

Two functionally irreplaceable and molecularly separable modules in cTAGE5 are both required for collagen VII export from the ER. The concentration of Sec12 induced by cTAGE5 serves for efficient production of activated Sar1 around ER exit sites, and the GTPase cycle of Sar1 seems to be required for collagen VII export from the ER.
Abstract
Two independent functions of cTAGE5 have been reported in collagen VII export from the endoplasmic reticulum (ER). cTAGE5 not only forms a cargo receptor complex with TANGO1, but it also acts as a scaffold to recruit Sec12, a guanine-nucleotide exchange factor for Sar1 GTPase, to ER exit sites. However, the relationship between the two functions remains unclear. Here we isolated point mutants of cTAGE5 that lost Sec12-binding ability but retained binding to TANGO1. Although expression of the mutant alone could not rescue the defects in collagen VII secretion mediated by cTAGE5 knockdown, coexpression with Sar1, but not with the GTPase-deficient mutant, recovered secretion. The expression of Sar1 alone failed to rescue collagen secretion in cTAGE5-depleted cells. Taken together, these results suggest that two functionally irreplaceable and molecularly separable modules in cTAGE5 are both required for collagen VII export from the ER. The recruitment of Sec12 by cTAGE5 contributes to efficient activation of Sar1 in the vicinity of ER exit sites. In addition, the GTPase cycle of Sar1 appears to be responsible for collagen VII exit from the ER.
INTRODUCTION
Collagens synthesized in the endoplasmic reticulum (ER) fold into trimers of long (>300 nm), rigid structures that are secreted to constitute the extracellular matrix (Ishikawa et al., 2015; Malhotra and Erlmann, 2015). The mechanism of collagen export from the ER seems distinct from conventional transport mechanisms because normal carriers budding from the ER as COPII-coated vesicles are ∼60–90 nm in diameter and too small to accommodate collagens (Miller and Schekman, 2013; Malhotra et al., 2015; Saito and Katada, 2015).
Several molecules specifically involved in collagen exit from the ER have been identified (Saito et al., 2009, 2011, 2014; Jin et al., 2012; Venditti et al., 2012; Nogueira et al., 2014; Santos et al., 2015). Among them, cTAGE5 forms a collagen VII cargo receptor complex with TANGO1. cTAGE5 and TANGO1 are both integral membrane proteins localized at ER exit sites and interact with each other by their cytoplasmic coiled-coil regions. C-terminal, proline-rich sequences of both proteins bind to the COPII inner-coat complex Sec23/Sec24, and the N-terminal SH3-like fold in TANGO1 interacts with collagen VII, indicating that these interactions modify the mechanism of conventional COPII transport to accommodate collagen VII into the carriers (Saito et al., 2009, 2011).
cTAGE5 has also been reported as a scaffold for Sec12, a guanine-nucleotide exchange factor (GEF) for Sar1 GTPase. This interaction seems required not only for concentration of Sec12 to the ER exit sites, but also for collagen VII to exit from the ER (Saito et al., 2014). However, why cTAGE5-mediated accumulation of Sec12 to the ER exit sites is necessary for collagen export and how cTAGE5 coordinates these two independent functions as a cargo receptor and a scaffold remain unclear.
In the present study, we isolated cTAGE5 mutants that lost Sec12 binding without affecting their interaction with TANGO1. By analyzing these mutants, we find that two functionally irreplaceable and molecularly separable modules in cTAGE5 are both required for collagen VII export from the ER. Proper localization of Sec12 induced by cTAGE5 serves for efficient production of activated Sar1 around ER exit sites, and the GTPase cycle of Sar1 seems to be required for collagen VII export from the ER.
