

SUMMARY
During class switch recombination (CSR), B cells replace the Igh Cμ or δ exons with another down-stream constant region exon (CH), altering the anti-body isotype. CSR occurs through the introduction of AID-mediated double-strand breaks (DSBs) in switch regions and subsequent ligation of broken ends. Here, we developed an assay to investigate the dynamics of DSB formation in individual cells. We demonstrate that the upstream switch region Sμ is first targeted during recombination and that the mechanism underlying this control relies on 53BP1. Surprisingly, regulation of break order occurs through residual binding of 53BP1 to chromatin before the introduction of damage and independent of its established role in DNA repair. Using chromosome conformation capture, we show that 53BP1 mediates changes in chromatin architecture that affect break order. Finally, our results explain how changes in Igh architecture in the absence of 53BP1 could promote inversional rearrangements that compromise CSR.
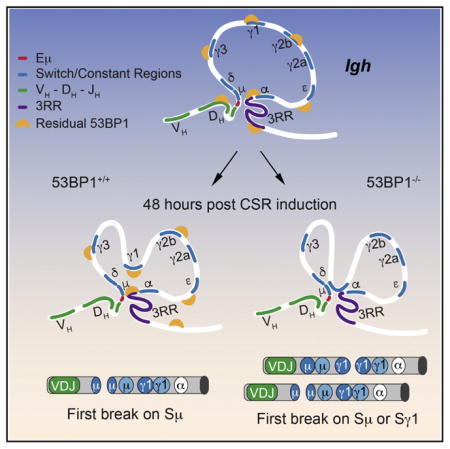
Graphical Abstract
INTRODUCTION
Class switch recombination (CSR) is dependent on the cytidine deaminase enzyme (AID), which initiates the formation of two double-strand breaks (DSBs) within the switch regions of Igh that precede each CH. The broken ends are then joined by members of the non-homologous end joining (NHEJ) pathway, placing a new CH exon in front of the V(D)J exons (Keim et al., 2013; Stavnezer and Schrader, 2014). This occurs through preferential joining of proximally located DSBs on the same chromosome (Gostissa et al., 2014). CSR is distinct from other recombination events that join two DSBs where ligation can either result in a deletional event or inversion of the intervening sequence (Dong et al., 2015). What makes CSR special is that somehow, through an unknown mechanism that is dependent on the DNA-damage pathway effector 53BP1, break repair is strongly biased toward deletional joining, thereby increasing the efficiency of the process (Di Noia, 2015; Dong et al., 2015).
The introduction of I-SceI sites in place of switch regions results in an increase in the frequency of inversional events. This demonstrates that the switch regions themselves are important for the bias toward deletional joining (Dong et al., 2015). Because I-SceI breaks are likely to occur simultaneously and at a similar frequency on the two sites, they do not reflect the dynamics of AID-mediated breaks on switch regions, which are presumed to occur at different rates and in a particular order, with the upstream Sμ site being targeted first (Chaudhuri et al., 2004). This suggests that break order might be an important determinant for successful deletional CSR. In addition, the fact that rare inter-chromosomal Igh rearrangements involving switch regions do not share a deletional bias (Dong et al., 2015) points to a role for chromatin architecture of the cis allele favoring deletional events. However, there have been no studies that examine break order or chromatin architecture of Igh and the impact of 53BP1 in either context.
The first studies investigating the dynamics of DSB formation during CSR indicate that AID targeting of Sμ occurs independently and at higher frequency than targeting of the downstream switch region (Dudley et al., 2002; Gu et al., 1993; Schrader et al., 2003; Zhang et al., 2010). Other studies using I-SceI-introduced DSBs in the Myc locus demonstrate that AID-induced translocations to the Igh Sμ region (Chiarle et al., 2011; Hu et al., 2014; Klein et al., 2011) occur at a 2-fold increased rate compared to downstream switch regions, a much lower frequency than that expected from mutation rate differences in each location (Dudley et al., 2002; Schrader et al., 2003). The discrepancy between these results might arise from the fact that these studies were based on analyses of populations of cells and, therefore, do not provide information about the dynamics of DSB introduction in single cells. To address this issue, we used a single-cell meta-phase-based fluorescence in situ hybridization (FISH) assay to study the dynamics of AID-mediated DSB introduction on Igh switch regions.
