

Fms-like tyrosine kinase 3 (FLT3) is one of the most commonly mutated genes in acute myeloid leukemia (AML) and as such has been a highly studied therapeutic target in the field. Indeed, highly potent FLT3 inhibitors such as quizartinib (AC220) have generated substantial antileukemic clinical effects among relapsed/refractory AML patients with FLT3-internal tandem duplication (ITD) mutations, as well as a smaller number of patients without documented FLT3 mutation.1 Importantly, nearly 70% of quizartinib-treated FLT3-ITD+ patients show objective antileukemic responses per the modified International Working Group response criteria,2 many of which allowed subsequent allogeneic hematopoietic cell transplantation and prolonged survival in some.3
Although initially FLT3 inhibition was believed to act on myeloblasts primarily via direct cytotoxicity,4 recent data suggest that in some AML patients FLT3 inhibition causes terminal differentiation of FLT3-ITD+ blasts.5,6 As FLT3 inhibition is a major area of ongoing preclinical and clinical investigation in the field of AML, we sought to define predictors of differentiation and cytotoxic effects of quizartinib therapy at a molecular and genetic level. We retrospectively performed an independent analysis of all marrow studies from the 21 patients treated with quizartinib at our center on a recent phase 2 clinical trial. Here we confirm that FLT3 inhibition by quizartinib generates potent antileukemic effects manifesting as two major patterns of drug response, namely differentiation and cytotoxic responses. We also provide evidence that cooperating mutations modify clinical response to FLT3 kinase inhibition.
Patients with relapsed/refractory AML after one to two prior lines of chemotherapy were enrolled in a phase 2 study of quizartinib monotherapy at the Hospital of the University of Pennsylvania (NCT00989261). We report here a correlative analysis of the entire institutional cohort that enrolled to the larger multicenter NCT00989261 phase 2 trial, internally termed the AC220-002, or ‘ACE’ trial. All patients provided written informed consent according to the Declaration of Helsinki both for trial participation as well as for institutional tissue banking. Bone marrow aspirates were collected at baseline and on treatment days 15, 29 and 56, unless a protocol-defined complete remission CRp, or CRi occurred. Marrow samples were additionally collected at study withdrawal or disease progression. All available histomorphologic, cytogenetic and molecular genetic materials were independently reviewed for publication. Comparison between patients was done at the best response time points defined by the lowest percent blasts in the bone marrow assessment. Bone marrow biopsies and aspirates were analyzed by light microscopy using a Leica DM 2500 clinical microscope (Buffalo Grove, IL, USA). Photographs were taken using an attached Leica DFC420 digital camera (Buffalo Grove, IL, USA). Fisher’s exact test was used in the analysis of contingency tables.
Twenty-one patient samples were analyzed, of which 19 were FLT3-ITD+ at initial diagnosis (the trial eventually expanded to enroll patients lacking FLT3-ITD). Among four patients with only wild-type (WT) FLT3 detected at study entry, two were previously FLT3-ITD+ but had low blast percentages at screening (<20% but >5% blasts consistent with early relapse). Therefore, the lack of FLT3-ITD detection in these cases may reflect sensitivity limitations of assays or loss of FLT3-ITD mutation at relapse.7
Objective antileukemic treatment effects were seen in 20/21 patients, evident by rapid clearance of peripheral blood blasts and extramedullary leukemia (for example, leukemia cutis), typically within 1 week, consistent with previously published trial data6 (data not shown). Primary resistance to quizartinib was only observed in one patient who in addition to a low-abundance FLT3-ITD (ratio of FLT3-ITD:WT of 1:14) had NRAS G13D, which has been shown to confer in vitro resistance to FLT3 inhibitors.8 On serial marrow testing, reductions in marrow blasts to <50% of baseline blast percentage were noted in 18/19 patients (one patient was not evaluable owing to <10% marrow blasts at baseline marrow; Figure 1a).
Figure 1.
