

Abstract
Large-scale 2D electron microscopy (EM), or nanotomy, is the tissue-wide application of nanoscale resolution electron microscopy. Others and we previously applied large scale EM to human skin pancreatic islets, tissue culture and whole zebrafish larvae1-7. Here we describe a universally applicable method for tissue-scale scanning EM for unbiased detection of sub-cellular and molecular features. Nanotomy was applied to investigate the healthy and a neurodegenerative zebrafish brain. Our method is based on standardized EM sample preparation protocols: Fixation with glutaraldehyde and osmium, followed by epoxy-resin embedding, ultrathin sectioning and mounting of ultrathin-sections on one-hole grids, followed by post staining with uranyl and lead. Large-scale 2D EM mosaic images are acquired using a scanning EM connected to an external large area scan generator using scanning transmission EM (STEM). Large scale EM images are typically ~ 5 - 50 G pixels in size, and best viewed using zoomable HTML files, which can be opened in any web browser, similar to online geographical HTML maps. This method can be applied to (human) tissue, cross sections of whole animals as well as tissue culture1-5. Here, zebrafish brains were analyzed in a non-invasive neuronal ablation model. We visualize within a single dataset tissue, cellular and subcellular changes which can be quantified in various cell types including neurons and microglia, the brain's macrophages. In addition, nanotomy facilitates the correlation of EM with light microscopy (CLEM)8 on the same tissue, as large surface areas previously imaged using fluorescent microscopy, can subsequently be subjected to large area EM, resulting in the nano-anatomy (nanotomy) of tissues. In all, nanotomy allows unbiased detection of features at EM level in a tissue-wide quantifiable manner.
Keywords: Developmental Biology, Issue 111, Large-scale electron microscopy, scanning EM, nanotomy, zebrafish, quantitative EM, correlated microscopy
Introduction
Recent technical developments have improved the versatility, applicability and quantitative nature of EM, leading to a revival of ultrastructural analysis. Advances include 3D EM, large scale 2D EM and improved methods and reagents for correlated light microscopy and electron microscopy (CLEM) to compare other modes of microscopic analysis directly to the EM level8-10. Large-scale 2D EM is particularly suitable to quantify or identify (novel) disease features for human pathology, study animal models for disease and tissue culture models. Due to the typically small field of view it is difficult to correlate changes at high magnification to a tissue wide scale, as well as to unbiasedly and quantitatively analyze ultrastructural features.
For pathological analysis of human tissue or the assessment of pathology in animal models, haematoxylin and eosin (H&E) colored sections of formaldehyde fixed paraffin embedded (FFPE) tissue is the standard. Besides simple H&E staining immunolabeling is also performed to identify pathological abnormalities. If such tissues could be analyzed at the EM level, specific cell types, and subcellular changes could be identified. The unbiased nature of large scale EM allows finding unexpected and novel features of disease. By large-scale EM areas up to square millimeters can be visualized. We and others previously applied nanotomy to rat pancreatic islets4, cell culture2, rat brain3, skin and mucosa7 and whole zebrafish larvae1,5 (www.nanotomy.org). Zebrafish are highly suitable for in vivo imaging, in particularly to visualize cell types which are difficult to access in mammalian tissues including brain immune cells11. Here the nanotomy procedure is described in detail, applied to coronal sections of zebrafish heads undergoing conditional neuronal ablation by conversion of metronidazole by neuronal expressed nitroreductase (www.nanotomy.org)5,12-14. All raw data is presented as zoomable HTML files, visualizing molecular to tissue scale changes. The presentation of the raw data allows unbiased analyses of the datasets from other angles by experts worldwide.
Protocol
All experiments with zebrafish larvae were approved by the Animal Experimentation Committee of the University of Groningen according to national and EU guidelines.
1. Sample Preparation
- Fixation and Embedding
- Fixation
- Anaesthetize fish larvae in tricaine (0.003%) added to the E3 (5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl2, 0.33 mM MgSO4, 10 mM HEPES, pH 7.6) zebrafish water and test for depth of anesthesia by poking gently with a 200 µl pipette tip under dissection microscope until motion is no longer detected (Figure 1A).