RESULTS
A Single-Cell System to Study the Order of DSB Formation during CSR
For our assay, we prepared metaphase spreads after 60–65 hr of B cell activation using αCD40 and IL4 to induce IgG1 switching. We used a mixture of four differentially labeled DNA probes, which included a mouse chromosome 12 paint (to identify the chromosome containing Igh), two locus-specific probes that hybridize to the region immediately surrounding the Igh locus (named 5′ and 3′ probes), and a probe that hybridizes to the region between Sμ and Sγ3 (named the Cμ probe) (Figure 1A). An Igh-switched allele was identified by loss of the Cμ signal on chromosome 12 (Figure 1B; Figure S1A). Using this system, we were able to accurately examine switching in an allele-specific manner, and we found that the level at which switching occurred was compatible with that detected by fluorescence-activated cell sorting (FACS) analysis (Table S1).
Figure 1. A Single-Cell System to Study the Dynamics of Break Order during CSR.

(A) Scheme of the Igh locus and location of Igh-specific probes used in the study. Switch and constant regions are represented as black and white ovals, respectively. Mouse chromosome 12 is identified by a yellow chromosome paint.
(B) Example of chromosome 12 with an unswitched (left) and switched Igh allele (right) is shown.
(C) Examples of Igh alleles with break first on Sμ (left), Sγ1 (center), or unknown (right). Full pictures of the cells where these examples were captured can be found in Figure S1.
The DNA probes we designed also can be used to detect CSR-related chromosomal abnormalities on the Igh locus. As a proof of principle, we activated splenic B cells in the presence of an inhibitor of ATM kinase (ATMi), which is known to increase chromosomal abnormalities in the Igh locus due to its contribution to DSB repair and activation of cell cycle checkpoints (Reina-San-Martin et al., 2004). As previously reported (Franco et al., 2006), the most frequent chromosomal abnormality on the Igh locus was the separation of its ends such that the Igh 5′ telomeric probe and the Igh 3′ signal were no longer found together on chromosome 12 (split ends) (Figure S1B; Table S2). In addition, we detected frequent Igh translocations with loci on other chromosomes as well as translocations between the two alleles of chromosome 12. The latter form dicentric chromosomes that retain the 3′ probe (Franco et al., 2006). Finally, we also detected frequent breaks on chromosome 12 that occurred centromeric to the Igh locus, as has been described previously in ATM-deficient B cells (Hu et al., 2014).
To investigate the order of DSBs introduced during CSR, we focused on cells where the 5′ and 3′ probes were no longer together at the telomeric end of chromosome 12 (Igh split ends category). It should be noted that this type of damage results from AID-induced breaks on switch regions, as it is not detected in AID-deficient cells (Aicda−/−) (Figure S1B). In cells activated to undergo IgG1 switching, chromosomes that retained the Cμ probe on chromosome 12 next to the Igh 3′ probe allowed us to infer that the break occurred at Sμ, while Sγ1 was still intact (Figure 1C, left and Figure S1C). On the other hand, if the Cμ probe was associated with the 5′ probe and both were separated from chromosome 12, we could conclude that the break occurred first on Sγ1 (Figure 1C, middle). Finally, in cells where the Cμ probe was separated from the 5′ probe and both were separated from chromosome 12, we could not identify the location of the first break and we classified these as unknown (Figure 1C, right). As the Cμ probe is located upstream of Sγ3, this same system also could be applied to lipopolysaccharide (LPS) activation, which induces switching to IgG3 and IgG2a, or to CH12 cells activated in the presence of αCD40, IL4, and TGF-β to undergo IgA switching (Figure S1A).
A 53BP1-Dependent Mechanism Ensures Targeting of Sμ
We analyzed the location of breaks during CSR in B cells derived from wild-type mice, ATM-deficient mice (Atm−/−), and wild-type B cells treated with an ATM kinase inhibitor. Across the three conditions, Sμ was the first switch region to be targeted in more than 95% of the chromosomes where break location could be determined (Figure 2A; Table S3). These results are in line with analysis of mutation rates that show that Sμ is targeted at a higher frequency than downstream switch regions (Dudley et al., 2002; Reina-San-Martin et al., 2007; Schrader et al., 2003).
Figure 2. 53BP1 Determines Break Order Independently of the DNA Damage Pathway.

(A and C) Igh alleles where location of the first break can be determined were divided into break-first-on-Sμ or -Sγ1 categories for the different mutants or treatments. Significance was calculated using Fisher’s exact test. Table S3 details the number of replicates and alleles analyzed. Plots represent the average frequency of each category across replicate experiments. N represents the amount of Igh alleles where break order could be determined across all replicates analyzed.
(B) Scheme shows the DSB repair pathway that is active during CSR.
