

A mutant syncytium with clustered nuclei can maintain normal growth and nucleocytoplasmic ratio control. As in the wild type, clustered nuclei exhibit cell cycle and transcriptional autonomy. Cyclin transcript enrichment near wild-type nuclei suggests a role in nuclear behavior; however, this spatial organization is dispensable for nuclear autonomy.
Abstract
Nuclei in syncytia found in fungi, muscles, and tumors can behave independently despite cytoplasmic translation and the homogenizing potential of diffusion. We use a dynactin mutant strain of the multinucleate fungus Ashbya gossypii with highly clustered nuclei to assess the relative contributions of nucleus and cytoplasm to nuclear autonomy. Remarkably, clustered nuclei maintain cell cycle and transcriptional autonomy; therefore some sources of nuclear independence function even with minimal cytosol insulating nuclei. In both nuclear clusters and among evenly spaced nuclei, a nucleus’ transcriptional activity dictates local cytoplasmic contents, as assessed by the localization of several cyclin mRNAs. Thus nuclear activity is a central determinant of the local cytoplasm in syncytia. Of note, we found that the number of nuclei per unit cytoplasm was identical in the mutant to that in wild-type cells, despite clustered nuclei. This work demonstrates that nuclei maintain autonomy at a submicrometer scale and simultaneously maintain a normal nucleocytoplasmic ratio across a syncytium up to the centimeter scale.
INTRODUCTION
Classic experiments demonstrated that cytoplasmic factors influence cell cycle progression (Johnson and Rao, 1971). However, nuclei of multinucleate tissue culture cells were later found to divide independently within one cell (Ghosh et al., 1978). Such nuclear-autonomous division also arises naturally in some filamentous fungi, such as Ashbya gossypii (Gladfelter et al., 2006). Nuclear autonomy has also been observed in other processes, including transcription in multinucleate skeletal muscle and placental cells (Bursztajn et al., 1989; Duca et al., 1998; Fogarty et al., 2011). This autonomy is surprising, given translation in a common cytoplasm, and has implications for syncytial development and growth.
In A. gossypii, there is evidence for division asynchrony arising from both nuclear-intrinsic variables and organization of the cytoplasm insulating neighboring nuclei. Division times of sister nuclei are positively correlated independent of the distance traveled from one to another, consistent with nuclear-intrinsic regulation (Anderson et al., 2013). However, regulated CLN3 (G1 cyclin) transcript cytoplasmic positioning and analysis of division times between nuclei that pass one another, as well as in mutants with slightly reduced internuclear distances, suggest that cytoplasmic signals also influence division (Anderson et al., 2013; Lee et al., 2013). Whether nuclear and cytoplasmic contributions to autonomy can function independently or are interconnected has been unclear.
To dissect the relative contributions of nucleus and cytoplasm to nuclear autonomy in A. gossypii, we analyzed the nuclear division cycle in a strain exhibiting severe nuclear spacing defects (Grava et al., 2011). We show that nuclei can behave autonomously even when highly clustered and yet maintain a consistent number of nuclei per unit volume of cytoplasm (#N/C).
RESULTS AND DISCUSSION
jnm1Δ cells maintain nuclear division autonomy
A. gossypii cells lacking the Jnm1 dynactin subunit exhibit a severe nuclear clustering phenotype with clusters 31.0 ± 9.2 μm apart (Figure 1, A and B, and Supplemental Figure S1C; Grava et al., 2011). Mutant nuclear diameter was the same as for wild type (WT; Figure 1C). Average #N/C was the same as for WT; however, mutant #N/C had a higher SD (Figure 1D). Hyphal growth rates were comparable between WT and jnm1Δ, and we observed no radial growth defects, in contrast with previous work (Supplemental Figure S1, A and B; Grava et al., 2011). This suggests that #N/C does not depend on local signals operating at the scale of single nuclei. Instead, there is integration across the entire cell for growth rate and #N/C regulation. In some plants, flow generated by cytoskeletal motor activity is believed to facilitate nutrient dispersal (Goldstein et al., 2008). Compromised dynein activity in jnm1Δ cells may reduce cytoplasmic mixing and result in local variations in these cell-wide signaling molecules, leading to greater variability among jnm1Δ #N/C values.
FIGURE 1:
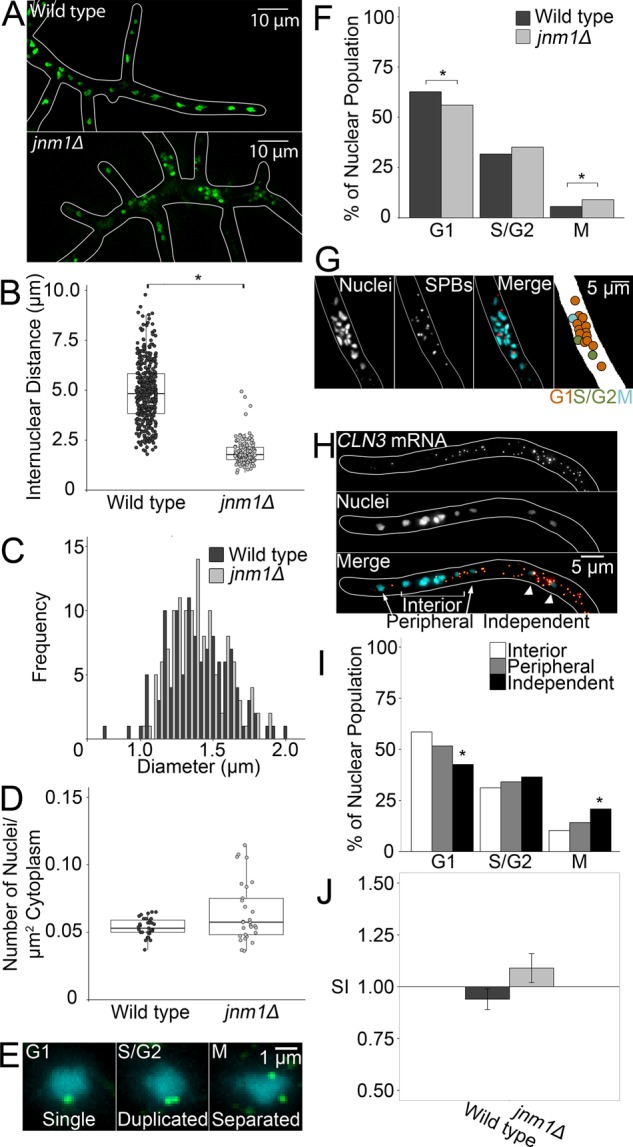
jnm1Δ cells grow similarly to WT A. gossypii but with decreased internuclear distance. *p < 0.05 by Kolmogorov–Smirnov (KS) test (A–E) or Z test (F–J). (A) Representative image of WT and jnm1Δ cells. Nuclei are shown in green and hyphal outline in white. (B) Internuclear distance in WT and jnm1Δ cells (n ≥ 199 nuclei, clusters average ∼30 μm apart). (C) Nuclear diameter in WT and jnm1Δ (n > 110). (D) Number of nuclei per square micrometer of cytoplasm (n ≥ 29 cells; SD: WT, 0.007; jnm1Δ, 0.022). (E) Representative images of SBPs in different cell cycle stages. SPBs are shown in green and nuclei in cyan. Images are presented as maximum-intensity projections for clarity; however, analyses were performed using Z-series. (F) Percentages of nuclei in each cell cycle state in WT and jnm1Δ (n ≥ 849 nuclei). (G) Representative image of cycle stages within a jnm1Δ nuclear cluster. (H) Representative image of jnm1Δ nuclei in different positions. (I) Percentage of jnm1Δ nuclei in each position in each cell cycle stage (n ≥ 54 interior, n ≥ 30 peripheral, n ≥ 24 interior nuclei). (J) SI of WT and jnm1Δ nuclei (n ≥ 775 interactions). Bars denote SE.
