

Abstract
Tissue culture has been used for over 100 years to study cells and responses ex vivo. The convention of this technique is the growth of anchorage dependent cells on the 2-dimensional surface of tissue culture plastic. More recently, there is a growing body of data demonstrating more in vivo-like behaviors of cells grown in 3-dimensional culture systems. This manuscript describes in detail the set-up and operation of a hollow fiber bioreactor system for the in vivo-like culture of mammalian cells. The hollow fiber bioreactor system delivers media to the cells in a manner akin to the delivery of blood through the capillary networks in vivo. The system is designed to fit onto the shelf of a standard CO2 incubator and is simple enough to be set-up by any competent cell biologist with a good understanding of aseptic technique. The systems utility is demonstrated by culturing the hepatocarcinoma cell line HepG2/C3A for 7 days. Further to this and in line with other published reports on the functionality of cells grown in 3-dimensional culture systems the cells are shown to possess increased albumin production (an important hepatic function) when compared to standard 2-dimensional tissue culture.
Keywords: Bioengineering, Issue 111, Hollow fiber bioreactor, tissue culture, cell culture, 3D cell culture, hepatocyte, HepG2/C3A, albumin secretion
Introduction
Tissue culture is an established technique for the growth and/or maintenance of cells that has been utilized for over 100 years1,2. The convenience of studying cells and responses ex vivo has far reaching advantages allowing experiments that would otherwise be extremely challenging if not impossible, for example the generation of genetically engineered cell lines and the use of reporter cells in high throughput screening assays3. More recently tissue culture has given rise to the field of tissue engineering, for the generation of in vitro models and for regenerative medicine. With these applications, interest in dynamic 3-dimensional (3D) culture systems has grown significantly.
3D culture methods (defined here as a 3D culture substrate and/or the introduction of directional dynamic flow) better recapitulate the architecture of the in vivo cellular environment, important for achieving a more physiological-like function. The ability to extract, grow, differentiate and transplant cells with the aim of repairing diseased and damaged tissue is a field of study that has a huge potential for patient benefit and commercial opportunity. For example the use of autologous keratinocytes for treating burns (see4) and the use of cell based therapies for the treatment of stroke (see5). Likewise, the market for in vitro models spans drug discovery to stratified medicine applications. The convention in tissue culture is the growth of adherent or anchorage dependent cell types on the 2-dimensional (2D) surface of a tissue culture flask. Whilst currently accepted as the gold standard in a research setting, recent interest in tissue engineering applications has highlighted the fact that current 2D tissue culture environment is inadequate for the required scale-up in cell production6.
For adherent cell types a scaffold is required, which will vary depending on end use, in terms of both the chemical composition and mechanical properties. Some systems employ scaffolds as well-plate inserts consisting of highly porous matrix formed from high internal phase emulsion templating (see7) or electrospun fibers (see8) requiring minimum adaptation from conventional 2D culture techniques. Cells can be seeded onto microcarriers of varying compositions and grown in stirred tanks that deliver nutrients and signaling molecules, and carry away waste products (mass transport) through a dynamic well mixed environment9. However, these systems are limited in their in vivo-like environment and further improvements can be made with regards to scale-up costs. Hollow fiber bioreactors (HFBs) are a 3D culture system that consist of fibers fixed into a module with cells typically seeded on the outside of the porous fibers and media delivered through the fiber lumen (reviewed in10) (Figure 1). HFBs offer an in vivo-like environment with the fibers mimicking blood capillaries and shielding the cells from the shear stresses associated with dynamic media delivery, while allowing defined shear to be applied to cells the via fluid flow through the side ports if desired. This creates a versatile culture system with superior mass transport in which high cell densities can be reached11. The HFB system is well suited for the maintenance of anchorage dependent cell types and has been used to culture a variety of cells including rat pancreatic islets of Langerhans12, mouse β-TC-3 insulinoma cell line12, primary human hepatocytes13, human bone marrow mononuclear cells14, Madin Darby canine kidney cells (MDCK)15 and Caco-2 cells16 to name a few.
In addition to the system advantages of mass transfer and scale-up, cells grown in 3D tissue culture systems tend to be more in vivo-like in morphology and more responsive to experimental cues. For example rat primary hepatocytes show a more cuboidal morphology, increased viability, greater induction of cytochrome-P450 enzyme activity and increased sensitivity to paracetamol toxicity when grown in a commercially available polystyrene scaffold compared to cells grown in 2D culture17. Using the same scaffold the hepatocarcinoma cell line HepG2 has also been shown to increase albumin production18 and show a more in vivo-like response to methotrexate compared to 2D cultured cells19. Primary human hepatocytes demonstrated delayed dedifferentiation, higher cytochrome-P450 activity and increased clearance for 4/5 compounds tested in a perfusion culture system20. Human neural stem cell derived neurons and glia cultured in a commercially available polystyrene scaffold exhibited both high (action potential) and low-frequency (local field potential) spontaneous activity whereas no neuronal activity was detected in the 2D cultured cells21. Caco-2 cells demonstrated enhanced differentiation in a HFB compared to 2D culture measured by increased alkaline phosphatase, γ-glutamyltransferase and P-glycoprotein activity and higher expression of F-actin and zona occludens-1 protein16. Despite the advantages, the routine culturing of cells in systems other than a 2D tissue culture flask surface is still not practiced in many laboratories, although the number of publications citing 3D cell culture is growing (8-fold increase in the last 10 years. Source: PubMed 'Results by Year' tool probed with '3D culture').
This manuscript describes the set-up and operating conditions of a HFB system for mammalian cell culture and demonstrates its utility in culturing the hepatocarcinoma cell line HepG2/C3A. The aim of this method is to culture the cells in a more in vivo-like culture system that retains enough simplicity to make it amenable to those who are new to 3D culture systems. The rationale behind using HFBs in the application describe here, which is to improve the predictability of liver models is that it is theoretically possible to mimic a hepatic sinusoid within the HFB environment22. This is not currently feasible with other culture systems.