RESULTS AND DISCUSSION
Construction of cTAGE5 point mutants lacking Sec12-binding activity
One of the two coiled-coil regions of cTAGE5 corresponding to amino acids (aa) 61–300 is reported to be responsible for interaction with Sec12 (Saito et al., 2014). To elucidate further the precise Sec12 interaction domain in cTAGE5, we coexpressed FLAG-tagged cTAGE5 deletion constructs with the hemagglutinin (HA)-tagged Sec12 cytoplasmic domain in 293T cells. Sec12 interacts with the aa 61–221 region in cTAGE5 but not with aa 115–300 (Figure 1A). This indicates that the interaction of cTAGE5 with Sec12 is mediated by aa 61–115, which is immediately C-terminus of the transmembrane region of cTAGE5 (Figure 1B). To ascertain further the importance of this region, we produced a cTAGE5 construct in which the aa 61–115 region is replaced with the corresponding region of TANGO1 (aa 1199–1253). As shown in Figure 1C, the mutant is still able to bind TANGO1, but not with Sec12, confirming the importance of aa 61–115 for the interaction with the GEF.
FIGURE 1:

Construction of cTAGE5 point mutants lacking Sec12-binding ability. (A) 293T cells were transfected with FLAG tag only, FLAG-tagged cTAGE5 coil1 (aa 61–300), cTAGE5 coil2 (aa 301–650), cTAGE5 (aa 61–221), or cTAGE5 (aa 115–300) with HA-tagged Sec12 (aa 1–386). Cell lysates were immunoprecipitated with anti-FLAG antibody and eluted with the FLAG peptide. Eluates and cell lysates were analyzed by SDS–PAGE followed by Western blotting with FLAG or HA antibodies. (B) Alignment of human cTAGE5 (aa 61–115) with corresponding regions from other species. Identical amino acids are shaded in black, and similar amino acids are shaded in gray. (C) 293T cells were transfected with FLAG tag only, FLAG-tagged cTAGE5 wild type, cTAGE5 (aa 61–300) T1, or cTAGE5 (aa 61–115) T1 with HA-tagged Sec12 or TANGO1. (D) 293T cells were transfected with FLAG tag only, FLAG-tagged cTAGE5 wild type, cTAGE5 R61A S65A, S68A R69A, Y71A, E75A K76A, K89A L93A, S97A, or L112A with HA-tagged Sec12. (E) 293T cells were transfected with FLAG tag only, FLAG-tagged cTAGE5 wild type, E75A K76A, K89A L93A, or S97A with HA-tagged TANGO1. (F) 293T cells were transfected with FLAG tag only, FLAG-tagged cTAGE5 wild type, K89A L93A, or K89A with HA-tagged Sec12 or TANGO1. (C–F) Cell lysates were immunoprecipitated with anti-FLAG antibody and eluted with the FLAG peptide. Eluates and cell lysates were analyzed by SDS–PAGE followed by Western blotting with FLAG or HA antibodies.
Next we sought to generate specific point mutants of cTAGE5 that are deficient in binding to Sec12. We compared sequence similarities of the region in cTAGE5 among different species and changed the conserved amino acids to alanine, as shown in Figure 1B. Although some of the point mutants were still capable of interacting with Sec12, several point mutants (E75A K76A, K89A L93A, and S97A) showed severe reduction of binding ability to Sec12 (Figure 1D). Moreover, these mutants still bound TANGO1, indicating that the mutations do not destroy the overall conformation of cTAGE5 (Figure 1E). The K89A single point mutation seemed to be enough to reduce binding to Sec12 (Figure 1F). These experiments allowed us to pinpoint critical residues in cTAGE5 involved in binding to Sec12.