We next assessed nuclear division in jnm1Δ cells, predicting that clustered nuclei should divide more synchronously due to their close proximity in shared cytosol. Based on spindle pole body (SPB) morphology, cell cycle proportions are strikingly similar between WT and jnm1Δ, and all stages are observed in one cluster (Figure 1, E–G). We noted a slight but significant decrease in the percentage of G1- and increase in M-phase jnm1Δ nuclei. To assess whether local cytoplasm volume influences cell cycle progression, we classified nuclei as within the cluster interior, at the periphery, or independent (Figure 1H). Deviation from WT cell cycle proportions was more pronounced among independent nuclei, with fewer G1- and more M-phase nuclei than interior or peripheral nuclei. However, clustered nuclei also differed from WT (Figure 1I). This suggests that even clustered nuclei might respond differently to their local cytoplasm. Therefore we next determined whether states of neighboring nuclei are correlated by calculating their synchrony index (SI; Nair et al., 2010). The SI compares the incidence of each type of interaction between neighbors (e.g., G1-G1, G1-S/G2, G1-M, etc.) to the number of interactions expected based on the proportions of each cell cycle state in the population. This can be evaluated using joint-count statistics (Moran, 1948). A value of 1 indicates that the incidence of neighboring nuclei in the same state is no different from that expected by chance. Values >1 indicate increased synchrony, and values <1 suggest anticorrelation of states between neighbors. As in the WT, the jnm1Δ SI is indistinguishable from 1, indicating that, remarkably, clustered nuclei are autonomous (Figure 1J). It is remarkable that jnm1Δ cells, which exhibit such striking nuclear position abnormalities, maintain normal growth and autonomous nuclear cycle progression.
A. gossypii nuclei are transcriptionally autonomous in WT and jnm1Δ
We speculated that transcription might be at the intersection of nuclear independence and cytoplasmic inputs into the division cycle. To assess the transcriptional state of each nucleus, we labeled transcripts encoding four cyclins (Supplemental Figure S1D) and, as a control, a septin (CDC12), using single-molecule RNA fluorescence in situ hybridization (smFISH; Figure 2, A and B, and Supplemental Figure S2). Bright, nuclear foci containing multiple transcripts were classified as active transcription sites (Figure 2C). The frequency of these signals was reduced by thiolutin, suggesting that these foci label active transcription (∼40% reduction after 20 min; Supplemental Figure S1D). Consistent with previous work, we detected these sites only in a subset of nuclei (Lee et al., 2013). The percentage of transcriptionally active nuclei varied from gene to gene (<10 to >80%; Figure 2E) and increased in jnm1Δ cells for all genes examined except CLN3. The maintenance of WT transcription levels of this cyclin may result from feedback regulation due to this gene product's pivotal role early in the cell cycle.
FIGURE 2:
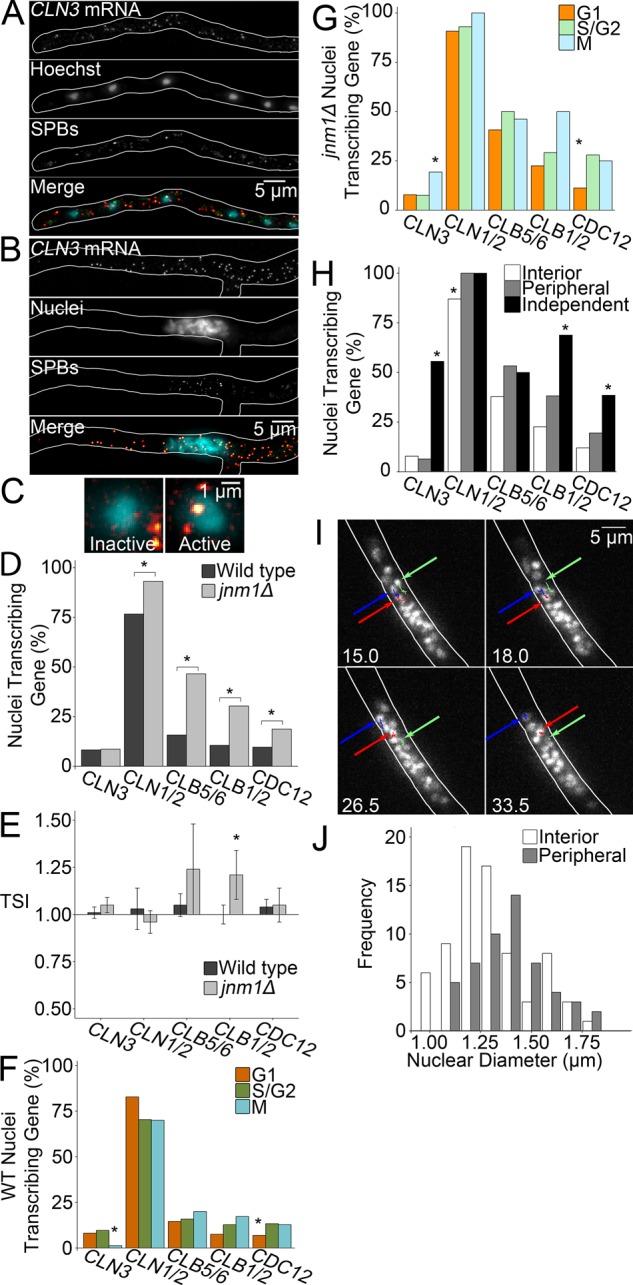
WT and jnm1Δ nuclei are transcriptionally autonomous. *p < 0.05 by Z test. (A, B) Representative images of smFISH and SPB labeling: (A) WT. (B) jnm1Δ. (C) Representative images of transcriptionally inactive and active nuclei by smFISH. (D) Percentage of transcriptionally active nuclei for each gene in WT and jnm1Δ (n ≥ 99 nuclei). (E) TSI for WT and jnm1Δ nuclei (n ≥ 96 interactions). Bars denote SE. (F) Percentage of WT nuclei in each cell cycle stage actively transcribing each gene (n ≥ 64 G1, n ≥ 54 S/G2, n ≥ 15 M nuclei). (G) Percentage of jnm1Δ nuclei in each cell cycle stage actively transcribing each gene (n ≥ 47 G1, n ≥ 27 S/G2, n ≥ 14 M nuclei). (H) Percentage of jnm1Δ nuclei in each position actively transcribing each gene (n ≥ 35 interior, n ≥ 12 peripheral, n ≥ 3 interior nuclei). (I) Frames from diSPIM time-lapse acquisition demonstrating jnm1Δ nuclear movement within clusters. Colored arrows mark the position of a specific nucleus in each frame; see also Supplemental Movie S1. Time is displayed in minutes. (J) Nuclear diameter of peripheral and interior jnm1Δ nuclei (n ≥ 53 nuclei, p < 0.05 by KS test).
