

Abstract
In the poultry industry, many efforts have been undertaken to further improve the growth performance of broilers and identification and modulation of body weight (BW)-related bacteria could be one of the strategies to improve productivity. However, studies regarding the relationship between microbiota and BW are scarce. The objective of the present study was to investigate the relationship between microbiota and BW in different sections of the gastrointestinal tract (GIT). A total of twenty 18-day-old birds were selected based on the BW, and samples were collected from the three different sections of the GIT, which included the crop, ileum and cecum. Bacterial genomic DNA was extracted from the samples, and the V4 region of 16S rRNA gene were amplified. Amplicons were sequenced on Illumina MiSeq, and microbial communities were analyzed by using QIIME. In principal coordinate analysis, bacterial communities were clustered into three groups, based on the sections of GIT. Several BW-related bacterial groups were identified from linear regression analysis. At the genus level, Streptococcus from the ileum as well as Akkermansia in both ileum and cecum, were negatively related to BW, whereas Bifidobacterium in the ileum and Lactococcus in the cecum showed a positive correlation. The results from the present study showed that particular bacterial communities in the GIT were related to BW, and the study has broadened the understanding of the intestinal microbial ecosystem in broiler chickens.
Electronic supplementary material
The online version of this article (doi:10.1186/s40064-016-2604-8) contains supplementary material, which is available to authorized users.
Keywords: Body weight, Broiler chickens, Gastrointestinal tract, Microbiota, Relationship
Background
In the broiler industry, productivity such as feed conversion ratio and growth rate has been improved for decades (Rubio et al. 2014); however, broiler chickens still seem to have a growth potential. Based on the consumer demand, strategies for increasing the market weight of broilers have been studied by many researchers, but an efficient approach has not yet been developed. Moreover, the use of antibiotics in animal feeds for growth promotion has been banned from January 2006 in the EU and from July 2011 in Korea, and this restriction has led to the development of efficient and safe antibiotic alternatives to enhance the growth performance of livestock animals, including broilers. In this current situation, development of efficient and economic strategies to improve the growth performance of broiler chickens are an important task in broiler industry, and understanding of the host-microbiota interaction is one of the possible strategies (Rinttila and Apajalahti 2013; Pedroso et al. 2012).
Development of high-throughput sequencing technology enabled culture-independent analysis of microbial communities, and several studies revealed that intestinal microbiota affects the host metabolism and physiology, such as metabolic homeostasis (Shin et al. 2011; Caricilli et al. 2011), angiogenesis (Reinhardt et al. 2012), obesity (Turnbaugh et al. 2009; Backhed et al. 2004), immune function (Ivanov and Littman 2011; Kuss et al. 2011; Ichinohe et al. 2011) and brain development (Diaz Heijtz et al. 2011). In the past decade, many studies were conducted to investigate the relationship between gut microbiota and body weight (BW). These studies revealed that in human and mice, certain phyla were positively correlated with BW (Delzenne and Cani 2011; Hildebrandt et al. 2009; Ley et al. 2006; Turnbaugh et al. 2009), while certain species were negatively correlated with BW (Everard et al. 2013; Santacruz et al. 2010; Everard et al. 2011; Santacruz et al. 2009). However, only a limited number of studies about relationship between intestinal microbiota and BW were conducted in livestock, especially in young animals. Identification and modulation of weight-related bacteria is one of the strategies to modulate BW, and it can potentially be a useful strategy to improve productivity in the broiler industry. Therefore, the objective of the present study was to investigate the relationship between the BW of broiler chickens and microbiota in different sections of the gastrointestinal tract (GIT).
Results
Microbial communities at different sections of GIT
We compared the microbial communities in three sections of the GIT—crop, ileum and cecum. A total of 950,771 (mean = 16,115 ± 6460) 16S rRNA reads were generated, with an average of 18,085 (±5788) reads per crop sample, 15,294 (±6676) reads per ileal sample and 15,064 (±6733) reads per cecal sample. PCoA based on unweighted and weighted UniFrac distances of the 16S rRNA revealed that samples were clustered into three distinct groups (Fig. 1). In PCoA plot, microbial communities were separated by sections of the GIT, and ileal samples were placed between the crop and cecal samples.
Fig. 1.
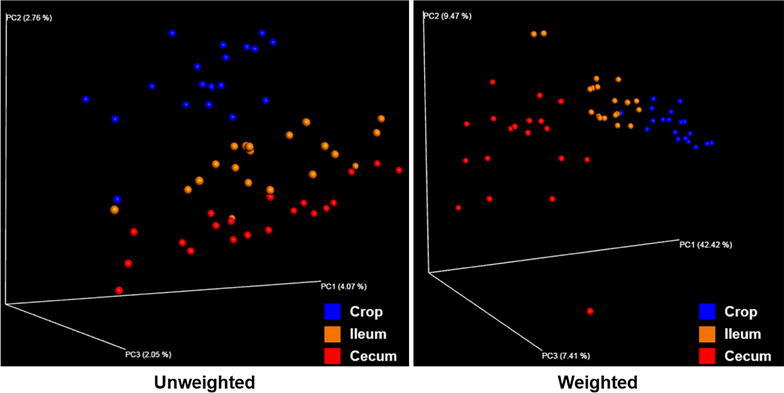
Principal coordinate analysis of unweighted and weighted UniFrac. Beta diversity patterns of crop samples (n = 19), ileal samples (n = 20) and cecal samples (n = 20) were explored using the principal coordinate analysis (PCoA). Subject color coding: blue crop samples; yellow ileal samples and red cecal samples
To determine which bacterial taxa contributed to separate microbial communities, relative abundance of taxa in each section is shown in Tables 1 and 2, and it is represented as a heat map (Additional file 1: Fig. S1). At the phylum level, Cyanobacteria and Proteobacteria were significantly more abundant in the crop, and Firmicutes was significantly more abundant in the ileum than in other sections (Additional file 1: Fig. S2). At the genus level, Bacillus was significantly more abundant in the crop (Additional file 1: Fig. S3A) and Prevotella was significantly more abundant in the ileum than in other sections (Additional file 1: Fig. S3F). Faecalibacterium, Ruminococcus and Akkermansia were significantly more abundant, and Lactobacillus and Streptococcus were significantly less abundant, in the cecum than in other sections (Additional file 1: Fig. S3). Especially, genus Bacteroides showed an incremental increase from the cranial to the caudal section (crop < ileum < cecum).