The point mutants of cTAGE5 lacking Sec12-binding ability fail to export collagen VII from the ER
A previous report showed that Sec12 localization to the ER exit sites is mediated by its interaction with cTAGE5 (Saito et al., 2014). To evaluate the ability of cTAGE5 point mutants to recruit Sec12 to the ER exit sites, we expressed mutants in cells in which endogenous cTAGE5 was depleted by small interfering RNA (siRNA) and examined the localization of Sec12. We previously showed that cTAGE5 knockdown leads to the severe reduction of Sec12 signals in methanol-fixed cells, although the protein expression level was unchanged. Further study revealed that Sec12 was dispersed throughout the ER under these conditions, whereas the localization and intensity of Sec16, a bona fide ER exit site marker, remained the same (Saito et al., 2014). In accordance with these reports, cTAGE5 knockdown led to the severe reduction of Sec12 signals from Sec16-positive structures (Figure 2A, cells with asterisks). The expression of wild-type cTAGE5 efficiently rescued localization of Sec12 to the ER exit sites in cTAGE5-depleted cells (Figure 2A, top left; Saito et al., 2014). The extent of Sec12 concentration at the ER exit sites was quantified as the signal of Sec12 divided by that of Sec16, as described in Materials and Methods (Figure 2B). Overall the mutants capable of binding to Sec12 efficiently recruited Sec12 to the correct localization, whereas the mutants that lost Sec12-binding ability failed to recruit the protein to the ER exit sites (Figure 2, A and B). Next we checked whether the mutants could promote collagen VII secretion from the ER. We quantified the signals of accumulated collagen VII within the ER as an index of its secretion (Saito et al., 2014). As shown in Figure 3A, the expression of mutants defective in Sec12 recruitment to the ER exit sites could not rescue the block of collagen VII secretion induced by cTAGE5 knockdown, although the wild type and mutants capable of interacting with Sec12 efficiently rescued collagen secretion.
FIGURE 2:

cTAGE5 mutants lacking Sec12-binding ability fail to recruit Sec12 to the ER exit sites. (A) HSC-1 cells were transfected with cTAGE5 siRNA and cultured for 24 h. cTAGE5-FLAG wild-type and mutant constructs were transfected, and the cells were cultured for another 24 h, fixed, and stained with Sec12 (clone 6B3), FLAG, and Sec16 antibodies. Scale bar, 10 μm. Typically, >90% of the cells showed efficient reduction of cTAGE5 expression by siRNA transfection (cells with asterisks). Less than 10% of the cells were transfected with FLAG-tagged constructs. Cells with asterisks indicate that the cells were efficiently depleted for cTAGE5, but FLAG-tagged constructs were not transfected. (B) Quantification of Sec12 immunofluorescence intensity at ER exit sites in A. A.U., arbitrary units. Fifty ER exit sites from 10 cells, n = 50 (analysis of variance). Error bars represent mean ± SEM; **p < 0.001 compared with wild-type expression; n.s., p > 0.05 compared with wild-type expression. The data shown are from a single representative experiment out of three repeats.