Given 53BP1’s unique role in biasing toward deletional recombination events, we next asked whether 53BP1 could influence DSB introduction. Indeed, in B cells deficient for 53BP1 (Trp53bp1−/−), the mechanism ensuring that breaks are introduced first on Sμ is lost and the first break is introduced randomly at Sμ and Sγ1 (Figure 2A). This mechanism is not specific for IgG1 switching, as the same effect was detected in cells activated with LPS (that switch to IgG3 and IgG2a) as well as in the CH12 cell line (where switching to IgA occurs) (Figure S2A).
53BP1 Impacts DSB Introduction Independently of the DNA Damage Pathway
Given our finding that 53BP1 is important for ensuring break order, we next asked whether proteins involved in 53BP1 recruitment also impact the dynamics of DSB introduction. Analysis of B cells deficient for H2AX (H2afx−/−) or expressing a hypomorphic NBS1 (Nbs1hypo) showed that these cells were able to maintain a wild-type phenotype with respect to break order (Figures 2B and 3C). This is a surprising result because γH2AX and the MRN complex (composed of MRE11, NBS1, and RAD50) are necessary for recruitment of 53BP1 following the introduction of DSBs on Igh (Panier and Boulton, 2014; Reina-San-Martin et al., 2004).
Figure 3. 53BP1 Impacts the Architecture of Igh during CSR.

(A) 4C-seq using a bait (black arrow) located on the Eμ enhancer. Lines represent the average 4C signal across replicates. Colored circles represent the 10-kb windows that have significant changes in contact frequency (adjusted p value < 0.05) from wild-type (WT) at 48 hr. For easier visualization, colored bars below lines represent consecutive windows that are statistically different from WT controls at 48 hr. For simplicity, only μ and γ1 switch regions are shown (blue boxes).
(B) Hi-C on the same region in CH12 cells. Dashed lines represent interactions between Eμ and 3RR. Hi-C data are from the CH12 in-situ-combined track described in Rao et al. (2014).
(C) Model shows changes in constant region locus conformation upon activation in WT and 53BP1-deficient cells and how these changes might favor inversional rearrangements.
To determine whether 53BP1’s role in inhibiting resection is an important factor in determining break order, we next analyzed metaphase spreads from RIF1-deficient B cells (Rif1−/−). 53BP1 recruits RIF1 in an ATM-dependent manner to inhibit resection of the switch regions and recruitment of BRCA1 (Panier and Boulton, 2014). Our results clearly indicate that B cells deficient in RIF1 did not recapitulate the defect in break order that was found in 53BP1 mutant B cells (Figures 2B and 2C). Finally, we examined activated B cells treated with a combination of ATMi and DNA PK inhibitors that prevent NHEJ repair (Callén et al., 2009). Once again, we found that the cells exhibited a wild-type phenotype for break order. Together, these data suggest that the mechanism by which 53BP1 impacts the choice of break order occurs independently of the MRN-ATM-γH2AX pathway and its known role in DNA repair.
Altered DSB Dynamics in the Absence of 53BP1 Is Not Due to an Accumulation of DSBs in Sγ1
Given that AID-mediated breaks are introduced during the G1 phase of the cell cycle, it could be argued that the defect in break order in 53BP1-deficient cells results from an accumulation of DSBs in Sγ1 that are more difficult to repair. However, if that were the case, slower repair would increase the chances of breaks in both switch regions, thereby increasing the number of cells in the unknown category, which we did not observe (Table S3). Moreover, cells deficient in proteins upstream (ATM, γH2AX, and MRN) or downstream (RIF1 and DNA PKcs) of 53BP1 in the DNA-damage repair pathway do not show the same phenotype as 53BP1-deficient cells. Thus, if the DSBs in Sγ1 were caused by an accumulation of difficult-to-repair breaks, the 53BP1 phenotype should be recapitulated in these other models. That this was not the case indicates that the 53BP1 phenotype is not an artifact caused by a defect in DNA repair. Additionally, we can rule out that the 53BP1 effect is related to an increase in resection, since we did not detect the same phenotype in RIF1-deficient cells. Moreover, inhibition of ATM in 53BP1-deficient cells, which prevents resection (Bunting et al., 2010; Dong et al., 2015), did not change the distribution of where breaks were introduced first (Figures 2B and 2C). Since ATMi blocks activation of cell cycle checkpoints, this result also suggests that the breaks we detected did not result from differences in the ability of the mutant cells to activate cell cycle checkpoints or a downstream accumulation of damage.
To further validate that the phenotype we observed in cells deficient for 53BP1 was not caused by the accumulation of Sγ1 breaks that are difficult to repair, we repeated the assay at different time points (48, 72, and 96 hr of activation) using wild-type B cells treated with an ATMi as well as B cells from 53BP1-deficient mice. At 48 hr, cells deficient for 53BP1 already displayed the same phenotype we observed at later time points, where the initial DSB could be found at the same frequency in Sμ and Sγ1 (Figure S2B). As AID-induced breaks only start to accumulate following 48 hr of activation (Schrader et al., 2005), this provides further support that the phenotype is not an artifact caused by the accumulation of breaks.
