP-E impairment (Figure 1A), as well as chromosomes that lag behind the separating masses of chromosomes during anaphase and telophase (Figure 1B). Polar chromosomes are missegregated in ∼25% of divisions in primary MEFs with reduced levels of CENP-E (Weaver et al., 2003), whereas lagging chromosomes are indicative of chromosome missegregation (Thompson and Compton, 2011). Consistent with earlier results (McEwen et al., 2001; Putkey et al., 2002; Weaver et al., 2003, 2007), reduction of CENP-E produced elevated levels of polar chromosomes (Figure 1, A and C). Truncation mutants of APC were reported to increase the percentage of lagging chromosomes (Fodde et al., 2001; Kaplan et al., 2001; Tighe et al., 2004). Accordingly, MEFs expressing the Min allele of Apc displayed lagging chromosomes at significantly higher frequency than wild-type or CENP-E+/− fibroblasts (Figure 1, B and C). Double-mutant MEFs had levels of polar chromosomes similar to those in CENP-E+/− cells and rates of lagging chromosomes akin to those in ApcMin/+ MEFs. Taken together, the double-mutant cells had a higher proportion of abnormal mitotic figures than either single mutant (Figure 1C). Thus, combining two insults, each of which produces low CIN, results in high CIN in this in vitro context.
FIGURE 1:

Reduction of CENP-E increases the rate of chromosome missegregation in ApcMin/+ cells and animals. CENP-E+/−;ApcMin/+ cells show higher rates of abnormal mitotic figures consistent with chromosome missegregation than either CENP-E+/− or ApcMin/+ singly heterozygous cells in vitro in primary MEFs (A–C) and in vivo in the mouse small intestine (D–F). (A) Polar chromosome (arrow) in primary MEF. (B) Lagging chromosome (arrow) in primary MEF. (C) Quantification of indicated mitotic defects; n > 100 metaphase and >150 total anaphase and telophase cells from each of three independent replicates. (D) Image of polar chromosomes (arrow) in murine small intestine. Right, enlargement of DNA in inset. (E) Lagging chromosome (arrow) in small intestine. Right, enlarged view of DNA in inset. (F) Quantification of mitotic defects in small intestine; n > 30 metaphases or anaphases and telophases from three mice of each genotype (four mice in ApcMin/+). *p < 0.05 vs. wild type, #p < 0.05 vs. CENP-E+/−;ApcMin/+.
To test whether the combination of heterozygous loss of CENP-E with mutation in Apc resulted in high CIN in vivo as well, we measured the frequency of abnormal mitotic figures in the crypts of 5-μm sections of murine small intestinal epithelium (Figure 1, D–F). CENP-E+/− and CENP-E+/−;ApcMin/+ doubly heterozygous intestines had increased levels of polar chromosomes (Figure 1, D and F). ApcMin/+ and CENP-E+/−;ApcMin/+ intestines showed an increased frequency of lagging chromosomes (Figure 1, E and F). Overall, double-mutant intestines had increased levels of both polar and lagging chromosomes, resulting in an elevated frequency of abnormal mitotic figures consistent with chromosome missegregation compared with Apc single mutants (Figure 1F). These data demonstrate that reduction of CENP-E in cells expressing an APC mutant increases the rate of mitotic defects and CIN in vitro and in vivo.
Increased cell death in CENP-E+/−;ApcMin/+ doubly heterozygous cells and animals
High rates of chromosome missegregation have been shown to result in cell death (Kops et al., 2004; Michel et al., 2004; Janssen et al., 2009; Silk et al., 2013). To determine whether high CIN caused by reduction of CENP-E in the presence of mutant APC produced inviable cells, we quantitated levels of apoptosis in primary MEFs, as well as in sections of normal and tumorous small intestine. High CIN caused by reduction of CENP-E in ApcMin/+ cells produced a marked increase in cleaved caspase-3 reactivity in primary MEFs (Figure 2, A and B). Consistent with this, CENP-E+/−;ApcMin/+doubly heterozygous MEFs exhibited a growth disadvantage compared with wild-type, CENP-E+/−, and ApcMin/+ singly heterozygous cells (Supplemental Figure S1A).
FIGURE 2:

High CIN increases cell death. (A) Image of cleaved caspase-3, a marker of apoptosis, in asynchronously cycling MEFS. Scale bar, 10 µm. (B) Quantification of cleaved caspase-3 showing that CENP-E+/−;ApcMin/+ cells have higher rates of apoptosis than wild-type, CENP-E+/−, and ApcMin/+ singly heterozygous cells with lower rates of CIN. n > 500 cells per genotype from each of three separate experiments. *p < 0.05 as compared to wild type. (C) Cleaved caspase-3 staining in ApcMin/+ and CENP-E+/−;ApcMin/+ adenomatous tumors. Scale bar, 50 µm. (D) Tumors from CENP-E+/−;ApcMin/+ animals show an increase in apoptotic cell death compared with tumors from ApcMin/+ singly heterozygous mice. Note that values from wild-type and CENP-E+/− animals are not shown because these animals do not develop tumors. n > 500 cells from 13 ApcMin/+ and 8 CENP-E+/−;ApcMin/+ tumors. *p < 0.05 as compared to ApcMin/+.
In the small intestine, daughter cells are generated in the base of the crypts and migrate to the villi, where they undergo apoptosis (Wright and Allison, 1985). Consistent with this, levels of cleaved caspase-3 reactivity were negligible in the crypts and elevated in the villi. Expression of the Min allele of Apc increased the levels of apoptosis (Supplemental Figure S1B). Of interest, in the polyps of the small intestine, which have elevated rates of cell division, the double-mutant animals had significantly increased levels of apoptosis relative to ApcMin/+ single mutants (Figure 2, C and D). Thus, high CIN increases the level of apoptosis in vitro and in vivo.
High CIN suppresses progression of intestinal tumors
Mice expressing the Min allele of Apc develop intestinal polyps by 90 d of age (Moser et al., 1990). Measurement of the size and number of these early-onset tumors provides information regarding effects on tumor initiation versus tumor progression. Suppression of tumor initiation produces a reduced number of tumors of similar size, whereas inhibition of tumor progression results in tumors of reduced size. Inhibition of tumor progression may also result in a reduction of observed tumor number, if polyps do not progress sufficiently to be detected.
To test whether high CIN suppresses tumor progression, we determined intestinal tumor sizes. Tumor polyps were significantly smaller in double-mutant animals than in ApcMin/+ animals in both the small intestine and the colon (Figure 3, A and B). Diet is known to affect polyp development in ApcMin/+ animals (Mutanen et al., 2000; Trasler et al., 2003). Similar results were obtained in animals housed in a separate animal facility receiving different feed (Supplemental Figure S2, A and B). These data demonstrate that high CIN due to reduction of CENP-E in ApcMin/+ mice suppresses tumor growth and progression in the small and large intestine and suggest that increasing the rate of CIN might be a successful chemotherapeutic strategy.