The increase in transcriptionally active nuclei might reflect increased transcriptional synchrony among jnm1Δ nuclei. We therefore assessed the frequency with which neighboring nuclei are in the same transcriptional state by adapting the SI to create a transcriptional synchrony index (TSI). As with the SI, the number of neighbor interaction types (inactive-inactive, active-active, or inactive-active) in cells was compared with that expected by chance. The WT TSI was statistically indistinguishable from chance for each cyclin examined (Figure 2E). However, WT nuclei transcribing the septin CDC12 were next to other nuclei transcribing CDC12 more frequently than by chance (Supplemental Figure S1F), suggesting that genes not involved in nuclear-autonomous processes may be more coordinated among nuclei than those that are. These CDC12-expressing nuclear pairs may be located near assembling septin structures—for example, new branch points or hyphal tips. Further research is required to assess this possibility. Nuclear-autonomous transcription was maintained in jnm1Δ cells, except for the M-phase cyclin CLB1/2, which might be caused by the greater percentage of M-phase jnm1Δ nuclei. Thus nuclei not only divide asynchronously but also maintain distinct transcriptional programs in both WT and mutants with clustered nuclei.
We next examined cyclin expression relative to the cell cycle and found that, in contrast with other species, only transcription of the CLN3 cyclin varied by cell cycle stage (Figure 2, F and G). This was true for both strains; however, in WT, fewer M-phase nuclei transcribe CLN3, and the opposite is true in jnm1Δ. This is also the only gene not transcribed by a larger percentage of jnm1Δ nuclei. Previous work showed that all cyclins except CLB5/6 are detectable within A. gossypii nuclei at all cell cycle stages (Hungerbuehler et al., 2007). This suggests that in the common cytoplasm of these cells, cyclin protein function is regulated via posttranscriptional mechanisms. We also observed cell cycle–dependent CDC12 transcription in WT and jnm1Δ, which was unexpected because septation is not known to be cell cycle regulated in A. gossypii. These data show that cyclin transcription is not strictly coupled to cell cycle progression in A. gossypii, and nuclei exhibit multiple, independent autonomous behaviors: division and transcription.
Transcription is sensitive to nuclear location within a cluster
We next looked at variables other than the cell cycle that might control cyclin transcription, such as local cytoplasm availability. For every gene, increased exposure to cytoplasm correlated with a greater percentage of active nuclei (Figure 2H). Nuclei may actively respond to their local cytoplasm, or, alternatively, a population of quiescent nuclei may remain in the cluster interior. To distinguish between these possibilities, we assessed nuclear movement in clusters with low photobleaching and high temporal resolution via dual-view light sheet microscopy (diSPIM; Wu et al., 2013; Kumar et al., 2014). Nuclei move within clusters (Figure 2I, Supplemental Video S1, and Supplemental Figure S1G), suggesting that transcriptional activity is dynamic and expression can change as nuclei experience different cluster positions, as well as indicating that nuclei can respond to cytoplasmic differences on very small scales. In other systems, nuclei enlarge when given more available cytoplasm, which has been linked to chromatin expansion and transcriptional up-regulation (Gurdon, 1976; Gurdon et al., 1976; Neumann and Nurse, 2007). Consistent with this, peripheral jnm1Δ nuclei are larger than interior nuclei, although the size distribution of the whole population resembles that of WT (Figures 1C and 2J).
mRNA is enriched near nuclei in both WT and jnm1Δ
How are nuclei both transcribing and cycling independently, as well as being sensitive to local cytoplasmic abundance and maintaining the same cellular #N/C? jnm1Δ cells must account for thousands of cubic micrometers of cytoplasm unoccupied by nuclei. Given the evidence for both local and global perception of cytoplasm, we examined the cytoplasmic localization of transcripts in jnm1Δ. We first assessed the cytoplasmic concentration of each transcript and found a greater average cytoplasmic density of each transcript in the mutant (Figure 3A). This, together with the increased proportion of transcriptionally active jnm1Δ nuclei in Figure 2D, suggests that transcripts may be overproduced in the mutant. It is also possible that decreased transcript degradation plays a role, but this has not been evaluated. There was also a greater variability of transcript densities in the mutant. CLN1/2 could not be measured at the level of single transcripts because of very high concentrations. We then examined transcript localization relative to nuclei, first focusing on WT. Previously CLN3 was demonstrated to be clustered in cytosol and restricted near nuclei (Lee et al., 2013). This previous study indicated that transcripts encoding other cyclins are not clustered relative to each other, but it was unclear whether the other cyclins and/or transcripts in general may be enriched near nuclei. Of note, we found enrichment for all cyclin genes in the vicinity of WT nuclei and less enrichment for CDC12, not known to be involved in nuclear-autonomous processes (Figure 3B).
FIGURE 3:
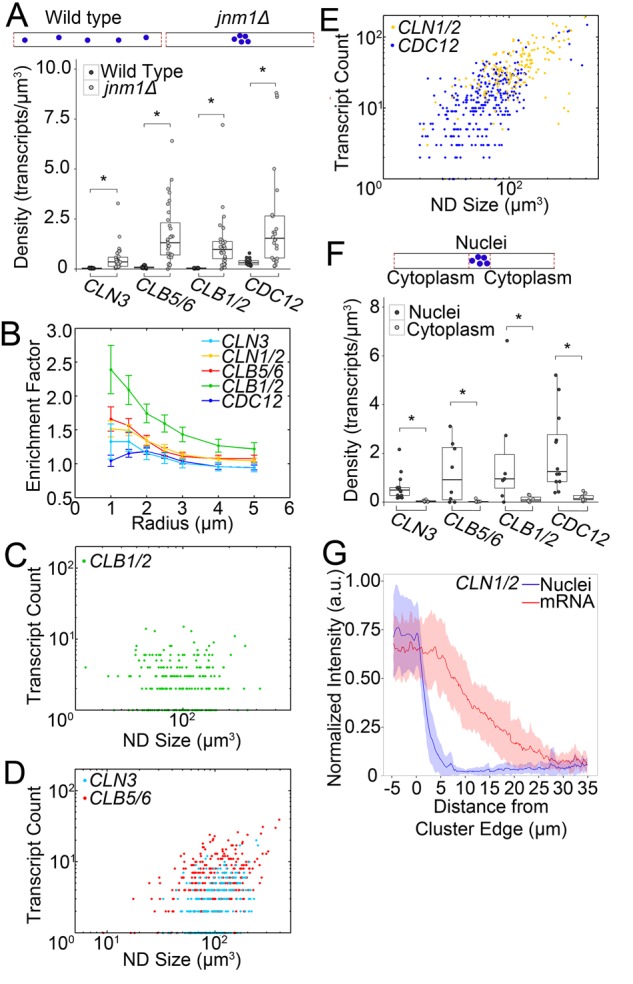
mRNA is enriched near A. gossypii nuclei. *p < 0.05 by KS test. (A) Top, cytoplasmic regions compared between WT and jnm1Δ. Bottom, cytoplasmic transcript density in WT and jnm1Δ (n ≥ 14 hyphae). (B) Enrichment scores for transcripts relative to WT nuclei (n ≥ 161 nuclei). Bars denote SE. (C–E) Number of transcripts per nucleus by ND size (n ≥ 161 nuclei, CLB1/2 r = −0.01, CLN3 r = 0.40, CLB5/6 r = 0.40, CLN1/2 r = 0.62, CDC12 r = 0.62). (F) Top, nuclear cluster region and nucleus-free regions of cytoplasm. Bottom, cytoplasmic density in jnm1Δ clusters and nuclear-free regions (n ≥ 7 hyphae). (G) CLN1/2 transcript signal intensity relative to nuclear clusters (0 indicates cluster edge; n = 30 cluster edges). Shaded region denotes SD.