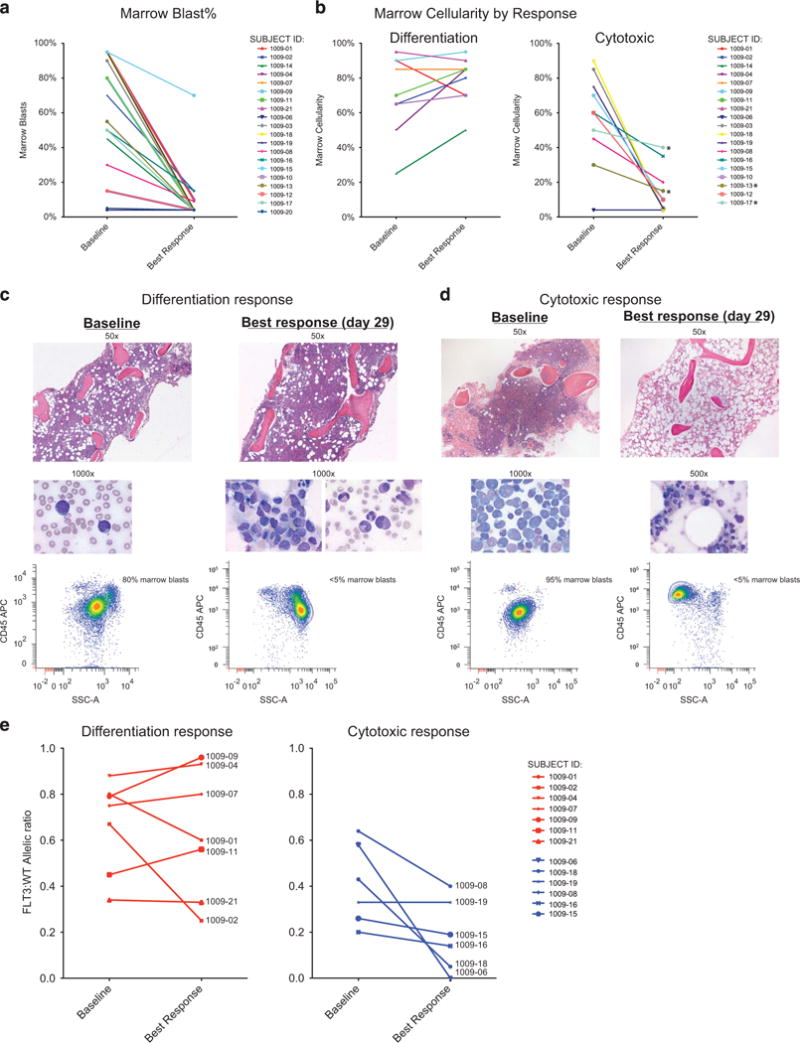
Quizartinib therapy reduces marrow blast burden by the induction of either differentiation or cytotoxicity responses. (a, b) Comparison of blast burden and marrow cellularity at baseline and at best response time points. (c, d) Representative photomicrographs of two patients with differentiation and cytotoxic responses. (e) Graph of FLT3-ITD:WT allelic ratio, organized by color according to response. The ratio has been converted to ITD frequency (ITD/ITD+WT) for ease of graphical display.
Reviewing the patients’ marrows, we noted that responses to quizartinib separated into two different groups. In what we term a cytotoxic response, quizartinib rapidly reduced marrow cellularity (to <40%) and peripheral counts showed limited, if any neutrophil recovery after blast clearance. In the contrasting response, which we term a differentiation response, patients retained or developed increased marrow cellularity and blast clearance from the blood was followed by robust neutrophil recovery, generally on the second month of therapy (Figure 1b). Representative photomicrographs of both responses in the bone marrow are shown in Figures 1c and d. Serial marrow sampling of patients with differentiation response generally showed hypercellularity that was preserved across 2–3 months of study therapy (Supplementary Table 1). Progression of leukemia or withdrawal from study therapy for allogeneic transplant limited longer follow-up of these marrow responses.
Importantly, in patients with the differentiation response, despite a marked reduction in marrow blast percentages, the allelic ratio of FLT3-ITD:WT remained relatively constant in many patients (Figure 1e), suggesting that regenerating hematopoiesis in these cases reflected differentiation of the leukemic clone. Indeed, before quizartinib, FLT3-ITD could only be demonstrated in the peripheral blood in the presence of circulating blasts. However, following clearance of circulating blasts in response to quizartinib, FLT3-ITD was readily detected in purified circulating neutrophils from patients with differentiation response (Supplementary Table 2). Allelic ratios of FLT3-ITD:WT from marrow obtained during differentiation response also generally showed preservation of FLT3-ITD allelic burden comparable to pretreatment levels, despite reduction in marrow blasts to <5%.