Protocol
1. Fibers
Manufacture the fibers by phase inversion spin casting (spinning). Details of this method can be found in23,24. NOTE: For this work the fibers are manufactured in-house using a non-biodegradable proprietary polymer, NMP as the solvent and H2O as the non-solvent. Details of other suitable polymers can be found in the discussion. The fibers used in the system described here are 1.05mm outer diameter with a 600-700 µm lumen diameter. The fibers are porous with pore diameters measuring 2.28 µm ± 1.5 µm (mean ± standard deviation). This is designed to separate cells from the media feed in the fiber lumen, replicating the liver sinusoid or vasculature of other tissues. Fibers can also be purchased from membrane suppliers such as Pall.
2. Module Fabrication
NOTE: The modules used in this study are made from 1 mm thick borosilicate glass with 2 side ports (Figure 2A). The fibers in the module described here have an outer surface area of 4.95 cm2 which is the equivalent of roughly half of a well of a 6-well plate.
Siliconize modules before first use by coating the inner surface with Sigmacote (Table 1) and allowing to dry in a fume hood. Autoclave (121 °C, 1 atm, 20 min) to increase the life of the treatment.
Using a scalpel cut 75 mm-long fibers and insert three fibers into each module leaving ~7 mm excess length at each end (Figure 3A).
Place ~0.5 ml of silicone glue (Table 1) into a weighing boat. Use a P200 pipette tip to pick up a small amount of silicone and work the glue into the ends of the module around the fibers to form a 3-5 mm plug (Figure 3B). Allow to dry for >3 hr.
Using a scalpel cut the silicone flush with the glass module ends (Figure 3C).
Wrap a small amount (~4 layers) of polytetrafluoroethylene (PTFE) tape around one side port.
3. System Setup and Sterilization
NOTE: Pump tubing and modules with fibers are not autoclaved and are sterilized using 70% ethanol. The authors recommend calibrating the pump tubing with the pump to be used. The procedure below is carried out in a laminar flow hood.
Autoclave (as section 2.1) all autoclavable components prior to set-up.
- Set-up
- Place 10 ml 70% ethanol into the reservoir bottle and set-up the reservoir bottle, Q-series cap, feed tube, pump and pump tubing (Table 1) as in Figure 4.
- Loosely place an end cap over the PTFE-taped side port. Slide the ends of the L/S16 module connectors over the module ends and free side port. Connect a 40 mm section of L/S13 tubing to the module connector nearest the capped side port (Table 1) as in Figure 2B.
- Connect the module to the pump tubing, ensuring to orientate the module so the capped side port is nearest the pump.
- Connect the permeate line and retentate line to the module connectors and the L/S14 of the Y-connector on the reservoir bottle (Table 1). Ensure that the set-up resembles the schematics in Figure 4.
- Sterilization
- Pump ethanol through the module at 800 µl/hr (267 µl/hr per fiber) for enough time to treat unautoclaved components for >30 min (adjust times if other sterilizing methods are used; see25).
- Wash
- To wash the ethanol out of the system, first turn off the pump and drain the tubing. First, detach the pump tubing from the module adaptor tubing. Hold the module aloft to drain the ethanol out of the fibers and retentate line, back into the reservoir bottle. Remove the side port end cap from the module to drain the ethanol from the module itself, and the permeate line. Reattach the side port end cap. Reverse the flow of media on the pump to drain the pump tubing and feed line of ethanol. Turn off the pump, and reattach the pump tubing to the module adaptor.
- Unscrew the ethanol bottle from the lid and replace with a bottle containing 10 ml of cell growth medium (such as EMEM, GMEM, DMEM or RPMI) without serum. Pump the medium through the system at 800 µl/hr until the retentate line is full of media. Clamp the retentate line to force permeation of the media through the fibers to wash the module. Wash for ~2 hr.
4. Seeding
NOTE: The media and supplements used in this protocol should be those that are established for the desired cell type. Please refer to the literature, the European Collection of Cell Cultures (ECACC) and American Type Culture Collection (ATCC) for further information. Prior to this method cells should be maintained according to established protocols for the desired cell type. For this work the hepatocarcinoma cell line HepG2/C3A subclone were maintained according to the distributors recommendations (ATCC).
NOTE: The seeding protocol presented below also serves to pre-culture the module with cell culture medium prior to cell growth. Should a more extensive pre-culture be required then this should be carried out prior to seeding the module by draining the wash media from the system, replacing with growth media and permeating this through the module for few hours. See section 7.5.2.1 for details on replacing media in the system.
- Prepare a single cell suspension by trypsinization according to established protocols for the desired cell type. A general protocol for a T75 culture is as follows: NOTE: The number of cells to use in the seeding step should be empirically determined for your desired cell type. The bioreactors described here are seeded at a 35-fold higher cells density (cell/cm2) than used in 2D tissue culture plastic for a 7 day culture.
- Wash the cells by adding 10 ml phosphate buffered saline (PBS), aspirate, then add 3 ml 0.05% trypsin ethylenediaminetetraacetic acid (EDTA) (enough to cover the cells) and incubate at 37 °C, 5% CO2 for 5 min.
- Harvest cells in 7 ml growth media supplemented with 10% fetal bovine serum (FBS) to neutralize the trypsin. Mix well, add 10 µl to the chamber of a hemocytometer and count the cells.
- Centrifuge at 200 x g for 5 min to pellet the cells.
- Aspirate the supernatant and resuspend cells at 4x106/ml in growth media supplemented as required for the desired cell type.
Turn off the pump and drain the feed tube and module as in 3.4.1.
Detach the module from the module connectors and attach module end caps (Table 1) pre-sterilized in 70% ethanol, leaving one side port free.
- Transfer ~500 µl of the cell suspension (2x106 cells) (4.1.4) to the module using an 18 G needle and 1 ml syringe, taking care to avoid bubble formation and not to damage the fibers.