FIGURE 3:
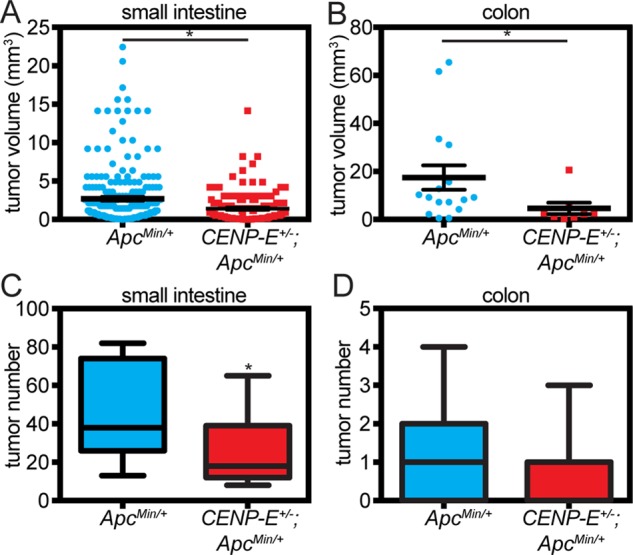
High CIN suppresses the growth and progression of intestinal tumors in ApcMin/+ mice. (A, B) High CIN due to reduction of CENP-E inhibits tumor progression, reducing the average size of tumors in the (A) small intestine and (B) colon of 91-d-old CENP-E+/−;ApcMin/+ mice compared with tumors in age matched ApcMin/+ littermates. In A, n = 237 tumors from eight ApcMin/+ mice and 190 tumors from nine CENP-E+/−;ApcMin/+ animals. In B, n = 16 and eight tumors from 15 ApcMin/+ and 15 CENP-E+/−;ApcMin/+ mice, respectively. (C, D) High CIN due to reduction of CENP-E suppresses tumor number in the small intestine (C) but not the colon (D) of 91-d-old ApcMin/+ mice; n = 15 mice of each genotype. Note that wild-type and CENP-E+/– mice are not shown because they do not develop tumors at this time point. The boxes in C and D show the 25th percentile, median, and 75th percentile of the data, and the whiskers show the range of the complete data set. The median line for the CENP-E+/−;ApcMin/+ genotype is not visible in D because it overlaps with the x-axis. These animals received Teklad Diet 8626. Similar results were obtained in an independent experiment in which animals were housed in a separate facility and received different chow (see Supplemental Figure S2). *p < 0.05.
High CIN does not inhibit the initiation of intestinal tumors
To determine whether high CIN inhibited tumor initiation as well as tumor progression, we examined tumor numbers. In the colon, tumor numbers were not significantly different in CENP-E+/−;ApcMin/+ versus ApcMin/+ mice in either of the animal cohorts receiving two different diets (Figure 3D and Supplemental Figure S2D), indicating that high CIN suppresses tumor progression without inhibiting tumor initiation in this context.
In the small intestine, doubly heterozygous animals developed significantly fewer adenomas than ApcMin/+ mice (Figure 3C and Supplemental Figure S2C). However, it was unclear whether this was due to suppression of tumor initiation or because tumor progression was suppressed so thoroughly that initiated tumors did not progress sufficiently to be detected. To address this question, we tested the ability of CENP-E+/−;ApcMin/+ and ApcMin/+ small intestine cells to become tumor-initiating cells. In the ApcMin/+ system, loss of the wild-type Apc gene converts ApcMin/+ cells into tumor-initiating cells. Intestinal cells that have lost APC function can be discriminated from those that retain it using three-dimensional organoid culture. Organoids recapitulate multiple features of the intact gut, including cell type composition and crypt-villus structures (Sato et al., 2011a, b; Fatehullah et al., 2013). Organoids containing wild- type Apc have a complex, highly branched morphology that mimics tissue organization in vivo (Sato et al., 2011a, b; Fatehullah et al., 2013, Figure 4A). Consistent with our data in MEFs and in mice showing that reduction of CENP-E increases cell death in ApcMin/+ cells, organoids from CENP-E+/−;APCMin/+ animals grew more slowly than those from ApcMin/+ littermates (Figure 4B). As in normal intestinal epithelial tissue (Figure 4C, white arrows), β-catenin is predominantly at the cell periphery in complex organoids expressing APC (Figure 4D, left). In contrast, organoids that have lost APC function have a distinctive, rounded, cyst-like morphology (Fatehullah et al., 2013; Onuma et al., 2013; Figure 4E). Like tumors of the small intestine in ApcMin/+ animals (Figure 4C, yellow arrowheads), cystic organoids have nuclear β-catenin staining, consistent with Apc loss (Onuma et al., 2013; Figure 4D, right).
FIGURE 4:

High CIN does not suppress tumor initiation in the small intestine. (A) Intestinal organoid with a branched, complex morphology typical of organoids retaining expression of wild- type APC. (B) Consistent with the elevated rates of cell death observed in CENP-E+/−;ApcMin/+ MEFs and animals, complex organoids from CENP-E+/−;ApcMin/+ animals increase in size more slowly than ApcMin/+ organoids; n = 30 ApcMin/+ and 40 CENP-E+/−;ApcMin/+ organoids. (C) The layer of normal intestinal epithelium (white arrows) surrounding an intestinal tumor (yellow arrowheads) exhibits cell surface β-catenin staining, whereas β-catenin also exhibits nuclear localization in the tumor cells. (D) As in intact intestine, β-catenin localizes predominantly to the cell surface in complex organoids (left). In cystic organoids (right), β-catenin also shows nuclear staining, consistent with Apc loss and constitutive activation of Wnt signaling. (E) Cystic organoids, which are characteristic of Apc loss (Fatehullah et al., 2013; Onuma et al., 2013), show a rounded appearance that is distinct from the complex, branched structure of organoids expressing APC, as shown in A. Cystic organoids from CENP-E+/−;ApcMin/− tumor-initiating cells are substantially larger than cystic ApcMin/− organoids derived from ApcMin/+ littermates on day 4 after plating. (F) A higher percentage of cells from CENP-E+/−;ApcMin/+ than ApcMin/+ small intestine form cystic organoids characteristic of Apc loss, demonstrating that high CIN does not inhibit tumor initiation in this context; n = 1782 organoids from ApcMin/+ and 1514 organoids from CENP-E+/−;ApcMin/+ animals. (G) Cystic organoids from tumor-initiating cells generated from CENP-E+/−;ApcMin/+ normal intestinal epithelium are substantially larger on day 4 after plating than cystic organoids from ApcMin/+ littermates. (H) In contrast, cystic organoids from mature CENP-E+/−;ApcMin/– tumor cells from adenomas of CENP-E+/−;ApcMin/+ animals show no such growth advantage, suggesting that tumor-initiating cells are less sensitive than mature tumor cells to high CIN. (I) Growth rates of cystic organoids from tumor-initiating cells for the 10 d after sizes were measured in G, showing that CENP-E+/−;ApcMin/- tumor-initiating cells rapidly lose their early growth advantage. *p < 0.05.