Cyclin enrichment near nuclei prompted further investigation of this localization. We examined how many transcripts are located near each WT nucleus relative to the volume of cytoplasm adjacent to that nucleus, which we refer to as the nuclear domain (ND). ND size is calculated by assigning each voxel in the hypha an index corresponding to the closest nucleus to that voxel (Supplemental Figure S2M). If transcripts are homogeneously distributed, ND size and transcript number will have a high correlation coefficient. However, we found low to moderate correlation coefficients between transcript count and ND size for most cyclins (Figure 3, C–E; r = −0.01–0.4), consistent with mRNA spatial restriction. The highest correlations were found for the septin CDC12 and the G1 cyclin CLN1/2, a cyclin involved in polarized growth in A. gossypii (both 0.62; Gladfelter et al., 2006; Hungerbuehler et al., 2007), suggesting that some transcripts are distributed throughout the cytoplasm rather than enriched near nuclei (Figure 3B). In jnm1Δ cells, regions containing the nuclear cluster had significantly higher transcript density than the adjacent, nucleus-free, cytoplasm (Figure 3F). For CLN1/2, we found that mRNA is also more concentrated near nuclei (Figure 3G). In WT cells, it appears that an individual nucleus sets up a cyclin mRNA gradient, whereas in jnm1Δ mutants, the nuclear collective establishes it. The patterning of cyclins relative to individual nuclei likely contributes to asynchrony; however, it is not strictly required, as clustered nuclei can still divide asynchronously.
Spatial distribution of transcripts relates to cell cycle and transcriptional state
To examine the enrichment trends of cyclins near nuclei, we determined the number of transcripts within a 1.5 μm region of interest (ROI; based on the peak enrichment distance in Figure 3B) based on cycle state (Figure 4A). We found no significant differences between the transcript numbers near nuclei in different states for any gene examined. However, previous work suggested that spatial distribution might be more important than transcript number (Lee et al., 2013). We therefore examined transcript enrichment relative to nuclei in each stage (Figure 4, B–F). Intriguingly, G1 cyclin CLN3 and M cyclin CLB1/2 exhibited enrichment changes throughout the cell cycle, based on enrichment curve shape. This indicates that mRNA spatial organization may change during the cell cycle, potentially to regulate translation, without significant changes in transcript number. Along with cyclin transcription uncoupled from cell cycle state and constitutive nuclear localization of most cyclins in A. gossypii (Hungerbuehler et al., 2007), this suggests that in these multinucleate cells, cyclin protein function is highly regulated at posttranscriptional levels.
FIGURE 4:
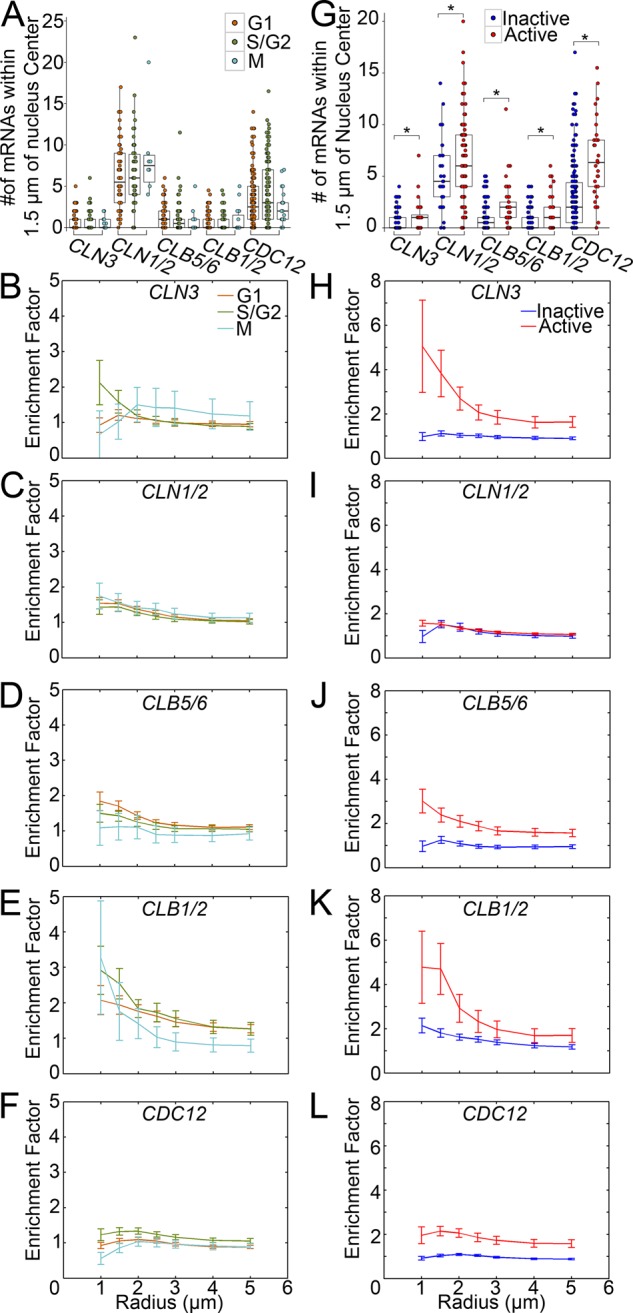
WT transcript localization correlates with nuclear autonomous behaviors. *p < 0.05 by Kolmogorov–Smirnov (KS) test. (A) Transcript number in ROI by cell cycle stage (n ≥ 64 G1, n ≥ 54 S/G2, n ≥ 10 M nuclei). (B–F) Transcript enrichment by cell cycle stage (n ≥ 161 nuclei). Bars denote SE. (G) Number of transcripts in ROI by transcriptional activity (n ≥ 28 active, n ≥ 30 inactive nuclei). (H–L) Transcript enrichment by transcriptional activity (n ≥ 161 nuclei). Bars denote SE.
We next performed the same analyses as before but based on transcriptional state. Transcriptionally active nuclei had significantly more transcripts and stronger transcript enrichment in the local cytoplasm than inactive nuclei for every gene (Figure 4, G–L). Nascent transcripts may be restricted to close proximity of their source nucleus, as they have not yet diffused away, been transported away, or been degraded, causing the increased enrichment for this population. This trend was most robust for the cyclins CLN3, CLB5/6, and CLB1/2 (Figure 4, H, J, and K). The polarity-involved cyclin, CLN1/2, and CDC12 (Figure 4, I and L) exhibited the least increase in enrichment near transcriptionally active nuclei, consistent with spatial homogenization of these transcripts involved in cell morphology.