Residual Binding of 53BP1 to Chromatin Independent of Damage Determines DSB Introduction
Our results indicate that 53BP1 ensures break order independently of its role in DNA repair. Residual binding of 53BP1 to chromatin has been shown to occur throughout the genome independent of DNA damage (Bekker-Jensen et al., 2005; Bohgaki et al., 2013; Bothmer et al., 2011; Santos et al., 2010). Binding relies on the histone mark H4K20me2, which is recognized by the TUDOR domain of 53BP1. This modification is deposited by the SUV4-20H1/H2 enzymes, and, in SUV4-20H1/H2-deficient cells, 90% of the methylated H4K20 is converted to the mono-methyl state (Schotta et al., 2008). CSR efficiency in these mutant B cells has been shown to be reduced by 50%, and there is accompanying damage detected on the Igh locus. Despite these findings, in mutant cells, recruitment of 53BP1 to sites of breaks occurs at high levels, albeit at a slower rate. Metaphase analysis of activated SUV4-20H1/H2-deficient splenic B cells (Suv4-20hdn) demonstrates that an absence of the di-methyl mark has no impact on the order of break introduction in the Sμ and Sγ1 switch regions (Figure S2C). This result suggests that the TUDOR domain of 53BP1 can efficiently recognize the H4K20me1 mark, a finding that supports previous observations (Botuyan et al., 2006; Oda et al., 2010).
The H4K20me1 mark is deposited by the enzyme PR-SET7, which is essential for proliferation and cellular viability (Oda et al., 2010). It is therefore not possible to assess whether loss of the mono-methyl mark on H4K20 contributes to the 53BP1 phenotype of disrupted break order. However, a single amino acid mutation on the TUDOR domain of 53BP1 (53BP1DR mutant mice) renders it unable to bind mono- or di-methylated H4K20 (Bekker-Jensen et al., 2005; Bothmer et al., 2011). Thus, analysis of activated splenic B cells isolated from these mice enabled us to address this question, and these cells fully recapitulated the phenotype of disrupted break order that we detected in 53BP1-deficient B cells (Figure S2C). Together these findings indicate that binding of 53BP1 to methylated H4K20 is important for determining break order, however, the di-methyl mark is dispensable for this function.
53BP1 Impacts the Architecture of Igh during CSR
Since changes in nuclear organization can have a dramatic impact on somatic recombination of antigen receptor loci (Chaumeil and Skok, 2012; Gerasimova et al., 2015), we asked whether a change in locus conformation could explain the changes in break order on switch regions during CSR. To determine the chromatin architecture of the Igh locus during CSR, we performed high-resolution circular chromosome conformation capture sequencing (4C-seq) in B cells activated to undergo IgG1 switching. For this we used the 4-bp cutter enzyme DpnII as our primary restriction enzyme. Use of this frequent cutter enzyme increases resolution of interactions along the Igh locus 10-fold compared to 4C-seq libraries prepared with 6-bp cutters (Jankovic et al., 2013). Furthermore, 4C allows expansion of previous chromosome conformation capture (3C) analyses investigating Igh chromatin conformation (Feldman et al., 2015; Sellars et al., 2009; Wuerffel et al., 2007), by providing an unbiased assessment of relative interaction frequencies of all sites from a single viewpoint.
For 4C-seq experiments, we designed a bait located on the Eμ enhancer, very close to Sμ (Figure 3A; Figure S3A). In resting cells (day 0), Eμ interacts predominantly with the 3′ regulatory region (3RR) enhancer, while the constant region exons between the two enhancers are extruded from this looping interaction. Analysis of Hi-C data, generated at high resolution in resting CH12-LX cells (Rao et al., 2014), confirms that, in resting cells, the Igh constant region is encompassed within a loop containing at its base the Eμ and 3RR enhancers (Figure 3B; Figure S3B). This structure resembles a topologically associated domain (TAD) (Nora et al., 2012) that is most likely to favor interactions between switch regions that are necessary for successful CSR.
According to our analysis, the Sγ1 region contacts the Eμ-3RR loop base at increased frequency 48 hr after activation (a time when most breaks are being introduced). This is in line with previous 3C studies on the Igh locus (Wuerffel et al., 2007). Interestingly, in 53BP1-deficient cells, interaction of Sμ with Sγ1 occurred at a significantly higher level (Figure 3A; Figure S3A). Importantly, cells with combined AID and 53BP1 deficiency (Trp53bp1−/−; Aicda−/−) displayed the same interaction profile as cells deficient in 53BP1 alone. These findings provide the first evidence that 53BP1 has an impact on chromatin structure. That this effect occurs upstream of DNA damage is consistent with our finding that 53BP1 alters break order independently of the MRN-ATM-γH2AX pathway.