Table 1.
Relative abundance of phyla found in each section of the GIT
| Phylum | Abundance (%) | SD1 | P value | ||
|---|---|---|---|---|---|
| Crop | Ileum | Cecum | |||
| Cyanobacteria | 12.89a | 3.41b | 1.34b | 3.27 | <0.001 |
| Bacteroidetes | 13.93a | 22.15b | 30.49c | 5.82 | <0.001 |
| Proteobacteria | 7.89a | 4.26b | 3.06b | 2.18 | <0.001 |
| Tenericutes | 0.85a | 0.94a | 1.63b | 0.51 | <0.001 |
| Firmicutes | 59.62ab | 64.15a | 58.37b | 6.85 | 0.027 |
| Euyarchaeota | 0.08a | 0.19b | 0.16ab | 0.12 | 0.033 |
| Verrucomicrobia | 0.09 | 0.06 | 0.22 | 0.21 | 0.057 |
| Synergistetes | 0.02 | 0.03 | 0.06 | 0.06 | 0.095 |
| Lentisphaerae | 0.02 | 0.04 | 0.03 | 0.02 | 0.118 |
| Actinobacteria | 1.18 | 0.95 | 1.05 | 0.35 | 0.133 |
| Fibrobacteres | 0.02 | 0.03 | 0.01 | 0.04 | 0.271 |
| Spirochaetes | 0.37 | 0.43 | 0.46 | 0.24 | 0.500 |
| Fusobacteria | 0.08 | 0.08 | 0.09 | 0.05 | 0.842 |
| n | 19 | 20 | 20 | ||
One-way ANOVA with Tukey’s post hoc test was used. Within a row, different superscript letters indicate significant difference (P < 0.05)
1Pooled standard deviation
Table 2.
Relative abundance of genera found in each section of the GIT
| Genus | Abundance (%) | SD1 | P value | ||
|---|---|---|---|---|---|
| Crop | Ileum | Cecum | |||
| Bacillus | 4.34a | 1.50b | 1.37b | 0.85 | <0.001 |
| Bacteroides | 4.22a | 7.54b | 12.59c | 2.66 | <0.001 |
| Oscillospira | 1.00a | 1.34a | 2.38b | 0.49 | <0.001 |
| Faecalibacterium | 1.14a | 1.47a | 4.62b | 1.30 | <0.001 |
| Blautia | 0.22a | 0.27a | 0.44b | 0.10 | <0.001 |
| Dorea | 0.21a | 0.30a | 0.45b | 0.12 | <0.001 |
| Ruminococcus | 0.99a | 1.24a | 1.85b | 0.43 | <0.001 |
| Desulfovibrio | 0.09a | 0.22b | 0.13a | 0.07 | <0.001 |
| Lactobacillus | 28.62a | 30.81a | 18.12b | 7.77 | <0.001 |
| Bilophila | 0.03a | 0.04a | 0.10b | 0.04 | <0.001 |
| Coprococcus | 0.17a | 0.23a | 0.34b | 0.11 | <0.001 |
| Staphylococcus | 1.56a | 1.18a | 0.67b | 0.60 | <0.001 |
| Streptococcus | 1.18a | 1.11a | 0.75b | 0.32 | <0.001 |
| Clostridium | 0.96a | 0.79ab | 0.60b | 0.27 | <0.001 |
| Pediococcus | 0.07a | 0.06a | 0.04b | 0.02 | 0.001 |
| Prevotella | 3.94a | 5.66b | 4.26a | 1.55 | 0.003 |
| Akkermansia | 0.05a | 0.04a | 0.20b | 0.20 | 0.033 |
| Methanobrevibacter | 0.08a | 0.16b | 0.15ab | 0.11 | 0.045 |
| Selenomonas | 0.10a | 0.09ab | 0.06b | 0.05 | 0.046 |
| Bifidobacterium | 0.84 | 0.61 | 0.77 | 0.33 | 0.077 |
| Enterococcus | 0.50 | 0.84 | 0.47 | 0.53 | 0.077 |
| Lactococcus | 0.09 | 0.05 | 0.03 | 0.16 | 0.489 |
| n | 19 | 20 | 20 | ||
One-way ANOVA with Tukey’s post hoc test was used. Within a row, different superscript letters indicate significant difference (P < 0.05)
1Pooled standard deviation
Relationship between alpha diversity and BW
In this study, the observed OTUs was used as a parameter of microbial diversity within the samples (alpha diversity). Relationship between observed OTUs and BW in each section was analyzed by linear regression analysis. Observed OTUs was positively correlated with BW in the crop (r = 0.75, P < 0.001) and the ileum (r = 0.39, P = 0.087) (Fig. 2a, b). Conversely, observed OTUs was negatively correlated with BW in the cecum (r = −0.67, P = 0.001) (Fig. 2c). These results suggest that microbial diversity positively correlates with BW in the crop and ileum, whereas it negatively correlates to BW in the cecum.
Fig. 2.