Next, we observed that nearly all 9 patients with the differentiation response pattern (8/9 FLT3-ITD+) had a normal karyotype, whereas conversely all 10 of the patients with a cytotoxic response (7/10 FLT3-ITD+ at study enrollment) had a baseline abnormal karyotype upon study enrollment (Table 1). We note that karyotypes listed reflect those measured at study entry and abnormal karyotypes seen then were in some cases normal at original AML diagnosis. We next tested whether the differentiation and cytotoxic responses differed with regard to particular cooperating mutations. As shown in Table 1, targeted next-generation sequencing of 33 hematologic malignancy-associated genes revealed that 8/9 patients with differentiation response to quizartinib had mutated DNMT3A (5 R882, 3 pathogenic non-R882 substitutions and 1 variant of unclear significance), and 7/9 additionally had an NPM1 mutation. By contrast, a diverse number of mutations were seen among the 8 patients with cytotoxic response who had samples available for mutational testing, including 2 with DNMT3A mutations, 1 of which also contained an NPM1 mutation. The latter patient had abnormal cytogenetics as shown. Noteworthy, at progression, 5/8 patients demonstrated new FLT3-TKD (D835) mutations by PCR and/or direct sequencing, 3 after cytotoxic response and 2 after differentiation response. No difference in time to progression was noted in the two groups of patients.
Table 1.
DNMT3A mutation status segregates patients into differentiation and cytotoxic responses
| Subjec | FLT3 Genotype | Best marrow blast (%) | Baseline karyotype response | Karyotype at response | Cooperating mutations |
|---|---|---|---|---|---|
| Differentiation response | |||||
| 1009-01 | FLT3-ITD | 10 | 46,XY | 46,XY | DNMT3A*, NPM1, ASXL1, IDH1 |
| 1009-02 | FLT3-ITD | 15 | 46,XX | 46,XX | DNMT3A, NPM1, TET2 |
| 1009-14 | FLT3-ITD | <5 | 46,XY | 46,XY | DNMT3A, NPM1 |
| 1009-04 | FLT3-ITD | <5 | 46,XX | 46,XX | DNMT3A, NPM1, WT1, ATM* |
| 1009-07 | FLT3-ITD | <5 | 46,XX | 46,XX | DNMT3A, NPM1 |
| 1009-09 | FLT3-ITD | <5 | 46,XY | 46,XY | DNMT3A, NPM1, TET2 |
| 1009-11 | FLT3-ITD | <5 | 46,XX | 46,XX | DNMT3A, NPM1, TET2 |
| 1009-10 | FLT3-WT | <5 | 46,XY | 46,XY | TET2 |
| 1009-21 | FLT3-ITD | 10 | 46,XY,+11 | 46,XY,+11 | DNMT3A, ASXL1 |
| Cytotoxic response | |||||
| 1009-06 | FLT3-ITD | <5 | Complex | 46,XY | Not evaluable |
| 1009-03 | FLT3-ITD | <5 | Hyperdiploid/complex | 46,XX | DNMT3A, RUNX1 |
| 1009-13 | FLT3-WT | <5 | Complex | 46,XY | TP53, NOTCH1 |
| 1009-12 | FLT3-WT | <5 | Complex | 46,XY | Not evaluable |
| 1009-17 | FLT3-WT | <5 | Complex | Complex | TP53, JAK2 |
| 1009-19 | FLT3-ITD | <5 | Complex | No growth | No mutations |
| 1009-08 | FLT3-ITD | <10 | Complex | Complex | RUNX1 |
| 1009-18 | FLT3-ITD | <5 | 46,XY, t(8;21),(q22;q22) with multiple additional abnormalities | 46,XY, t(8;21),(q22;q22) with multiple additional abnormalities | TET2* |
| 1009-16 | FLT3-ITD | 15 | 46,XY,del(5)(q23q33) | 46,XY,del(5)(q23q33) | ATM* |
| 1009-15 | FLT3-ITD | 70 | 47,XX,+8,del(16)(q13) | 47,XX,+8,del(16)(q13) | DNMT3A, NPM1 |
Abbreviations: FLT3, Fms-like tyrosine kinase 3; ITD, internal tandem duplication. Genetic and cytogenetic studies at baseline and best response are shown. Responding patients with normal karyotype uniformly showed differentiation response. Patients with differentiation response had a statistically significant increase in the incidence of NPM1 mutations (odds ratio (OR) = 24.5, P<0.01) and/or DNMT3A (OR = 10.5, P<0.05, Fisher’s exact test). One subject with rapidly progressive leukemia and one with unclassifiable response due to low baseline blast content are not shown. FLT3-ITD and D835 mutations were detected using fluorescent primers and multiplexed PCR amplification followed by capillary electrophoresis and/or EcoRV digestion resistance assay by published methods. Allelic ratios of FLT3-ITD to the WT sequence were calculated using peak area (GeneMapper