- Cap the side port using an end cap. Incubate the cells at 37 °C, 5% CO2 for 2-4 hr with manual turning of the module by 180° every 5 min. Alternatively the modules can be attached to a tube rotator with an intermittent mixing setting (Table 1).
- Following seeding, attach an end cap onto an injection port (Table 1) pre-sterilized in 70% ethanol, and attach this to the PTFE-taped side port. Remove the other side port end cap and slowly drain the cells by injecting air into the attached injection port using a 27 G needle and 1 ml syringe.
- Replace the injection port with an end cap. Slowly fill the module with media using the free side port and an 18 G needle with a 1 ml syringe. Remove the module end caps and attach the module to the tubing using the module connectors.
Replace the wash media bottle with one containing 50 ml growth media and supplements following the procedure detailed in 7.5.2.1. Pump the growth media through the system at 800 µl/hr.
5. Proliferation
NOTE: The fibers used in the research system described here are set to permeate at ~80 µl/hr, with an 800 µl/hr feed rate.
Use the fibers to grow the cells for periods of up to 7 days in a humidified incubator set at 37 °C, 5% CO2. NOTE: Monitoring of nutrients and metabolites during the growth phase can provide useful information on the proliferation, metabolic uptake and output of the cells and nutrient and metabolite levels in the media. For example glucose usage and lactate production. Kits are available from various suppliers that can quantitate these factors from media (see Table 1 for those used in this study). Injection ports can be added to the permeate and retentate tubing and media sampled via a 27 G needle and syringe, and media can be sampled from the media reservoir bottle. This gives nutrient and metabolism information for the input and both output streams. Sampling should be carried out in a laminar flow hood. Sterilize injection ports before sampling by holding an ethanol soaked blue roll against the port for >30 sec.
6. Excision
NOTE: Fibers can be excised from the module at the end of an experiment for analysis.
Unplug and drain the HFB.
Insert a scalpel/micro knife (Table 1) blade between the glass and silicone. Turn the module so as to cut away the silicone from the glass. Repeat this procedure at both ends of the module.
Using the blade hook out the silicone plug from one end and gently pull. Ensure that the fibers come with it.
7. Cell Analysis
- Cell numbers NOTE: For the C3A cells used in this study any time points within the 7 day growth period are suitable for use in this calculation as growth rates do not substantially change over the cell densities achieved in this timeframe.
- After excision (Section 6) dip the fibers into PBS to wash and cut them into a 1.5 ml tube containing 0.5 ml Tris EDTA (TE) buffer. Subject this to two freeze-thaw cycles in a -80 °C freezer. Measure the DNA content using PicoGreen and determine cell numbers by comparing this value to a standard curve made with the desired cell type26.
- Cell proliferation rates
- Using the cell numbers calculated at two different time points calculate the specific growth rate µ (Equation 1) where Ln(X1) is the natural log of the cell number at the first time point and Ln(X2) is the natural log of the cell number at the second time point. µ=(Ln(X2)-Ln(X1))/time(hr) (1) From this calculate the population doubling times (dT) (Equation 2) where µ is the specific growth rate. dT=Ln2/µ (2)
- Cell viability
- After excision (Section 6) dip the fibers into PBS to wash and cut them into a 1.5 ml tube containing 500 µl 0.05% trypsin ethylenediaminetetraacetic acid (EDTA). Incubate at 37 °C for 10 min.
- Mix and add 10 µl of cell suspension to 10 µl trypan blue. Load 10 µl onto a haemocytometer and count the number of dead (blue) and alive cells.
- Imaging
- After excision dip the fibers into PBS to wash and use scissors to cut them into smaller lengths into a 24-well plate. Add 400 µl 4% paraformaldehyde (in PBS) and incubate at RT for 20 min.
- Wash with PBS by pipetting 400 µl on and off. Repeat this step with fresh PBS.
- Add 400 µl 4',6-diamidino-2-phenylindole (DAPI) diluted in PBS to approx. 100 ng/ml and incubate at RT for 20 min. Protect from light.
- Wash with PBS twice (as 7.3.2) and once with H2O. Add a fluorescence mounting medium to cover the fiber and image immediately to collect data before the samples dry (DAPI ex/em; 359/461 nm).
- Take images at different focal planes and use 'focus stacking' software (e.g., stack focuser plugin for ImageJ, below) to make a composite image showing a greatly expanded depth of field. This is required as the fibers are not flat.
- Download ImageJ (http://imageJ.nih.gov/ij/) and the ‘stack-focuser’ plugin (http://rsb.info.nih.gov/ij/plugins/stack-focuser.html).
- In ImageJ open the images to be stacked. Then in the 'Image' menu go to 'Stacks'-'Images to Stack'. In the 'Plugins' menu go to 'Stack Focuser'. Specify an n for nxn kernel. Trial and error with 'n' may be required in order to generate an image with little 'noise'. Values between 11 & 77 tend to work well.
- Albumin secretion NOTE: This is a hepatocyte cell function test and not a general test of cell function.
- Seed cells (HepG2/C3A) onto 2D tissue culture plastic at 10,667/cm2 and grown for 6 days. Seed HFBs as described in Section 4 and grow for 6 days.
- Following this proliferation period change the culture media (EMEM + 10% FBS, 1x glutamine & 1x penicillin/streptomycin) for the tissue culture plastic and HFB to a serum free Williams E media supplemented with 1x glutamine and 1x penicillin/streptomycin for 24 hr:
- Drain the tubing and HFB module following the steps set out in 3.4.1. Unscrew the reservoir bottle and replace this with a bottle containing the Williams E media. Pump this through the HFB at 800 µl/hr.
- Following 24 hr, take media samples. Quantify secreted albumin by ELISA according to the manufacturer's instructions (Table 1). Dilute media samples 1 in 10 to 1 in 40 before use in order to bring albumin concentrations within the range of the standard curve.