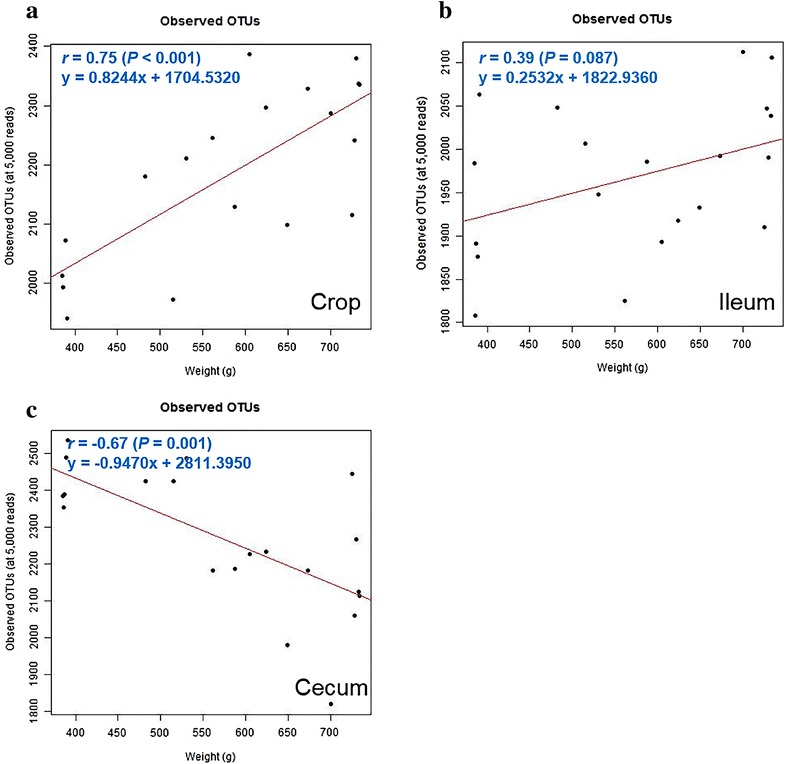
The relationship between BW and observed OTUs in chickens. The relationship was assessed by Pearson’s correlation coefficient (r) and P values from simple linear regression in the crop (a), ileum (b) and cecum (c)
BW related bacteria at each section of the GIT
A linear regression analysis was performed to determine which bacterial taxa were related to BW at each section of the GIT in the chicks. The overall significant results (r and P values) of analysis at the three sections are represented in Table 3. First, in the crop, weight related bacterial groups were explored (Additional file 1: Table S2). At the phylum level, Bacteroidetes (r = 0.66, P = 0.002) and Euryarchaeota (r = 0.52, P = 0.023) were positively related with BW, while Actinobacteria (r = −0.65, P = 0.003) was negatively related with BW. At the genus level, Ruminococcus (r = 0.72, P < 0.001) and Faecalibacterium (r = 0.65, P = 0.002) were positively related with BW, while Bifidobacterium (r = −0.64, P = 0.003) and Lactobacillus (r = −0.39, P = 0.099) were negatively related with BW.
Table 3.
The relationship between BW and bacterial relative abundance in each section of the GIT
| Crop | Ileum | Cecum | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Abundance (%) | r | P | Abundance (%) | r | P | Abundance (%) | r | P | |
| Phylum | |||||||||
| Actinobacteria | 1.18 | −0.65 | 0.003 | 0.95 | 0.08 | 0.729 | 1.05 | 0.00 | 0.990 |
| Bacteroidetes | 13.93 | 0.66 | 0.002 | 22.15 | 0.30 | 0.192 | 30.49 | <0.01 | 0.990 |
| Euryarchaeota | 0.08 | 0.52 | 0.023 | 0.19 | 0.52 | 0.018 | 0.16 | 0.14 | 0.558 |
| Firmicutes | 59.62 | −0.21 | 0.392 | 64.15 | −0.36 | 0.119 | 58.37 | 0.06 | 0.812 |
| Proteobacteria | 7.89 | −0.17 | 0.482 | 4.26 | 0.10 | 0.672 | 3.06 | −0.17 | 0.472 |
| Spirochaetes | 0.37 | 0.33 | 0.172 | 0.43 | 0.47 | 0.035 | 0.46 | 0.23 | 0.323 |
| Verrucomicrobia | 0.09 | 0.30 | 0.208 | 0.06 | −0.10 | 0.690 | 0.22 | −0.41 | 0.073 |
| Genus | |||||||||
| Akkermansia a | 0.05 | 0.09 | 0.706 | 0.04 | −0.51 | 0.023 | 0.08 | −0.55 | 0.022 |
| Anaerovibrio | 0.35 | 0.20 | 0.413 | 0.44 | −0.20 | 0.398 | 0.29 | −0.81 | <0.001 |
| Bacteroides | 4.22 | 0.54 | 0.016 | 7.54 | 0.05 | 0.834 | 12.59 | −0.34 | 0.142 |
| Bifidobacterium | 0.84 | −0.64 | 0.003 | 0.61 | 0.49 | 0.029 | 0.77 | 0.01 | 0.981 |
| Faecalibacterium | 1.14 | 0.65 | 0.003 | 1.47 | 0.23 | 0.335 | 4.62 | 0.32 | 0.164 |
| Lactobacillus | 28.62 | −0.39 | 0.099 | 30.81 | −0.32 | 0.172 | 18.12 | 0.02 | 0.924 |
| Lactococcus | 0.09 | 0.24 | 0.315 | 0.05 | 0.16 | 0.490 | 0.03 | 0.59 | 0.006 |
| Methanobrevibacter | 0.08 | 0.52 | 0.024 | 0.16 | 0.56 | 0.010 | 0.15 | 0.18 | 0.435 |
| Prevotella | 3.94 | 0.15 | 0.542 | 5.66 | 0.34 | 0.145 | 4.26 | −0.59 | 0.006 |
| Ruminococcus | 0.99 | 0.72 | <0.001 | 1.24 | 0.08 | 0.746 | 1.85 | −0.35 | 0.132 |
| Streptococcus | 1.18 | 0.03 | 0.913 | 1.11 | −0.81 | <0.001 | 0.75 | −0.06 | 0.787 |
r is Pearson’s correlation coefficient
aThree outliers were identified using Grubb’s test and removed from the cecum data
Next, in the ileum, weight related bacterial groups were explored (Additional file 1: Table S3). At the phylum level, Euryarchaeota (r = 0.52, P = 0.018) and Spirochaetes (r = 0.47, P = 0.035) were positively related with BW. At the genus level, Methanobrevibacter (r = 0.56, P = 0.010) and Bifidobacterium (r = 0.49, P = 0.029) were positively related with BW, while Akkermansia (r = −0.51, P = 0.023) was negatively related with BW (Fig. 3b). Especially, Streptococcus (r = −0.81, P < 0.001) showed a strong negative correlation with BW (Fig. 3a).