FIGURE 3:
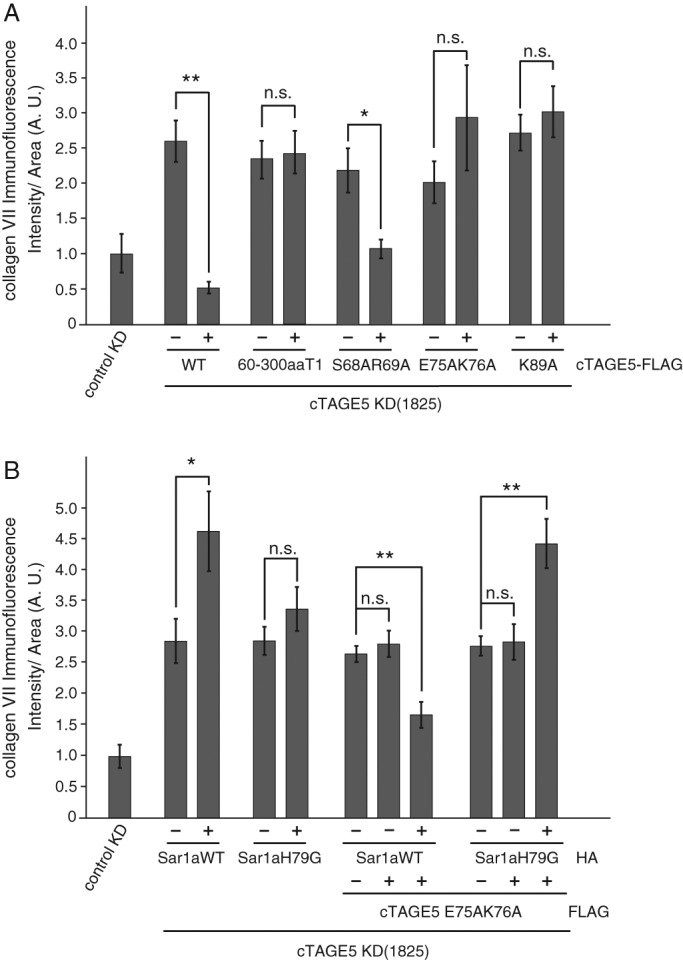
Sar1 coexpression with cTAGE5 mutant recovers collagen VII secretion from the ER. HSC-1 cells were treated with control or cTAGE5 siRNA and cultured for 24 h. For cTAGE5 siRNA-treated cells, cTAGE5-FLAG wild type or mutants (A) or cTAGE5-FLAG constructs together with HA-Sar1a constructs (B) were transfected and further cultured for 24 h. The cells were fixed and stained with collagen VII and FLAG (A) or collagen VII, FLAG, and HA antibodies (B). Collagen VII immunofluorescence signal per cell (A.U., arbitrary units) were quantified in each cell category as described later. The cells positively stained with FLAG or HA antibodies were categorized as the constructs expressed, and the surrounding unstained cells were categorized as nontransfected counterparts. Within each well, cells transfected with constructs are labeled as +, and nontransfected cells are labeled as –. Analysis of variance. Error bars represent mean ± SEM; **p < 0.001; *p < 0.05; n.s., p > 0.05. The data shown are from a single representative experiments out of three repeats. (A) Cells treated with control siRNA (n = 78); cells treated with cTAGE5 siRNA and wild type– (n = 140); wild type+ (n = 49); 60-300aaT1– (n = 111); 60-300aaT1+ (n = 49); S68A R69A– (n = 131); S68A R69A+ (n = 50); E75A K76A– (n = 114); E75A K76A+ (n = 48); and K89A– (n = 167); K89A+ (n = 51). (B) Cells treated with control siRNA (n = 75); cells treated with cTAGE5 siRNA and HA-Sar1aWT– (n = 62); HA-Sar1aWT+ (n = 12); HA-Sar1aH79G– (n = 135); HA-Sar1aH79G+ (n = 37); E75AK76A–, Sar1aWT– (n = 358); E75AK76A+, Sar1aWT– (n = 74); E75AK76A+, Sar1aWT+ (n = 54); E75AK76A–, Sar1aH79G– (n = 272); E75AK76A+, Sar1aH79G– (n = 67); and E75AK76A+, Sar1aH79G+ (n = 54).
Sar1 coexpression with cTAGE5 mutant lacking Sec12-binding ability recovers secretion of collagen VII from the ER
The foregoing results strongly suggest that cTAGE5-mediated concentration of Sec12 to ER exit sites is necessary for collagen VII secretion, independent of cTAGE5 formation of the cargo–receptor complex with TANGO1. However, the biological meaning of the concentration of Sec12 to specific sites has not been fully addressed. Because Sec12 is a GEF for Sar1 GTPase, we hypothesized that Sec12 accumulation at ER exit sites is responsible for the efficient production of activated Sar1 in the vicinity of ER exit sites. Thus we overexpressed Sar1 GTPase together with the cTAGE5 double mutant in cTAGE5-depleted cells. As shown in Figure 3B, expression of Sar1 GTPase alone had no effect or even increase accumulation of collagen VII within the ER, whereas the expression of both cTAGE5 mutant and Sar1 GTPase markedly recovered the secretion of collagen VII in cTAGE5-depleted cells. Of interest, expression of the GTPase-deficient activated form of Sar1 (Sar1 H79G) together with cTAGE5 mutant failed to rescue the accumulation of collagen VII (Figure 3B).