To determine the rate at which tumor-initiating cells were generated, we scored the proportion of cystic organoids that formed from crypts of ApcMin/+ and CENP-E+/−;APCMin/+ mice. A previous study showed that increasing the rate of CIN due to reduced expression of the mitotic checkpoint gene Bub1 in p53+/− mice accelerated loss of the chromosome containing the wild- type copy of p53 (Baker et al., 2009). This suggested that the formation of cystic organoids should be higher from CENP-E+/−;APCMin/+ than ApcMin/+ animals due to increased loss of the chromosome containing the wild-type allele of Apc. Consistent with this, the percentage of cystic organoids that formed from CENP-E+/−;APCMin/+ animals was higher than that from ApcMin/+ littermates (Figure 4F). These data demonstrate that, although fewer adenomas are detected in the small intestine of CENP-E+/−;APCMin/+ animals, high CIN accelerates the formation of tumor-initiating cells.
One potential caveat to the conclusion that high CIN suppresses tumor growth is that tumors that grew at the same rate in CENP-E+/−;APCMin/+ and ApcMin/+ animals would be smaller in CENP-E+/−;APCMin/+ animals if tumor initiation were delayed in this genotype. The findings that tumor initiation is not inhibited in the small intestine or colon (Figure 3, C and D, and Supplemental Figure 2, C and D), and that loss of wild-type Apc occurs earlier in organoids from CENP-E+/−;APCMin/+ than from APCMin/+ crypts (Figure 4F) argue against this. However, if this were the case, two populations of tumors would be expected to arise in CENP-E+/−;APCMin/+ animals—a population of large tumors that arose due to somatic recombination, as occurs in APCMin/+ mice with a wild-type complement of CENP-E (Luongo et al., 1994; Haigis et al., 2002; Haigis and Dove, 2003), and a population of small tumors that arose later due to loss of the copy of chromosome 18 containing the wild-type copy of Apc. The lack of a population of large tumors in the CENP-E+/−;APCMin/+ animals (Figure 3, A and B, and Supplemental Figure 2, A and B) further supports the conclusion that high CIN due to reduction of CENP-E suppresses the growth and progression of tumors in APCMin/+ mice.
Tumor-initiating cells are less sensitive than mature tumor cells to high CIN
Because high CIN suppresses tumor progression without inhibiting tumor initiation, this raises the question of whether tumor-initiating cells are more tolerant of high CIN than mature tumor cells. To test this, we isolated crypts from adenomas, as well as from normal small intestine. Organoids generated from normal intestine require a new or relatively recent loss of wild-type Apc to grow with a cystic morphology and thus provide a source of tumor-initiating cells. In contrast, cystic organoids from adenomas contain more mature tumor cells in which loss of the wild-type Apc gene occurred earlier. Remarkably, cystic organoids from CENP-E+/−;ApcMin/- tumor-initiating cells were substantially larger than cystic organoids from ApcMin/- tumor-initiating cells 4 d after seeding (Figure 4, E and G). This was not true of the cystic organoids formed from the mature cells from adenomas, which were equivalent in size in both genotypes (Figure 4H). Of interest, however, the tolerance of the tumor-initiating cells to high CIN did not persist, as cystic organoids from CENP-E+/−;ApcMin/– tumor-initiating cells did not outgrow cystic organoids from ApcMin/– tumor-initiating cells over the next 10 d (Figure 4I). These data suggest that tumor-initiating cells are indeed less sensitive to cell death caused by high rates of chromosome missegregation. However, this insensitivity is rapidly lost.
Increasing CIN as chemotherapy
Therapeutically exploiting tumor suppression caused by high CIN to treat preexisting tumors requires that the inhibitory effects of high CIN be exerted on tumor growth and progression as opposed to tumor initiation. Here we show that converting low CIN into high CIN substantially inhibits tumor progression in the small intestine and colon. Moreover, our data suggest that relatively modest increases in chromosome missegregation can have substantial effects on cell death and tumor outcome. This is true not only when increases in chromosome missegregation occur early in tumor growth; tumors that have reached a size sufficient for diagnosis are also likely to be susceptible to small increases in the rate of CIN if they have a preexisting low rate of chromosome missegregation. We recently found that concentrations of the chemotherapy drug paclitaxel (Taxol) found in breast tumors are too low to cause mitotic arrest and instead induce chromosome missegregation on multipolar spindles (Zasadil et al., 2014). It appears that this commonly used chemotherapeutic agent kills cells in patient tumors by causing an increase in the rate of chromosome missegregation, and is perhaps particularly effective in cells that have a preexisting low rate of CIN. Consistent with this, although patients with tumors classified as CIN have poorer outcomes than those with chromosomally stable tumors (Carter et al., 2006), when CIN tumors are subdivided based on the level of expression of CIN genes, patients with breast, ovarian, gastric, and lung cancer whose tumors have the highest level of CIN have improved recurrence- or metastasis-free survival relative to patients with tumors containing a lower rate of CIN (Birkbak et al., 2011). Similarly, breast cancer patients with high CIN tumors, based on clonal variability in fluorescent in situ hybridization analysis, outlived patients with tumors exhibiting low CIN (Roylance et al., 2011). Thus, multiple human data sets substantiate our mouse models showing that increasing the rate of chromosome missegregation might be a useful therapeutic strategy, particularly in tumors that already exhibit a low rate of CIN.