Fig. 3.
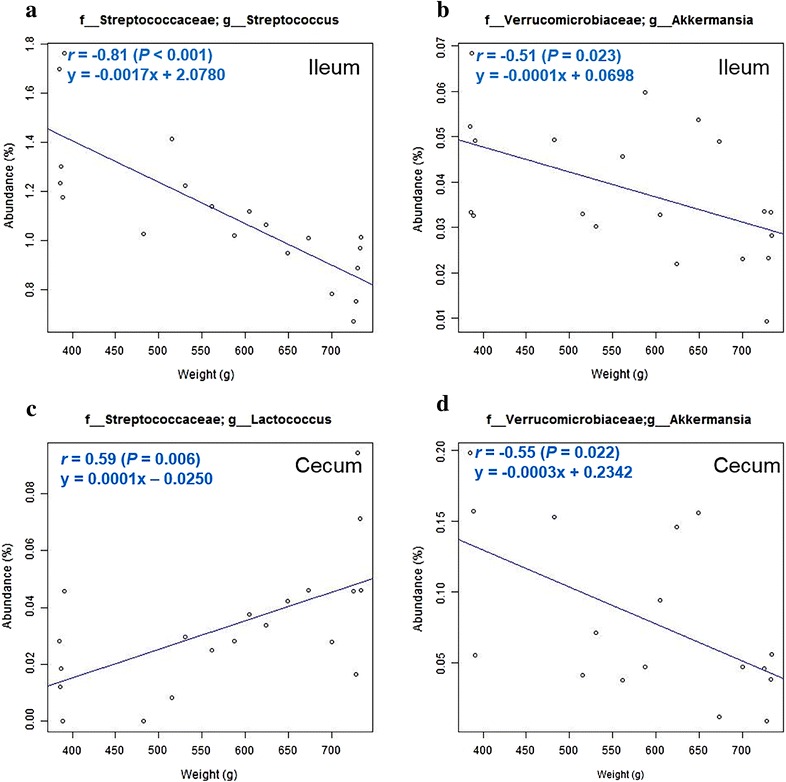
Weight related genera in the chicken GIT. Streptococcus (a) and Akkermansia (b) were significantly correlated with BW in the ileum. Lactococcus (c) and Akkermansia (d) were significantly correlated with BW in the cecum. d Three outliers were identified using Grubb’s test and removed from the dataset. The relationship between abundance of microbial taxa and BW was assessed by Pearson’s correlation coefficient (r) and P values from simple linear regression
Finally, weight related bacterial groups were explored in the cecum (Additional file 1: Table S4). At the phylum level, Lentisphaerae (r = −0.50, P = 0.023) and Verrucomicrobia (r = −0.41, P = 0.073) were negatively related with BW. At the genus level, Lactococcus (r = 0.59, P = 0.006) was positively related with BW (Fig. 3c), while Anaerovibrio (r = −0.81, P < 0.001), Prevotella (r = −0.59, P = 0.006) and Akkermansia (r = −0.55, P = 0.022) were negatively related with BW (Fig. 3d).
Discussion
In this study, we observed that microbial communities were clearly separated in different sections of the GIT in broiler chickens. Similar results were suggested by several previous researchers (Looft et al. 2014; Kamada et al. 2013; Sekelja et al. 2012; Videnska et al. 2013); for example, Looft et al. reported that ileum, cecum, mid-colon and feces have different microbial communities in swine at the phylum and genus level (Looft et al. 2014). Sekelja et al. also reported that a clear separation of microbial composition was seen between the upper gut (crop and gizzard), ileum and lower gut (cecum and colon) in broiler chickens (Sekelja et al. 2012). That these distinctions may be due to different nutrient requirements, is a critical factor for colonizing the commensal bacteria because each section has different nutrient factors (Deusch et al. 2015). The crop flora may be affected mainly by the microbial composition in the feed because the crop temporarily stores feed before digestion and is related to the breakdown of starch, whereas the ileal flora may be affected mainly by the nutrient composition of the ingested feed because ileum has a role involving the nutritional absorption of digested feed. The cecum is an anaerobic environment and plays an important role in recycling urea, absorption of water and digestion of plant structural carbohydrates, such as cellulose and hemicellulose. These varied features of each section of the GIT may result in the microbial community distinction.
Previous studies have revealed that the intestinal microbial diversity was negatively related with BW gain (Clarke et al. 2014; Turnbaugh et al. 2009). In our study, similar results were observed in the cecum, although the reverse was seen in the crop and ileum. Several studies suggested that reduced gut microbial diversity is correlated with several diseases such as inflammatory bowel disease and obesity-associated diabetes (Chang et al. 2008; Michail et al. 2012; Murri et al. 2013; Turnbaugh et al. 2009). While several other studies concluded that microbial diversity is not related with these diseases (Mejia-Leon et al. 2014; de Goffau et al. 2014; Walters et al. 2014), these results are controversial as of now, and thus, more studies are needed.
