

Endophilin is a founding member of the membrane-bending protein family. It promotes rapid endocytosis on a milliseconds-to-seconds time scale. The short duration of rapid endocytosis requires endophilin to act quickly. High-resolution kinetic measurements show the sequential action that endophilin takes to quickly deform membranes.
Abstract
Numerous proteins act in concert to sculpt membrane compartments for cell signaling and metabolism. These proteins may act as curvature sensors, membrane benders, and scaffolding molecules. Here we show that endophilin, a critical protein for rapid endocytosis, quickly transforms from a curvature sensor into an active bender upon membrane association. We find that local membrane deformation does not occur until endophilin inserts its amphipathic helices into lipid bilayers, supporting an active bending mechanism through wedging. Our time-course studies show that endophilin continues to drive membrane changes on a seconds-to-minutes time scale, indicating that the duration of endocytosis events constrains the mode of endophilin action. Finally, we find a requirement of coordinated activities between wedging and scaffolding for endophilin to produce stable membrane tubules in vitro and to promote synaptic activity in vivo. Together these data demonstrate that endophilin is a multifaceted molecule that precisely integrates activities of sensing, bending, and stabilizing curvature to sculpt membranes with speed.
INTRODUCTION
Membrane-bending proteins play a key role in shaping intracellular compartments (McMahon and Gallop, 2005; Zimmerberg and Kozlov, 2006; Frost et al., 2009; Shibata et al., 2009). Steady-state analyses suggest several mechanisms to explain how these proteins impose unique shapes on flat membranes (McMahon and Gallop, 2005; Zimmerberg and Kozlov, 2006; Frost et al., 2009; Prinz and Hinshaw, 2009; Shibata et al., 2009; Baumgart et al., 2011; Bigay and Antonny, 2012; Stachowiak et al., 2012). For example, these proteins may act as curvature sensors that stabilize spontaneous membrane structures or function as active benders that physically force membranes to curve. Many membrane-bending proteins engage in both sensing and bending activities, suggesting that these activities are functionally linked and not mutually exclusive (Peter et al., 2004; Zimmerberg and Kozlov, 2006; Prinz and Hinshaw, 2009). Although previous studies provided significant insights, the mechanistic understanding of membrane-bending remains in the form of snapshots. It is unknown how multiple mechanisms act together to promote membrane remodeling. Here we use high-resolution kinetic analyses to explore the action of the membrane-bending protein endophilin.
Endophilin promotes synaptic vesicle (SV) endocytosis at nerve terminals. Inactivation of endophilin produces profound synaptic defects, including depletion of SVs, accumulation of endocytic intermediates, and subsequent failure in synaptic transmission (Ringstad et al., 1999; Gad et al., 2000; Guichet et al., 2002; Rikhy et al., 2002; Schuske et al., 2003; Verstreken et al., 2003; Bai et al., 2010; Milosevic et al., 2011). To support neuronal activity, endophilin must act rapidly because SV endocytosis is one of the fastest membrane recycling processes—for example, SV endocytosis can take place in ≤1 s (von Gersdorff and Matthews, 1994; Hsu and Jackson, 1996; Hook and Thoreson, 2012; Watanabe et al., 2013a, b). Of interest, endophilin is only recruited to endocytic intermediates at a late stage before scission (Ferguson et al., 2009; Liu et al., 2009), which further increases the demand for speed in its action.
The endophilin Bin–Amphiphysin–Rvs domain (EndoBAR) is a founding member of the membrane-remodeling BAR family (Frost et al., 2009). EndoBAR expression in endophilin-knockout neurons fully or partially rescues endocytic defects at synapses, demonstrating the functional importance of this domain (Bai et al., 2010; Milosevic et al., 2011; Dong et al., 2015). The ability of EndoBAR to stabilize/produce membrane curvature is well documented. It converts flat membranes in vitro into bilayer tubules of various diameter, as well as into small, highly curved vesicles and cylindrical micelles (Farsad et al., 2001; Gallop et al., 2006; Masuda et al., 2006; Mizuno et al., 2010; Suresh and Edwardson, 2010; Boucrot et al., 2012; Mim et al., 2012). When overexpressed in cells, EndoBAR disturbs the plasma membranes and produces deep membrane invaginations (Masuda et al., 2006; Boucrot et al., 2012).
EndoBAR dimers are the active units for shaping membranes (Weissenhorn, 2005; Gallop et al., 2006; Mim et al., 2012). Each EndoBAR dimer has a crescent shape with a basic, membrane-binding surface at its concave surface (Peter et al., 2004; Weissenhorn, 2005; Gallop et al., 2006; Masuda et al., 2006). Several models have been proposed to describe how endophilin senses and bends membranes. First, the “scaffold” model predicts that dimeric EndoBAR uses its crescent shape as a rigid mold to impose curvature on membranes (Gallop et al., 2006; Masuda et al., 2006; Mim et al., 2012). Second, the “wedging” model suggests that the bending property of EndoBAR is powered by insertion of two amphipathic helices (H0 and HI) into membranes, which induces an asymmetric distribution of lipids in the bilayer to produce curved membranes (Gallop et al., 2006; Masuda et al., 2006). Third, cryo–electron microscopy (EM) and simulation studies suggest that EndoBAR forms highly organized lattice structures on membrane tubules, suggesting that intermolecular interactions between neighboring EndoBAR dimers facilitate curvature generation (Mim et al., 2012; Cui et al., 2013; Simunovic and Voth, 2015). Finally, recent studies suggest that the insertion depth of amphipathic helices modulates the scaffolding effects (Ambroso et al., 2014; Isas et al., 2015), switching endophilin action from tubulation to vesiculation. Although it is known that multiple mechanisms are engaged, how these activities coordinate to promote endophilin’s function in rapid endocytosis remains elusive.
Here we demonstrate with time-resolved kinetic analyses that endophilin is a multifaceted molecule that integrates activities of sensing, bending, and stabilizing curvature to sculpt membranes. We show that wedging and scaffolding activities are highly coordinated to ensure endophilin function in vivo.
RESULTS
To understand how endophilin rapidly shapes membranes, we used stopped-flow rapid-mixing apparatus and fluorescence methods to investigate the early action that endophilin takes on membranes.
First, we used a Förster resonance energy transfer (FRET) method to study endophilin–membrane association. The donor fluorophore (Pacific Blue) was covalently linked to the single cysteine residue (Cys-108) in EndoBAR (Capraro et al., 2013). We incorporated a trace amount of 7-nitrobenz-2-oxa-1,3-diazole–labeled phosphoethanolamine (NBD-PE) into liposomes as the acceptor fluorophores (Figure 1A). Binding between EndoBAR and the liposomes brings the donor and acceptor fluorophores into close proximity, leading to increased NBD fluorescence and decreased Pacific Blue fluorescence due to nonradiative energy transfer from donor to acceptor (Figure 1B; see controls in Supplemental Figure S1). Electron microscopy and cosedimentation studies show that NBD-PE (5% of total lipids) does not impair liposome morphology and EndoBAR–membrane association (Supplemental Figure S2).
FIGURE 1:

EndoBAR senses membrane curvature to promote rapid association. (A) Schematic diagram of the FRET assay (Capraro et al., 2013). (B) FRET signals report the EndoBAR–membrane association. Steady-state emission spectra were collected using an excitation wavelength of 400 nm, with Pacific Blue–labeled EndoBAR (1 μM) in the presence of liposomes (0.5 mM total lipids; liposomes were prepared using extrusion through polycarbonate filters with 1-μm pore size [designated 1μm filter] and 50-nm pore size [designated 50nm filter]). PS/PC, liposomes harboring 25% DOPS, 70% POPC, and 5% NBD-DPPE. PC only, liposomes harboring 95% POPC and 5% NBD-DPPE. The donor fluorophore Pacific Blue was covalently linked to the native Cys-108 residues of EndoBAR. BSA was used as a control for nonspecific interactions (Supplemental Figure S1). (C) Stopped-flow analyses show that EndoBAR binds more rapidly to liposomes with higher degree of curvature. Liposomes (0.5 mM total lipids) of various diameters and Pacific Blue–labeled EndoBAR (1 μM) were rapidly mixed using the SX-20 stopped-flow spectrometer. NBD fluorescence was collected using a 515-nm long-pass filter. The raw data traces were fitted with a single-exponential function to obtain the rate constant kobs. (D) EndoBAR binds tighter to liposomes with a higher degree of curvature. Average data from three independent experiments. The dissociation constant (Kd) for EndoBAR–liposome interactions is calculated as koff/kon. Rate constant (kobs) is plotted as a function of [liposome] to determine kon and koff. Error bars indicate SD.
Real-time kinetic analyses using the stopped-flow apparatus show that EndoBAR rapidly binds membranes in a curvature-dependent manner (Figure 1). EndoBAR binds large liposomes (prepared by extrusion through filters with 1-μm pores [designated “1-μm filter”]; 0.5 mM total lipids, 25% phosphatidylserine [PS]/70% phosphatidylcholine [PC]/5% NBD-PE) with a rate constant of 70 ± 3 s−1. The association rates were significantly accelerated (150 ± 19 s−1) on high-curvature membranes (liposomes extruded through filters with 50-nm pores [designated “50-nm filter”]; Figure 1C). This translates into ∼15-fold higher affinity for small liposomes than for large ones (Figure 1D). Similar results were obtained using full-length endophilin, although association rates were slightly but significantly slower than those for EndoBAR (Supplemental Figure S3). These data strongly suggest that endophilin has a preference for highly curved membranes, consistent with previously reported curvature-sensing ability (Peter et al., 2004; Gallop et al., 2006; Bhatia et al., 2009; Baumgart et al., 2011; Sorre et al., 2012).