It is also interesting that, in contrast to previous studies (Feldman et al., 2015; Sellars et al., 2009; Wuerffel et al., 2007), we detected a slight reduction in the interaction between Eμ and the 3RR following 48 hr of activation. The interaction frequency was still high between Eμ and 3RR, but lower than that seen for Eμ-Cγ1. This discrepancy could result from different activation conditions: αCD40 + IL4 versus LPS + IL4. When we used the same 4C primers in an experiment in which B cells were activated with LPS + IL4, we also found an increase in the interaction between Eμ and the 3RR (Thomas-Claudepierre et al., 2016). It is known that activation under these conditions results in differences in targeting of switch regions: LPS with IL4 induces DSBs at Sγ3, Sγ1, and Sε, while αCD40 + IL4 results in targeting of Sγ1 and Sε. Therefore, it is plausible that different activation conditions lead to differences in the activation rate of the 3RR enhancer region and, therefore, interfere with chromatin dynamics.
Our results suggest a possible mechanism by which 53BP1 could influence break order during CSR. Residual binding of 53BP1 to chromatin independently of DNA-damage controls Igh chromatin architecture ensuring a restricted contact between the Sγ1 region and the loop base containing Sμ and the two Igh enhancers (Eμ and 3RR). In the absence of 53BP1, increased contact of Sγ1 with the enhancer hub is linked to the first break occurring randomly in Sμ or Sγ1. We propose that this change in chromosome looping exposes Sγ1 to the same high concentration of AID to which Sμ is exposed, resulting in the first break being introduced as frequently in Sγ1 as in Sμ (Figure 3C).
DISCUSSION
In this study, we have developed an assay to investigate the order in which AID-mediated DSBs are introduced on Igh switch regions. Using this assay, we have shown that class switching B cells rely on a 53BP1-dependent mechanism that ensures the first break is introduced on the most upstream switch region (Sμ). Proteins involved in DSB repair and recruitment of 53BP1 to sites of damage do not contribute to break order regulation, suggesting that 53BP1 has a role in orchestrating CSR outside of its well-characterized function in DNA repair. It is tempting to speculate that failure to orchestrate appropriate break order could, in part, explain why 53BP1 mutant B cells have the lowest levels of switching compared to B cells deficient in other repair factors (Di Virgilio et al., 2013; Dong et al., 2015).
It is important to note that our assay cannot distinguish between breaks that would result in successful CSR from breaks that would be repaired within the same switch region and cause small internal deletions. We, therefore, assume that both these events have similar dynamics when it comes to targeting of switch regions by AID. We also cannot determine whether the amount of breaks in each location is altered in the absence of 53BP1 or simply a change in the location of the first break. Analysis of AID-induced mutations in Sμ suggests that AID targeting is not affected by an absence of 53BP1 (Reina-San-Martin et al., 2007). This is expected, as the absence of 53BP1 does not impact AID expression nor does it impact the level of switch region sterile transcripts (Reina-San-Martin et al., 2007). To measure differences in DSB formation, the J. Stavnezer lab used ligation-mediated PCR (LM-PCR) and showed that an absence of 53BP1 does not affect the level of DSBs introduced at Sμ (Khair et al., 2014). Furthermore, Sγ3 is targeted at the same frequency in wild-type versus 53BP1-deficient B cells undergoing switching to IgG3 in the presence of LPS. Unfortunately, assessment of breaks in Sγ1 is hindered by its length and repetitiveness. These findings suggest that, although 53BP1 is important for determining where the first break is introduced, the level of DSBs at these sites is not altered.
A recent study has shown that CSR is intrinsically biased toward productive deletional events with non-productive inversional events being suppressed (Dong et al., 2015). As in our study, the mechanism underlying orientation-dependent recombination depends on 53BP1 and is independent of the ATM/γH2AX pathway and resection via RIF1. The authors of this paper propose that nuclear organization of the switch regions may be responsible for establishing this mechanism. Our data support this model, as they identify a role for 53BP1 in altering the architecture of the Igh constant region, leading to changes in break order and the outcome of CSR.