This work indicates that nuclear-intrinsic autonomy in A. gossypii can generate independent nuclear behavior even in the absence of internuclear cytoplasmic regions. It appears that nuclear-intrinsic variability is associated with transcriptional autonomy and generates differences between NDs. In the WT, nuclear spacing promotes division autonomy between neighbors via independent NDs. The positive correlation of division times between nuclei that bypass each other suggests that exchange of ND components can influence cell cycle progression (Anderson et al., 2013). In cells with mild spacing phenotypes, we suggest that such domains surround groups of nuclei instead of individuals, which share cytoplasmic components, increasing division synchrony (Anderson et al., 2013). Within the large jnm1Δ nuclear clusters, independent NDs cannot be efficiently constructed, and nuclear-intrinsic sources of autonomy appear to dominate for reasons that are not yet clear and result in independent cell cycle progression and transcription.
Within a viscous, flowing cytosol, macromolecules would tend to stay together as they flow throughout the cell (Roper et al., 2015). Previously cytoplasmic flow within filamentous fungal cells was shown to generate distinct cytoplasmic regions (Pieuchot et al., 2015). In addition, it has been hypothesized that individual nuclei within syncytia can maintain a distinct endoplasmic reticulum, which may serve to restrict certain signals to the ND of specific nuclei (Baum and Baum, 2014). Our data on nuclear size, cell cycle state, and transcriptional activity in peripheral versus interior nuclei indicate that nuclei dynamically adapt to their local cytoplasm. This may enable nuclei to integrate cellular cues in order to respond in a manner appropriate for each autonomous nucleus but, remarkably, also for the cell at large.
Of importance, nuclear autonomy is not unique to fungi, and mechanisms of cytoplasmic organization and nuclear-intrinsic variability may be widely conserved. For example, only a subset of nuclei within multinucleate mammalian muscle cells transcribe genes necessary for neuromuscular junction formation, and not all nuclei of placental syncytia are transcriptionally active (Bursztajn et al., 1989; Duca et al., 1998; Fogarty et al., 2011). Nuclear autonomy within a common cytoplasm is an excellent readout for the functional compartmentalization of a cell.
MATERIALS AND METHODS
Strain construction
Oligonucleotides and plasmids and strains used are listed in Supplemental Table S1. All restriction enzymes are from New England Biolabs (Ipswich, MA). All oligonucleotides were synthesized at Integrated DNA Technologies (Coralville, IA), except AGO234 and AGO235, which were synthesized by Invitrogen (Carlsbad, CA). All sequencing was performed at the Dartmouth College Core Facility (Hanover, NH).
To generate plasmid pUC19-mCherry:GEN (AGB180), AGB048 and AGB021 were digested with BamHI and SapI to yield 3435– and 2029–base pair fragments, respectively. Fragments were gel-extracted using a QIAquick gel extraction kit (Qiagen, Redwood City, CA) and ligated with T4 Ligase (New England Biolabs). The resulting plasmid was isolated from transformants using a Qiagen QIAprep Spin miniprep kit. The correct product was verified by digestion with EcoRV and PvuI. To generate the plasmid pRS416 AgTUB4-mCherry:GEN (AGB182), the mCherry:GEN cassette was PCR amplified from AGB180 with AGO 400 and AGO 401 using Roche Expand high-fidelity polymerase plus dimethyl sulfoxide (Thermo Fisher Scientific, Waltham, MA). The 3045-nucleotide product was gel-extracted using a QIAquick gel extraction kit. This fragment was cotransformed into yeast with AGB144 to yield AGB182. Plasmid rescue from yeast was performed using a Qiagen QIAprep Spin miniprep kit. The plasmid was verified by digestion with BglII and NheI and sequenced with AGO318, 404, 406, and 407.
To generate AgTUB4-mCherry:GEN (AG430), plasmid AGB182 was digested with BglI and SacII. The 5485-nucleotide fragment was isolated with a Qiagen gel extraction kit and transformed into AG416. Genomic DNA was isolated using a Qiagen DNeasy Plant miniprep kit. Heterokaryons were verified by PCR with oligonucleotides AGO404, 406, 407, and 509 using New England Biolabs standard Taq polymerase. Spores were isolated and grown on selection to produce homokaryons as described previously (Alberti-Segui et al., 2001). Homokaryons were verified using the same oligonucleotides as the heterokaryons.
To generate jnm1Δ strains AG690.1 and AG733.2, the selection marker was amplified from AGB009 (NAT) or AGB021 (GEN), respectively, flanked by 45–base pair homology to the termini of AgJNM1 with primers AGO1138 and AGO1139 using Roche Diagnostics (Indianapolis, IN) Expand high-fidelity polymerase plus 5 M betaine from Sigma-Aldrich (St. Louis, MO). This PCR product was transformed by electroporation into the appropriate A. gossypii parent strain (AG430 and AG128 for AG690.1 and AG733.2, respectively) to produce heterokaryotic transformants as previously described (Alberti-Segui et al., 2001). AgJNM1 deletion was verified by two methods: PCR from genomic DNA, and examination of internuclear distance by microscopy. For PCR verification, genomic DNA was isolated from heterokaryons using the Qiagen Plant DNeasy Kit. This genomic DNA was used as a PCR template with oligonucleotides AGO234, 235, 1125, 1126, 1127, and 1128 with New England Biolabs standard Taq polymerase to verify deletion of the AgJNM1 gene and presence of the selection marker.
Microscope setup and growth conditions
Unless otherwise specified, all images were acquired using a Zeiss Axioimage-M1 upright light microscope (Carl Zeiss, Jena, Germany) equipped with a Plan-Apochromat 63×/1.4 numerical aperture oil objective and an Exfo X-Cite 120 lamp. For fluorescence imaging, the following filter cubes were used: Zeiss 38HE (GFP), Chroma 41002B (TAMRA), Zeiss 49 (Hoechst), and Chroma 41043 (mCherry; Chroma Technology, Brattleboro, VT). Images were acquired on an Orca-AG charge-coupled device (CCD) camera (C4742-80-12AG; Hamamatsu, Bridgewater, NJ) driven by μManager (Edelstein et al., 2014). Iterative deconvolution was performed using the appropriated calculated point-spread functions in Volocity (PerkinElmer-Cetus, Waltham, MA). Subsequent analyses were performed using Fiji (Schindelin et al., 2012) and Matlab (MathWorks, Natick, MA).
For imaging, Ashbya were grown in 10- to 20-ml Ashbya Full Medium (AFM) with ampicillin (100 μg/ml) and appropriate antibiotics, that is, G418 (200 μg/ml) and/or CloNAT (50 μg/ml), for ∼16-h shaking at 30°C. To image H4-GFP–labeled nuclei, cells were washed into 2× low-fluorescence minimal medium and mounted on thin 2% agarose gel pads with a 3:1 (vol/vol) ratio of 2× low-fluorescence minimal medium and AFM with appropriate selection, and edges were sealed with Valap. H4-GFP z-series were acquired using 40% neutral density, 50-ms exposure, and 0.5-μm interplane spacing through the cell (approximately eight Z-planes) and processed with iterative deconvolution (40 iterations). Internuclear distance was defined as the average distance between the center of a nucleus and the center of each neighboring nucleus. The cross-sectional area of the hypha was determined for the widest part of the hypha based on phase images, and the number of nuclei in this region was divided by this area to produce #N/C. Nuclear diameter was determined by measuring the distance across the nucleus in two perpendicular directions and averaging these lengths. Measurements were taken for both H4-GFP labeled nuclei and Hoechst-stained nuclei (see later description).