We next incorporated a trace amount of the lipophilic fluorescent dye 6-dodecanoyl-2-dimethylaminonaphthalene (laurdan) into membranes to study how endophilin deforms lipid bilayers. Laurdan fluorescence is highly sensitive to changes at the water–membrane interface (Figure 2A; Parasassi and Gratton, 1992; Zhang et al., 2006; Ionescu and Ganea, 2012). Membrane deformation leads to an increase in membrane water content that consequently quenches laurdan fluorescence and shifts its spectrum, making laurdan an excellent indicator for detecting early and local membrane deformation. As expected, the addition of EndoBAR induced a significant reduction in fluorescence intensity and a slight shift of the laurdan spectrum (Figure 2B; 0.25% laurdan). The shift in fluorescence spectrum became more apparent when the laurdan concentration was increased to 2% (unpublished data). The incorporation of trace amounts of laurdan does not affect EndoBAR–membrane association (Supplemental Figure S4, A–C) and endophilin-induced tubulation (Supplemental Figure S4, D and E). Mutant versions of EndoBAR that fail to generate membrane tubules in EM studies (Gallop et al., 2006; Masuda et al., 2006) display attenuated laurdan changes (Supplemental Figure S5A), consistent with reduced bending ability in these mutants. To further confirm that laurdan detects membrane deformation for tubulation, we used the FCHO-1 F-BAR domain. Unlike endophilin, which has both tubulation and vesiculation activities, F-BAR proteins mainly generate membrane tubules (Boucrot et al., 2012). We found that FCHO1 F-BAR efficiently quenches laurdan fluorescence (Supplemental Figure S6), suggesting that laurdan detects changes that lead to membrane tubules.
FIGURE 2:

Membrane deformation and association are distinct processes. (A) Schematic representation showing that laurdan fluorescence decreases upon membrane deformation. (B) Laurdan fluorescence was significantly quenched in the presence of EndoBAR (1 μM). Liposomes (0.5 mM total lipids) were composed of 25% DOPS, 5%DPPE, 70% POPC, and trace amount (0.25%) of laurdan. Laurdan was excited at 365 nm. Solid and dashed lines indicate conditions with and without EndoBAR, respectively. (C) Membrane deformation occurs after EndoBAR–membrane association. Time course of laurdan quenching (blue) and EndoBAR-membrane FRET (orange) were collected using the SX-20 stopped-flow spectrometer. FRET signal was inverted for clarity in comparison. In both kinetic analyses, [EndoBAR] was 1.5 μM, and liposomes were composed of 0.5 mM total lipids. Note that liposomes with different fluorescent reporters (laurdan vs. NBD-PE) were used in these experiments. (D) EndoBAR–membrane association becomes faster when protein concentration increases. Representative traces show FRET kinetics at various [EndoBAR]. (E) Laurdan quenching traces obtained with various [EndoBAR] are plotted. Laurdan quenching kinetics does not vary with [EndoBAR]. (F) Rate constants (kobs) of membrane association and deformation obtained by single-exponential fits are plotted vs. [EndoBAR]. Average data from three independent experiments. Error bars indicate SD. (G) EndoBAR deforms membranes containing short-tail lipids (05:0 PC(1,2-dipentanoyl-sn-glycero-3-phosphocholine)) with faster rates. Liposomes are composed of 25% DOPS, 5% DPPE, and 70% PC (the fractions of 5-carbon PC are indicated; the rest of lipids were POPC). All liposomes were prepared by extrusion (filters with pores 1 μm in diameter) and contain a trace amount (0.25%) of laurdan. kobs is plotted vs. fraction of five-carbon PC in the liposomes.
To further validate that laurdan quenching is due to membrane changes rather than interactions between EndoBAR and laurdan, we performed control experiments using smaller liposomes (Zhang et al., 2006). Because EndoBAR binds smaller liposomes with much higher affinity (Bhatia et al., 2009), there is an increased chance that EndoBAR will encounter laurdan on these liposomes. If laurdan fluorescence is quenched by unexpected EndoBAR–laurdan interactions, we expect increased degrees of laurdan quenching in samples with smaller liposomes. In contrast, if membrane changes are the reason for laurdan quenching, we expect less quenching with smaller liposomes, as these membranes begin with more water exposure. Indeed, we found that laurdan changes do not correlate with endophilin–membrane association. The extent of laurdan fluorescence drop is significantly reduced when highly curved liposomes are used (Supplemental Figure S5, B–D), despite the fact that endophilin binds these liposomes with increased affinity (Figure 1). Together these results indicate that laurdan quenching is not due to direct interactions between laurdan and EndoBAR. Instead, laurdan reports changes in lipid bilayers, as expected (Zhang et al., 2006).
To understand the speed of EndoBAR-induced membrane deformation, we monitored the time course of laurdan quenching using a stopped-flow rapid-mixing apparatus. Our kinetic results show that membrane deformation occurs in two stages: a delayed phase (∼34 ms) with little decrease of laurdan fluorescence (<14%), followed by a rapid phase of large fluorescence drop that takes place after the initial delay (Figure 2C). Compared with membrane association (kobs = 85 ± 2 s−1), the speed of membrane deformation is slower (kbending = 21 ± 1 s−1; Figure 2C). Similar results were observed with full-length endophilin (Supplemental Figure S7), although membrane deformation is slightly accelerated (26 ± 5 s−1 for full-length and 21 ± 1 s−1 for EndoBAR). These data show that the protein–membrane association and early membrane deformation are temporally separable events.
We next asked whether membrane association and bending are mechanistically distinct. We found that membrane association rate is positively correlated with endophilin concentration (Figure 2, D–F), indicating that membrane association is dominated by the collision frequency between endophilin and membranes. In contrast, the rates of membrane deformation reported by laurdan fluorescence do not vary with endophilin concentration (Figure 2, E and F, and Supplemental Figure S7C), suggesting that mechanisms other than collision set the rate-limiting step of membrane deformation. These data indicate that distinct mechanisms are used for membrane association and membrane deformation.
Previous studies showed that membrane fluidity significantly affects endocytosis (Pinot et al., 2014) and the membrane-bending ability of endocytic proteins (Pinot et al., 2014; Vanni et al., 2014). To confirm that laurdan quenching is limited by membrane rigidity, we incorporated short-chain lipids (five carbons) into liposomes to increase membrane fluidity. We found that EndoBAR bends these “more fluidic” liposomes at higher rates (Figure 2G; Vanni et al., 2014).
To further investigate the mechanisms for endophilin-induced membrane bending, we studied the role of the amphipathic helices (H0 and HI) of endophilin, which have been indicated as the wedges that generate asymmetric distribution of lipids in the bilayer (Gallop et al., 2006; Masuda et al., 2006; Jao et al., 2010; Boucrot et al., 2012; Cui et al., 2013; Ambroso et al., 2014). We placed a single tryptophan (Trp) residue in either EndoBAR H0 (F10W) or HI (M70W) to monitor membrane insertion (Figure 3A). To test whether these Trp reporters impair endophilin function, we carried out cosedimentation, flotation experiments, EM analyses, behavior assays, and electrophysiological recordings. We found that expression of Trp mutant EndoBAR in endophilin-knockout worms fully recovers the defects in locomotion rates and synaptic transmission (Supplemental Figure S8). Using the sucrose flotation assay, we observed that Trp-mutant proteins bind membranes to similar levels as wild-type EndoBAR (Supplemental Figure S9A). Finally, EM analyses confirmed that Trp mutations do not alter the shape distribution of membrane tubules (Supplemental Figure S9, B and C). These results suggest that EndoBARs with Trp reporters remain functional in vivo and in vitro.
FIGURE 3:

Insertion of amphipathic helices is a postbinding event that initiates local membrane deformation. (A) Schematic diagram showing the structure of an EndoBAR dimer (modified from PDB ID 1ZWW; Weissenhorn, 2005). Two amphipathic helices (H0 in green, HI in purple) in each monomeric EndoBAR are highlighted. Because these helices were not resolved in the crystal structure, cartoons are used to illustrate helices. (B, C) Trp fluorescence changes indicate that the H0 and the HI helices have inserted into membranes. A single Trp residue was placed into the H0 (F10W) and the HI (M70W), respectively. EndoBAR (1 μM) was incubated with liposomes (1 μm in diameter and 0.5 mM total lipids). PS/PC, 25% DOPS, 5% DPPE, and 70% POPC. PC, 95% POPC and 5% DPPE. Trp fluorescence was excited at 285 nm. (D–F) The H0 and HI helices penetrate into the hydrophobic core of membranes. The depth of helix insertion was calculated using parallax analysis (Kaiser and London, 1998; Bai et al., 2000). The fluorescence quencher groups (bromide; Br) were immobilized onto the lipid carbon tail at positions 6/7 and 9/10. Trp fluorescence was measured using an excitation wavelength of 285 nm. The degree of Trp fluorescence quenching was quantified using ln(F/F0), where F and F0 indicate Trp fluorescence intensity at 350 nm in the presence and absence of Br2-PC liposomes, respectively (Bai et al., 2000). Average data from three independent experiments. (F) Diagram indicating the depth of penetration of H0 and HI helices. For wild-type EndoBAR, the H0 and HI helices penetrate a similar depth into membranes (∼8 ± 2 Å from the center of the lipid bilayer). (G) Kinetics of Trp fluorescence showing that H0 inserts into the membrane with a significant delay, ∼34 ms. Rate constant and delay time are presented as mean ± SD. (H) The speed of membrane insertion of H0 does not vary with protein concentration. Average data from three independent experiments. Error bars indicate SD. (I) Membrane insertion occurs at the same time as laurdan fluorescence abruptly drops. Representative traces of membrane association (FRET), membrane deformation (laurdan), and H0 insertion are aligned at time 0. Trp fluorescence and FRET traces are inverted for clarity in comparison. In all conditions, EndoBAR (1.5 μM) was incubated with liposomes (extruded by polycarbonate filters with 1-μm-diameter pores, 0.5 mM lipids).