The major concern regarding the intentional generation of aneuploidy and CIN is their potential to initiate additional tumors. However, multiple animal models with elevated levels of aneuploidy and CIN are not tumor prone. These include animals that express a kinase-dead version of the Bub1 mitotic checkpoint protein (Ricke et al., 2012), a dominant-negative fragment of Bub1 (Cowley et al., 2005), or ∼75% of the endogenous level of Bub1 (Jeganathan et al., 2007; Baker et al., 2009; Li et al., 2010), as well as those with hypomorphic expression of the anaphase-promoting complex/cyclosome cofactor Cdc20 (Malureanu et al., 2010). In contrast, other animal models that exhibit aneuploidy and CIN do develop increased levels of spontaneous, carcinogen-induced and/or genetically driven tumors. Animals with similar levels of aneuploidy can have no tumor burden, a slightly increased tumor incidence, or a striking phenotype in which the majority of animals develop tumors (reviewed in Ricke et al., 2008; Holland and Cleveland, 2009; Zasadil et al., 2013). In addition, mutation of genes resulting in aneuploidy and CIN can promote tumors in some contexts but suppress them in others. One example of this is heterozygous loss of BubR1, which increases colon tumor formation but decreases the incidence of small intestine tumors in ApcMin/+ mice (Rao et al., 2005). Although the reason for this is not clear, most genes whose mutation leads to aneuploidy and CIN, including BubR1, have been implicated in functions outside of chromosome segregation that could reasonably be expected to affect tumor phenotype, including cell death pathways and the DNA damage response (Shin et al., 2003; Baek et al., 2005; Fang et al., 2006; Zasadil et al., 2013). It may be that the nonmitotic roles of BubR1 are tissue specific. Although there is striking diversity in the tumor phenotypes of animals that exhibit aneuploidy and CIN, it is clear that aneuploidy and CIN are not sufficient to cause an increase in tumor burden.
A second concern regarding the therapeutic use of strategies to induce CIN is that increasing CIN may not be effective against chromosomally stable tumor cells, potentially including tumor-initiating cells. Chromosomally stable tumor cells may benefit from combination therapy in which one drug that causes CIN is used to sensitize tumor cells to treatment with a second CIN-inducing drug, such as paclitaxel. In this case, tumor-specific targeting may be necessary for at least one of the treatments to prevent lethality in rapidly dividing, normal cells.
Intriguingly, treatments that cause chromosome missegregation in diploid cells do not necessarily produce persistently aneuploid cell populations. Experimentally inducing CIN in chromosomally stable cells produces only a transient increase in aneuploidy (Thompson and Compton, 2008, 2010). Chromosomally stable cancer cell lines have also been shown to exhibit aberrant mitotic divisions expected to produce aneuploid progeny, yet they maintain a relatively stable consensus karyotype over many generations (Roschke et al., 2002).
Even if aneuploidy does result from pharmacologically increasing CIN, aneuploidy has been shown to cause a growth defect in yeast and murine cells under optimal conditions (Torres et al., 2007; Williams et al., 2008). Similarly, human skin fibroblasts trisomic for chromosome 21 grow more slowly than diploid skin fibroblasts (Segal and McCoy, 1974). However, several studies show that specific aneuploid karyotypes can confer a growth advantage in response to certain stresses. In yeast, preexisting aneuploidy can result in accelerated growth in response to particular environmental stresses, and specific aneuploidies can evolve to overcome functional insufficiencies or adapt to environmental challenges (Rancati et al., 2008; Pavelka et al., 2010; Kaya et al., 2015; Millet et al., 2015). In pathogenic fungi, development of aneuploidy has been implicated as an adaptive response to confer azole resistance (Selmecki et al., 2006, 2009). Similarly, aneuploidy is believed to confer a selective advantage in liver, as mice heterozygous for a gene on chromosome 16 that confers sensitivity to fumarylacetoacetate hydrolase (Fah) deficiency developed injury-resistant aneuploid liver nodules that had lost one copy of chromosome 16 after liver damage in the absence of Fah (Duncan et al., 2012). In addition, human colonic cells grown in serum-free conditions acquire a third copy of chromosome 7 and outgrow diploid cells in serum-free media (Ly et al., 2011). These studies show that whereas aneuploidy often confers an overall growth disadvantage, specific aneuploidies can allow cells of various species to proliferate under conditions that are suboptimal for euploid cell growth. In addition, in some cases, what was believed to be stable aneuploidy has been reported to cause CIN. Aneuploid yeast strains often show an increase in karyotypic variation and genomic instability (Pavelka et al., 2010; Sheltzer et al., 2011; Zhu et al., 2012). Human chromosomally stable colorectal cancer cell lines (DLD1) with an extra copy of chromosome 7 or 13 show an increase in chromosome missegregation rates, as do amniotic fibroblasts that are trisomic for chromosome 13. Of interest, these cells predominantly missegregate the trisomic chromosome, suggesting that a portion of them revert to the diploid state (Nicholson et al., 2015).
Thus, although it is a concern, it is not a foregone conclusion that treatments aimed at inducing high CIN would produce aneuploidy in normal tissues or would increase the likelihood of secondary tumors later in life. We conclude that intentional induction of high CIN merits further consideration as a therapeutic strategy, at least in adult patients. It is now of interest to define the threshold of chromosome missegregation that is necessary to result in cell death, the types of cell death induced by high CIN, and the extent to which p53 function is required for tumor suppression mediated by high CIN.
MATERIALS AND METHODS
Intestinal sample collection, tumor counts, and measurements
Mice were maintained in a C57BL/6 genetic background. Mouse experiments were conducted after approval by the University of Wisconsin–Madison Institutional Animal Care and Use Committee. Intestines from 90- or 91-d-old mice were collected and flushed with ice-cold phosphate-buffered saline (PBS), opened lengthwise, further washed with ice-cold PBS, and laid flat on bibulous paper and then fixed in 10% buffered Formalin overnight. Fixed tissues were then washed with 70% EtOH and stored in 70% EtOH until embedding. Counts and measurements of intestinal polyps were performed using a dissecting scope before embedding for histological analysis.
Cell culture
MEFs were isolated from E14.5 embryos and maintained in DMEM (Life Technologies, Carlsbad, CA) containing 200 mM l-glutamine, 50 μg/ml penicillin-streptomycin, 10 mM nonessential amino acids, 100 mM sodium pyruvate, 1 mM β-mercaptoethanol, and 15% fetal calf serum (FCS) at 10% CO2, 3% O2, and 37°C. For fixed-cell analysis and videomicroscopy, cells were grown in media containing 10% FCS for 48 h before fixation or the start of time-lapse acquisition. Growth curves were seeded directly into media containing 10% FCS.