Representative Results
Cell seeding is a critical step. The ability of cells to adhere to the fiber in a 3D set-up is considerably less than that seen for 2D tissue culture plastic. This is likely due to the reduced contact time between cell and substrate. In the HFB the cells fall through the module during the static phases of seeding. These are considerably shorter in time compared to the one continuous static phase used in 2D. Cells have to make connections to the substrate in this time. Additionally, due to the curved nature of the fibers there is no flat surface for the cells to rest and make connections like there is in 2D culture. Whilst this can be compounded by the chemistry of the fiber employed we know this not to be the case for the polymer employed in this study (data not shown). Increasing the seeding time and cell seeding density leads to the formation of cell aggregates that fall through the module during the static phases at a faster rate than single cells and further reduces the contact time between cells and substrate. Increasing the time also leads to yellowing of the media as there is a high density of cells and no media exchange during this step. Using the conditions described in Section 4 cell seeding rates of ~15% are consistently achieved with the HepG2/C3A cells (Figure 5A). Whilst this may be considered a low fraction there are enough cells to generate well-populated fibers in a matter of days (Figures 5B and 6); following a seven-day proliferation period the cell density reached in the HFB is approaching 3x105/cm2. This is an appropriate density for many assays utilizing hepatocytes and can be considered to be reaching confluence.
The proliferation rates achieved in the HFB are slower when compared to that achieved in 2D cell culture (Figure 5C). This is unlikely to be due to loss of cells in the dynamic flow of the system as cell counts in the media are low (average ± SEM: 20,343 cells ± 3,674 cells per 24 hr at confluence, which is 4% of the total cells) and this does not increase when the permeation rate is turned up to 400 µl/hr (data not shown). It is also unlikely to be due to a decrease in viability and subsequent loss of cells (see Figure 5D and below). It may be explained, at least in part by the fact this cell line was clonally derived from HepG2 and selected on its ability to show contact inhibition of cell proliferation. The cells in the HFB are seeded at 6x the density of the 2D cells which may lead to slower proliferation rates.
Viability remains high throughout the HFB with cells exhibiting >90%. Whilst there is a slight decrease in viability at the outlet end compared to the inlet this was not found to be significant (t-test p=0.22).
Monitoring of glucose consumption and lactic acid production has been used to tune the media feed rate and media volume in the system. Using 800 µl/hr and a total media volume of 50 ml the glucose and lactic acid levels are maintained above and below (respectively) those seen in standard tissue culture plastic culture.
Albumin secretion is a key liver function carried out by hepatocytes. It is secreted into the serum where it plays several roles in transport and homeostasis. The albumin secretion by cells grown in the HFB is 15-fold higher than that in cells grown on 2D (Figure 7). This demonstrates the cells are functional in the HFB and at least in the case of albumin secretion, this function is elevated in the HFB.
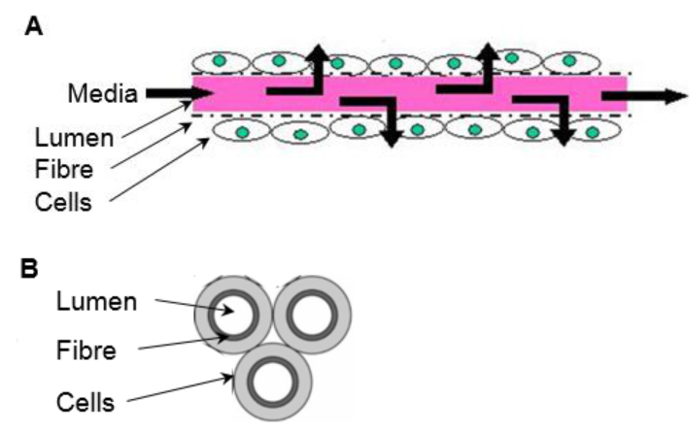 Figure 1. The in vivo-like environment of a HFB. Cells are seeded onto the outside of the porous fibers. Media is delivered through the fiber lumen, mimicking a blood capillary. (A) Longitudinal section of a fiber (not to scale). (B) Cross section of a 3 fiber reactor. Please click here to view a larger version of this figure.
Figure 1. The in vivo-like environment of a HFB. Cells are seeded onto the outside of the porous fibers. Media is delivered through the fiber lumen, mimicking a blood capillary. (A) Longitudinal section of a fiber (not to scale). (B) Cross section of a 3 fiber reactor. Please click here to view a larger version of this figure.
 Figure 2. The HFB module. (A) The dimensions of the module used in this study. The dimensions were chosen to fit 3 fibers and fulfil the needs of the current research project. Different sizes can be manufactured and tailored for individual systems and fibers. (B) A photograph of the module with attached end cap and module connectors. Please click here to view a larger version of this figure.
Figure 2. The HFB module. (A) The dimensions of the module used in this study. The dimensions were chosen to fit 3 fibers and fulfil the needs of the current research project. Different sizes can be manufactured and tailored for individual systems and fibers. (B) A photograph of the module with attached end cap and module connectors. Please click here to view a larger version of this figure.
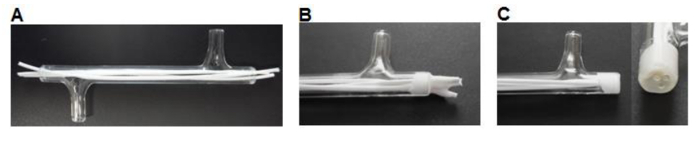 Figure 3. Module fabrication. (A) Fibers are cut to size and inserted into the module. (B) Fibers are glued into the module, allowed to dry. (C) The ends are cut flush with the glass. Please click here to view a larger version of this figure.
Figure 3. Module fabrication. (A) Fibers are cut to size and inserted into the module. (B) Fibers are glued into the module, allowed to dry. (C) The ends are cut flush with the glass. Please click here to view a larger version of this figure.