Our results show that both Trp probes exhibit increased fluorescence in the presence of PS/PC liposomes (compared with PC liposomes; Figure 3, B and C), suggesting that these Trp residues are inserted into hydrophobic regions of membranes containing negatively charged lipids (Gallop et al., 2006; Masuda et al., 2006; Jao et al., 2010; Ambroso et al., 2014). Membrane insertion of these amphipathic helices is further confirmed using membrane-embedded bromide quenchers, which silence Trp fluorescence through physical contacts with the bromide groups in membranes (Figure 3, D and E). We find that both Trp residues insert into the lipid core to similar depth (∼12 ± 2 Å below the lipid phosphates, or ∼8 ± 2 Å away from the center of the lipid bilayer; Figure 3F). These data are in agreement with recent findings showing that the F10 residue locates at ∼11 Å below the lipid phosphates, and the M70 residue reaches ∼8 Å below the lipid phosphates on membrane tubules (Ambroso et al., 2014).
Our kinetics analyses show that Trp fluorescence does not increase during the initial period (∼34 ms), which is identical to the delay observed in the laurdan assay and is drastically different from membrane association kinetics (Figures 2C and 3, G and H). The speed of H0 and HI insertion is significantly slower than membrane association (Figure 3, G–I; unpublished data), indicating that H0 and HI do not insert until EndoBAR is fully associated with membranes. Similar to membrane deformation, the rates of helix insertion remain identical at various protein concentrations (Figure 3I), showing that protein–membrane collision is not the rate-limiting step for insertion. These results show that the membrane association and helix insertion are kinetically and mechanistically different. It is worth noting that the structure of H0 and HI was not well resolved in crystallographic studies due to high degrees of flexibility (Masuda et al., 2006; Jao et al., 2010). Previous studies suggest that the H0 and HI regions undergo conformational changes on membranes (Gallop et al., 2006; Masuda et al., 2006; Jao et al., 2010). Such intramolecular rearrangements may explain the delay and the concentration-independent nature of Trp fluorescence kinetics.
Because helix insertion occurs more rapidly than membrane deformation (Figure 3I), we speculated that laurdan quenching is due to the wedging effect after helix insertion. To test whether helix insertion actively works against membrane rigidity, we compared Trp and laurdan fluorescence changes using rigid membranes harboring saturated lipids with long carbon chains (dimyristoyl-phosphatidylcholine [DMPC], 14:0; Dimova et al., 2006; Kummrow and Helfrich, 1991; Marsh, 2006). Of interest, we found that whereas EndoBAR H0 inserts into rigid membranes (Supplemental Figure S10A), laurdan fluorescence remains largely unquenched (Supplemental Figure S10B). These results indicate that helix insertion does not directly quench laurdan fluorescence. Instead, membrane rigidity serves as a limiting factor that restricts the effect of helix insertion on membrane deformation. Together our results indicate that endophilin binds membranes through collision-dependent curvature-sensing mechanisms. After binding, a collision-independent mechanistic transition prepares endophilin for insertion and subsequently drives membrane deformation.
To estimate the time course of large-scale membrane deformation, we used electron microscopy to analyze liposome samples that were incubated with EndoBAR for 3 s, 2 min, and 30 min, respectively (Figure 4). It has been reported that EndoBAR has both tubulation and vesiculation activities. We found that these activities take place on different time scales. At 3 s after incubation, liposomes largely retain their morphology. After 2 min, >40% of liposomes have tubules on them (Figure 4B). By contrast, vesiculation at this time point is not apparent, as the diameter distribution of liposome/vesicles remains unchanged (Kolmogorov–Smirnov [KS] test, nonsignificant; Figure 4C, middle). After 30 min of incubation, the fraction of liposomes with tubules did not increase further. However, the diameter of liposomes/vesicles became significantly smaller (Figure 4C, right), indicating the formation of small vesicles. Although our data suggest that vesiculation happens later than tubulation, we do not know whether these events are sequential or parallel. Nonetheless, EM studies suggest that large-scale membrane deformation occurs on the seconds-to-minutes time scale.
FIGURE 4:

Large-scale membrane deformation induced by EndoBAR occurs on seconds-to-minutes time scale. (A) EndoBAR-induced membrane deformation was examined by EM. Representative images were collected from liposomes without proteins (blank) and liposomes incubated with EndoBAR (1 μM) for 3 s, 2 min, and 30 min, respectively. Reaction was stopped by fixing with 0.5× Karnovsky’s solution. Liposomes contained 25% DOPS, 5% DPPE, and 70% POPC and were prepared by extrusion through filters with 1-μm pore size. (B) Membrane tubulation appeared after 3 s of incubation. Fraction of tubulation = number of liposomes with extended protrusions/number of total liposomes. Samples were collected in three independent experiments. Student’s t test was used for statistical analysis. Error bars indicate SD. (C) Vesiculation became significant after 2 min of incubation. To measure the vesicle diameter, ImageJ was used to draw a small circle that covers each vesicle/liposome. The diameter of the circle was used in these analyses. The distribution of vesicle diameter is represented as histograms. Size distribution of blank liposomes (pink) is plotted as control. Data from samples with EndoBAR are shown in light green. The number of analyzed images is 25 (blank), 28 (3 s), 34 (2 min), and 27 (30 min). Vesicle diameter values that were greater or equal to 20 nm were binned to 10-nm width. The KS test was used for statistical analysis. ##Unequal distributions.
Next we examined the scaffolding role of EndoBAR dimers in membrane bending. We introduced two hydrophilic serine residues to replace M97 and F110 at the EndoBAR dimer interface (Weissenhorn, 2005; Gallop et al., 2006). Free equilibrium simulations suggest that mutant EndoBAR adopts a more curved dimer surface, with an average radius of gyration of 116 ± 5 Å, compared with 138 ± 6 Å for wild-type EndoBAR (Figure 5A). The changes in scaffold shape are due to a sliding motion at the interface of EndoBAR dimers rather than to changes of EndoBAR monomer conformation. To test whether there are changes in the dimer scaffold, we performed cross-linking experiments using Bis-MAL-dPEG11, which has two cysteine-reactive maleimide groups separated by a ∼3-nm polyethylene glycol (PEG) linker (Figure 5B). Previous studies showed that Bis-MAL-dPEG11 does not cross-link wild-type EndoBAR dimers (Figure 5B, top right; Capraro et al., 2013), as the two C108 residues (one in each monomer) are located ∼6 nm away from each other (Weissenhorn, 2005). Of interest, we found that Bis-MAL-dPEG11cross-links the mutant (M97, F110) EndoBAR (Figure 5B, bottom right). Although cross-linking experiments provide only low-resolution information, these results are consistent with the computational findings suggesting a change in the EndoBAR dimer scaffold.
FIGURE 5:

Mutations that alter the dimer scaffold disrupt the tubulation property. (A) Free equilibrium simulations show that a mutant (M97S, F110S; green) EndoBAR adopts a more curved crescent surface than wild-type EndoBAR (purple). (B) Left, schematic for cross-linking experiments using Bis-MAL-dPEG11 (∼3 nm in length). The distance between two Cys-108 residues in the EndoBAR dimer is ∼6 nm. Right, wild-type (wt) and mutant EndoBAR (5 μM) were incubated with cross-linker (15 μM) at 4°C for various time as indicated and samples were analyzed on nonreducing SDS–PAGE gels. EndoBAR monomers (∼28 kDa) and cross-linked dimers (∼58 kDa) are indicated by arrowheads. (C) Transmission electron micrographs for liposomes (prepared using filters with 1-μm pore size) incubated with wild-type EndoBAR (left) and mutant M97S, F110S EndoBAR (right). (D) Distribution of tubule width. Membrane tubules induced by mutant EndoBAR are thinner (three independent repeats). The KS test was used for statistical analysis. ##Unequal distributions. (E) Mutant EndoBAR produced shorter membrane tubules than the wild-type EndoBAR (three independent repeats). Liposomes and proteins were incubated for 30 min before fixation. The KS test was used for statistical analysis.
To determine the membrane-bending ability of mutant (M97S, F110S) EndoBAR, we used electron microscopy to observe large-scale membrane deformation. We quantified tubule length and width from liposome samples that were incubated with either wild type or the mutant EndoBAR for 30 min (Figure 5C). Both the length and the diameter distribution histograms are significantly different from those obtained from wild-type EndoBAR samples (Figure 5, D and E; KS test). In samples mixed with the mutant EndoBAR, membrane tubules became thin and short (Figure 5, D and E), suggesting that these tubules have defects in stability.