Software, Applied Biosystems, Grand Island, NY, USA). DNA was additionally extracted from cryopreserved blast samples and analyzed by next-generation sequencing for 23 genes commonly mutated in AML.15 Briefly, sequencing was done using an amplicon-based capture protocol (Luminex, Grand Island, NY, USA) with sequencing using MiSeq. Average depth of coverage was over 2000X and minimum depth was 250X. Sensitivity for mutations was 4% for all mutations other than FLT3-ITD, which was sensitive to a variant allele frequency of 1%. With the exception of variants of unclear significance (VOUS)—as indicated by asterisk—all gene mutations listed are recurrent pathogeneic sequence alterations as referenced by public databases, including dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), the Catalog of Somatic Mutations in Cancer (http://cancer.sanger.ac.uk/cosmic) and the 1000 Genomes Project (http://www.1000genomes.org). Statistical analyses did not include VOUS.
To summarize, AC220 induces two different patterns of clinical response to therapy. Patients demonstrating a differentiation response are frequently characterized by normal cytogenetics and the presence of FLT3-ITD, DNMT3A, and NPM1 mutations. Overall, our data suggest that FLT3-ITD potentially serves different functions in leukemogenesis based on the context of various cooperating mutations. Mutational analysis by the Cancer Genome Atlas revealed a subgroup of AML that contain mutations in FLT3, DNMT3A and NPM1.9 The significance of this grouping of mutations remains unexplained. DNMT3A catalyzes the methylation of cytosine residues at 5-carbon sites, thereby epigenetically altering gene expression. Recurrent mutations in DNMT3A disrupt its methyltransferase activity, occur early in leukemogenesis, and expand the hematopoietic stem cell pool; however, functional effects upon differentiation are controversial.10,11 NPM1 contributes to ribosome biogenesis, cell division and DNA damage response/repair; however, its precise function in normal hematopoiesis or leukemogenesis remains incompletely understood.12,13 Our data provide the first demonstration that this group of patients have a unique differentiation response to AC220, which may suggest the underlying biology of this triple-mutant leukemia affecting myeloid differentiation.14 Our study sample was highly enriched for the mutation composite of FLT3, DNMT3A and NPM1. Of note, patients with this triple mutational profile appear to have an increased likelihood of relapse, which may explain their frequency in our cohort of relapsed/refractory patients (Papaemmanuil E, et al. EHA abstracts 2015, and personal communication). Because of this enrichment, effects generated by NPM1 or DNMT3A alone could be missed, as is the potential to detect other genotypes associated with differentiation response. This may have the potential to bias our results, and further study is needed to validate our observations.
Taken together, data from our cohort provide additional mechanistic insight into the antileukemic effects of potent FLT3 inhibition in AML patients by providing a cytogenetic and molecular correlation between the differential cytotoxic and differentiation effects experienced by patients on quizartinib. In addition, the data may generate discussion regarding possibly redefining treatment responses to FLT3 and other molecularly targeted inhibitors in AML. There is precedent for this in other tumor types where clinical benefit to molecularly targeted agents has been noted in the absence of traditional metrics. This is exemplified by ibrutinib therapy of chronic lymphocytic leukemia, where therapy-induced lymphocytosis is characteristic of response rather than resistance. Although determining clinical benefit to FLT3 inhibitors is beyond the scope of this report, our data nonetheless provide a framework for predicting and understanding patient responses to FLT3 inhibition both as monotherapy and future combination trials.