Dual function of cTAGE5 in collagen export from the ER
In this study, we identified specific and critical residues in the Sec12-binding region of cTAGE5 (E75A K76A, K89A, and S97A) and confirmed that cTAGE5-mediated Sec12 accumulation at ER exit sites is required for collagen VII secretion. In our previous study, we used a cTAGE5 construct in which one of the coiled-coil domains was swapped with that of TANGO1 to show the importance of Sec12 recruitment for collagen VII secretion (Saito et al., 2014). However, this mutant might be structurally inactive or could abrogate not only Sec12 binding but also other, unidentified functions required for collagen transport due to the extensive alterations of amino acids. Of note, the constructs used in the present study are only single or double point mutants, so that block of collagen secretion should be directly mediated by defects in Sec12 recruitment. Because involvement of ubiquitination in collagen secretion has been reported (Jin et al., 2012), we examined whether Sec12 binding to cTAGE5 might also be regulated by ubiquitination. Although K89 is a candidate residue to be ubiquitinated within the Sec12-binding domain, the ubiquitination status of cTAGE5 is not changed with the K89A point mutation, indicating that this residue is not critical for ubiquitination (Supplemental Figure S1). During the preparation of the manuscript, we also found that cTAGE5 can form a homodimer, and so we examined whether the point mutant can also still homodimerize. As shown in Supplemental Figure S2, HA-tagged cTAGE5 was efficiently immunoprecipitated with FLAG-tagged cTAGE5, as well as in the mutant lacking Sec12-binding affinity. These results indicate that the cTAGE5 mutant retained these properties of wild-type cTAGE5, except for its affinity for Sec12. The detailed characterization of cTAGE5 dimerization will be reported elsewhere. The dimeric properties of cTAGE5 have no influence on the interpretation of the present characterization, as the cTAGE5 rescue experiment replaces cTAGE5 within the cells, and thus the properties observed in this article are derived solely from expressed cTAGE5.
The most important feature of the present study is that the defect in Sec12 recruitment of the cTAGE5 mutant can be rescued by overexpression of Sar1 GTPase. In yeast, the phenotype of the Sec12 ts mutant can be rescued by the addition of Sar1, indicating that overexpressed Sar1 is functional in the absence of efficient GEF activity (Nakano and Muramatsu, 1989; Oka et al., 1991). However, of note, the conditions in this study are slightly different. As shown previously, cTAGE5 knockdown leads to the dispersion of Sec12 throughout the ER, but the proteins are indeed present within the ER. Moreover, conventional cargo can be effectively transported with the scattered Sec12, and only when Sec12 was depleted by siRNA was conventional cargo secretion blocked in addition to that of collagen (Saito et al., 2014). Thus not only is the rescue of collagen VII secretion probably achieved by the production of activated Sar1, but its local concentration around ER exit sites should also be critical. These functions of cTAGE5 may cooperate with that of Sec16 to stabilize Sar1-GTP around ER exit sites (Kung et al., 2012; Yorimitsu and Sato, 2012; Bharucha et al., 2013). Coexpression of the activated form of Sar1 with the double mutant failed to rescue collagen VII secretion. This is in good agreement with the case in yeast (Oka et al., 1991). It is also interesting to note that sedlin has been isolated as being essential for collagen I transport and reported to be involved in the inactivation of Sar1 GTPase (Venditti et al., 2012). These results suggest that not only the activation, but also the inactivation process of Sar1 GTPase would be critical for collagen export from the ER.
A summary of our findings is presented schematically in Figure 4. Knockdown of cTAGE5 renders the cargo receptor incompetent and causes inefficient production of activated Sar1 in the vicinity of ER exit sites (Figure 4, scheme 2). The expression of the cTAGE5 double mutant alone (Figure 4, scheme 3) or Sar1a (Figure 4, scheme 4) is not sufficient to rescue the blockage of collagen VII secretion, but the expression of both constructs is required to sufficiently substitute cTAGE5 functions for collagen VII secretion (Figure 4, scheme 5). Therefore we conclude that cTAGE5 has two distinct and irreplaceable roles in collagen VII export from the ER. cTAGE5 might act as a core factor for coordinating these two functions.