To ask how these mutations (M97S, F110S) affect early membrane deformation, we examined the extent and the kinetics of laurdan quenching by mutant EndoBAR. Surprisingly, the laurdan assay showed that the mutant EndoBAR has enhanced ability to induce local membrane deformation. Both the extent and the speed of laurdan quenching were increased in the presence of the mutant EndoBAR (Supplemental Figure S11, A and B).
Because amphipathic helices H0 and HI have an important role in early membrane deformation, we next asked whether amphipathic helices of the mutant EndoBAR are properly positioned in membranes. We found that in the mutant EndoBAR, the H0 Trp probe stays outside membranes, evidenced by the lack of fluorescence enhancement by PS/PC liposomes (Figure 6A) and by the absence of fluorescence quenching in the presence of membrane-embedded bromide labels (Figure 6C). These data suggest that the H0 helix in the mutant EndoBAR fails to insert into membranes. However, it is also possible that the H0 helix has become heavily tilted in such a way that part of the helix remains in the bilayer, whereas the F10W residue stays outside of the lipid core. We next examined the insertion depth of HI and found that the penetration depth of HI became shallower (Figure 6D and Supplemental Figure S11C). Together these results show that mutations at the dimer interface lead to both scaffolding and wedging defects.
FIGURE 6:

The H0 helix of mutant EndoBAR fails to insert into membranes. Trp fluorescence was monitored as described in Figure 3. EndoBAR (1 μM) was incubated with liposomes (prepared by extrusion through filters with 1-μm pores; 0.5 mM total lipids). PS/PC, 25% DOPS, 5% DPPE, and 70% POPC. PC, 95% POPC and 5% DPPE. Trp fluorescence was excited at 285 nm. The H0 helix of the mutant EndoBAR fails to insert (green) (A), whereas the HI insertion remains intact (magenta) (B). (C) Membrane-embedded bromide does not quench F10W (H0) fluorescence. Liposomes contain 25% (6,7)-Br2-PC, 25% DOPS, 5% DPPE, and 45% POPC. (D) Schematic showing altered H0 and HI insertion of the mutant EndoBAR. Parallax analysis was used to determine the position of the HI helix in membranes (M97S, F110S mutant, ∼11 Å from the center of bilayer; EndoBAR WT, ∼8 Å; three independent repeats; p < 0.05, Student’s t test).
To test the function of the mutant EndoBAR (M97S, F110S) in vivo, we expressed single-copy transgenes encoding either wild-type or mutant versions of EndoBAR in the nervous system of C. elegans. Mutant worms that lack the endophilin unc-57 gene exhibit severe defects in the recycling of SVs, leading to a smaller pool of SVs and a corresponding decrease in synaptic transmission (Schuske et al., 2003; Bai et al., 2010). We used both locomotion assays and electrophysiological recordings at neuromuscular junctions as readouts to monitor endophilin’s function in vivo. As previously reported, expression of mCherry-tagged mouse endophilin BAR domain (mEndoBAR::mCherry) under the control of a panneuronal promoter (Prab-3) fully restored locomotion rates, endogenous excitatory postsynaptic current (EPSC) frequency, and evoked EPSC amplitudes in unc-57 mutant worms (Figure 7, A–F; Bai et al., 2010; Dong et al., 2015). By contrast, although mutant (M97S, F110S) EndoBAR has a similar distribution pattern (Supplemental Figure S12), it failed to rescue behavioral and synaptic defects. In particular, failure in restoring endogenous EPSC frequency indicates that mutant EndoBAR has lost its synaptic activity. These data suggest that proper scaffolding and wedging are necessary for endophilin function in vivo. Surprisingly, we found that the mutant (M97S, F110S) EndoBAR introduced two additional defects to the unc-57 mutant worms. First, the amplitudes of endogenous EPSCs became smaller, indicating reduced acetylcholine signaling from individual SV exocytotic events (Figure 7D). This phenotype is often associated with smaller SV size or defects in loading neurotransmitters into SVs. Second, when compared with mutant worms lacking unc-57 endophilin, the amplitudes of evoked EPSCs were further reduced (Figure 7, E and F). These data indicate that the mutant EndoBAR has gained a new role in inhibiting an endophilin-independent SV recycling pathway, possibly by competing with other membrane-remodeling proteins such as amphiphysin and syndapin. However, further studies are required to fully address the inhibitory role of mutant EndoBAR.
FIGURE 7:

The mutant (M97S, F110S) endophilin exhibits functional defects in vivo. A panneuronal promoter Prab-3 was used to drive transgene expression. Single-copy transgenes encoding mCherry-tagged mouse EndoBAR variants (Prab-3::mEndoBAR::mCherry) were introduced into unc-57(e406) mutant (A–F) and wild-type N2 worms (G and H). (A) C. elegans locomotion is restored by neuronal expression of mEndoBAR wild type (BAR wt) but not the mutant M97S,F110S mEndoBAR. Representative trajectories (15 animals) of 30-s locomotion are shown for each genotype. The starting points for each trajectory are aligned for clarity. (B–E) Electrophysiological recordings show that mutant mEndoBAR failed to recover synaptic defects in unc-57 mutant worms. Representative traces (B, E) and summary data for endogenous EPSC rates (C) and amplitudes (D) and for evoked EPSC amplitudes (F) are shown for the indicated genotypes. ***p < 0.001 and *p < 0.05 vs. wild-type controls. ###p < 0.001 and #p < 0.05 vs. unc-57 mutants. The number of worms analyzed for each genotype is indicated in the bar graphs. Error bars represent SEM. (G, H) The mutant EndoBAR inhibits synaptic activities in wild-type worms. Amplitudes of evoked EPSCs were significantly reduced when the mutant EndoBAR was expressed in the wild-type C. elegans. **p < 0.01 vs. wild-type mEndoBAR controls. The number of worms analyzed for each genotype is indicated in the bar graphs. Error bars represent SEM.
Taken together, our results suggest that dimeric endophilin serves as a rigid scaffold to coordinate membrane insertion of amphipathic helices, determine membrane curvature, and stabilize curvature on membrane tubules.
DISCUSSION
Endophilin plays a crucial role in endocytosis, which occurs over different time scales. In particular, endophilin is essential for rapid recycling of synaptic vesicles (Ringstad et al., 1999; Guichet et al., 2002; Schuske et al., 2003; Bai et al., 2010; Milosevic et al., 2011). The short duration of SV endocytosis requires endophilin to act quickly. Although multiple mechanisms have been proposed, steady-state analyses have limited power in revealing the immediate action that endophilin takes to remodel membranes. Our kinetic measurements show that endophilin uses sequential actions of sensing and wedging to sculpt membranes; both steps are mediated by distinct mechanisms. Different rate-limiting steps of sensing and bending indicate that these activities could be individually tuned. Of interest, we observed that after local membrane deformation, endophilin continues to drive membrane changes on the seconds-to-minutes time scale, which suggests that large-scale membrane changes develop slowly and require more time. We therefore propose that the kinetic property of various endocytosis events constrains the model of endophilin action. Taken together, our data highlight the early action that endophilin takes to remodel membranes and provide mechanistic insights in particular into how endophilin meets kinetic requirements for rapid endocytosis.
A mechanistic transition from a curvature sensor to a membrane bender
Our results show that endophilin takes about 30 ms to transform from a curvature sensor into an active bender after binding membranes. The initial curvature sensing and the subsequent bending activities are mechanistically distinct.
Endophilin binds membranes through collision-dependent, curvature-sensing mechanisms. After binding, a collision-independent mechanism is engaged to prepare endophilin for insertion and deformation. These findings indicate that endophilin does not immediately deform membranes upon its arrival. We propose that the bending function is activated only after helices become ready for insertion. This model is consistent with previous findings showing that the H0 and the HI regions are disordered in crystal structures (Weissenhorn, 2005; Gallop et al., 2006; Masuda et al., 2006). They become helical only after binding membranes (Löw et al., 2008; Jao et al., 2010). Slow conformational changes could explain the collision-independent nature of initial membrane deformation.
A lag of 30 ms in endophilin bending action is an important factor to consider when studying endophilin’s role in SV endocytosis because the duration of an endocytic event at a synapse can be as short as 50 ms (Watanabe et al., 2013a, b). The delay between association and membrane deformation indicates that endophilin takes multiple steps to morph into a membrane bender. The distinct nature of endophilin binding and bending activities may allow differential regulation of protein targeting and membrane remodeling by posttranslational modifications such as phosphorylation (Matta et al., 2012; Ambroso et al., 2014; Arranz et al., 2015).
An active role of helix insertion in promoting membrane deformation
Previous studies suggested that H0 insertion is essential for curvature sensing, that is, deletion and mutation in the H0 helix disrupt curvature-dependent enrichment of EndoBAR on liposomes (Bhatia et al., 2009). Based on these findings, it was proposed that the curvature-sensing property is mediated by H0 insertion. Of interest, our kinetic results show that the curvature-dependent association reached completion before insertion began, indicating that early sensing ability during association is not due to helix insertion. These data suggest that alternative mechanisms are involved during the initial sensing step. For example, helices may loosely attach onto membranes without entering the hydrophobic core, which could subsequently change the off-rate of EndoBAR–membrane complexes. In addition, positively charged residues located on the concave dimer surface may also contribute to initial sensing ability (Peter et al., 2004; Weissenhorn, 2005; Gallop et al., 2006).