- When processing samples for correlative EM after acquiring in vivo imaging data using confocal or 2 photon imaging in 1.8% agarose as described,5,15 fix larvae in agarose using 4% PFA in PBS containing Triton-X-100 (0.05%).
- Cut larvae from agarose and process for large-scale EM5.
- Fix larvae in fresh 4% paraformaldehyde in PBS containing Triton-X-100 (0.05%) for 2 hr at room temperature in plastic tubes. Note: Cells grown in culture (in vitro) can be directly fixed in glutaraldehyde, as described in the next step (1.1.1.5).
- Remove solution and add 100 µl 0.5% PFA, 2% glutaraldehyde (GA), and 0.1 M sodium cacodylate (pH 7.4). Store overnight at 4 °C. Rinse samples twice in 0.1 M sodium cacodylate.
- Cut larval heads rostrally to the hindbrain to facilitate penetration of osmium. Use fine syringes/forceps.
- Postfix larvae in 1% osmium tetroxide (OsO4)/1.5% potassium ferrocyanide (K4[Fe(CN)6]) in 0.1 M sodium cacodylate on ice for 2 hr and rinse 3 x 5 min in double distilled H2O.
- Dehydrate in ethanol by 20 min incubations in increasing ethanol concentrations (50%, 70%, 3 times 100%).
- Embedding
- Prepare epoxy resin according to standard recipes used in EM labs. Note: We use epoxy resin as follows: mix 19.8 g glycid ether 100, 9.6 g 2-dodecenylsuccinic acid anhydride and 11.4 g methylnadic anhydride using a magnetic stirrer. Add 0.5 g DMP-30 (tris-(dimethylaminomethyl) phenol) and mix again.
- Incubate larvae overnight in 50% epoxy resin in ethanol (v/v) while rotating (RT).
- Replace diluted epoxy-resin with pure resin. Incubate for 30 min, add fresh resin again and incubate for another 30 min. Again refresh the resin and then incubate 3 hr at RT, followed by 15 min at 58 °C and another 1 hr at RT under low pressure (200 mbar).
- Orient the heads in commercially available silicon flat embedding molds under a dissection microscope using a needle or toothpick with the anterior facing the side of the mold or as needed(Figure 1B).
- Polymerize epoxy resin overnight at 58 °C. If epoxy resin is still too soft allow another day to polymerize and harden at 58 °C. Test stiffness by applying pressure using forceps. If this easily leaves an indentation allow another day to polymerize and harden at 58 °C. When specimen is fully hardened proceed to trimming and sectioning.
- Trim away excess resin from the stub using razor blades (Figure 1B).
- Sectioning, Contrasting, and Mounting
- Sectioning
- Section epoxy resin block using an ultramicrotome.
- Use a glass knife or a diamond histoknife to cut semithin sections (500 nm) for toluidine blue/basic fuchsin (Figure 1D) staining to detect the right positioning.
- Transfer semi-thin sections on microscopic slides by picking them up with a glass Pasteur pipet whose tip has been closed by melting in a flame. Dry on a hot plate until no water is left. Stain for 10 sec with 1% toluidine blue in water on a hot plate and after rinsing with water, stain for 10 sec with 0.05% basic fuchsin in 1% sodium tetraborate. Examine with a normal light microscope at 10X to 40X.
- When the proper site/orientation within the brain is reached, continue sectioning the epoxy resin block with a diamond knife to cut ultrathin sections (~ 70 nm; Figure 1E)
- Use easily identifiable anatomic structures in and around the brain, including olfactory pits, eyes, grey-white matter boundaries, to identify the region of interest during ultrathin sectioning and to adjust the sectioning angle when the sample is tilted.
- Mount section on single slot L2 x 1 copper grids (Figure 1F), to allow acquisition uninterrupted by grid bars. Use Formvar coated grids to support the sections.