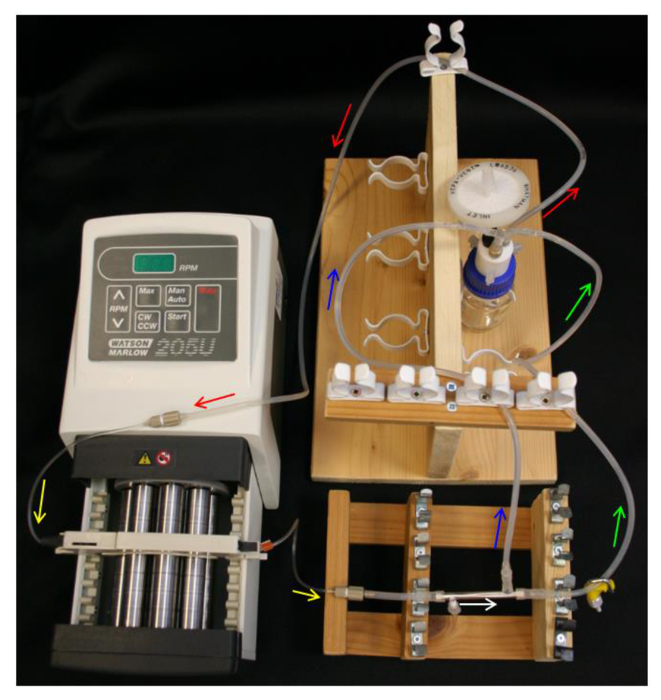 Figure 4. HFB system set-up. Arrows indicate direction of media flow. Red = feed tube. Yellow = pump tube. White = HFB module. Green = retentate tube with clamp. Blue = permeate tube. Please click here to view a larger version of this figure.
Figure 4. HFB system set-up. Arrows indicate direction of media flow. Red = feed tube. Yellow = pump tube. White = HFB module. Green = retentate tube with clamp. Blue = permeate tube. Please click here to view a larger version of this figure.
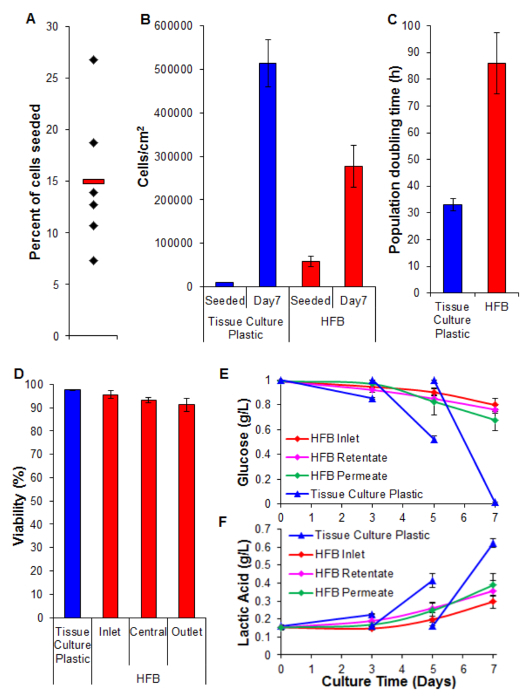 Figure 5. Seeding, proliferation, viability and biochemical analysis. (A) Percent of cells seeded. Black diamonds represent individual HFBs. The red bar is the mean. (B) Cells densities at seeding and after a 7 day proliferation period. 2D cell numbers were determined by trypsinization and counting using a haemocytometer. HFB cell numbers were determined using the PicoGreen assay and a standard curve produced from C3A cells26. n=5-6. Bars = SEM. (C) Population doubling times. n=5-7. Bars = SEM. (D) Viability determined by trypan blue exclusion at the end of a 7 day proliferation. Inlet, central and outlet represent regions within the HFB. n=3-5. Bars=SEM. (E&F) Glucose consumption and lactic acid production. Levels were monitored at the reservoir bottle (inlet) as well as the retentate and permeate outlets and compered to routine culture on tissue culture plastic (media changed on day 3&5). n=3-5. Bars=SEM. Please click here to view a larger version of this figure.
Figure 5. Seeding, proliferation, viability and biochemical analysis. (A) Percent of cells seeded. Black diamonds represent individual HFBs. The red bar is the mean. (B) Cells densities at seeding and after a 7 day proliferation period. 2D cell numbers were determined by trypsinization and counting using a haemocytometer. HFB cell numbers were determined using the PicoGreen assay and a standard curve produced from C3A cells26. n=5-6. Bars = SEM. (C) Population doubling times. n=5-7. Bars = SEM. (D) Viability determined by trypan blue exclusion at the end of a 7 day proliferation. Inlet, central and outlet represent regions within the HFB. n=3-5. Bars=SEM. (E&F) Glucose consumption and lactic acid production. Levels were monitored at the reservoir bottle (inlet) as well as the retentate and permeate outlets and compered to routine culture on tissue culture plastic (media changed on day 3&5). n=3-5. Bars=SEM. Please click here to view a larger version of this figure.
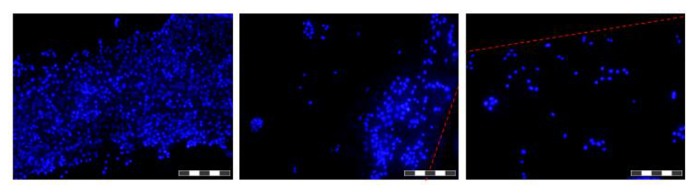 Figure 6. Images of cells grown on fibers in a HFB. Cells were seeded and grown for 48 hr before fibers were excised, washed in PBS, fixed in 4% paraformaldehyde, washed in PBS and nuclei stained with DAPI. Each image is a composite of 12 'focus stacked' images in order to increase the depth of field of the resulting image. Areas of higher and lower cell density are found along the fiber at this time point and are shown. Where present fiber boundaries are denoted with a red dashed line. Images were taken on an inverted fluorescence microscope. Image = 10X objective. Bar = 200 µm. Please click here to view a larger version of this figure.
Figure 6. Images of cells grown on fibers in a HFB. Cells were seeded and grown for 48 hr before fibers were excised, washed in PBS, fixed in 4% paraformaldehyde, washed in PBS and nuclei stained with DAPI. Each image is a composite of 12 'focus stacked' images in order to increase the depth of field of the resulting image. Areas of higher and lower cell density are found along the fiber at this time point and are shown. Where present fiber boundaries are denoted with a red dashed line. Images were taken on an inverted fluorescence microscope. Image = 10X objective. Bar = 200 µm. Please click here to view a larger version of this figure.