FIGURE 4:

Schematic representation of collagen receptor complex under conditions used in this study. In wild-type cells, cTAGE5/TANGO1/Sec12 acts as a cargo receptor for collagen VII and also produces activated Sar1 in the vicinity of ER exit sites (scheme 1). cTAGE5 knockdown renders cargo receptor incompetent and leads to Sec12 dispersion, which inhibits the production of activated Sar1 around ER exit sites. Therefore collagen VII secretion is impaired (scheme 2). The expression of cTAGE5 mutants in cTAGE5-depleted cells rescues the formation of cargo receptor complex, but production of activated Sar1 remains inefficient. Collagen VII cannot be secreted (scheme 3). The expression of only Sar1 is not efficient for the formation of cargo–receptor complex, thereby inhibiting collagen VII secretion (scheme 4). The coexpression of Sar1 and cTAGE5 mutants rescues the formation of the cargo–receptor complex and the amount of activated Sar1 around ER exit sites. Thus collagen VII secretion can be rescued (scheme 5).
The components required for collagen VII export have begun to emerge. Additional work is required to reveal the temporal order and special regulation of these players to achieve collagen VII secretion from the ER.
MATERIALS AND METHODS
Antibodies
Anti–collagen VII monoclonal antibody (NP-185) was kindly provided by Lynn Sakai (Oregon Health and Science University). Other antibodies were used as described previously (Saito et al., 2009, 2011, 2014).
Cell culture and transfection
HeLa, HSC-1, and 293T cells were cultured in DMEM supplemented with 10% fetal bovine serum. Lipofectamine RNAi max (Thermo Fisher Scientific, Waltham, MA) was used for transfecting siRNA. For plasmid transfection, polyethylenimine MAX (Polysciences, Warrington, PA) or FuGENE 6 (Promega, Madison, WI) was used.
Immunoprecipitation and Western blotting
The experiments were essentially performed as described previously (Saito et al., 2014). Briefly, extracted cells were centrifuged at 100,000 × g for 30 min at 4°C. Cell lysate was immunoprecipitated with FLAG M2 antibodies. The beads were washed five times with Tris-buffered saline/0.1% Triton X-100 and processed for sample preparation.
Immunofluorescence microscopy
Immunofluorescence microscopy analysis was performed as described previously (Saito et al., 2009, 2011). Cells grown on coverslips were washed with phosphate-buffered saline (PBS), fixed with methanol (6 min, −20°C), washed with PBS, and blocked in blocking solution (5% bovine serum albumin in PBS with 0.1% Triton X-100 for 30 min). After blocking, cells were stained with primary antibody (1 h at room temperature), followed by incubation with Alexa Fluor–conjugated secondary antibody (1 h at room temperature). Images were acquired with confocal laser scanning microscopy (Zeiss LSM700, Plan-Apochromat 63×/1.40 numerical aperture (NA) oil immersion objective lens). The acquired images were processed with Zeiss Zen 2009 software (Carl Zeiss, Oberkochen, Germany). Sec12 and Sec16 immunofluorescence intensities were quantified by the profile mode. Because the signals of each marker should be proportional to the size of ER exit sites in nontreated cells, Sec12 intensity was normalized to that of Sec16 (Farhan et al., 2008).
Quantification of collagen VII staining
Quantification of collagen VII accumulation was essentially performed as described previously (Saito et al., 2011, 2014). Stained cells were analyzed by Zeiss Axio Imager M1 microscopy (EC Plan-Neofluar 40×/ 0.75 NA objective lens) and processed with AxioVision software (Carl Zeiss). Area calculation and intensity scanning were done by ImageJ software (National Institutes of Health, Bethesda, MD).
Supplementary Material
Acknowledgments
We thank Lynn Sakai for providing the NP185 antibody. We also thank members of the Katada lab for valuable discussions. This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (K.S. and T.K.) and the Japan Society for the Promotion of Science (K.S. and T.K.).
Abbreviations used:
- aa
amino acids
- A.U.
arbitrary units
- ER
endoplasmic reticulum
- GEF
guanine-nucleotide exchange factor
- HA
hemagglutinin
- PBS
phosphate-buffered saline
- siRNA
small interfering RNA.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-03-0180) on May 11, 2016.