Tip growth assay
Cells grown for ∼14 h at 30°C were washed with 50 mM Tris, pH 7.5, and 150 mM NaCl and resuspended in the same buffer with 25 μg/ml fluorescein isothiocyanate–concanavalin A (FITC-ConA; EY Laboratories, San Mateo, CA). Cells were incubated at room temperature for 10 min, washed into AFM with appropriate selection, and returned to 30°C for 1 h. Cells were subsequently fixed using 3.7% formaldehyde as described and mounted using Prolong Gold mounting medium. Z-series were acquired with a 1.0-μm step size through the volume of the hypha. FITC-ConA signal was acquired using 10% neutral density and 10-ms exposure, and phase images were acquired using 50-ms exposure. Growth after FITC-ConA labeling was quantified by measuring the length of the tip region unlabeled with FITC. Measurements were taken in Fiji.
Radial growth assay
AFM agarose plates with appropriate selection were inoculated with 10 μl of clean spores in triplicate for each strain. Plates were imaged every 24 h for 10 d using a ChemiDoc XRS+ Molecular Imager system with ImageLab 5.1 software (Bio-Rad Laboratories, Hercules, CA). Mycelial area was calculated in Fiji.
diSPIM imaging
For volumetric time-lapse acquisition of jnm1Δ nuclear clusters with high temporal resolution and minimal photobleaching and phototoxicity, cells were imaged on a diSPIM system (Wu et al., 2013; Kumar et al., 2014). Cells grown for 15 h at 30°C were washed into 2× low- fluorescence minimal medium and mounted on very thin 2% agarose gel pads with 2× low-fluorescence minimal medium and appropriate selection. Mounted cells were incubated in a humid chamber at 30°C for 45 min to adhere to the gel pad. Gel pads were examined, and an area with cells visible to the eye was selected for imaging. A gel piece of ~1 × 0.5 cm with these cells was excised, transferred onto the center of a 50 × 24 mm coverslip, and sealed down with Valap on all gel edges. The coverslip was fixed in the diSPIM imaging chamber under 8–10 ml of 2× low-fluorescence medium with appropriate selection. We acquired 30- to 40-μm volumes with 1-μm interplane spacing every 30 s using both arms with 80% laser power (~250 μW) and 20-ms exposures. Images were registered and deconvolved using MIPAV (mipav.cit.nih.gov/) as described previously (Wu et al., 2013).
RNA smFISH and immunofluorescence
RNA smFISH labeling was performed as previously described (Lee et al., 2013, 2016). Briefly, cells grown overnight at 30°C in AFM were fixed using 3.7% (vol/vol) formaldehyde (Fisher Scientific) and then washed twice with ice-cold buffer B (1.2 M sorbitol and 0.1 M potassium phosphate, pH 7.5). Cells were resuspended in 1 ml of spheroplasting buffer (10 ml buffer B and 2 mM vanadyl ribonucleoside complex from New England Biolabs) and digested using 1.5 mg/ml Zymolyase (MP Biomedicals, Santa Ana, CA) for 40 min at 37°C. Cells were washed twice with ice-cold buffer B, resuspended in 70% ethanol, and incubated at 4°C overnight. Cells were then washed twice with wash buffer (20× saline sodium citrate [SSC], 10% [vol/vol] deionized formamide from Ambion, Thermo Fisher Scientific). Tetramethyl rhodamine (TAMRA)–conjugated RNA FISH probes (Biosearch Technologies, Novato, CA) complementary to each transcript of interest were initially resuspended in 20 μl of TE buffer (10 mM Tris-Cl, 1 mM EDTA, pH 8.0) and diluted 1:10 to yield the working stock. One microliter of working stock was diluted into 100 μl of hybridization buffer (1 g of dextran sulfate from Sigma-Aldrich, 10 mg of Escherichia coli tRNA from Roche Diagnostics, 2 mM vanadyl ribonucleoside complex, 2 mg of bovine serum albumin (BSA) from Sigma-Aldrich, 20× SSC, and 10% [vol/vol] deionized formamide), and this hybridization solution was used to resuspend the washed cells. This solution was incubated in the dark overnight at 37°C. Cells were washed once with wash buffer, incubated for 30 min in wash buffer at 37°C, and washed again with wash buffer. Cells were resuspended in 500 μl of wash buffer with 5 μg/ml Hoechst (Invitrogen) and incubated at room temperature for 30 min. Cells were washed a final time with wash buffer, mounted on glass slides with 20 μl of Prolong Gold mounting medium (Invitrogen), sealed with nail polish, and imaged.
For SPB counterstaining, after probe hybridization and the 30-min wash buffer incubation, cells were washed twice with 1× phosphate-buffered saline PBS and blocked in 200 μl of 1× PBS with 1 mg/ml BSA for 1 h at room temperature. Cells were washed once in 1× PBS and resuspended in 200 μl of 1× PBS with 1 mg/ml BSA and 1:200 GFP Booster Atto 488 (ChromoTek, Planegg-Martinsried, Germany) and incubated at 4°C overnight. Cells were washed three times with 500 μl of 1× PBS, and nuclear counterstaining was performed by resuspending cells in 500 μl of 1× PBS with 5 μg/ml Hoechst and incubating at room temperature for 30 min. Cells were washed three times in 1× PBS, resuspended in mounting medium, mounted, and sealed as described. Z-series were acquired with a 0.3-μm step size through the entire volume of the hypha (∼4 μm), and exposure settings were as follows: TAMRA smFISH probes, 100% neutral density 100 ms; GFP Booster SPBs, 100% neutral density, 100 ms; and Hoechst-labeled nuclei, 40% neutral density 50 ms. Iterative deconvolution was performed using Volocity as follows: 40 iterations for nuclei, 100 iterations for smFISH signal, and SPB signal not deconvolved.
Synchrony indices
The cell cycle synchrony index was determined as described previously (Nair et al., 2010). For both cell cycle and transcriptional synchrony indices, the incidence of each type of interaction between neighbors (e.g., G1 neighboring G1; G1 neighboring S/G2; transcriptionally active neighboring transcriptionally active; etc.) was quantified. Nuclear neighbors were defined as nuclei whose NDs directly contact each other. The quantities of each interaction were used to generate the ratio of observed/chance counts based on the proportions of each category in the population (either cell cycle state or transcriptional state), which enables comparison of results between populations of different proportions, that is, genes that vary in expression penetrance. This ratio can be evaluated using joint-count statistics (Moran, 1948). Values close to 1 indicate that the incidence of neighboring nuclei in the same state is no different from expected by chance. Values >1 occur when neighbors are more commonly in the same state than expected by chance, which suggests coordination between neighbors. A value <1 indicates that pairs of nuclei are in different states with a higher frequency than by chance, and the behaviors of neighboring nuclei are potentially anticorrelated due to interference between neighbors.
smFISH analyses
For assessing the cycle state of each nucleus, a single SPB was denoted as “G1,” duplicated SPBs as “S/G2,” and separated SPBs as “M” (Figure 1E), a categorization based on the behavior of Saccharomyces cerevisiae SPBs (Pringle and Hartwell, 1981; Simmons Kovacs et al., 2008) due to the high degree of conservation between A. gossypii and S. cerevisiae (>90% syntenic homology; Dietrich et al., 2004).