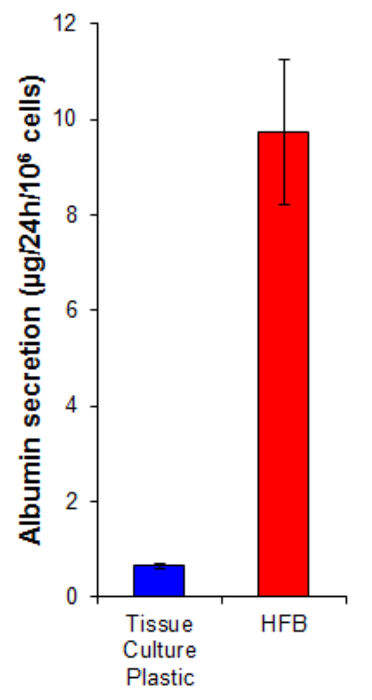 Figure 7. Albumin secretion. Cells were seeded onto tissue culture plastic at 10,667/cm2 and in the HFB as described in Section 4. Cells were proliferated for 6 days. Following this proliferation period the tissue culture plastic and HFB culture media was changed for a serum free Williams E media supplemented with glutamine and penicillin/streptomycin for 24 hr. Media samples were taken and an albumin quantified by ELISA according to the manufactures instructions (Table 1). n=5-6. Bars = SEM. Please click here to view a larger version of this figure.
Figure 7. Albumin secretion. Cells were seeded onto tissue culture plastic at 10,667/cm2 and in the HFB as described in Section 4. Cells were proliferated for 6 days. Following this proliferation period the tissue culture plastic and HFB culture media was changed for a serum free Williams E media supplemented with glutamine and penicillin/streptomycin for 24 hr. Media samples were taken and an albumin quantified by ELISA according to the manufactures instructions (Table 1). n=5-6. Bars = SEM. Please click here to view a larger version of this figure.
| Section | Name of Equipment | Company | Cat. No. | Notes | Images |
| 2 | Glass HFB Module | Soham Scientific | --- | Custom Item. | |
| 2.1 | Sigmacote® | Sigma-Aldrich | SL2 | ||
| 2.3 | Silicoset 151 | Intertronics | ACCSS151 | Silicone Glue. | |
| 2.5 | PTFE tape | Sigma-Aldrich | Z104388 | ||
| 3.2.1 | Reservoir bottle | Fisher | 11972619 | ||
| Q-series cap | Kinesis | 00932Q-3V | PTFE the screw threads of the adapters and fitting nut. Attach to the Q-series cap. Attach an 8.5 cm section of the supplied PTFE tubing under the 1 mm adapter and a 4 cm section under the 3 mm adapter. |

|
|
| Adapter, Male, 1.0 mm ID | Kinesis | 008NB10-KD5L | |||
| Adapter, Male, 3.0 mm ID | Kinesis | 008NB30-KD5L | |||
| Fitting Nut | Kinesis | U-350 | |||
| Neoprene tubing | Fisher | 10366344 | Attach the Hepa filter to 6 cm of neoprene tubing and attach this to the 'fitting nut'. Attach a 2x 30 mm sections of L/S14 tubing to the top two barbs of the Y-connector and a 3 cm section of L/S16 tubing to the bottom. Attach this to the barb of the 3 mm ID adapter. (Section 3.2.1) |
|
|
| HEPA-vent | Fisher | 11374634 | |||
| Y-connector, barbed | Cole Parmer | OU-06295-10 | |||
| L/S16 Silicone tubing | Cole Parmer | OU-96410-16 | |||
| L/S14 Silicone tubing | Cole Parmer | WZ-96410-14 | |||
| L/S13 Silicone tubing | Cole Parmer | OU-96410-13 | 80 cm to connect the 1.0 mm barbed adapter on the Q-series cap to the pump tubing = Feed tube. | ||
| WM 205U/CA pump | Fisher | 1248-6300 | |||
| WM pump tubing, PVC, blue-orange, 0.25 mm bore | Fisher | 12416310 | PTFE the screw thread of the male adapter and connect the female adapter. Work the pump tubing over one of the barbs. Repeat this set-up at the other end of the tubing. (Section 3.2.1) |

|
|
| Adapter, Male, 1.0 mm ID | Kinesis | 008NB10-KD5L | |||
| Adapter, Female, 1.0 mm ID | Kinesis | 008NB10-KD2L | |||
| 3.2.2 | Female Luer cap | Cole Parmer | WZ-45508-64 | Side port end caps. |
|
| L/S13 Silicone tubing | Cole Parmer | OU-96410-13 | 40 mm section to connect the pump tubing to a module connector. | ||
| L/S16 Silicone tubing | Cole Parmer | OU-96410-16 | 3x 30 mm of L/S16 fitted to 3x reducers = module connectors. (Section 3.2.2) | ||
| Barbed reducer 1/8" x 1/16" | Cole Parmer | 30616-43 | |||
| 3.2.4 | L/S13 Silicone tubing | Cole Parmer | OU-96410-13 | 55 cm section to connect the retentate to the L/S14 of the Y-connector on the Q-series cap. |
|
| L/S13 Silicone tubing | Cole Parmer | OU-96410-13 | 45 cm section to connect the permeate to the L/S14 of the Y-connector on the Q-series cap. | ||
| Straight barbed union | Cole Parmer | WZ-30612-43 | Attach to the end of the L/S13 that will connect with the L/S14 of the Y-connector. | ||
| 3.4.2 | Clamp | VWR | 229-0609 | ||
| 4.3 | 4 mm Silicone tubing | Fisher | FB68858 | Fold over a 40 mm section of tubing and secure with a cable tie = Module end cap. (Section 4.3) |

|
| Cable tie | Fisher | 12326377 | |||
| 4.4 | MACSmix tube rotator | Miltenyi Biotech | 130-090-753 | An adaptation may be required to attach the modules. | |
| 4.5 | Leur Injection port | Thistle Scientific | IB-10820 | Attach the end cap to the injection port. (Section 4.5) | |
| Female Luer cap | Cole Parmer | WZ-45508-64 | |||
| 5 | L-lactic acid kit | Megazyme | K-LATE | ||
| 5 | D-glucose kit | Megazyme | K-GLUC | ||
| 6.2 | Scalpel / micro knife | InterFocus | 10315-12 | ||
| 7.4.3 | Albumin ELISA | Bethyl Labs | E80-129 |
Table 1. Components of the HFB set-up. The text section relevant to each component is given in column 1.