It is unclear at this point whether the change in location of the first break has any impact on whether recombination occurs via a deletional rather than an inversional event or whether this bias is solely due to the change in chromatin configuration. However, we speculate that alterations in locus conformation that lead to higher frequency Sμ-Sγ1 interactions in the absence of 53BP1 favor the ligation events needed for inversional recombination. This requires that two pairs of broken ends find each other in 3D space, in contrast to deletional events that rely on only one pair of ends coming together (Figure 3C). Clearly, inversional joining events favor the introduction of breaks occurring in close proximity, and this is indeed what we detected in 53BP1-deficient cells. Thus, the four broken ends are in more frequent contact compared to wild-type cells.
Here, we have uncovered an unanticipated function for 53BP1 in controlling chromatin conformation of Igh independently of damage during CSR. This finding raises the question of whether 53BP1 has similar functions in altering chromatin conformation outside of the Igh locus, where it could protect against AID targeting and other types of DNA damage. Likely candidate regions are repeats enriched with H4K20me2 marks that act as substrates for residual binding of 53BP1. Hi-C experiments will show whether the effect on chromatin conformation that we detected on the Igh locus expands to such repeat regions.
EXPERIMENTAL PROCEDURES
For further description see the Supplemental Experimental Procedures.
Metaphase FISH Assays
Metaphase FISH analyses were performed and analyzed as previously described (Rocha et al., 2012). The probe identifying the 5′ end of Igh was generated by labeling of BAC RP24-386J17 and the 3′ of Igh was labeled using BAC CT7-199M11. The Cμ probe was designed using the webfish tool (Nedbal et al., 2012) (webfish2.org). These probes were amplified by PCR from genomic DNA, mixed together at equimolar amounts, and then labeled by nick translation.
4C-Seq
4C-seq was performed and analyzed as previously described (Raviram et al., 2016; Rocha et al., 2012). The DpnII and Csp6 enzymes were used for digestions. Mice deficient for AID (Aicda−/−), 53BP1 (Trp53bp1−/−), as well as double-deficient mice (Trp53bp1−/−;Aicda−/−) were generated by breeding double heterozygous Trp53bp1+/−; Aicda+/− mice. The 53bp1+/+; Aicda+/+ mice also were generated from this breeding scheme and used as controls. 4C-seq libraries were produced by PCR amplification using primers containing Illumina single-read adaptors, barcodes for lane multiplexing, and the following Igh Eμ-specific sequence: 5′–TCTGTCCTAAAGGCTCTGAGATC and 5′–GAACACAGAAGTATGTGTATGGA. A total of 1 μg DNA was amplified per sample using 30 PCR cycles.
Mapping was performed using Bowtie2 to a reduced genome consisting of all unique 24-nt-long regions surrounding DpnII sites, allowing for zero mismatches. As can be seen in Figure S3C, our strategy allows mapping of 4C reads to the whole constant region of Igh including the switch regions. Samples containing less than 0.6 million mapped reads (following removal of undigested and self-ligated fragments) were discarded. Additionally, samples with fewer than 0.2 cis/trans ratio of mapped reads and with fewer than 40% read coverage for all DpnII sites in the 200 kb surrounding the bait region were discarded, as these are considered important criteria for high-quality 4C-seq datasets.
Analysis of 4C-seq signal was performed by building successive 10-kb windows with 95% overlap and calculating the total number of reads per window. The following mm10 coordinates were used for this analysis Chr12: 113175000–113475000. For comparison between genotypes and/or conditions, DESeq2 1.10.0 with default parameters was used to normalize total read count per window between samples and to identify the windows with significant 4C signal differences, using an FDR-adjusted p value cutoff of 0.05.
Supplementary Material
Highlights.
53BP1 is essential for preferential targeting of the upstream switch region
53BP1’s absence results in increased interaction frequency between switch regions
Alterations in switch region targeting and locus architecture impact CSR
53BP1’s action is independent of its role in the double-strand break repair pathway
Acknowledgments
We thank members of the J.A.S. lab for discussions, Alexander Nubla and New York University Medical Center (NYUMC) Division of Laboratory Animal Resources (DLAR) for mouse breeding, the NYU High Performance Computing (HPC) (Shenglong Wang) for computing technical support, and the NYUMC Denome Technology Center (GTC) core. We are also thankful to the following people who shared material with us: Frederick Alt, Michel Nussenzweig, and Barry Sleckman. This work was supported by grants from the NIH (GM59413 and P30 CA008748 to J.P. and GM086852 and GM112192 to J.A.S.) and the Deutsche Forschungsgemeinschaft (SFB684) to G.S. J.A.S. is a Leukemia Lymphoma Society Scholar. P.P.R. is a National Cancer Center and American Society of Hematology fellow.
Footnotes
ACCESSION NUMBERS
The accession number for the 4C-seq data reported in this paper is GEO: GSE81180.