Immunofluorescence and immunohistochemistry
Primary MEFs were washed with MTSB (100 mM 1,4-piperazinediethanesulfonic acid, pH 6.9, 30% glycerol, 1 mM ethylene glycol tetraacetic acid, and 1 mM MgSO4) and permeabilized in MTSB plus 0.05% Triton X-100 for 45 s at room temperature. Fixation was performed in MTSB plus 4% formaldehyde and 0.1% glutaraldehyde for 10 min at room temperature. For cleaved caspase-3 detection, glutaraldehyde was not included in the fixative solution. Coverslips were blocked overnight in Triton Block (0.2 M glycine, 2.5% fetal bovine serum [FBS], and 0.1% Triton X-100 in 1× PBS). Primary antibody incubation (α-tubulin YL1/2, 1:500, Serotec, Hercules, CA; cleaved caspase-3, 1:200, Cell Signaling, Danvers, MA) was performed for 1 h at room temperature in blocking solution. Coverslips were washed 3× in PBS plus 0.1% Triton X-100 and incubated in Alexa Fluor–conjugated secondary antibodies diluted 1:200 in PBS for 45 min at room temperature. Coverslips were washed 3× in PBS plus 0.1% Triton X-100, incubated for 3 min in 1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) in PBS, rinsed 2× in PBS, and mounted in Vectashield (Vector Labs, Burlingame, CA) mounting medium.
Paraffin-embedded sections of 5-μm thickness were first deparaffinized in xylenes 3 × 10 min, rinsed in 100% ethanol, and hydrated in a series of 100, 95, 80, and 50% ethanol for 1 min each, followed by 5 min in double-distilled H2O. Antigen retrieval was performed in a pressure cooker set to 250°F for 4 min in citrate buffer (10 mM citric acid plus 0.05% Tween-20). Slides were then washed in PBS and blocked for 1 h at room temperature in PBS plus 10% FBS. Primary antibody incubation (α-tubulin DM1α, 1:200; cleaved caspase-3, 1:200, Cell Signaling) was performed overnight at 4°C in a humidified chamber. Slides were washed 3 × 5 min in PBS plus 0.1% Triton X-100 and incubated in secondary antibodies (Alexa Fluor, 1:200 in PBS) for 1 h at room temperature. After three subsequent washes, slides were incubated in 5 μg/ml DAPI in PBS for 10 min, washed 2× in PBS, and mounted using Vectashield (Vector Labs).
Images were acquired using a Nikon Eclipse Ti-E inverted microscope with a Hamamatsu ORCA Flash 4.0 camera using 10× (0.3 numerical aperture [NA]), 40× (0.75 NA), or 100× (1.4 NA) objectives. Image acquisition, analysis, and processing were performed using Nikon Elements AR. Autoquant was used for deconvolution. Images shown are maximum projections of z-stacks except in Figures 1E and 4 where single z-planes are shown.
Organoids
Intestines were harvested from 65-d-old mice, and crypt fractions were isolated as described (Sato and Clevers, 2013). Crypts were seeded in Matrigel (9.2 mg/ml; BD). Adenomas were digested with 75 U/ml collagenase for 30 min at 37°C. Cultures were fed DMEM/F12 Advanced (Life Technologies) media containing 40 ng/ml epidermal growth factor (PeproTech, Rocky Hill, NJ), 50 ng/ml Noggin (PeproTech), and 250 ng/ml R-spondin (R&D, Minneapolis, MN) every other day. For growth curves, saved coordinates for each plate were visited every day (days 4–14), and images were acquired using a 10× (0.3 NA) objective and Hamamatsu Orca FLASH 4.0 camera driven by Nikon Elements software.
For immunofluorescence, organoids were fixed with 4% formaldehyde in MTSB for 30 min at room temperature and transferred to sieve wells in MTSB plus 0.05% Triton. Sieve wells consist of two adjacent wells that are open on the top and separated by a microporous membrane. The sieve well halves, designed to hold ∼100 μl each, are micromilled (PCNC 770; Tormach, Waunakee, WI) into separate pieces of polystyrene (PS; 52420098; MSC Industrial Supply, Melville, NY). With a microporous membrane (1221420; Maine Manufacturing, Sanford, ME) sandwiched between them, the PS halves are solvent bonded via acetonitrile (271004; Sigma-Aldrich, St. Louis, MO). The top of the bonded device is coated with a film of wax, and the outer faces of the bonded device are sealed with an optical adhesive (04729757001; Roche, Basel, Switzerland). Organoids were blocked for 1 h in PBS + 10% FBS and stained with β-catenin antibody (Cell Signaling) overnight at 4°C. Secondary antibody staining was done with Alexa Fluor 555 anti-rabbit for 1 h at room temperature. Organoids were imaged through No. 1.5 coverslips on a Nikon Ti-E inverted microscope using a 100× (1.4 NA) objective and a Hamamatsu Orca FLASH4 camera driven by Nikon Elements software.
Statistical analysis
Statistical analysis was performed using MSTAT 6.1.4 software (mcardle.wisc.edu/mstat/). Outliers were determined using Grubbs’ method and excluded from analysis. Differences between samples were tested using the Wilcoxon rank sum test (tumor numbers, organoid analysis), chi-squared test (cell death, polar and lagging chromosomes), and the Sen–Adichie test (rates of growth/increase). Error bars represent SEM unless otherwise indicated.
Supplementary Material
Acknowledgments
We thank our colleagues for helpful discussions and suggestions. This work was supported in part by National Institutes of Health Grants R01CA140458 (B.W.), P30CA014520 (D.J.B.), and T32CA009135 (L.Z., E.B.).
Abbreviations used:
- APC
adenomatous polyposis coli
- CENP-E
centromere-associated protein E
- CIN
chromosomal instability
- MEF
murine embryonic fibroblast
- Min
multiple intestinal neoplasia.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-10-0747) on May 4, 2016.