- Contrasting
- Stain ultrathin sections on grids for 2 min in 2% uranyl acetate in methanol followed by Reynolds lead citrate for another 2 min 16.
2. Image Acquisition
- Large Scale STEM Note: We use scanning EM with an external scan generator capable of acquiring multiple large fields of view at high resolution3,5,7,17 using STEM detection. Typically, one image generated this way is equivalent to the fields of view of ~ 100 Transmission Electron Microscope (TEM) images, significantly reducing the amount of stitching.
- Mounting Sample in Microscope
- Place grid with section in the multiple grid sample holder and transfer into the chamber of the SEM.
- Alignment of the STEM Detector
- Put the grid with the sample under the beam and move it up so it will be approx. 5 mm from the lens pole. Acquire an image at 20 kV using the in-lens detector.
- Focus on grid edge and adjust working distance to 4.0 mm.
- Move the sample holder to a safe position and bring in the STEM detector.
- Center the hole of the STEM detector in the image-field by turning the adjustments screws while imaging at maximum speed with the in-lens detector.
- Carefully bring back the sample holder which will just fit between the lens pole and the detector.
- Acquire an image with the in-lens detector to be sure to be on the section area.
- Switch to STEM detection (gain medium and all quadrants set on 'minus') and acquire an image and select the area to be scanned.
- Raise acceleration voltage to 21 kV and wait for stabilization of the beam (stable image again) and then go to 29 kV in steps of 2 kV.
- Acquiring Images
- Pre-irradiate the sample (to prevent brightness changes caused by the e-beam) by zooming out so the complete area to be scanned fits the image window.
- Change aperture to 120 µm (to keep the proper dynamic range the detector gains have to be reduced one step).
- De-focus so the image is blurred.
- Make the scanned area as tight as possible using the reduced area scan option.
- Set scan speed such that a frame is scanned in approx. 1 - 2 sec. Note: Depending on the size and tissue this typically takes ½ hr (100 x 100 µm FOV) to up to 3 hr (1,000 x 500 µm).
- Zoom in at least 100X and scan a small area for 10 sec. Note: When this area does not change brightness compared to its surrounding, pre-irradiation is sufficient.
- Bring back the 30 µm aperture (and STEM gain on medium)
- Focus the sample.
- Align aperture using the wobbler (at ~ 40,000X magnification).
- While in focus select the lightest area/feature, set scan speed such that details are visible.
- In the microscope software adjust brightness and contrast by carefully watching the histogram to keep all pixels in the dynamic range. Do the same for the darkest area/features.
- Go back to the bright area and check again. Be on the safe side so there is some space on both sides of the histogram.
- Zoom out so the complete area to be scanned fits the imaging window, and start the large area acquisition program.
- Use the wizard option to set up a mosaic by selecting an area from the screen. Use pixel size 2 - 5 nm depending on what details are necessary. Use dwell time 3 µs for STEM.
- Select option "autofocus on previous tile", use the large scale acquisition software autofocus with default company settings, check box "verify setting prior to execution" and define where to save the data.
- Press "optimize" to check microscope settings. Note: Now the time needed will be shown. This might be up to 72 hr for 1 x 0.5 mm areas.
- In window "maximum tile resolution" type 20,000 so individual tiles will not be bigger than 20k x 20k, preventing blunt edge effects. Press "execute".
- When asked for checking focus and Brightness/Contrast (B/C), switch off external scan generator and carefully adjust focus and astigmatism in the microscope software. Do not change B/C. Note: The stage can be moved and magnification changed, but magnification has to be set back to the desired pixel size.
- Switch on external scan generator again and press "continue".
3. Data Analysis
Open large-scale EM file viewer program and open the file "mosaic info". This will open all the tiled tif-files. This preferably is performed on another workstation to not obstruct new acquisitions.
Choose option 'auto stitch entire mosaic (parameters overlap mode on half, stitching threshold 0.90, noise reduction automatic. In case stitching criteria cannot be fulfilled, zoom in and manually place tile in position and click 'continue as is').