Several weight related bacterial groups in various region of the GIT were explored in this study. Especially, the genus Streptococcus showed a significant negative correlation with BW in the ileum. The genus Streptococcus can be divided into six groups, on the basis of 16S rRNA gene sequences—S. anginosus group, S. bovis group, S. mitis group, S. mutans group, S. pyogenes group and S. salivarius group (Kawamura et al. 1995). Many species of Streptococcus are known normal gut flora, however, some species are responsible for many diseases. For example, the S. anginosus group bacteria are associated with infections at multiple body sites, and abscess formation (Ruoff 1988; Belko et al. 2002; Bert et al. 1998). S. mutans group bacteria (Loesche 1986; Simon-Soro and Mira 2015) and S. mitis group bacteria (Mitchell 2011; Catto et al. 1987) are known pathogens of the buccal cavity. In humans, S. acidominius is a known pathogen that causes invasive diseases (Wu et al. 2014) such as pneumonia (Baker and Carlson 2008; Akaike et al. 1988), meningitis (Finkelstein et al. 2003) and brain abscess (Cone et al. 2007). Our data is consistent with the hypothesis that pathogenic Streptococcus might affect BW yet a more detailed analysis of the Streptococcus community is needed.
Recently many researchers revealed that Akkermansia muciniphila, the mucin degrading bacterium belonging to the genus Akkermansia, was negatively related to weight gain and obesity in mice and humans (Everard et al. 2013; Santacruz et al. 2010; Everard et al. 2011). Shin et al. also, reported that A. muciniphila has anti-diabetic potential against type 2 diabetes in high-fat diet fed mice (Shin et al. 2014). Similar results were obtained in our study. Akkermansia was inversely correlated with BW in the ileum and cecum, in spite of all birds having ingested the same feed. This suggests that the abundance of Akkermansia is more related to the BW, than the feed composition.
Many researchers reported that Firmicutes/Bacteroidetes (F/B) ratio increases when body mass index is increased, and F/B ratio is higher in the obese group than in the lean group (Delzenne and Cani 2011; Hildebrandt et al. 2009; Ley et al. 2006), although Clarke et al. suggested an opposing result (Clarke et al. 2014). In our study however, F/B ratio showed no significant relationship with BW in the ileum and cecum, although a negative relationship was observed in the crop (Additional file 1: Fig. S4). Moreover, Firmicutes and Bacteroidetes showed no significant correlation with BW in all parts of the GIT. More studies about the relationship between F/B ratio and BW are needed to explain these results.
Conclusions
In this study, microbial communities were explored in various regions of the GIT, and several weight related bacterial groups were identified from linear regression analysis, such as the genus Streptococcus and genus Akkermansia. These results broaden our understanding of the microbial ecosystem and show that certain bacterial groups affect the BW of broiler chickens. Although more studies about the relationship between microbiota and BW are needed, these bacterial groups will be initial targets for improving the growth performance of broiler chickens.
Methods
Birds and sample preparation
A total of 545, day-old, male, Ross 308 broiler chicks (initial mean body weight = 38.5 g) were purchased from a local hatchery (Yangji hatchery, Pyeongtaek, Republic of Korea). The protocol for this experiment was reviewed and approved by the Institutional Animal Care and Use Committee at Chung-Ang University (IACUC #: 14-0005). Birds were provided with water and feed ad libitum from day 0 to day 17, housed in battery cages (76 cm × 78 cm × 45 cm = width × length × height for each cage). The cages were environmentally controlled, with continuous light. On day 0, birds were weighed and tagged, and fed the standard commercial starter diet until day 17 (Table 4). On day 17, a total of 20 birds were selected out of the 545 birds. For the selection, each of the five heaviest and lightest birds were selected, and from the remaining 535 birds, 10 birds were selected by BW spaced at equal intervals, with the BW ranging from 482 to 700 g (Additional file 1: Table S1). The total BW ranged from 385 to 734 g (mean ± SD = 575.9 ± 134.7 g) for the 20 selected chicks.
Table 4.
Chemical composition of standard commercial starter diet
| Item | Contents per kg |
|---|---|
| Metabolizable energy | 3120 kcal |
| Crude protein | 215 g |
| Fat (ether extract) | 85.1 g |
| Crude fiber | 32.0 g |
| Calcium | 9.0 g |
| Phosphorous | 6.7 g |
| Lysine | 13.1 g |
| Sulfur-containing amino acids | 10.3 g |
On day 17, the 20 selected birds were euthanized by CO2 asphyxiation, and the digesta of the crop, ileum, and ceca were collected. The ileal digesta were collected from the distal two-third section of ileum from the Meckel’s diverticulum, to about 2 cm proximal to the ileocecal junction, through squeezing of the intestinal tract. All the samples collected from the birds were kept in a freezer at −20 °C for further analysis.
DNA extraction and sequencing
DNA was extracted from the crop (n = 19, one sample was missing), ileal (n = 20) and cecal (n = 20) digesta (~250 mg) using NucleoSpin®Soil Kit (Macherey–Nagel, Düren, Germany), according to the manufacturer’s instructions, and stored at −20 °C. In this study, the V4 region of bacterial 16S rRNA gene was amplified from the total extracted DNA, because this region, which is commonly used in microbial community analysis, provides sufficient phylogenetic richness (Zhao et al. 2013; Caporaso et al. 2011). PCR amplification was performed using the Takara Ex-taq polymerase (Takara Bio, Shiga, Japan) and universal primers (forward: 5′-GGACTACHVGGGTWTCTAAT-3′, reverse: 5′-GTGCCAGCMGCCGCGGTAA-3′). The amplification program consisted of one cycle of 94 °C for 3 min; 40 cycles of 94 °C for 45 s, 55 °C for 1 min, and 72 °C for 1.5 min; and finally one cycle of 72 °C for 10 min. Amplicons were separated by gel electrophoresis and purified using QIAquick Gel Extraction Kit (Qiagen, CA, USA).