REFERENCES
- Abrieu A, Kahana JA, Wood KW, Cleveland DW. CENP-E as an essential component of the mitotic checkpoint in vitro. Cell. 2000;102:817–826. doi: 10.1016/s0092-8674(00)00070-2. [DOI] [PubMed] [Google Scholar]
- Baek KH, Shin HJ, Jeong SJ, Park JW, McKeon F, Lee CW, Kim CM. Caspases-dependent cleavage of mitotic checkpoint proteins in response to microtubule inhibitor. Oncol Res. 2005;15:161–168. doi: 10.3727/096504005776367906. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Jin F, Jeganathan KB, van Deursen JM. Whole chromosome instability caused by Bub1 insufficiency drives tumorigenesis through tumor suppressor gene loss of heterozygosity. Cancer Cell. 2009;16:475–486. doi: 10.1016/j.ccr.2009.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkbak NJ, Eklund AC, Li Q, McClelland SE, Endesfelder D, Tan P, Tan IB, Richardson AL, Szallasi Z, Swanton C. Paradoxical relationship between chromosomal instability and survival outcome in cancer. Cancer Res. 2011;71:3447–3452. doi: 10.1158/0008-5472.CAN-10-3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boveri T. Ueber mehrpolige Mitosen als Mittel zur Analyse des Zellkerns. Vehrhandl Phys Med Ges Wurzburg NF. 1902;35:67–90. English translation available at http://10e.devbio.com/article.php?id=24. [Google Scholar]
- Boveri T. Zur Frage der Entstehung maligner Tumoren, Jena, Germany: Gustav Fischer. English translation, The Origin of Malignant Tumors (1929) Baltimore: Williams and Wilkins; 1914. [Google Scholar]
- Brown KD, Coulson RM, Yen TJ, Cleveland DW. Cyclin-like accumulation and loss of the putative kinetochore motor CENP-E results from coupling continuous synthesis with specific degradation at the end of mitosis. J Cell Biol. 1994;125:1303–1312. doi: 10.1083/jcb.125.6.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter SL, Eklund AC, Kohane IS, Harris LN, Szallasi Z. A signature of chromosomal instability inferred from gene expression profiles predicts clinical outcome in multiple human cancers. Nat Genet. 2006;38:1043–1048. doi: 10.1038/ng1861. [DOI] [PubMed] [Google Scholar]
- Chesnokova V, Kovacs K, Castro AV, Zonis S, Melmed S. Pituitary hypoplasia in Pttg-/- mice is protective for Rb+/- pituitary tumorigenesis. Mol Endocrinol. 2005;19:2371–2379. doi: 10.1210/me.2005-0137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley DO, Muse GW, Van Dyke T. A dominant interfering Bub1 mutant is insufficient to induce or alter thymic tumorigenesis in vivo, even in a sensitized genetic background. Mol Cell Biol. 2005;25:7796–7802. doi: 10.1128/MCB.25.17.7796-7802.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donangelo I, Gutman S, Horvath E, Kovacs K, Wawrowsky K, Mount M, Melmed S. Pituitary tumor transforming gene overexpression facilitates pituitary tumor development. Endocrinology. 2006;147:4781–4791. doi: 10.1210/en.2006-0544. [DOI] [PubMed] [Google Scholar]
- Duncan AW, Hanlon Newell AE, Bi W, Finegold MJ, Olson SB, Beaudet AL, Grompe M. Aneuploidy as a mechanism for stress-induced liver adaptation. J Clin Invest. 2012;122:3307–3315. doi: 10.1172/JCI64026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, Liu T, Wang X, Yang YM, Deng H, Kunicki J, Traganos F, Darzynkiewicz Z, Lu L, Dai W. BubR1 is involved in regulation of DNA damage responses. Oncogene. 2006;25:3598–3605. doi: 10.1038/sj.onc.1209392. [DOI] [PubMed] [Google Scholar]
- Fatehullah A, Appleton PL, Näthke IS. Cell and tissue polarity in the intestinal tract during tumourigenesis: cells still know the right way up, but tissue organization is lost. Philos Trans R Soc Lond B Biol Sci. 2013;368:20130014. doi: 10.1098/rstb.2013.0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fodde R, Kuipers J, Rosenberg C, Smits R, Kielman M, Gaspar C, van Es JH, Breukel C, Wiegant J, Giles RH, Clevers H. Mutations in the APC tumour suppressor gene cause chromosomal instability. Nat Cell Biol. 2001;3:433–438. doi: 10.1038/35070129. [DOI] [PubMed] [Google Scholar]
- Fong MY, Farghaly H, Kakar SS. Tumorigenic potential of pituitary tumor transforming gene (PTTG) in vivo investigated using a transgenic mouse model, and effects of cross breeding with p53 (+/-) transgenic mice. BMC Cancer. 2012;12:532. doi: 10.1186/1471-2407-12-532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM, Caya JG, Reichelderfer M, Dove WF. Intestinal adenomas can develop with a stable karyotype and stable microsatellites. Proc Natl Acad Sci USA. 2002;99:8927–8931. doi: 10.1073/pnas.132275099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haigis KM, Dove WF. A Robertsonian translocation suppresses a somatic recombination pathway to loss of heterozygosity. Nat Genet. 2003;33:33–39. doi: 10.1038/ng1055. [DOI] [PubMed] [Google Scholar]
- Holland AJ, Cleveland DW. Boveri revisited: chromosomal instability, aneuploidy and tumorigenesis. Nat Rev Mol Cell Biol. 2009;10:478–487. doi: 10.1038/nrm2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Papadopoulos N, McKinley AJ, Farrington SM, Curtis LJ, Wyllie AH, Zheng S, Willson JK, Markowitz SD, Morin P, et al. APC mutations in colorectal tumors with mismatch repair deficiency. Proc Natl Acad Sci USA. 1996;93:9049–9054. doi: 10.1073/pnas.93.17.9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwanaga Y, Chi YH, Miyazato A, Sheleg S, Haller K, Peloponese JM, Jr, Li Y, Ward JM, Benezra R, Jeang KT. Heterozygous deletion of mitotic arrest-deficient protein 1 (MAD1) increases the incidence of tumors in mice. Cancer Res. 2007;67:160–166. doi: 10.1158/0008-5472.CAN-06-3326. [DOI] [PubMed] [Google Scholar]
- Jallepalli PV, Waizenegger IC, Bunz F, Langer S, Speicher MR, Peters JM, Kinzler KW, Vogelstein B, Lengauer C. Securin is required for chromosomal stability in human cells. Cell. 2001;105:445–457. doi: 10.1016/s0092-8674(01)00340-3. [DOI] [PubMed] [Google Scholar]
- Janssen A, Kops GJ, Medema RH. Elevating the frequency of chromosome mis-segregation as a strategy to kill tumor cells. Proc Natl Acad Sci USA. 2009;106:19108–19113. doi: 10.1073/pnas.0904343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeganathan K, Malureanu L, Baker DJ, Abraham SC, van Deursen JM. Bub1 mediates cell death in response to chromosome missegregation and acts to suppress spontaneous tumorigenesis. J Cell Biol. 2007;179:255–267. doi: 10.1083/jcb.200706015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan KB, Burds AA, Swedlow JR, Bekir SS, Sorger PK, Nathke IS. A role for the Adenomatous Polyposis Coli protein in chromosome segregation. Nat Cell Biol. 2001;3:429–432. doi: 10.1038/35070123. [DOI] [PubMed] [Google Scholar]