Discussion
This manuscript describes the set-up and operation of a hollow fiber bioreactor (HFB) system for mammalian cell culture and its utility is demonstrated in proliferating the hepatocyte cell line HepG2/C3A. The system is designed to fit onto the shelf of a standard incubator and its set-up is simple enough to be carried out by any competent cell biologist familiar with aseptic technique.
The fibers used in the research system described here are manufactured in house by phase inversion spin casting (spinning) using a non-biodegradable proprietary polymer. It is possible to make fibers by spinning from a variety of materials suitable for cell culture, both biodegradable and non-biodegradable, for example; polycaprolactone (PCL)27, poly-L-lactide acid (PLLA)28, poly(lactic-co-glycolic acid) (PLGA)29, polysulfone (PSU)12 and polyetheretherketone (PEEK)13. Each has different properties and should be chosen based on the needs of the system. Check the compatibility of the employed fiber with the ethanol used in the sterilization step. PLGA is known to plasticize with ethanol necessitating an alternative treatment such as antibiotic/antimycotic solution25.
The dimensions of the glass modules used here were chosen based on the needs of the current research. Different sizes can be made by any reputable glass blowing company. A consideration in module size is the number of cells, which is linked to the number of fibers in the module and likely flow rates. The more cells there are in the module the higher the flow rates will need to be in order to maintain favorable culture conditions at the outflow end of the bioreactor. This will reach a limit as some point and some trial and error may be required with monitoring of media conditions in the module permeate. Mathematical modelling may provide some insights into the required module dimensions and flow rates22.
The dimensions of the apparatus used here is designed to fit onto an incubator shelf. The length of the tubing is dictated by the length needed to reach between the connectors whilst also allowing enough movement of components to allow for set-up and operation. If time course sampling is required, for example in the monitoring of media conditions in the module permeate then injection ports can be added to the retentate and permeate lines to facilitate this.
A prerequisite for any cell culture system is to keep cells alive and in most cases growing. In the light of studies demonstrating a more in vivo-like phenotype in cells grown in 3D culture systems it also seems important to provide an environment that closely mimics the in vivo environment encountered by the cells. This last point is often neglected in 2D cell culture in favor of the convenience this culture system offers. The HFB mimics in vivo capillary networks by delivering nutrients to the cells through the lumen of fibers. The waste products are also removed from the system by the dynamic flow. This creates an in vivo-like system for cell culture and one that closely mimics the in vivo environment seen by hepatocytes, making this system a better choice compared to 2D tissue culture plastic for culturing these cells. This is corroborated by the fact the cells secrete 15-fold the amount of albumin, an important hepatic function, in the HFB culture system compared to those grown on 2D tissue culture plastic.
Whilst the HFB system is suited to most if not all anchorage dependent cell types, the example here is for hepatocytes because there is a real need to be able to culture functional, more in vivo-like hepatocytes for use in drug development by the pharmaceutical industry and in bioartificial liver devices for the extracorporeal support of liver failure patients. The need for more functional cells extends beyond these examples, particularly as the field of regenerative medicine enters a phase of translational work. The advantages of a more in vivo-like culture environment should not be overlooked.
Disclosures
MJE is a founder and director of Cellesce Ltd which aims to provide bioprocessing solutions for tissue engineering-based research and industry.
Acknowledgments
This work was funded by the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs) CRACK IT funding.
References
- Lewis WH. Experimental evidence in support of the theory of outgrowth of the axis cylinder. American J Anat. 1906;6(1):461–471. [Google Scholar]
- Harrison RG. The development of peripheral nerve fibers in altered surroundings. Wilhelm Roux Arch Entwickl Mech Org. 1910;30(2):15–33. [Google Scholar]
- Ding L, et al. A genome-scale RNAi screen for Oct4 modulators defines a role of the Paf1 complex for Embryonic Stem Cell identity. Cell Stem Cell. 2009;4(5):403–415. doi: 10.1016/j.stem.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Groeber F, Holeiter M, Hampel M, Hinderer S, Schenke-Layland K. Skin tissue engineering -In vivo and in vitro applications. Adv Drug Deliv Rev. 2011;63(4):352–366. doi: 10.1016/j.addr.2011.01.005. [DOI] [PubMed] [Google Scholar]
- Sinden JD, Vishnubhatla I, Muir KW. Prospects for stem cell-derived therapy in stroke. Prog Brain Res. 2012;201:119–167. doi: 10.1016/B978-0-444-59544-7.00007-X. [DOI] [PubMed] [Google Scholar]
- Ouyang A, Yang S-T. A two-stage perfusion fibrous bed bioreactor system for mass production of embryonic stem cells. Expert Opin Biol Ther. 2008;8(7):895–909. doi: 10.1517/14712598.8.7.895. [DOI] [PubMed] [Google Scholar]