For Illumina sequencing, DNA library was constructed using NEBNext Ultra DNA Library Prep Kit for Illumina (New England BioLabs, MA, USA), with some modifications of the manufacturer’s instructions. The size selection of adaptor-ligated DNA and cleanup of PCR amplification steps were replaced with PCR purification using a QIAquick PCR Purification Kit (Qiagen, CA, USA). Adaptor ligation and index primer addition were performed using NEBNext Multiplex Oligos for Illumina (New England BioLabs, MA, USA). DNA library construction was confirmed by agarose gel electrophoresis, and amplicons were purified using QIAquick Gel Extraction Kit. They were then sequenced on Illumina MiSeq platform (NICEM, SNU, Seoul, Republic of Korea). The 16S rRNA gene sequences obtained from MiSeq were deposited into NCBI’s Sequence Read Archive (SRA) database with accession number SRP065823.
Microbial community analysis
Microbial community was analyzed by using Quantitative Insights Into Microbial Ecology (QIIME) version 1.9.0 (http://qiime.org). Raw sequence reads were quality filtered and demultiplexed. The sequence reads were clustered into operational taxonomic units (OTUs) at 97 % similarity using the Greengene database. Microbial diversity was assessed within samples (alpha diversity) or between samples (beta diversity) using QIIME. Alpha diversity (observed OTUs) was calculated through rarefaction with ten iterations. Beta diversity was calculated on the sequence reads based on weighted and unweighted UniFrac distance matrices. Principal coordinate analysis (PCoA) was performed based on UniFrac distances and visualized with EMPeror Software (Vazquez-Baeza et al. 2013).
Statistical analysis
Statistical analysis was performed with R statistical package (version 3.0.3) (R Foundation for Statistical Computing, Vienna, Austria). Abundance of microbial taxa was expressed as percentage of total 16S rRNA gene sequences. One-way analysis of variance (ANOVA) and post hoc Tukey’s HSD test for multiple mean comparisons were used to find significant differences in microbial taxa among each section of the GIT. Significance was assumed at P < 0.05. The relationship between abundance of microbial taxa and BW was assessed by Pearson’s correlation coefficient (r) and P values from simple linear regression.
Authors’ contributions
GGH carried out microbial community analysis, performed statistical analysis and drafted the manuscript. EBK carried out microbial community analysis, performed the statistical analysis and participated in the design of the study. JL carried out animal experiment, collected the intestinal digesta samples and helped to draft the manuscript. JYL carried out microbial community analysis. GJ and JP carried out sample preparation for DNA sequencing. CSH and IKK contributed to manuscript preparation. DYK participated in animal experiment. YJC participated in the design of the study and contributed to manuscript preparation. CK participated in the design of the study, carried out animal experiment and helped to draft the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This study was supported by the Strategic Initiative for Microbiomes in Agriculture and Food, Ministry o Agriculture, Food and Rural Affairs, Republic of Korea (914005-04). This paper was supported by the KU Research Professor Program of Konkuk University.
Competing interests
The authors declare that they have no competing interests.
Additional file
10.1186/s40064-016-2604-8 Heat map of the relative abundance of taxa in each sample at the phylum level (A) and at the genus level (B). A range of colors, from green to red, indicates the prevalence of each taxon. Taxa are sorted in ascending order by P values from one-way ANOVA test. Figure S2. Relative abundance of phyla found in each section of the GI tract. (A) Cyanobacteria, (B) Bacteroidetes, (C) Proteobacteria, (D) Firmicutes. Different superscript letters indicate statistical significance (P < 0.05). One-way ANOVA with Tukey’s post hoc test was used to find significant differences of relative abundance. Figure S3. Relative abundance of genera found in each section of the GI tract. (A) Bacillus, (B) Bacteroides, (C) Faecalibacterium, (D) Ruminococcus, (E) Lactobacillus, (F) Prevotella, (G) Streptococcus, (H) Akkermansia. Different superscript letters indicate statistical significance (P < 0.05). One-way ANOVA with Tukey’s post hoc test was used to find significant differences of relative abundance. Figure S4. The relationship between body weight and Firmicutes/Bacteroidetes (F/B) ratio. (A) Crop, (B) Ileum, (C) Cecum. The relationship between abundance of microbial taxa and BW was assessed by Pearson’s correlation coefficient (r) and P values from simple linear regression. Table S1. Sample information in this study. Table S2. The relationship between body weight and bacterial abundance in crop. Table S3. The relationship between body weight and bacterial abundance in ileum. Table S4. The relationship between body weight and bacterial abundance in cecum.
Footnotes
Changsu Kong and Yun-Jaie Choi contributed equally to this work
Geon Goo Han, Eun Bae Kim and Jinyoung Lee contributed equally to this work
Contributor Information
Geon Goo Han, Email: geongoo1@snu.ac.kr.
Eun Bae Kim, Email: itanimal@kangwon.ac.kr.
Jinyoung Lee, Email: jylee920130@gmail.com.
Jun-Yeong Lee, Email: akirus86@snu.ac.kr.
Gwideuk Jin, Email: lbmicrobiota@kangwon.ac.kr.
Jongbin Park, Email: jong9412@kangwon.ac.kr.
Chul-Sung Huh, Email: cshuh@snu.ac.kr.
Ill-Kyong Kwon, Email: ikkwon@kangwon.ac.kr.
Dong Yong Kil, Email: dongyong@cau.ac.kr.
Yun-Jaie Choi, Email: cyjcow@snu.ac.kr.
Changsu Kong, Email: changsukong@gmail.com.