For assessing the transcriptional activity of each nucleus, the integrated density of a single, cytoplasmic mRNA was calculated for each image. Any smFISH foci determined to be at least two times the value of a single mRNA and located within a nucleus (based on Hoechst signal) was identified as an active transcription site. Nuclei containing active transcription sites were classified as transcriptionally active for that gene. To confirm that large, nuclear foci indicate sites of active transcription, cells were treated with 3 μg/ml thiolutin for 20 min before fixation and smFISH processing.
For determining the number of transcripts within a 1.5-μm ROI centered on each nucleus, nuclei and mRNAs were detected, and the integrated density value for a single mRNA was determined using Fiji as previously described (Lee et al., 2016). A custom MATLAB script was used to calculate the number of transcripts detected within 1.5 μm of each nuclear center. Similarly, MATLAB scripts were used to determine the transcript density within the entire hypha (WT) or specific hyphal regions (jnm1Δ). To compare the transcript density between WT and jnm1Δ, the number of nuclei within a mutant nuclear cluster was determined. This value was multiplied by the average internuclear distance in the WT in order to determine the cytoplasmic area that number of nuclei would occupy, given wild-type spacing. The density of transcripts was determined in this region for comparison with wild type. MATLAB was also used to determine transcript density in areas of the hypha containing the nuclear cluster or cluster-free regions of cytoplasm. For assessment of CLN1/2 smFISH signal, the signal intensity was determined by tracing the hypha with a segmented line of the same width as the hypha. The line trace tool in Fiji was used to determine the average signal intensity across the width of this line. Cluster edges were registered in these line traces, and intensity was normalized based on the dynamic range for each hypha (i.e., maximum signal is represented by 1 and cytoplasmic background by 0).
Three-dimensional hyphal reconstruction for Voronoi volume (ND) and enrichment analyses
The edge of each hypha was found from a single, phase microscopy image from the center of the z-series. In contrast, the nuclei and transcripts detected with Fiji each have three-dimensional (3D) position data (Supplemental Figure S2I). The first analysis step is to reconstruct the 3D shape of the hypha from the two-dimensional (2D) phase image. The center surface of the hypha was estimated by fitting a multinomial function (cubic in x, linear in y) to the x, y, z-coordinates of all of the detected mRNAs and nuclei. This fit minimizes the sum of squares of the z-distances between the central surface and the detected mRNAs and nuclei and creates a ribbon that approximates the center surface of the hypha by associating with each x, y-pixel within the hyphal outline a z-coordinate (Supplemental Figure S2J). For each x, y-coordinate, the distance of the pixel to the nearest hyphal edge was measured within the 2D projection. A 3D object was then built out from the 2D ribbon, creating a 3D grid of voxels. Each voxel is a cube whose dimensions are equal to the x, y-pixel size of the microscope images (i.e., 0.1 × 0.1 × 0.1 μm3; the z-steps are rescaled so that voxels are isotropic). Each x, y, z-point in the ribbon is replaced by a sphere whose radius is equal to the distance of the corresponding x, y-point from the nearest hyphal edge (Supplemental Figure S2J). It was necessary to regularize the distribution of sphere radii used for volume reconstruction if the hyphal image contained a branch point. The center of a branch point may be much further from the nearest nonhyphal pixel than the typical hyphal radius. Replacing this point by a sphere creates a large spherical bulge in the hyphal. There is no way, using the combination of 2D and 3D data available for hyphal reconstruction, to rule out that such spheres do occur, but they are not observed using other imaging methods. Accordingly, we use the fact that if a hypha were a cylinder, then the distribution of distances between interior and exterior points would be linear. We found that real distances conformed closely to a linear distribution, except for bulge artifacts. Therefore real distributions were regularized by fitting them to a straight line. Sphere radii that exceeded the maximum radius allowed by the linear fit were reduced to this maximum radius. This regularization method produced 3D reconstructions that resembled the known extruded circular shapes of real hypha and did not bulge at branching points (Supplemental Figure S2K). This method builds a 3D model for the hyphal volume that encloses the detected mRNAs and nuclei. Small numbers (<5%) of mRNA signals were detected outside of this volume. It is likely that these signals represent background noise, since most mRNAs detected outside of the hyphal volume were found far from other mRNAs or nuclei and as much as 10 μb from the reconstructed hyphal boundary.
Voronoi volume (ND) assignment
The hyphal volume was partitioned into nuclear neighborhoods using a discretized Voronoi method. Specifically, each voxel in the reconstructed hypha was assigned an index corresponding to the single closest nucleus to that voxel. Regions of hypha that are assigned the same index form the Voronoi neighborhood of each nucleus. Voronoi neighborhoods divide the hyphal volume by slices, with each slice consisting of the plane of points that is exactly equidistant between neighboring nuclei (Supplemental Figure S2L). Because our method assigns each voxel a neighborhood index, we can assign mRNAs to nuclei by finding the index in the nearest voxel to the mRNA (Supplemental Figure S2M).
Enrichment analyses
The total number of mRNAs contained within control spheres of different radii (1, 1.5, 2, 2.5,…, 5 μm) centered on each nucleus was counted. These spheres were constrained from overlapping from each other or from leaving the reconstructed hyphal interior (only the part of the sphere contained in the Voronoi neighborhood of each nucleus is considered). The volume of the nucleus is also excluded (i.e., remove a 1-μm-diameter sphere centered on the detected nuclear center) because mRNAs detected within this region contribute to transcription activity counts and not to estimates of mRNA abundance. mRNA count data are presented as enrichment factors, calculated by dividing the observed number of mRNAs by the number that would be expected if all mRNAs were uniformly dispersed through the entire available cytoplasmic volume. In Figures 3 and 4, the colored curves represent the mean enrichment factors for all control spheres, and the error bars represent the uncertainty in this mean (i.e., the SD in the enrichment factor between different nuclei divided by the square root of the number of nuclei counted). Poisson noise in the counts of randomly distributed mRNAs can produce an additional source of variation in enrichment factors. However, we showed, using Monte Carlo simulations of randomly distributed mRNAs, that this noise negligibly affects the uncertainty in the mean enrichment factor per control sphere.
Supplementary Material
Acknowledgments
We thank all Gladfelter lab members and Charles Cole, Erik Griffin, James Moseley, Roger Sloboda, and Hugh Roberts for discussions and critical reading of the manuscript. This work was supported by National Institutes of Health R01-GM081506 (A.S.G., S.E.R., and P.O.), the National Science Foundation GK-12 Program and the Neukom Institute at Dartmouth College (S.E.R.), the Alfred P. Sloan Foundation and National Science Foundation DMS-1351860 (M.R. and S.-S.C.), a National Institutes of Health Ruth L. Kirschstein National Research Service Award (T32-GM008185; S.-S.C.), and the Intramural Research Programs of the National Institutes of Health National Institute of Biomedical Imaging and Bioengineering Whitman Investigator and Grass Foundation Programs at the Marine Biological Laboratory at Woods Hole (A.K. and H.S.)