- Carnachan RJ, Bokhari M, Przyborski SA, Cameron NR. Taloring the morphology of emulsion-templated porous polymers. Soft Matter. 2006;2(7):608–616. doi: 10.1039/b603211g. [DOI] [PubMed] [Google Scholar]
- Deshpande P, et al. Simplifying corneal surface regeneration using a biodegradable synthetic membrane and limbal tissue explants. Biomaterials. 2013;34(21):5088–5106. doi: 10.1016/j.biomaterials.2013.03.064. [DOI] [PubMed] [Google Scholar]
- Storm MP, Orchard CB, Bone HK, Chaudhuri JB, Welham MJ. Three-dimensional culture systems for the expansion of pluripotent embryonic stem cells. Biotechnol Bioeng. 2010;107(4):683–695. doi: 10.1002/bit.22850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wung N, Acott SM, Tosh D, Ellis MJ. Hollow fibre membrane bioreactors for tissue engineering applications. Biotechnol Lett. 2014;36(12):2357–2366. doi: 10.1007/s10529-014-1619-x. [DOI] [PubMed] [Google Scholar]
- Ellis M, Jarman-Smith M, Chaudhuri JB. Bioreactor systems for tissue engineering: A four-dimensional challenge. In: Chaudhuri J, Al-Rubeai , editors. Bioreactors for tissue engineering; principles, design and operation. Springer; 2005. pp. 1–18. Chapter 1. [Google Scholar]
- Silva AI, Mateus M. Development of a polysulfone hollow fibre vascular bio-artificial pancreas device for in vitro studies. J Biotechnol. 2009;139(3):236–249. doi: 10.1016/j.jbiotec.2008.12.004. [DOI] [PubMed] [Google Scholar]
- De Bartolo L, et al. Human hepatocyte functions in a crossed hollow fiber membrane bioreactor. Biomaterials. 2009;30(13):2531–2543. doi: 10.1016/j.biomaterials.2009.01.011. [DOI] [PubMed] [Google Scholar]
- Schmelzer E, Finoli A, Nettleship I, Gerlach JC. Long-term three-dimensional perfusion culture of human adult bone marrow mononuclear cells in bioreactors. Biotechnol Bioeng. 2015;112(4):801–810. doi: 10.1002/bit.25485. [DOI] [PubMed] [Google Scholar]
- Tapia F, et al. Production of high-titer human influenza A virus with adherent and suspension MDKC cells cultured in a single-use hollow fiber bioreactor. Vaccine. 2014;32(8):1003–1011. doi: 10.1016/j.vaccine.2013.11.044. [DOI] [PubMed] [Google Scholar]
- Deng X, Zhang G, Shen C, Yin J, Meng Q. Hollow fiber culture accelerates differentiation of Caco-2 cells. Appl Microbiol Biotechnol. 2013;97(15):6943–6955. doi: 10.1007/s00253-013-4975-x. [DOI] [PubMed] [Google Scholar]
- Schutte M, et al. Rat primary hepatocytes show enhanced performance and sensitivity to acetaminophen during three-dimensional culture on a polystyrene scaffold designed for routine use. Assay Drug Dev Technol. 2011;9(5):475–486. doi: 10.1089/adt.2011.0371. [DOI] [PubMed] [Google Scholar]
- Bokhari M, Carnachan RJ, Cameron NR, Przyborski SA. Novel cell culture device enabling three-dimensional cell growth and improved cell function. Biochem Biophys Res Commun. 2007;354(4):1095–1100. doi: 10.1016/j.bbrc.2007.01.105. [DOI] [PubMed] [Google Scholar]
- Bokhari M, Carnachan RJ, Cameron NR, Przyborski SA. Culture of HepG2 liver cells on three dimensional polystyrene scaffolds enhances cell structure and function during toxicological challenge. J Anat. 2007;211(4):567–576. doi: 10.1111/j.1469-7580.2007.00778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivares A, et al. Morphological behaviour and metabolic capacity of cryopreserved human primary hepatocytes cultivated in a perfused multiwell device. Xenobiotica. 2015;45(1):29–44. doi: 10.3109/00498254.2014.944612. [DOI] [PubMed] [Google Scholar]
- Smith I, et al. Human neural stem cell-derived cultures in three-dimensional substrates form spontaneously functional neuronal networks. J Tissue Eng Regen Med. 2015. Epub ahead of print. [DOI] [PubMed]
- Davidson AJ, Ellis MJ, Chaudhuri JB. A theoretical approach to Zonation in a bioartificial liver. Biotechnol Bioeng. 2012;109(1):234–243. doi: 10.1002/bit.23279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder M. The basic principles of membrane technology. 2nd ed. Kluwer Academic Publishers; 1996. Chapter 3 section 4. [Google Scholar]
- Ellis MJ, Chaudhuri JB. Poly(lactic-co-glycolic acid) hollow fibre membranes for use as a tissue engineering scaffold. Biotechnol Bioeng. 2007;96(1):177–187. doi: 10.1002/bit.21093. [DOI] [PubMed] [Google Scholar]
- Shearer H, Ellis MJ, Perera SP, Chaudhuri JB. Effects of common sterilization methods on the structure and properties of poly(D,L lactic-co-glycolic acid) scaffolds. Tissue Eng. 2006;12(10):2717–2727. doi: 10.1089/ten.2006.12.2717. [DOI] [PubMed] [Google Scholar]
- Forsey RW, Chaudhuri JB. Validity of DNA analysis to determine cell numbers in tissue engineering scaffolds. Biotechnol Lett. 2009;31(6):819–823. doi: 10.1007/s10529-009-9940-5. [DOI] [PubMed] [Google Scholar]
- Williamson MR, Coombes AGA. Gravity spinning of polycaprolactone fibres for applications in tissue engineering. Biomaterials. 2004;25(3):459–465. doi: 10.1016/s0142-9612(03)00536-2. [DOI] [PubMed] [Google Scholar]
- El-Salmawy A, et al. Preparation and properties of pronectin F-coated biodegradable hollow fibres. J Artif Organs. 2005;8(4):245–251. doi: 10.1007/s10047-005-0314-z. [DOI] [PubMed] [Google Scholar]
- Meneghello G, et al. Fabrication and characterization of poly(lactic-co-glycolic acid)/polyvinyl alcohol blended hollow fibre membranes for tissue engineering applications. J Memb Sci. 2009;344(1-2):55–61. [Google Scholar]