Our data from Trp fluorescence and laurdan quenching experiments indicate that helix insertion plays an active role in membrane deformation. First, there is a tight time coincidence between helix insertion and the initiation of membrane deformation, suggesting that helix insertion serves as a rate-limiting step for local membrane deformation. Second, experiments using DMPC liposomes indicate that helix insertion works against membrane rigidity to disturb lipid bilayers. Indeed, simulation findings show that, when amphipathic helices become folded on lipid bilayers, membrane defects are enlarged to accommodate the additional turns of hydrophobic faces of the helices (Cui et al., 2011). After helix insertion, the formation of endophilin linear aggregates on membranes may facilitate further changes in membranes (Simunovic et al., 2013; Simunovic and Voth, 2015).
Time scales of large-scale membrane-remodeling events
EndoBAR remodels membranes into various structures, including bilayer tubules of various diameter, highly curved vesicles, and cylindrical micelles (Farsad et al., 2001; Mizuno et al., 2010; Mim et al., 2012; Ambroso et al., 2014). Our results suggest that these large-scale morphological changes occur on longer time scales, ranging from seconds to minutes. Under our experimental conditions, tubulation becomes visible at between 3 s and 2 min, whereas vesiculation happens after 2 min. Our data outline a time frame for endophilin to produce morphological effects. Of greater importance, these results suggest that endophilin’s action depends on how much time it has on endocytic membranes. All endocytosis events end with scission, although the duration of endocytic events varies significantly across different cell types. It is plausible that during rapid endocytosis, endophilin engages in only early actions due to time restriction, whereas it engages in additional activity during slower events. We propose that the temporal behavior of endophilin is an important aspect to consider when studying endophilin’s contributions to cellular activities.
A fine balance between curvature stability and membrane fission
Transient membrane intermediates are often energetically unstable to sustain tension due to their high degree of curvature. Fission disrupts the membrane continuity by pinching off remodeling membranes from their origins and subsequently creates new membrane compartments. Endophilin has a role in regulating tubule stability and membrane fission. Previous findings showed that BAR protein coats provide mechanical support for membrane tubules (Sorre et al., 2012). A recent study identified a new role for endophilin in the fission events during clathrin-independent endocytosis of Shiga and cholera toxin (Renard et al., 2015). Here our results suggest that a delicate balance between scaffolding and wedging is critical for tubule stability.
The mutant (M97S, F110S) endophilin exhibits both scaffolding and wedging defects, that is, a more curved scaffold interface, an H0 helix that fails to insert, and an HI helix that inserts less deeply. These data suggest that these mutations strongly affect how scaffolding and wedging mechanisms are integrated. Of interest, a mechanism for wedging–scaffolding integration has recently been proposed (Ambroso et al., 2014; Isas et al., 2015). These studies show that amphipathic helices insert deeply into membrane tubules, whereas on vesicles, they sit at much shallower positions in lipid bilayers. Deep insertion of helices brings the concave protein surface closer to membranes, which subsequently enhances the scaffolding effect (Isas et al., 2015). Therefore it is possible that a wedging–scaffolding switch controls the decision between tubulation and vesiculation (Isas et al., 2015). Our results are consistent with this model. The mutant EndoBAR HI helix inserts less deeply into the bilayer, and the H0 helix remains outside of the lipid core. These defects are likely to reduce the scaffolding effect and switch EndoBAR into a state promoting vesiculation. Consequently, weak scaffolding and shallower insertion may lead to increased curvature and reduced tubule stability (Campelo et al., 2008; Zemel et al., 2008; Ambroso et al., 2014; Isas et al., 2015).. In addition, changes in the dimer scaffold of mutant EndoBAR may lead to misplacement of amphipathic helices in membranes. The amphipathic helix H0 has been proposed to mediate intermolecular interactions between endophilin dimers (Mim et al., 2012; Cui et al., 2013), which hold endophilin in a position to form a stable lattice to promote the growth of membrane tubules. These models suggest that a coordinated integration of scaffolding and wedging activities is critical for tubule stability.
Finally, our results show that single-copy expression of the mutant (M97S, F110S) endophilin fails to restore miniature EPSC (mEPSC) frequency, suggesting that curvature stability is required for endophilin to support SV endocytosis in vivo. Surprisingly, expression of the mutant endophilin leads to a small but significant decrease in individual synaptic signals. These data suggest that the mutant endophilin not only loses its ability to support SV endocytosis, but it also gains an inhibitory role in endophilin-independent SV recycling—for example, slow clathrin-mediated endocytosis. Because several endophilin-like proteins have been implicated in clathrin-mediated endocytosis in neurons, we speculate that the mutant endophilin becomes “toxic” by interfering with other membrane-bending proteins. A reduction in mEPSC amplitude may be due to defects in the recycling of vesicular acetylcholine transporter or, alternatively, precocious pinching and smaller SVs.
Overall this study provides a dynamic view of how endophilin acts on the milliseconds-to-seconds time scale to support rapid endocytosis. It shows that endophilin is a multifaceted molecule that performs sensing and bending activities in a sequential manner to sculpt membranes. Delicate integration of multiple mechanisms may equip endophilin with functional specificity, which potentially explains why endophilin cannot be substituted by other membrane-bending BAR-domain proteins at synapses (Bai et al., 2010).
MATERIALS AND METHODS
Materials
Lipids 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine (DPPE), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), and NBD-DPPE were obtained from NOF America Corporation (White Plains, NY). The 05:0 PC (1,2-dipentanoyl-sn-glycero-3-phosphocholine), 16:0–18:0 (6-7BR) PC (1-palmitoyl-2-(6,7-dibromo)stearoyl-sn-glycero-3-phosphocholine), and 16:0–18:0 (9-10BR) PC (1-palmitoyl-2-(9,10-dibromo)stearoyl-sn-glycero-3-phosphocholine) were purchased from Avanti Polar Lipids (Alabaster, AL). Pacific Blue C5-maleimide and laurdan were purchased from Life Technologies (Carlsbad, CA). Other reagents and chemicals were purchased from Sigma-Aldrich unless otherwise noted.
Plasmids
cDNA encoding mouse endophilinA1 was amplified as previously described (Bai et al., 2010) and used for all analyses in this study. DNA fragments encoding his6-SUMO was inserted into the NcoI and BamHI sites of PET28A vector (Novagen, Madison, WA), which subsequently results in the plasmid BJP-A03. To construct plasmids for recombinant protein expression, DNA fragments encoding endophilin variants were ligated into the BamHI and NotI sites of BJP-A03. All mutations were generated using the overlapping primer method.
Recombinant proteins
Recombinant endophilinA1 variants were expressed as hexahistidine (his6)-SUMO–tagged fusion proteins in the BL21(DE3) Escherichia coli strain and purified using standard protocols. Briefly, the bacteria culture was grown in Luria broth medium at 37°C. When OD600 reached 0.6, isopropyl–d-thiogalactopyranoside (0.2 mM) was added to induce protein expression. The bacteria culture was further shaken at 200 rpm overnight at 15°C for protein production. Bacteria were harvested by centrifugation and lysed by sonication in the lysis buffer (50 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid [HEPES], pH 7.8, 300 mM NaCl, 15 mM imidazole). Proteins were purified using Ni-nitriloacetic acid (NTA)-agarose beads (Qiagen, Valencia, CA), and then eluted with lysis buffer plus 200 mM imidazole. Fusion proteins were cleaved by his6–ubiquitin-like-specific protease 1, and his6-SUMO fragments were removed using Ni-NTA-agarose beads (Qiagen). The resulting endophilin fragments have the native sequence without additional residues from the SUMO tag. Purified proteins were dialyzed against HEPES buffer (50 mM HEPES, pH 7.4, 150 mM NaCl) plus 2 mM dithiothreitol.
Fluorescence labeling of proteins
Native cysteine residue (C108) of EndoBAR was labeled by incubation of proteins with a 10-fold molar excess of Pacific Blue C-maleimide (Life Technologies) at 4°C overnight in HEPES buffer (50 mm HEPES, 150 mm NaCl, pH 7.4) with 5 mm Tris(2-carboxyethyl)phosphine). Unbound fluorophores were removed by dialyzing against 1 l of HEPES buffer for 4 h. Pacific Blue concentration was determined using molar extinction coefficient (35,000 cm−1 M−1 at 401 nm). Protein concentrations were measured using Coomassie blue staining of proteins in SDS–PAGE gels using bovine serum albumin (BSA) as standard. For all labeling experiments, the labeling ratio was >0.8–0.9.
Liposome preparation
Phospholipids were dried under compressed nitrogen for at least 3 h and then lyophilized in Labconco Free Zone 2.5 lyophilizer for 1.5 h to remove residue chloroform. Dried lipids were reconstituted in HEPES buffer, followed by a brief sonication to homogenize the mixture. Large unilamellar liposomes with various diameters were prepared by extrusion at least 15 times through polycarbonate membranes, using Mini extruders (Avanti Polar Lipids).