Supplemental Information includes Supplemental Experimental Procedures, three figures, and three tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2016.05.073.
AUTHOR CONTRIBUTIONS
P.P.R., Y.F., J.K., V.L., A.A., E.S., A.P., and A.B. performed experiments. P.P.R., R.R., and J.A.S. analyzed the data. E.R.M., R.B., J.P., and G.S. contributed with reagents and analytical tools. P.P.R. and J.A.S. conceived the project and wrote the manuscript with help from all authors.
References
- Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. Dynamic assembly and sustained retention of 53BP1 at the sites of DNA damage are controlled by Mdc1/NFBD1. J Cell Biol. 2005;170:201–211. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohgaki M, Bohgaki T, El Ghamrasni S, Srikumar T, Maire G, Panier S, Fradet-Turcotte A, Stewart GS, Raught B, Hakem A, Hakem R. RNF168 ubiquitylates 53BP1 and controls its response to DNA double-strand breaks. Proc Natl Acad Sci USA. 2013;110:20982–20987. doi: 10.1073/pnas.1320302111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callén E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, Nussenzweig M. Essential role for DNA-PKcs in DNA double-strand break repair and apoptosis in ATM-deficient lymphocytes. Mol Cell. 2009;34:285–297. doi: 10.1016/j.molcel.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430:992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- Chaumeil J, Skok JA. The role of CTCF in regulating V(D)J recombination. Curr Opin Immunol. 2012;24:153–159. doi: 10.1016/j.coi.2012.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell. 2011;147:107–119. doi: 10.1016/j.cell.2011.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM. Molecular biology: Unequal opportunity during class switching. Nature. 2015;525:44–45. doi: 10.1038/nature15209. [DOI] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Panchakshari RA, Zhang T, Zhang Y, Hu J, Volpi SA, Meyers RM, Ho YJ, Du Z, Robbiani DF, et al. Orientation-specific joining of AID-initiated DNA breaks promotes antibody class switching. Nature. 2015;525:134–139. doi: 10.1038/nature14970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley DD, Manis JP, Zarrin AA, Kaylor L, Tian M, Alt FW. Internal IgH class switch region deletions are position-independent and enhanced by AID expression. Proc Natl Acad Sci USA. 2002;99:9984–9989. doi: 10.1073/pnas.152333499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldman S, Achour I, Wuerffel R, Kumar S, Gerasimova T, Sen R, Kenter AL. Constraints contributed by chromatin looping limit recombination targeting during Ig class switch recombination. J Immunol. 2015;194:2380–2389. doi: 10.4049/jimmunol.1401170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franco S, Gostissa M, Zha S, Lombard DB, Murphy MM, Zarrin AA, Yan C, Tepsuporn S, Morales JC, Adams MM, et al. H2AX prevents DNA breaks from progressing to chromosome breaks and translocations. Mol Cell. 2006;21:201–214. doi: 10.1016/j.molcel.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Gerasimova T, Guo C, Ghosh A, Qiu X, Montefiori L, Verma-Gaur J, Choi NM, Feeney AJ, Sen R. A structural hierarchy mediated by multiple nuclear factors establishes IgH locus conformation. Genes Dev. 2015;29:1683–1695. doi: 10.1101/gad.263871.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gostissa M, Schwer B, Chang A, Dong J, Meyers RM, Marecki GT, Choi VW, Chiarle R, Zarrin AA, Alt FW. IgH class switching exploits a general property of two DNA breaks to be joined in cis over long chromosomal distances. Proc Natl Acad Sci USA. 2014;111:2644–2649. doi: 10.1073/pnas.1324176111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Zou YR, Rajewsky K. Independent control of immuno-globulin switch recombination at individual switch regions evidenced through Cre-loxP-mediated gene targeting. Cell. 1993;73:1155–1164. doi: 10.1016/0092-8674(93)90644-6. [DOI] [PubMed] [Google Scholar]