References
- Akaike T, Suga M, Ando M, Ando Y, Araki S, Fujise R. Streptococcus acidominimus infections in a human. Jpn J Med. 1988;27(3):317–320. doi: 10.2169/internalmedicine1962.27.317. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, Gordon JI. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004;101(44):15718–15723. doi: 10.1073/pnas.0407076101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker L, Carlson R. Streptococcus acidominimus isolated from a multiloculated empyema in a critically ill adult man with pneumonia: case report and review of literature. Heart Lung. 2008;37(4):308–310. doi: 10.1016/j.hrtlng.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Belko J, Goldmann DA, Macone A, Zaidi AK. Clinically significant infections with organisms of the Streptococcus milleri group. Pediatr Infect Dis J. 2002;21(8):715–723. doi: 10.1097/00006454-200208000-00002. [DOI] [PubMed] [Google Scholar]
- Bert F, Bariou-Lancelin M, Lambert-Zechovsky N. Clinical significance of bacteremia involving the “Streptococcus milleri” group: 51 cases and review. Clin Infect Dis. 1998;27(2):385–387. doi: 10.1086/514658. [DOI] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4516–4522. doi: 10.1073/pnas.1000080107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caricilli AM, Picardi PK, de Abreu LL, Ueno M, Prada PO, Ropelle ER, Hirabara SM, Castoldi A, Vieira P, Camara NOS, Curi R, Carvalheira JB, Saad MJA. Gut microbiota is a key modulator of insulin resistance in TLR 2 knockout mice. PLoS Biol. 2011 doi: 10.1371/journal.pbio.1001212. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Catto BA, Jacobs MR, Shlaes DM. Streptococcus mitis. A cause of serious infection in adults. Arch Intern Med. 1987;147(5):885–888. doi: 10.1001/archinte.1987.00370050081014. [DOI] [PubMed] [Google Scholar]
- Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, Young VB. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J Infect Dis. 2008;197(3):435–438. doi: 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- Clarke SF, Murphy EF, O’Sullivan O, Lucey AJ, Humphreys M, Hogan A, Hayes P, O’Reilly M, Jeffery IB, Wood-Martin R, Kerins DM, Quigley E, Ross RP, O’Toole PW, Molloy MG, Falvey E, Shanahan F, Cotter PD. Exercise and associated dietary extremes impact on gut microbial diversity. Gut. 2014;63(12):1913–1920. doi: 10.1136/gutjnl-2013-306541. [DOI] [PubMed] [Google Scholar]
- Cone LA, Etebar S, Waterbor RB. Brain abscess due to Streptococcus acidominimus: first case report. Surg Neurol. 2007;67(3):296–297. doi: 10.1016/j.surneu.2006.06.062. [DOI] [PubMed] [Google Scholar]
- de Goffau MC, Fuentes S, van den Bogert B, Honkanen H, de Vos WM, Welling GW, Hyoty H, Harmsen HJM. Aberrant gut microbiota composition at the onset of type 1 diabetes in young children. Diabetologia. 2014;57(8):1569–1577. doi: 10.1007/s00125-014-3274-0. [DOI] [PubMed] [Google Scholar]
- Delzenne NM, Cani PD. Interaction between obesity and the gut microbiota: relevance in mutrition. Annu Rev Nutr. 2011;31:15–31. doi: 10.1146/annurev-nutr-072610-145146. [DOI] [PubMed] [Google Scholar]
- Deusch S, Tilocca B, Camarinha-Silva A, Seifert J. News in livestock research—use of Omics-technologies to study the microbiota in the gastrointestinal tract of farm animals. Comput Struct Biotechnol J. 2015;13:55–63. doi: 10.1016/j.csbj.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz Heijtz R, Wang S, Anuar F, Qian Y, Bjorkholm B, Samuelsson A, Hibberd ML, Forssberg H, Pettersson S. Normal gut microbiota modulates brain development and behavior. Proc Natl Acad Sci USA. 2011;108(7):3047–3052. doi: 10.1073/pnas.1010529108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Lazarevic V, Derrien M, Girard M, Muccioli GG, Neyrinck AM, Possemiers S, Van Holle A, Francois P, de Vos WM, Delzenne NM, Schrenzel J, Cani PD. Responses of gut microbiota and glucose and lipid metabolism to prebiotics in genetic obese and diet-induced leptin-resistant mice. Diabetes. 2011;60(11):2775–2786. doi: 10.2337/db11-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, Guiot Y, Derrien M, Muccioli GG, Delzenne NM, de Vos WM, Cani PD. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA. 2013;110(22):9066–9071. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein Y, Marcus N, Mosseri R, Bar-Sever Z, Garty BZ. Streptococcus acidominimus infection in a child causing Gradenigo syndrome. Int J Pediatr Otorhinolaryngol. 2003;67(7):815–817. doi: 10.1016/S0165-5876(03)00088-0. [DOI] [PubMed] [Google Scholar]
- Hildebrandt MA, Hoffmann C, Sherrill-Mix SA, Keilbaugh SA, Hamady M, Chen YY, Knight R, Ahima RS, Bushman F, Wu GD (2009) High-fat diet determines the composition of the murine gut microbiome independently of obesity. Gastroenterology 137(5):1716–1724 e1711–1712. doi:10.1053/j.gastro.2009.08.042 [DOI] [PMC free article] [PubMed]
- Ichinohe T, Pang IK, Kumamoto Y, Peaper DR, Ho JH, Murray TS, Iwasaki A. Microbiota regulates immune defense against respiratory tract influenza A virus infection. Proc Natl Acad Sci USA. 2011;108(13):5354–5359. doi: 10.1073/pnas.1019378108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Littman DR. Modulation of immune homeostasis by commensal bacteria. Curr Opin Microbiol. 2011;14(1):106–114. doi: 10.1016/j.mib.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada N, Chen GY, Inohara N, Nunez G. Control of pathogens and pathobionts by the gut microbiota. Nat Immunol. 2013;14(7):685–690. doi: 10.1038/ni.2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura Y, Hou XG, Sultana F, Miura H, Ezaki T. Determination of 16S rRNA sequences of Streptococcus mitis and Streptococcus gordonii and phylogenetic relationships among members of the genus Streptococcus. Int J Syst Evol Microbiol. 1995;45(2):406–408. doi: 10.1099/00207713-45-2-406. [DOI] [PubMed] [Google Scholar]