Export as an html file, or alternatively as a single TIF. Note that for TIF export a downscaling might be needed. Include the final pixel size in the file name enabling measurements later.
Perform annotations in the TIF file by opening it in an image editing program. In parallel, open the zoomable html file in a web browser for optimal resolution observation.
Representative Results
Large-scale EM dataset of coronal sections of the head of 1-week-old zebrafish larvae shows many tissue and cellular features in a single large scale image (www.nanotomy.org).
Nanotomy of control brain sections reveals typical ultrastructural features of neural tissue of the rostral forebrain including olfactory fiber bundles, neuronal nuclei and neuronal sub-compartments including synapses (Figure 2A, www.nanotomy.org). Cell types in the head that can be distinguished include chondrocytes with large vacuoles in the lower jaw, osmiophilic myelin of the olfactory nerve cells, endothelial cells of a small vessel, structures include the meninx (the zebrafish counterpart of the meninges, the membranous layer encasing the mammalian brain). Subcellular structures include the glycocalyx of the epidermis, different nuclear morphologies and foci in different cell types and organelles including Golgi apparatus, endoplasmic reticulum (ER), mitochondria and synaptic vesicles and post-synaptic densities.
Unexpectedly the nuclei of cells lining the ventricle are very electron dense compared to other cells dispersed more laterally in the brain. Additionally, these ventricular nuclei show dark foci that are absent in other cells in the brain and appear different in size and structure from the heterochromatin, which is found in nuclei of phagocytic microglia (Figure 2). Cells lining the ventricle in developing zebrafish include mostly radial glial cells and neuronal progenitors, which may reflect an altered chromatin state than more differentiated cells. As stem cells in tissue are generally quite rare, nanotomy to correlate tissue labeling for stem cell markers with tissue scale EM could therefore be used to identify ultrastructural features of stem cells.
Large-scale EM (www.nanotomy.org) of a coronal section in a zebrafish larva undergoing neuronal ablation reveals many specific tissues, and cellular and molecular features not found in control animals. Cellular features include phagocytic microglia and vacuoles the size of cells, likely representing dying cells (Figure 2B, www.nanotomy.org). Previous EM studies have identified microglia in vertebrates18,19. Characteristic features of microglia include elongated nuclei, clumps of patchy chromatin next to the nuclear envelope, prominent Golgi apparatus, free polyribosomes, granular endoplasmic reticulum (ER) with long narrow cisternae, relatively dark/dense cytoplasm, and numerous inclusions, such as phagosomes, lipid droplets and lysosomes. All these hallmarks, attributed to microglia in mammalian tissues, were also found in these cells in zebrafish brains (Figure 2B, www.nanotomy.org).
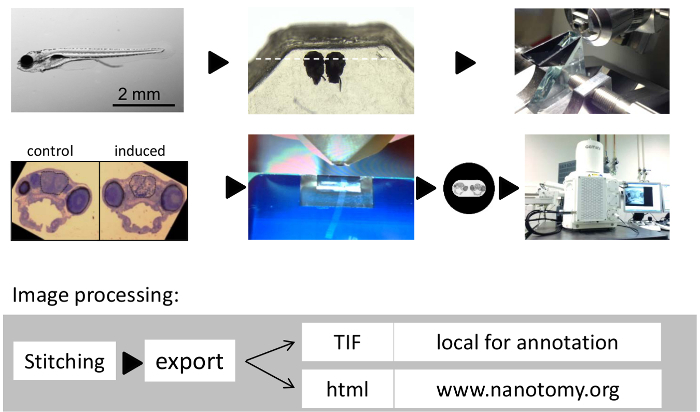 Figure 1: Workflow of Large-scale Tissue Electron Microscopy on Zebrafish Brains. (A) 7 day-old zebrafish larva. (B) Zebrafish larval heads embedded side by side in epoxy resin. (C) Glass knife for semi -thin sectioning and orienting the specimen. (D) Representative semithin toluidine blue stained section showing two side-by-side embedded zebrafish larvae. (E) Diamond knife to cut ultrathin (70 nm) sections. (F) Edited image in (D) of whole zebrafish larval brain section on one-hole grid to indicate positioning. (G) Microscopy system used for large scale 2D EM in this study. (H) Flow of image processing. Please click here to view a larger version of this figure.