Steady-state fluorescence measurements
Steady-state fluorescence measurements were carried out using a Cary eclipse fluorescence spectrophotometer (Agilent, Santa Clara, CA). Pacific Blue fluorescence was excited at 400 nm, laurdan fluorescence was excited at 365 nm, and Trp fluorescence was excited at 285 nm. For all steady-state measurements, 1 μM protein was incubated with liposomes (0.5 mM total lipids with indicated composition) for 5 min before measurements. All experiments were carried out at room temperature.
Stopped-flow kinetic measurements
Stopped-flow measurements were carried out using a SX-20 stopped-flow spectrometer (Applied Photophysics, Leatherhead, UK) at room temperature. Laurdan was excited at 365 nm, Trp was excited at 285 nm, and Pacific Blue was excited at 400 nm. Laurdan emission fluorescence was collected using a 395-nm long-pass filter, NBD fluorescence was collected using a 515-nm long-pass filter, and Trp fluorescence was collected using 295-nm long-pass filter. All of the filters were supplied by Applied Photophysics. The dead time of the instrument was ∼1 ms. The on-rate (kon) and off-rate (koff) for the interaction of endophilin with liposomes was calculated using pseudo-first-order kinetics (kobs = [liposome]kon + koff).
Depth measurements
The depth measurements of Trp probes were carried out using parallax analysis (McIntosh and Holloway, 1987; Kaiser and London, 1998; Bai et al., 2000) The distance of the Trp residue from the bilayer center (ZCF) was calculated from the equation
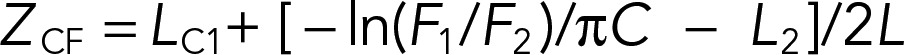 |
where LC1 represents the distance from the bilayer center to the shallow quencher (11 Å for 6,7-Br2-PC), C is the mole fraction of the quencher divided by the lipid area (70 Å2), F1 and F2 are the relative fluorescence intensities of the shallow (6,7-Br2-PC) and deep (9,10-Br2-PC) quenchers, respectively, and L is the difference in the depth of the two quenchers (0.9 Å per CH2 or CBr2 group). For these brominated lipids, the thickness of the hydrophobic region is ∼29 Å.
Negative staining and transmission electron microscopy
Sample preparation for negative stain was carried out as previously described, with modifications (Farsad et al., 2001). Briefly, nickel grids were glow discharged for 40 s, after which grids were incubated with samples for their respective times. Samples were fixed using 0.5× Karnovsky’s fix at 3 s, 2 min, and 30 min. Samples were then washed with one drop of 0.1 M cacodylate buffer, followed by a four-drop H2O wash. One drop of 1% uranyl acetate was touched on the sample. The grids were then carefully dragged through dry filter papers and put in a desiccator overnight to dry. All images were collected in a JEOL TEM 1400 transmission electron microscope. All measurements were made using ImageJ.
Simulation setup
The BAR-domain dimer was modeled based on Protein Data Bank (PDB) ID 2CO8 (Weissenhorn, 2005). The VMD Molefacture plug-in (www.ks.uiuc.edu/Research/vmd/plugins/) was used to model the H0 helices in helix conformation. The VMD Mutator plug-in was used to introduce mutations. Sodium and chloride ions were added to neutralize the simulated systems and to reach an ion concentration of 100 mM. The simulations used a hybrid-resolution model, PACE (Han and Schulten, 2012, 2013; Yu and Schulten, 2013; Qi et al., 2014; Gamini et al., 2014), and yet the results rival those with all-atom simulations. The BAR domain was placed in the center of the water box with 20-Å margins in the x-direction and 25 Å of water margins in the y- and z-directions on each side. The resulting models contained ∼180,000 particles. The simulations described a constant-temperature, constant-pressure ensemble; temperature was maintained at 310 K through a Langevin thermostat with damping coefficient γ = 0.5 ps−1; pressure was maintained at 1 atm with a Langevin-piston barostat (Cui et al., 2011). Short-range nonbonded interactions were cut off smoothly between 10 and 12 Å; electrostatic interactions were modeled by effective potentials with shift from 0 to 12 Å; simulations were performed with an integration time step of 5 fs in NAMD 2.9 (Phillips et al., 2005).
All trajectories were aligned to PDB structures based on helix residues before analysis. The final 50-ns trajectories were clustered with 6-Å cutoff. The cluster central structure of the most populated cluster with >80% structure population was chosen as representative structure. The protein curvature was calculated as previously reported (Arkhipov et al., 2008; Yin et al., 2009; Yu and Schulten, 2013). The principal axis of the BAR domain defines an xz-plane, with the xz-axes being defined by the largest and smallest dimensions of the principal axis at time t = 0. The radius of curvature of the BAR domain was calculated by least-squared fitting of a circle to the helical residue profile (residues 29–58 and 90–250, H0 and HI helices not included) in the xz-plane. All visualization and analysis were performed in VMD.
Liposome flotation assay
Experiments were carried out using published protocols with minor modifications (Wragg et al., 2013). Briefly, protein (1 μM) and liposomes (2 mM total lipids; 70% PC, 25% PS, and 5% NBD-PE) were incubated at room temperature for 5 min. A sucrose gradient with respective percentages was prepared in a buffer containing 50 mM HEPES (pH 7.4) and 150 mM NaCl. Gradients were prepared to a final of 30 and 20% by diluting stock solution of 60% sucrose. Proteins and liposomes were loaded together into the 30% sucrose fraction. The sucrose gradient was set up using 500 μl of 30% sucrose solution and 500 μl of buffer separated by 4 ml of 20% sucrose solution. Samples were centrifuged in a SW-55-Ti rotor (Beckman) at 55,000 rpm for 2 h. A 50-μl sample was collected from the bottom. The top fraction was collected to include NBD-containing liposomes and concentrated to 50 μl. Samples (30 μl) were loaded onto 10% SDS–PAGE gels, followed by Western blotting. EndoBAR was detected using a mouse monoclonal antibody against endophilin A1 (55702; Abcam) and a horseradish peroxidase–conjugated goat anti-mouse secondary antibody (GenScript) and visualized using enhanced chemiluminescence.
Liposome cosedimentation assay
Experiments were performed as previously described (Hui et al., 2006). Protein (1 μM) and liposomes (2 mM total lipids; 70% PC, 25% PS, and 5% NBD-PE) were incubated at room temperature for 30 min. Samples were centrifuged in a TlA100.1 at 75,000 rpm for 30 min. Twenty microliters of the supernatant and pellet was resuspended in SDS sample buffer and loaded onto 10% SDS–PAGE gels. Proteins were visualized using Coomassie blue stain.
Electrophysiology
Young adult worms were immobilized on Sylgard-coated coverslips with cyanoacrylate glue (Histoacryl Blue; Aesculap). Animals were dissected in extracellular solution via a dorsolateral incision, and gonad and intestines were removed to reveal the underlying ventral nerve cord and body-wall-muscle quadrants as previously described (Richmond and Jorgensen, 1999; Madison et al., 2005; Dong et al., 2015). Then the worm prep was mounted onto a fixed-stage upright microscope (BX51WI; Olympus) equipped with a 60× water-immersion objective lens.
Whole-cell patch clamp recordings were carried out at 20°C. A body-wall muscle cell was voltage clamped at −60 mV to record postsynaptic currents. Evoked EPSC responses were induced by applying a 0.4-ms, 30-μA pulse, generated by a stimulus isolator (A365; WPI), though a borosilicate pipette (∼2 MΩ) placed in close apposition to the ventral nerve cord. Series resistance was compensated to 70% for the evoked EPSC recording. The currents were amplified using EPC-10 (HEKA). The signals were sampled at 10 kHz using Patchmaster (HEKA), after low-pass filtering at 2 kHz. Patch pipettes (2–5 MΩ) were pulled using borosilicate glass and fire polished. The extracellular solution contained (in mM) 150 NaCl, 5 KCl, 1 CaCl2, 5 MgCl2, 10 glucose, and 10 HEPES, titrated to pH 7.3 with NaOH, 330 mOsm with sucrose. The internal solution contained (in mM) 135 CH3O3SCs, 5 CsCl, 5 MgCl2, 5 ethylene glycol tetraacetic acid, 0.25 CaCl2, 10 HEPES, and 5 Na2ATP, adjusted to pH 7.2 using CsOH. All chemicals were purchased from Sigma-Aldrich.
Electrophysiological data were analyzed using Igor Pro 6 (Wavemetrics) with custom-written software. Average values are reported as SEM. Statistical analysis was performed using Igor Pro 6. The p values were generated using Student’s t test or one-way analysis of variance followed by Dunnett’s test. p < 0.05 was considered to be significant.
Supplementary Material
Acknowledgments
We thank Harmit Malik, Gerald Smith, and members of the Bai lab for comments. We thank Anbang Dai, Peter Rupert, Roland Strong, and the Fred Hutchinson Cancer Research Center shared resources for technical assistance. This work was supported by grants from the National Institutes of Health (R01-NS085214 to J.B. and P41-RR005969, R01-GM067887, and U54-GM087519 to K.S.), a National Institutes of Health Interdisciplinary Training Grant (T32CA080416 to K.P.), and an American Heart Association Fellowship (14POST18230006 to K.P.). Computer time was provided by the Texas Advanced Computing Center through Grant MCA93S028 allocated by the Extreme Science and Engineering Discovery Environment Program funded by the National Science Foundation.