REFERENCES
- Bharucha N, Liu Y, Papanikou E, McMahon C, Esaki M, Jeffrey PD, Hughson FM, Glick BS. Sec16 influences transitional ER sites by regulating rather than organizing COPII. Mol Biol Cell. 2013;24:3406–3419. doi: 10.1091/mbc.E13-04-0185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farhan H, Weiss M, Tani K, Kaufman RJ, Hauri HP. Adaptation of endoplasmic reticulum exit sites to acute and chronic increases in cargo load. EMBO J. 2008;27:2043–2054. doi: 10.1038/emboj.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa Y, Boudko S, Bachinger HP. Ziploc-ing the structure: triple helix formation is coordinated by rough endoplasmic reticulum resident PPIases. Biochim Biophys Acta. 2015;1850:1983–1993. doi: 10.1016/j.bbagen.2014.12.024. [DOI] [PubMed] [Google Scholar]
- Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M. Ubiquitin-dependent regulation of COPII coat size and function. Nature. 2012;482:495–500. doi: 10.1038/nature10822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung LF, Pagant S, Futai E, D’Arcangelo JG, Buchanan R, Dittmar JC, Reid RJ, Rothstein R, Hamamoto S, Snapp EL, et al. Sec24p and Sec16p cooperate to regulate the GTP cycle of the COPII coat. EMBO J. 2012;31:1014–1027. doi: 10.1038/emboj.2011.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra V, Erlmann P. The pathway of collagen secretion. Annu Rev Cell Dev Biol. 2015;31:109–124. doi: 10.1146/annurev-cellbio-100913-013002. [DOI] [PubMed] [Google Scholar]
- Malhotra V, Erlmann P, Nogueira C. Procollagen export from the endoplasmic reticulum. Biochem Soc Trans. 2015;43:104–107. doi: 10.1042/BST20140286. [DOI] [PubMed] [Google Scholar]
- Miller EA, Schekman R. COPII - a flexible vesicle formation system. Curr Opin Cell Biol. 2013;25:420–427. doi: 10.1016/j.ceb.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano A, Muramatsu M. A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol. 1989;109:2677–2691. doi: 10.1083/jcb.109.6.2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira C, Erlmann P, Villeneuve J, Santos AJ, Martinez-Alonso E, Martinez-Menarguez JA, Malhotra V. SLY1 and Syntaxin 18 specify a distinct pathway for Procollagen VII export from the endoplasmic reticulum. eLife. 2014;3:e02784. doi: 10.7554/eLife.02784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Nishikawa S, Nakano A. Reconstitution of GTP-binding Sar1 protein function in ER to Golgi transport. J Cell Biol. 1991;114:671–679. doi: 10.1083/jcb.114.4.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R, Malhotra V. TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell. 2009;136:891–902. doi: 10.1016/j.cell.2008.12.025. [DOI] [PubMed] [Google Scholar]
- Saito K, Katada T. Mechanisms for exporting large-sized cargoes from the endoplasmic reticulum. Cell Mol Life Sci. 2015;72:3709–3720. doi: 10.1007/s00018-015-1952-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Yamashiro K, Ichikawa Y, Erlmann P, Kontani K, Malhotra V, Katada T. cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell. 2011;22:2301–2308. doi: 10.1091/mbc.E11-02-0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Yamashiro K, Shimazu N, Tanabe T, Kontani K, Katada T. Concentration of Sec12 at ER exit sites via interaction with cTAGE5 is required for collagen export. J Cell Biol. 2014;206:751–762. doi: 10.1083/jcb.201312062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos AJ, Raote I, Scarpa M, Brouwers N, Malhotra V. TANGO1 recruits ERGIC membranes to the endoplasmic reticulum for procollagen export. eLife. 2015;4:e10982. doi: 10.7554/eLife.10982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, Beznoussenko GV, Mironov AA, Mironov A, Jr, Zelante L, et al. Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science. 2012;337:1668–1672. doi: 10.1126/science.1224947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorimitsu T, Sato K. Insights into structural and regulatory roles of Sec16 in COPII vesicle formation at ER exit sites. Mol Biol Cell. 2012;23:2930–2942. doi: 10.1091/mbc.E12-05-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.