- Kuss SK, Best GT, Etheredge CA, Pruijssers AJ, Frierson JM, Hooper LV, Dermody TS, Pfeiffer JK. Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science. 2011;334(6053):249–252. doi: 10.1126/science.1211057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–1023. doi: 10.1038/4441022a. [DOI] [PubMed] [Google Scholar]
- Loesche WJ. Role of Streptococcus mutans in human dental decay. Microbiol Rev. 1986;50(4):353–380. doi: 10.1128/mr.50.4.353-380.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Looft T, Allen HK, Cantarel BL, Levine UY, Bayles DO, Alt DP, Henrissat B, Stanton TB. Bacteria, phages and pigs: the effects of in-feed antibiotics on the microbiome at different gut locations. ISME J. 2014;8(8):1566–1576. doi: 10.1038/ismej.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia-Leon ME, Petrosino JF, Ajami NJ, Dominguez-Bello MG, de la Barca AM. Fecal microbiota imbalance in Mexican children with type 1 diabetes. Sci Rep. 2014;4:3814. doi: 10.1038/srep03814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michail S, Durbin M, Turner D, Griffiths AM, Mack DR, Hyams J, Leleiko N, Kenche H, Stolfi A, Wine E. Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm Bowel Dis. 2012;18(10):1799–1808. doi: 10.1002/ibd.22860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell J. Streptococcus mitis: walking the line between commensalism and pathogenesis. Mol Oral Microbiol. 2011;26(2):89–98. doi: 10.1111/j.2041-1014.2010.00601.x. [DOI] [PubMed] [Google Scholar]
- Murri M, Leiva I, Gomez-Zumaquero JM, Tinahones FJ, Cardona F, Soriguer F, Queipo-Ortuno MI. Gut microbiota in children with type 1 diabetes differs from that in healthy children: a case–control study. BMC Med. 2013 doi: 10.1186/1741-7015-11-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedroso AA, Maurer J, Cheng Y, Lee MD. Remodeling the intestinal ecosystem toward better performance and intestinal health. J Appl Poult Res. 2012;21(2):432–443. doi: 10.3382/japr.2011-00401. [DOI] [Google Scholar]
- Reinhardt C, Bergentall M, Greiner TU, Schaffner F, Ostergren-Lunden G, Petersen LC, Ruf W, Backhed F. Tissue factor and PAR1 promote microbiota-induced intestinal vascular remodelling. Nature. 2012;483(7391):627–631. doi: 10.1038/nature10893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinttila T, Apajalahti J. Intestinal microbiota and metabolites-implications for broiler chicken health and performance. J Appl Poult Res. 2013;22(3):647–658. doi: 10.3382/japr.2013-00742. [DOI] [Google Scholar]
- Rubio LA, Msuya JM, Mutalib MS, Khaza’ai H, Hui LS, Ismail N, Khorami H, Montague J, Wilcox J, Harmon AH. Modulation of the intestinal microbiota composition and productive parameters in productive animals and birds. Austin J Nutr Food Sci. 2014;2(2):1011. [Google Scholar]
- Ruoff KL. Streptococcus anginosus (“Streptococcus milleri”): the unrecognized pathogen. Clin Microbiol Rev. 1988;1(1):102–108. doi: 10.1128/CMR.1.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santacruz A, Marcos A, Warnberg J, Marti A, Martin-Matillas M, Campoy C, Moreno LA, Veiga O, Redondo-Figuero C, Garagorri JM, Azcona C, Delgado M, Garcia-Fuentes M, Collado MC, Sanz Y, Grp ES. Interplay between weight loss and gut microbiota composition in overweight adolescents. Obesity. 2009;17(10):1906–1915. doi: 10.1038/oby.2009.112. [DOI] [PubMed] [Google Scholar]
- Santacruz A, Collado MC, Garcia-Valdes L, Segura MT, Martin-Lagos JA, Anjos T, Marti-Romero M, Lopez RM, Florido J, Campoy C, Sanz Y. Gut microbiota composition is associated with body weight, weight gain and biochemical parameters in pregnant women. Br J Nutr. 2010;104(1):83–92. doi: 10.1017/S0007114510000176. [DOI] [PubMed] [Google Scholar]
- Sekelja M, Rud I, Knutsen SH, Denstadli V, Westereng B, Naes T, Rudi K. Abrupt temporal fluctuations in the chicken fecal microbiota are explained by its gastrointestinal origin. Appl Environ Microbiol. 2012;78(8):2941–2948. doi: 10.1128/AEM.05391-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SC, Kim SH, You H, Kim B, Kim AC, Lee KA, Yoon JH, Ryu JH, Lee WJ. Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science. 2011;334(6056):670–674. doi: 10.1126/science.1212782. [DOI] [PubMed] [Google Scholar]
- Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, Lee MS, Bae JW. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut. 2014;63(5):727–735. doi: 10.1136/gutjnl-2012-303839. [DOI] [PubMed] [Google Scholar]
- Simon-Soro A, Mira A. Solving the etiology of dental caries. Trends Microbiol. 2015;23(2):76–82. doi: 10.1016/j.tim.2014.10.010. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. Giga Sci. 2013;2(1):16. doi: 10.1186/2047-217X-2-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videnska P, Faldynova M, Juricova H, Babak V, Sisak F, Havlickova H, Rychlik I. Chicken faecal microbiota and disturbances induced by single or repeated therapy with tetracycline and streptomycin. BMC Vet Res. 2013;9:30. doi: 10.1186/1746-6148-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters WA, Xu Z, Knight R. Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 2014;588(22):4223–4233. doi: 10.1016/j.febslet.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Liang B, Gong Y, Zhang L, Zou Y, Ge J. Streptococcus acidominimus causing invasive disease in humans: a case series. J Med Case Rep. 2014;8:57. doi: 10.1186/1752-1947-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Wang G, Siegel P, He C, Wang H, Zhao W, Zhai Z, Tian F, Zhao J, Zhang H, Sun Z, Chen W, Zhang Y, Meng H. Quantitative genetic background of the host influences gut microbiomes in chickens. Sci Rep. 2013;3:1163. doi: 10.1038/srep01163. [DOI] [PMC free article] [PubMed] [Google Scholar]