- Kaya A, Gerashchenko MV, Seim I, Labarre J, Toledano MB, Gladyshev VN. Adaptive aneuploidy protects against thiol peroxidase deficiency by increasing respiration via key mitochondrial proteins. Proc Natl Acad Sci USA. 2015;112:10685–10690. doi: 10.1073/pnas.1505315112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerr SE, Thomas CB, Thibodeau SN, Ferber MJ, Halling KC. APC germline mutations in individuals being evaluated for familial adenomatous polyposis: a review of the Mayo Clinic experience with 1591 consecutive tests. J Mol Diagn. 2013;15:31–43. doi: 10.1016/j.jmoldx.2012.07.005. [DOI] [PubMed] [Google Scholar]
- Kim Y, Heuser JE, Waterman CM, Cleveland DW. CENP-E combines a slow, processive motor and a flexible coiled coil to produce an essential motile kinetochore tether. J Cell Biol. 2008;181:411–419. doi: 10.1083/jcb.200802189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kops GJ, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci USA. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Baker DJ, Guo L, Gao X, Wei Z, Han S, van Deursen JM, Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc Natl Acad Sci USA. 2010;107:14188–14193. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Fang X, Wei Z, York JP, Zhang P. Loss of spindle assembly checkpoint-mediated inhibition of Cdc20 promotes tumorigenesis in mice. J Cell Biol. 2009;185:983–994. doi: 10.1083/jcb.200904020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–5952. [PubMed] [Google Scholar]
- Ly P, Eskiocak U, Kim SB, Roig AI, Hight SK, Lulla DR, Zou YS, Batten K, Wright WE, Shay JW. Characterization of aneuploid populations with trisomy 7 and 20 derived from diploid human colonic epithelial cells. Neoplasia. 2011;13:348–357. doi: 10.1593/neo.101580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malureanu L, Jeganathan KB, Jin F, Baker DJ, van Ree JH, Gullon O, Chen Z, Henley JR, van Deursen JM. Cdc20 hypomorphic mice fail to counteract de novo synthesis of cyclin B1 in mitosis. J Cell Biol. 2010;191:313–329. doi: 10.1083/jcb.201003090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BF, Chan GK, Zubrowski B, Savoian MS, Sauer MT, Yen TJ. CENP-E is essential for reliable bioriented spindle attachment, but chromosome alignment can be achieved via redundant mechanisms in mammalian cells. Mol Biol Cell. 2001;12:2776–2789. doi: 10.1091/mbc.12.9.2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Burrell RA, Endesfelder D, Novelli MR, Swanton C. Cancer chromosomal instability: therapeutic and diagnostic challenges. EMBO Rep. 2012;13:528–538. doi: 10.1038/embor.2012.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel L, Diaz-Rodriguez E, Narayan G, Hernando E, Murty VV, Benezra R. Complete loss of the tumor suppressor MAD2 causes premature cyclin B degradation and mitotic failure in human somatic cells. Proc Natl Acad Sci USA. 2004;101:4459–4464. doi: 10.1073/pnas.0306069101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet C, Ausiannikava D, Le Bihan T, Granneman S, Makovets S. Cell populations can use aneuploidy to survive telomerase insufficiency. Nat Commun. 2015;6:8664. doi: 10.1038/ncomms9664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247:322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- Mutanen M, Pajari AM, Oikarinen SI. Beef induces and rye bran prevents the formation of intestinal polyps in Apc(Min) mice: relation to beta-catenin and PKC isozymes. Carcinogenesis. 2000;21:1167–1173. doi: 10.1093/carcin/21.6.1167. [DOI] [PubMed] [Google Scholar]
- Nicholson JM, Macedo JC, Mattingly AJ, Wangsa D, Camps J, Lima V, Gomes AM, Dória S, Ried T, Logarinho E, et al. Chromosome mis-segregation and cytokinesis failure in trisomic human cells. Elife. 2015;4 doi: 10.7554/eLife.05068. doi: 10.7554/eLife.05068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onuma K, Ochiai M, Orihashi K, Takahashi M, Imai T, Nakagama H, Hippo Y. Genetic reconstitution of tumorigenesis in primary intestinal cells. Proc Natl Acad Sci USA. 2013;110:11127–11132. doi: 10.1073/pnas.1221926110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature. 2010;468:321–325. doi: 10.1038/nature09529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. APC mutations occur early during colorectal tumorigenesis. Nature. 1992;359:235–237. doi: 10.1038/359235a0. [DOI] [PubMed] [Google Scholar]
- Putkey FR, Cramer T, Morphew MK, Silk AD, Johnson RS, McIntosh JR, Cleveland DW. Unstable kinetochore-microtubule capture and chromosomal instability following deletion of CENP-E. Dev Cell. 2002;3:351–365. doi: 10.1016/s1534-5807(02)00255-1. [DOI] [PubMed] [Google Scholar]
- Rancati G, Pavelka N, Fleharty B, Noll A, Trimble R, Walton K, Perera A, Staehling-Hampton K, Seidel CW, Li R. Aneuploidy underlies rapid adaptive evolution of yeast cells deprived of a conserved cytokinesis motor. Cell. 2008;135:879–893. doi: 10.1016/j.cell.2008.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, Jhanwar-Uniyal M, Dai W. Colonic tumorigenesis in BubR1+/-ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci USA. 2005;102:4365–4370. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, Jeganathan KB, Malureanu L, Harrison AM, van Deursen JM. Bub1 kinase activity drives error correction and mitotic checkpoint control but not tumor suppression. J Cell Biol. 2012;199:931–949. doi: 10.1083/jcb.201205115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, Jeganathan KB, van Deursen JM. Bub1 overexpression induces aneuploidy and tumor formation through Aurora B kinase hyperactivation. J Cell Biol. 2011;193:1049–1064. doi: 10.1083/jcb.201012035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, van Ree JH, van Deursen JM. Whole chromosome instability and cancer: a complex relationship. Trends Genet. 2008;24:457–466. doi: 10.1016/j.tig.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roschke AV, Stover K, Tonon G, Schäffer AA, Kirsch IR. Stable karyotypes in epithelial cancer cell lines despite high rates of ongoing structural and numerical chromosomal instability. Neoplasia. 2002;4:19–31. doi: 10.1038/sj.neo.7900197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roylance R, Endesfelder D, Gorman P, Burrell RA, Sander J, Tomlinson I, Hanby AM, Speirs V, Richardson AL, Birkbak NJ, et al. Relationship of extreme chromosomal instability with long-term survival in a retrospective analysis of primary breast cancer. Cancer Epidemiol Biomarkers Prev. 2011;20:2183–2194. doi: 10.1158/1055-9965.EPI-11-0343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, Clevers H. Primary mouse small intestinal epithelial cell cultures. Methods Mol Biol. 2013;945:319–328. doi: 10.1007/978-1-62703-125-7_19. [DOI] [PubMed] [Google Scholar]
- Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, Van Houdt WJ, Pronk A, Van Gorp J, Siersema PD, et al. Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology. 2011a;141:1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011b;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaar BT, Chan GK, Maddox P, Salmon ED, Yen TJ. CENP-E function at kinetochores is essential for chromosome alignment. J Cell Biol. 1997;139:1373–1382. doi: 10.1083/jcb.139.6.1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schvartzman JM, Duijf PH, Sotillo R, Coker C, Benezra R. Mad2 is a critical mediator of the chromosome instability observed upon Rb and p53 pathway inhibition. Cancer Cell. 2011;19:701–714. doi: 10.1016/j.ccr.2011.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, McCoy EE. Studies on Down’s syndrome in tissue culture. I. Growth rates and protein contents of fibroblast cultures. J Cell Physiol. 1974;83:85–90. doi: 10.1002/jcp.1040830112. [DOI] [PubMed] [Google Scholar]
- Selmecki AM, Dulmage K, Cowen LE, Anderson JB, Berman J. Acquisition of aneuploidy provides increased fitness during the evolution of antifungal drug resistance. PLoS Genet. 2009;5:e1000705. doi: 10.1371/journal.pgen.1000705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmecki A, Forche A, Berman J. Aneuploidy and isochromosome formation in drug-resistant Candida albicans. Science. 2006;313:367–370. doi: 10.1126/science.1128242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer JM, Blank HM, Pfau SJ, Tange Y, George BM, Humpton TJ, Brito IL, Hiraoka Y, Niwa O, Amon A. Aneuploidy drives genomic instability in yeast. Science. 2011;333:1026–1030. doi: 10.1126/science.1206412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin HJ, Baek KH, Jeon AH, Park MT, Lee SJ, Kang CM, Lee HS, Yoo SH, Chung DH, Sung YC, et al. Dual roles of human BubR1, a mitotic checkpoint kinase, in the monitoring of chromosomal instability. Cancer Cell. 2003;4:483–497. doi: 10.1016/s1535-6108(03)00302-7. [DOI] [PubMed] [Google Scholar]
- Silk AD, Zasadil LM, Holland AJ, Vitre B, Cleveland DW, Weaver BA. Chromosome missegregation rate predicts whether aneuploidy will promote or suppress tumors. Proc Natl Acad Sci USA. 2013;110:E4134–E4141. doi: 10.1073/pnas.1317042110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Hernando E, Diaz-Rodriguez E, Teruya-Feldstein J, Cordon-Cardo C, Lowe SW, Benezra R. Mad2 overexpression promotes aneuploidy and tumorigenesis in mice. Cancer Cell. 2007;11:9–23. doi: 10.1016/j.ccr.2006.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo R, Schvartzman JM, Socci ND, Benezra R. Mad2-induced chromosome instability leads to lung tumour relapse after oncogene withdrawal. Nature. 2010;464:436–440. doi: 10.1038/nature08803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, Gould KA, Dove WF. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256:668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Examining the link between chromosomal instability and aneuploidy in human cells. J Cell Biol. 2008;180:665–672. doi: 10.1083/jcb.200712029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SL, Compton DA. Chromosome missegregation in human cells arises through specific types of kinetochore-microtubule attachment errors. Proc Natl Acad Sci USA. 2011;108:17974–17978. doi: 10.1073/pnas.1109720108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tighe A, Johnson VL, Taylor SS. Truncating APC mutations have dominant effects on proliferation, spindle checkpoint control, survival and chromosome stability. J Cell Sci. 2004;117:6339–6353. doi: 10.1242/jcs.01556. [DOI] [PubMed] [Google Scholar]
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- Trasler J, Deng L, Melnyk S, Pogribny I, Hiou-Tim F, Sibani S, Oakes C, Li E, James SJ, Rozen R. Impact of Dnmt1 deficiency, with and without low folate diets, on tumor numbers and DNA methylation in Min mice. Carcinogenesis. 2003;24:39–45. doi: 10.1093/carcin/24.1.39. [DOI] [PubMed] [Google Scholar]
- von Hansemann D. Ueber asymmetrische Zelltheilung in Epithelkrebsen und deren biologische Bedeutung. Virchow’s Arch Pathol Anat. 1890;119:299–326. [Google Scholar]
- Wang Z, Yu R, Melmed S. Mice lacking pituitary tumor transforming gene show testicular and splenic hypoplasia, thymic hyperplasia, thrombocytopenia, aberrant cell cycle progression, and premature centromere division. Mol Endocrinol. 2001;15:1870–1879. doi: 10.1210/mend.15.11.0729. [DOI] [PubMed] [Google Scholar]
- Weaver BA, Bonday ZQ, Putkey FR, Kops GJ, Silk AD, Cleveland DW. Centromere-associated protein-E is essential for the mammalian mitotic checkpoint to prevent aneuploidy due to single chromosome loss. J Cell Biol. 2003;162:551–563. doi: 10.1083/jcb.200303167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008;322:703–709. doi: 10.1126/science.1160058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood KW, Lad L, Luo L, Qian X, Knight SD, Nevins N, Brejc K, Sutton D, Gilmartin AG, Chua PR, et al. Antitumor activity of an allosteric inhibitor of centromere-associated protein-E. Proc Natl Acad Sci USA. 2010;107:5839–5844. doi: 10.1073/pnas.0915068107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright N, Allison M. The Biology of Epithelial Cell Populations. New York: Oxford University Press; 1985. [Google Scholar]
- Yao X, Anderson KL, Cleveland DW. The microtubule-dependent motor centromere-associated protein E (CENP-E) is an integral component of kinetochore corona fibers that link centromeres to spindle microtubules. J Cell Biol. 1997;139:435–447. doi: 10.1083/jcb.139.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen TJ, Compton DA, Wise D, Zinkowski RP, Brinkley BR, Earnshaw WC, Cleveland DW. CENP-E, a novel human centromere-associated protein required for progression from metaphase to anaphase. EMBO J. 1991;10:1245–1254. doi: 10.1002/j.1460-2075.1991.tb08066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Lu W, Chen J, McCabe CJ, Melmed S. Overexpressed pituitary tumor-transforming gene causes aneuploidy in live human cells. Endocrinology. 2003;144:4991–4998. doi: 10.1210/en.2003-0305. [DOI] [PubMed] [Google Scholar]
- Zasadil LM, Andersen KA, Yeum D, Rocque GB, Wilke LG, Tevaarwerk AJ, Raines RT, Burkard ME, Weaver BA. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci Transl Med. 2014;6:229ra243. doi: 10.1126/scitranslmed.3007965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasadil LM, Britigan EM, Weaver BA. 2n or not 2n: aneuploidy, polyploidy and chromosomal instability in primary and tumor cells. Semin Cell Dev Biol. 2013;24:370–379. doi: 10.1016/j.semcdb.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Pavelka N, Bradford WD, Rancati G, Li R. Karyotypic determinants of chromosome instability in aneuploid budding yeast. PLoS Genet. 2012;8:e1002719. doi: 10.1371/journal.pgen.1002719. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.