Abbreviations used:
- Cys
cysteine
- EndoBAR
Endophilin Bin Amphiphysin Rvs domain
- PS/PC
phosphatidylserine/phosphocholine
- SV
synaptic vesicle
- Trp
tryptophan
- WT
wild type.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-04-0264) on May 11, 2016.
REFERENCES
- Ambroso MR, Hegde BG, Langen R. Endophilin A1 induces different membrane shapes using a conformational switch that is regulated by phosphorylation. Proc Natl Acad Sci USA. 2014;111:6982–6987. doi: 10.1073/pnas.1402233111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkhipov A, Yin Y, Schulten K. Four-scale description of membrane sculpting by BAR domains. Biophys J. 2008;95:2806–2821. doi: 10.1529/biophysj.108.132563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arranz AM, Delbroek L, Kolen KV, Guimarães MR, Mandemakers W, Daneels G, Matta S, Calafate S, Shaban H, Baatsen P, et al. LRRK2 functions in synaptic vesicle endocytosis through a kinase-dependent mechanism. J Cell Sci. 2015;128:541–552. doi: 10.1242/jcs.158196. [DOI] [PubMed] [Google Scholar]
- Bai J, Earles CA, Lewis JL, Chapman ER. Membrane-embedded synaptotagmin penetrates cis or trans target membranes and clusters via a novel mechanism. J Biol Chem. 2000;275:25427–25435. doi: 10.1074/jbc.M906729199. [DOI] [PubMed] [Google Scholar]
- Bai J, Hu Z, Dittman JS, Pym ECG, Kaplan JM. Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell. 2010;143:430–441. doi: 10.1016/j.cell.2010.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgart T, Capraro BR, Zhu C, Das SL. Thermodynamics and mechanics of membrane curvature generation and sensing by proteins and lipids. Annu Rev Phys Chem. 2011;62:483–506. doi: 10.1146/annurev.physchem.012809.103450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia VK, Madsen KL, Bolinger P-Y, Kunding A, Hedegård P, Gether U, Stamou D. Amphipathic motifs in BAR domains are essential for membrane curvature sensing. EMBO J. 2009;28:3303–3314. doi: 10.1038/emboj.2009.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigay J, Antonny B. Curvature, lipid packing, and electrostatics of membrane organelles: defining cellular territories in determining specificity. Dev Cell. 2012;23:886–895. doi: 10.1016/j.devcel.2012.10.009. [DOI] [PubMed] [Google Scholar]
- Boucrot E, Pick A, Çamdere G, Liska N, Evergren E, McMahon HT, Kozlov MM. Membrane fission is promoted by insertion of amphipathic helices and is restricted by crescent BAR domains. Cell. 2012;149:124–136. doi: 10.1016/j.cell.2012.01.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campelo F, McMahon HT, Kozlov MM. The hydrophobic insertion mechanism of membrane curvature generation by proteins. Biophys J. 2008;95:2325–2339. doi: 10.1529/biophysj.108.133173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capraro BR, Shi Z, Wu T, Chen Z, Dunn JM, Rhoades E, Baumgart T. Kinetics of endophilin N-BAR domain dimerization and membrane interactions. J Biol Chem. 2013;288:12533–12543. doi: 10.1074/jbc.M112.435511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Lyman E, Voth GA. Mechanism of membrane curvature sensing by amphipathic helix containing proteins. Biophys J. 2011;100:1271–1279. doi: 10.1016/j.bpj.2011.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui H, Mim C, Vázquez FX, Lyman E, Unger VM, Voth GA. Understanding the role of amphipathic helices in N-BAR domain driven membrane remodeling. Biophys J. 2013;104:404–411. doi: 10.1016/j.bpj.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova R, Aranda S, Bezlyepkina N, Nikolov V, Riske KA, Lipowsky R. A practical guide to giant vesicles. Probing the membrane nanoregime via optical microscopy. J Phys Condens Matter. 2006;18:S1151. doi: 10.1088/0953-8984/18/28/S04. [DOI] [PubMed] [Google Scholar]
- Dong Y, Gou Y, Li Y, Liu Y, Bai J. Synaptojanin cooperates in vivo with endophilin through an unexpected mechanism. eLife. 2015;4:e05660. doi: 10.7554/eLife.05660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farsad K, Ringstad N, Takei K, Floyd SR, Rose K, Camilli PD. Generation of high curvature membranes mediated by direct endophilin bilayer interactions. J Cell Biol. 2001;155:193–200. doi: 10.1083/jcb.200107075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Raimondi A, Paradise S, Shen H, Mesaki K, Ferguson A, Destaing O, Ko G, Takasaki J, Cremona O, et al. Coordinated actions of actin and BAR proteins upstream of dynamin at endocytic clathrin-coated pits. Dev Cell. 2009;17:811–822. doi: 10.1016/j.devcel.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost A, Unger VM, De Camilli P. The BAR domain superfamily: membrane-molding macromolecules. Cell. 2009;137:191–196. doi: 10.1016/j.cell.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gad H, Ringstad N, Löw P, Kjaerulff O, Gustafsson J, Wenk M, Di Paolo G, Nemoto Y, Crun J, Ellisman MH, et al. Fission and uncoating of synaptic clathrin-coated vesicles are perturbed by disruption of interactions with the SH3 domain of endophilin. Neuron. 2000;27:301–312. doi: 10.1016/s0896-6273(00)00038-6. [DOI] [PubMed] [Google Scholar]
- Gallop JL, Jao CC, Kent HM, Butler PJG, Evans PR, Langen R, McMahon HT. Mechanism of endophilin N-BAR domain-mediated membrane curvature. EMBO J. 2006;25:2898–2910. doi: 10.1038/sj.emboj.7601174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamini R, Han W, Stone JE, Schulten K. Assembly of Nsp1 nucleoporins provides insight into nuclear pore complex gating. PLoS Comput Biol. 2014;10:e1003488. doi: 10.1371/journal.pcbi.1003488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guichet A, Wucherpfennig T, Dudu V, Etter S, Wilsch-Bräuniger M, Hellwig A, González-Gaitán M, Huttner WB, Schmidt AA. Essential role of endophilin A in synaptic vesicle budding at the Drosophila neuromuscular junction. EMBO J. 2002;21:1661–1672. doi: 10.1093/emboj/21.7.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Schulten K. Further optimization of a hybrid united-atom and coarse-grained force field for folding simulations: improved backbone hydration and interactions between charged side chains. J Chem Theory Comput. 2012;8:4413–4424. doi: 10.1021/ct300696c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W, Schulten K. Characterization of folding mechanisms of Trp-Cage and WW-domain by network analysis of simulations with a hybrid-resolution model. J Phys Chem B. 2013;117:13367–13377. doi: 10.1021/jp404331d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook MJV, Thoreson WB. Rapid synaptic vesicle endocytosis in cone photoreceptors of salamander retina. J Neurosci. 2012;32:18112–18123. doi: 10.1523/JNEUROSCI.1764-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu SF, Jackson MB. Rapid exocytosis and endocytosis in nerve terminals of the rat posterior pituitary. J Physiol. 1996;494:539–553. doi: 10.1113/jphysiol.1996.sp021512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Bai J, Chapman ER. Ca2+-triggered simultaneous membrane penetration of the tandem C2-domains of synaptotagmin I. Biophys J. 2006;91:1767–1777. doi: 10.1529/biophysj.105.080325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionescu D, Ganea C. A study of quercetin effects on phospholipid membranes containing cholesterol using Laurdan fluorescence. Eur Biophys J. 2012;41:307–318. doi: 10.1007/s00249-011-0786-0. [DOI] [PubMed] [Google Scholar]
- Isas JM, Ambroso MR, Hegde PB, Langen J, Langen R. Tubulation by amphiphysin requires concentration-dependent switching from wedging to scaffolding. Structure. 2015;23:873–881. doi: 10.1016/j.str.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao CC, Hegde BG, Gallop JL, Hegde PB, McMahon HT, Haworth IS, Langen R. Roles of amphipathic helices and the bin/amphiphysin/rvs (BAR) domain of endophilin in membrane curvature generation. J Biol Chem. 2010;285:20164–20170. doi: 10.1074/jbc.M110.127811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser RD, London E. Determination of the depth of BODIPY probes in model membranes by parallax analysis of fluorescence quenching. Biochim Biophys Acta. 1998;1375:13–22. doi: 10.1016/s0005-2736(98)00127-8. [DOI] [PubMed] [Google Scholar]
- Kummrow M, Helfrich W. Deformation of giant lipid vesicles by electric fields. Phys Rev A. 1991;44:8356–8360. doi: 10.1103/physreva.44.8356. [DOI] [PubMed] [Google Scholar]
- Liu J, Sun Y, Drubin DG, Oster GF. The mechanochemistry of endocytosis. PLoS Biol. 2009;7:e1000204. doi: 10.1371/journal.pbio.1000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Löw C, Weininger U, Lee H, Schweimer K, Neundorf I, Beck-Sickinger AG, Pastor RW, Balbach J. Structure and dynamics of helix-0 of the N-BAR domain in lipid micelles and bilayers. Biophys J. 2008;95:4315–4323. doi: 10.1529/biophysj.108.134155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, Kaplan JM. UNC-13 interaction with syntaxin is required for synaptic transmission. Curr Biol. 2005;15:2236–2242. doi: 10.1016/j.cub.2005.10.049. [DOI] [PubMed] [Google Scholar]