Acknowledgments
This study was supported by the National Institutes of Health, the National Cancer Institute (NCI; grant 1K23CA141054 to AEP) and the Leukemia and Lymphoma Society (NPS, CCS and AEP). NPS was supported by a grant from the National Cancer Institute (1R01 CA166616-01) and MC is supported by R21CA198621 also from the NCI. We thank Cezary Swider, PhD for technical expertise and assistance with cell processing.
Footnotes
Results were presented in part at the 54th annual meeting of the American Society of Hematology meeting.
CONFLICT OF INTEREST
AEP has served as a consultant to Ambit Biosciences, Astellas Pharmaceuticals and Arog Pharmaceuticals. CCS received clinical trial grant funding from Astellas. The remaining authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
GEN designed research, collected data, performed research, performed histopathologic analysis of clinical samples and wrote the paper; JC analyzed data and wrote the paper; DR collected data; JDM and CDW performed research NPS and CCS performed research; AB performed histopathologic analysis of clinical samples; MC and AEP designed research, analyzed data and wrote the paper. All authors provided critical review of the paper and approved the submission.
Supplementary Information accompanies this paper on the Leukemia website (http://www.nature.com/leu)
References
- 1.Cortes JE, Perl AE, Dombret H, Kayser S, Steffen B, Rousselot P, et al. Final results of a phase 2 open-label, monotherapy efficacy and safety study of quizartinib (AC220) in patients >= 60 years of age with FLT3 ITD positive or negative relapsed/refractory acute myeloid leukemia. Blood. 2012;120:48. [Google Scholar]
- 2.Cheson BD, Bennett JM, Kopecky KJ, Buchner T, Willman CL, Estey EH, et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol. 2003;21:4642–4649. doi: 10.1200/JCO.2003.04.036. [DOI] [PubMed] [Google Scholar]
- 3.Smith CC, Wang Q, Chin CS, Salerno S, Damon LE, Levis MJ, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260–263. doi: 10.1038/nature11016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, et al. A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood. 2002;99:3885–3891. doi: 10.1182/blood.v99.11.3885. [DOI] [PubMed] [Google Scholar]
- 5.Fathi AT, Le L, Hasserjian RP, Sadrzadeh H, Levis M, Chen YB. FLT3 inhibitor-induced neutrophilic dermatosis. Blood. 2013;122:239–242. doi: 10.1182/blood-2013-01-478172. [DOI] [PubMed] [Google Scholar]
- 6.Sexauer A, Perl A, Yang X, Borowitz M, Gocke C, Rajkhowa T, et al. Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AML. Blood. 2012;120:4205–4214. doi: 10.1182/blood-2012-01-402545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cloos J, Goemans BF, Hess CJ, van Oostveen JW, Waisfisz Q, Corthals S, et al. Stability and prognostic influence of FLT3 mutations in paired initial and relapsed AML samples. Leukemia. 2006;20:1217–1220. doi: 10.1038/sj.leu.2404246. [DOI] [PubMed] [Google Scholar]
- 8.Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007;109:1643–1652. doi: 10.1182/blood-2006-05-023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller CA, Wilson RK, Ley TJ. Genomic landscapes and clonality of de novo AML. N Engl J Med. 2013;369:1473. doi: 10.1056/NEJMc1308782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. DNMT3A is essential for hematopoietic stem cell differentiation. Nat Genet. 2012;44:23–31. doi: 10.1038/ng.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506:328–333. doi: 10.1038/nature13038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Falini B, Bolli N, Liso A, Martelli MP, Mannucci R, Pileri S, et al. Altered nucleophosmin transport in acute myeloid leukaemia with mutated NPM1: molecular basis and clinical implications. Leukemia. 2009;23:1731–1743. doi: 10.1038/leu.2009.124. [DOI] [PubMed] [Google Scholar]
- 13.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 14.Galanis A, Levis M. Inhibition of c-Kit by tyrosine kinase inhibitors. Haematologica. 2015;100:e77–e79. doi: 10.3324/haematol.2014.117028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sehgal AR, Gimotty PA, Zhao J, Hsu JM, Daber R, Morrissette JD, et al. DNMT3A mutational status affects the results of dose-escalated induction therapy in acute myelogenous leukemia. Clin Cancer Res. 2015;21:1614–1620. doi: 10.1158/1078-0432.CCR-14-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]