Abbreviations used:
- #N/C
number of nuclei per unit volume of cytoplasm
- ND
nuclear domain.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-02-0129) on May 18, 2016.
REFERENCES
- Alberti-Segui C, Dietrich F, Altmann-Jöhl R, Hoepfner D, Philippsen P. Cytoplasmic dynein is required to oppose the force that moves nuclei towards the hyphal tip in the filamentous ascomycete Ashbya gossypii. J Cell Sci. 2001;114:975-986. doi: 10.1242/jcs.114.5.975. [DOI] [PubMed] [Google Scholar]
- Anderson CA, Eser U, Korndorf T, Borsuk ME, Skotheim JM, Gladfelter AS. Nuclear repulsion enables division autonomy in a single cytoplasm. Curr Biol. 2013;23:1999–2010. doi: 10.1016/j.cub.2013.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum DA, Baum B. An inside-out origin for the eukaryotic cell. BMC Biol. 2014;12:76. doi: 10.1186/s12915-014-0076-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bursztajn S, Berman SA, Gilbert W. Differential expression of acetylcholine receptor mRNA in nuclei of cultured muscle cells. Proc Natl Acad Sci USA. 1989;86:2928–2932. doi: 10.1073/pnas.86.8.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich FS, Voegeli S, Brachat S, Lerch A, Gates K, Steiner S, Mohr C, Pöhlmann R, Luedi P, Choi S, et al. The Ashbya gossypii genome as a tool for mapping the ancient Saccharomyces cerevisiae genome. Science. 2004;304:304–307. doi: 10.1126/science.1095781. [DOI] [PubMed] [Google Scholar]
- Duca KA, Chiu KP, Sullivan T, Berman SA, Bursztajn S. Nuclear clustering in myotubes: a proposed role in acetylcholine receptor mRNA expression. Biochim Biophys Acta. 1998;1401:1–20. doi: 10.1016/s0167-4889(97)00118-3. [DOI] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using μManager software. J Biol Methods. 2014;1:e10. doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogarty NME, Mayhew TM, Ferguson-Smith AC, Burton GJ. A quantitative analysis of transcriptionally active syncytiotrophoblast nuclei across human gestation. J Anat. 2011;219:601–610. doi: 10.1111/j.1469-7580.2011.01417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Paweletz N, Ghosh I. Mitotic asynchrony of multinucleate cells in tissue culture. Chromosoma. 1978;65:293–300. doi: 10.1007/BF00286409. [DOI] [PubMed] [Google Scholar]
- Gladfelter AS, Hungerbuehler AK, Philippsen P. Asynchronous nuclear division cycles in multinucleated cells. J Cell Biol. 2006;172:347–362. doi: 10.1083/jcb.200507003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein RE, Tuval I, van de Meent J-W. Microfulidics of cytoplasmic streaming and its implications for intracellular transport. Proc Natl Acad Sci USA. 2008;105:3663-3667. doi: 10.1073/pnas.0707223105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grava S, Keller M, Voegeli S, Seger S, Lang C, Philippsen P. Clustering of nuclei in multinucleated hyphae is prevented by dynein-driven bidirectional nuclear movements and microtubule growth control in Ashbya gossypii. Eukaryot Cell. 2011;10:902–915. doi: 10.1128/EC.05095-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurdon JB. Injected nuclei in frog oocytes: fate, enlargement, and chromatin dispersal. J Embryol Exp Morphol. 1976;36:523–540. [PubMed] [Google Scholar]
- Gurdon JB, De Robertis EM, Partington G. Injected nuclei in frog oocytes provide a living cell system for the study of transcriptional control. Nature. 1976;260:116–120. doi: 10.1038/260116a0. [DOI] [PubMed] [Google Scholar]
- Hungerbuehler AK, Philippsen P, Gladfelter AS. Limited functional redundancy and oscillation of cyclins in multinucleated Ashbya gossypii fungal cells. Eukaryot Cell. 2007;6:473–486. doi: 10.1128/EC.00273-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RT, Rao PN. Nucleo-cytoplasmic interactions in the acheivement of nuclear synchrony in DNA synthesis and mitosis in multinucleate cells. Biol Rev Camb Philos Soc. 1971;46:97–155. doi: 10.1111/j.1469-185x.1971.tb01180.x. [DOI] [PubMed] [Google Scholar]
- Kumar A, Wu Y, Christensen R, Chandris P, Gandler W, McCreedy E, Bobinsky A, Colón-Ramos DA, Bao Z, McAuliffe M, et al. Dual-view plane illumination microscopy for rapid and spatially isotropic imaging. Nat Protoc. 2014;9:2555–2573. doi: 10.1038/nprot.2014.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Roberts SE, Gladfelter AS. Quantitative spatial analysis of transcripts in multinucleate cells using single-molecule FISH. Methods 98, 124–133. 2016. [DOI] [PMC free article] [PubMed]
- Lee C, Zhang H, Baker AE, Occhipinti P, Borsuk ME, Gladfelter AS. Protein aggregation behavior regulates cyclin transcript localization and cell-cycle control. Dev Cell. 2013;25:572–584. doi: 10.1016/j.devcel.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran PAP. Interpretation of statistical maps. J R Stat Soc. 1948;10:243–251. [Google Scholar]
- Nair DR, D’Ausilio CA, Occhipinti P, Borsuk ME, Gladfelter AS. A conserved G1 regulatory circuit promotes asynchronous behavior of nuclei sharing a common cytoplasm. Cell Cycle. 2010;9:3771–3779. doi: 10.4161/cc.9.18.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann FR, Nurse P. Nuclear size control in fission yeast. J Cell Biol. 2007;179:593–600. doi: 10.1083/jcb.200708054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieuchot L, Lai J, Loh RA, Leong FY, Chiam KH, Stajich J, Jedd G. Cellular subcompartments through cytoplasmic streaming. Dev Cell. 2015;34:410–420. doi: 10.1016/j.devcel.2015.07.017. [DOI] [PubMed] [Google Scholar]
- Pringle JR, Hartwell LH. The Saccharomyces cerevisiae cell cycle. In: Strathern JD, Jones EW, Broach JR, editors. The Molecular Biology of the Yeast Saccharomyces: Life Cycle and Inheritance. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1981. pp. 97–142. [Google Scholar]
- Roper M, Lee C, Hickey PC, Gladfelter AS. Life as a moving fluid: fate of cytoplasmic macromolecules in dynamic fungal syncytia. Curr Opin Microbiol. 2015;26:116–122. doi: 10.1016/j.mib.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Priebisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons Kovacs LA, Nelson CL, Haase SB. Intrinsic and cyclin-dependent kinase-dependent control of spindle pole body duplication in budding yeast. Mol Biol Cell. 2008;19:3243–3253. doi: 10.1091/mbc.E08-02-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Wawrzusin P, Senseney J, Fischer RS, Chirstensen R, Santella A, York AG, Winter PW, Waterman CM, Bao Z, et al. Spatially isotropic four-dimensional imaging with dual-view plane illumination microscopy. Nat Biotechnol. 2013;31:1032–1038. doi: 10.1038/nbt.2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.