- Marsh D. Elastic curvature constants of lipid monolayers and bilayers. Chem Phys Lipids. 2006;144:146–159. doi: 10.1016/j.chemphyslip.2006.08.004. [DOI] [PubMed] [Google Scholar]
- Masuda M, Takeda S, Sone M, Ohki T, Mori H, Kamioka Y, Mochizuki N. Endophilin BAR domain drives membrane curvature by two newly identified structure-based mechanisms. EMBO J. 2006;25:2889–2897. doi: 10.1038/sj.emboj.7601176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta S, Van Kolen K, da Cunha R, van den Bogaart G, Mandemakers W, Miskiewicz K, De Bock P-J, Morais VA, Vilain S, Haddad D, et al. LRRK2 controls an EndoA phosphorylation cycle in synaptic endocytosis. Neuron. 2012;75:1008–1021. doi: 10.1016/j.neuron.2012.08.022. [DOI] [PubMed] [Google Scholar]
- McIntosh TJ, Holloway PW. Determination of the depth of bromine atoms in bilayers formed from bromolipid probes. Biochemistry. 1987;26:1783–1788. doi: 10.1021/bi00380a042. [DOI] [PubMed] [Google Scholar]
- McMahon HT, Gallop JL. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature. 2005;438:590–596. doi: 10.1038/nature04396. [DOI] [PubMed] [Google Scholar]
- Milosevic I, Giovedi S, Lou X, Raimondi A, Collesi C, Shen H, Paradise S, O’Toole E, Ferguson S, Cremona O, et al. Recruitment of endophilin to clathrin-coated pit necks is required for efficient vesicle uncoating after fission. Neuron. 2011;72:587–601. doi: 10.1016/j.neuron.2011.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mim C, Cui H, Gawronski-Salerno JA, Frost A, Lyman E, Voth GA, Unger VM. Structural basis of membrane bending by the N-BAR protein endophilin. Cell. 2012;149:137–145. doi: 10.1016/j.cell.2012.01.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno N, Jao CC, Langen R, Steven AC. Multiple Modes of endophilin-mediated conversion of lipid vesicles into coated tubes. Implications for synaptic endocytosis. J Biol Chem. 2010;285:23351–23358. doi: 10.1074/jbc.M110.143776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasassi T, Gratton E. Packing of phospholipid vesicles studied by oxygen quenching of Laurdan fluorescence. J Fluoresc. 1992;2:167–174. doi: 10.1007/BF00866931. [DOI] [PubMed] [Google Scholar]
- Peter BJ, Kent HM, Mills IG, Vallis Y, Butler PJG, Evans PR, McMahon HT. BAR domains as sensors of membrane curvature: the amphiphysin BAR structure. Science. 2004;303:495–499. doi: 10.1126/science.1092586. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinot M, Vanni S, Pagnotta S, Lacas-Gervais S, Payet L-A, Ferreira T, Gautier R, Goud B, Antonny B, Barelli H. Polyunsaturated phospholipids facilitate membrane deformation and fission by endocytic proteins. Science. 2014;345:693–697. doi: 10.1126/science.1255288. [DOI] [PubMed] [Google Scholar]
- Prinz WA, Hinshaw JE. Membrane-bending proteins. Crit Rev Biochem Mol Biol. 2009;44:278–291. doi: 10.1080/10409230903183472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Cheng X, Han W, Jo S, Schulten K, Im W. CHARMM-GUI PACE CG Builder for solution, micelle, and bilayer coarse-grained simulations. J Chem Inf Model. 2014;54:1003–1009. doi: 10.1021/ci500007n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renard H-F, Simunovic M, Lemière J, Boucrot E, Garcia-Castillo MD, Arumugam S, Chambon V, Lamaze C, Wunder C, Kenworthy AK, et al. Endophilin-A2 functions in membrane scission in clathrin-independent endocytosis. Nature. 2015;517:493–496. doi: 10.1038/nature14064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richmond JE, Jorgensen EM. One GABA and two acetylcholine receptors function at the C. elegans neuromuscular junction. Nat Neurosci. 1999;2:791–797. doi: 10.1038/12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rikhy R, Kumar V, Mittal R, Krishnan KS. Endophilin is critically required for synapse formation and function in Drosophila melanogaster. J Neurosci. 2002;22:7478–7484. doi: 10.1523/JNEUROSCI.22-17-07478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Gad H, Löw P, Di Paolo G, Brodin L, Shupliakov O, De Camilli P. Endophilin/SH3p4 is required for the transition from early to late stages in clathrin-mediated synaptic vesicle endocytosis. Neuron. 1999;24:143–154. doi: 10.1016/s0896-6273(00)80828-4. [DOI] [PubMed] [Google Scholar]
- Schuske KR, Richmond JE, Matthies DS, Davis WS, Runz S, Rube DA, van der Bliek AM, Jorgensen EM. Endophilin is required for synaptic vesicle endocytosis by localizing synaptojanin. Neuron. 2003;40:749–762. doi: 10.1016/s0896-6273(03)00667-6. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Hu J, Kozlov MM, Rapoport TA. Mechanisms shaping the membranes of cellular organelles. Annu Rev Cell Dev Biol. 2009;25:329–354. doi: 10.1146/annurev.cellbio.042308.113324. [DOI] [PubMed] [Google Scholar]
- Simunovic M, Srivastava A, Voth GA. Linear aggregation of proteins on the membrane as a prelude to membrane remodeling. Proc Natl Acad Sci USA. 2013;110:20396–20401. doi: 10.1073/pnas.1309819110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simunovic M, Voth GA. Membrane tension controls the assembly of curvature-generating proteins. Nat Commun. 2015;6:7219. doi: 10.1038/ncomms8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorre B, Callan-Jones A, Manzi J, Goud B, Prost J, Bassereau P, Roux A. Nature of curvature coupling of amphiphysin with membranes depends on its bound density. Proc Natl Acad Sci USA. 2012;109:173–178. doi: 10.1073/pnas.1103594108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stachowiak JC, Schmid EM, Ryan CJ, Ann HS, Sasaki DY, Sherman MB, Geissler PL, Fletcher DA, Hayden CC. Membrane bending by protein–protein crowding. Nat Cell Biol. 2012;14:944–949. doi: 10.1038/ncb2561. [DOI] [PubMed] [Google Scholar]
- Suresh S, Edwardson JM. The endophilin N-BAR domain perturbs the structure of lipid bilayers. Biochemistry. 2010;49:5766–5771. doi: 10.1021/bi100760e. [DOI] [PubMed] [Google Scholar]
- Vanni S, Hirose H, Barelli H, Antonny B, Gautier R. A sub-nanometre view of how membrane curvature and composition modulate lipid packing and protein recruitment. Nat Commun. 2014;5:4916. doi: 10.1038/ncomms5916. [DOI] [PubMed] [Google Scholar]
- Verstreken P, Koh T-W, Schulze KL, Zhai RG, Hiesinger PR, Zhou Y, Mehta SQ, Cao Y, Roos J, Bellen HJ. Synaptojanin is recruited by endophilin to promote synaptic vesicle uncoating. Neuron. 2003;40:733–748. doi: 10.1016/s0896-6273(03)00644-5. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature. 1994;370:652–655. doi: 10.1038/370652a0. [DOI] [PubMed] [Google Scholar]
- Watanabe S, Liu Q, Davis MW, Hollopeter G, Thomas N, Jorgensen NB, Jorgensen EM. Ultrafast endocytosis at Caenorhabditis elegans neuromuscular junctions. eLife. 2013a;2:e00723. doi: 10.7554/eLife.00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Rost BR, Camacho-Pérez M, Davis MW, Söhl-Kielczynski B, Rosenmund C, Jorgensen EM. Ultrafast endocytosis at mouse hippocampal synapses. Nature. 2013b;504:242–247. doi: 10.1038/nature12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissenhorn W. Crystal structure of the endophilin-A1 BAR domain. J Mol Biol. 2005;351:653–661. doi: 10.1016/j.jmb.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Wragg RT, Snead D, Dong Y, Ramlall TF, Menon I, Bai J, Eliezer D, Dittman JS. Synaptic vesicles position complexin to block spontaneous fusion. Neuron. 2013;77:323–334. doi: 10.1016/j.neuron.2012.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Arkhipov A, Schulten K. Simulations of membrane tubulation by lattices of amphiphysin N-BAR domains. Structure 1993. 2009;17:882–892. doi: 10.1016/j.str.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H, Schulten K. Membrane sculpting by F-BAR domains studied by molecular dynamics simulations. PLoS Comput Biol. 2013;9:e1002892. doi: 10.1371/journal.pcbi.1002892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemel A, Ben-Shaul A, May S. Modulation of the spontaneous curvature and bending rigidity of lipid membranes by interfacially adsorbed amphipathic peptides. J Phys Chem B. 2008;112:6988–6996. doi: 10.1021/jp711107y. [DOI] [PubMed] [Google Scholar]
- Zhang Y-L, Frangos JA, Chachisvilis M. Laurdan fluorescence senses mechanical strain in the lipid bilayer membrane. Biochem Biophys Res Commun. 2006;347:838–841. doi: 10.1016/j.bbrc.2006.06.152. [DOI] [PubMed] [Google Scholar]
- Zimmerberg J, Kozlov MM. How proteins produce cellular membrane curvature. Nat Rev Mol Cell Biol. 2006;7:9–19. doi: 10.1038/nrm1784. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.