- Hu J, Tepsuporn S, Meyers RM, Gostissa M, Alt FW. Developmental propagation of V(D)J recombination-associated DNA breaks and translocations in mature B cells via dicentric chromosomes. Proc Natl Acad Sci USA. 2014;111:10269–10274. doi: 10.1073/pnas.1410112111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic M, Feldhahn N, Oliveira TY, Silva IT, Kieffer-Kwon KR, Yamane A, Resch W, Klein I, Robbiani DF, Casellas R, Nussenzweig MC. 53BP1 alters the landscape of DNA rearrangements and suppresses AID-induced B cell lymphoma. Mol Cell. 2013;49:623–631. doi: 10.1016/j.molcel.2012.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev. 2013;27:1–17. doi: 10.1101/gad.200014.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khair L, Guikema JE, Linehan EK, Ucher AJ, Leus NG, Ogilvie C, Lou Z, Schrader CE, Stavnezer J. ATM increases activation-induced cytidine deaminase activity at downstream S regions during class-switch recombination. J Immunol. 2014;192:4887–4896. doi: 10.4049/jimmunol.1303481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell. 2011;147:95–106. doi: 10.1016/j.cell.2011.07.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedbal J, Hobson PS, Fear DJ, Heintzmann R, Gould HJ. Comprehensive FISH probe design tool applied to imaging human immunoglobulin class switch recombination. PLoS ONE. 2012;7:e51675. doi: 10.1371/journal.pone.0051675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Hübner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol Cell. 2010;40:364–376. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panier S, Boulton SJ. Double-strand break repair: 53BP1 comes into focus. Nat Rev Mol Cell Biol. 2014;15:7–18. doi: 10.1038/nrm3719. [DOI] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raviram R, Rocha PP, Müller CL, Miraldi ER, Badri S, Fu Y, Swanzey E, Proudhon C, Snetkova V, Bonneau R, Skok JA. 4C-ker: A Method to Reproducibly Identify Genome-Wide Interactions Captured by 4C-Seq Experiments. PLoS Comput Biol. 2016;12:e1004780. doi: 10.1371/journal.pcbi.1004780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen HT, Nussenzweig A, Nussenzweig MC. ATM is required for efficient recombination between immunoglobulin switch regions. J Exp Med. 2004;200:1103–1110. doi: 10.1084/jem.20041162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reina-San-Martin B, Chen J, Nussenzweig A, Nussenzweig MC. Enhanced intra-switch region recombination during immunoglobulin class switch recombination in 53BP1−/− B cells. Eur J Immunol. 2007;37:235–239. doi: 10.1002/eji.200636789. [DOI] [PubMed] [Google Scholar]
- Rocha PP, Micsinai M, Kim JR, Hewitt SL, Souza PP, Trimarchi T, Strino F, Parisi F, Kluger Y, Skok JA. Close proximity to Igh is a contributing factor to AID-mediated translocations. Mol Cell. 2012;47:873–885. doi: 10.1016/j.molcel.2012.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos MA, Huen MS, Jankovic M, Chen HT, López-Contreras AJ, Klein IA, Wong N, Barbancho JL, Fernandez-Capetillo O, Nussenzweig MC, et al. Class switching and meiotic defects in mice lacking the E3 ubiquitin ligase RNF8. J Exp Med. 2010;207:973–981. doi: 10.1084/jem.20092308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callén E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22:2048–2061. doi: 10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Bradley SP, Vardo J, Mochegova SN, Flanagan E, Stavnezer J. Mutations occur in the Ig Smu region but rarely in Sgamma regions prior to class switch recombination. EMBO J. 2003;22:5893–5903. doi: 10.1093/emboj/cdg550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrader CE, Linehan EK, Mochegova SN, Woodland RT, Stavnezer J. Inducible DNA breaks in Ig S regions are dependent on AID and UNG. J Exp Med. 2005;202:561–568. doi: 10.1084/jem.20050872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellars M, Reina-San-Martin B, Kastner P, Chan S. Ikaros controls isotype selection during immunoglobulin class switch recombination. J Exp Med. 2009;206:1073–1087. doi: 10.1084/jem.20082311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J, Schrader CE. IgH chain class switch recombination: mechanism and regulation. J Immunol. 2014;193:5370–5378. doi: 10.4049/jimmunol.1401849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Claudepierre AS, Robert I, Rocha PP, Raviram R, Schiavo E, Heyer V, Bonneau R, Luo VM, Reddy JK, Borggrefe T, et al. Mediator facilitates transcriptional activation and dynamic long-range contacts at the IgH locus during class switch recombination. J Exp Med. 2016;213:303–312. doi: 10.1084/jem.20141967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuerffel R, Wang L, Grigera F, Manis J, Selsing E, Perlot T, Alt FW, Cogne M, Pinaud E, Kenter AL. S-S synapsis during class switch recombination is promoted by distantly located transcriptional elements and activation-induced deaminase. Immunity. 2007;27:711–722. doi: 10.1016/j.immuni.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Franklin A, Boboila C, McQuay A, Gallagher MP, Manis JP, Khamlichi AA, Alt FW. Downstream class switching leads to IgE antibody production by B lymphocytes lacking IgM switch regions. Proc Natl Acad Sci USA. 2010;107:3040–3045. doi: 10.1073/pnas.0915072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.