Figure 1: Workflow of Large-scale Tissue Electron Microscopy on Zebrafish Brains. (A) 7 day-old zebrafish larva. (B) Zebrafish larval heads embedded side by side in epoxy resin. (C) Glass knife for semi -thin sectioning and orienting the specimen. (D) Representative semithin toluidine blue stained section showing two side-by-side embedded zebrafish larvae. (E) Diamond knife to cut ultrathin (70 nm) sections. (F) Edited image in (D) of whole zebrafish larval brain section on one-hole grid to indicate positioning. (G) Microscopy system used for large scale 2D EM in this study. (H) Flow of image processing. Please click here to view a larger version of this figure.
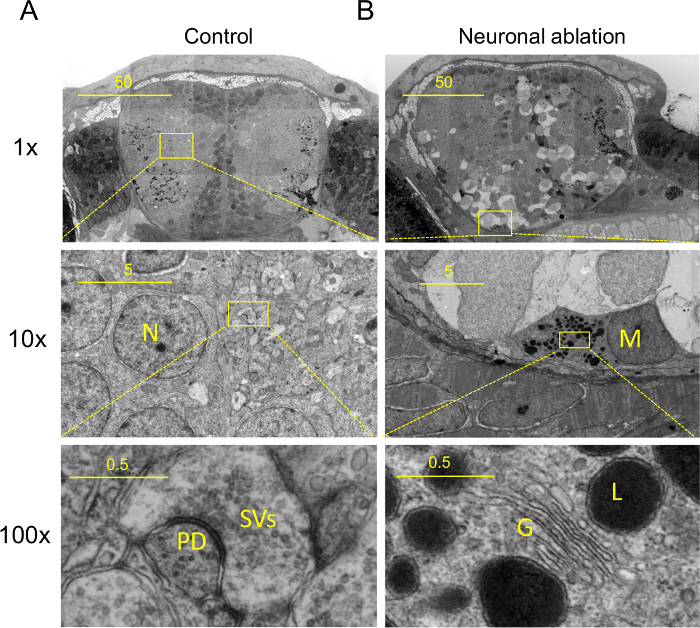 Figure 2:Nanotomy Results: From Macromolecule to Tissue.(A) Nanotomy of brains of 7 days post fertilization control and (B) treated (neuronal ablation; Figure 1), showing features specific to the neuronal ablated degenerative brain. (B) These include phagocytic microglia, dark cells undergoing cell death and spongy appearance of neural tissue ((A) and (B)). Upper panels (adapted from5 ): 10X magnified view of region indicated in middle panels. Middle panels: 10X magnified view of region indicated in lower panels. (A, middle panel) High magnification view of neuropil showing synapse in (A, lower panel), synaptic vesicles (SVs) and postsynaptic density (PD). (B, Middle panel) High magnification view of amoeboid microglia or possibly a macrophage (middle panel, M) showing typical amoeboid microglial features (lower panel) including prominent Golgi apparatus (G), inclusions including lysosomal vacuoles (L). Scale bars: 50 µm (top), 5 µm (middle) and 0.5 µm (bottom). This figure has been modified from5. Please click here to view a larger version of this figure, or visit the full data online (nanotomy.org).
Figure 2:Nanotomy Results: From Macromolecule to Tissue.(A) Nanotomy of brains of 7 days post fertilization control and (B) treated (neuronal ablation; Figure 1), showing features specific to the neuronal ablated degenerative brain. (B) These include phagocytic microglia, dark cells undergoing cell death and spongy appearance of neural tissue ((A) and (B)). Upper panels (adapted from5 ): 10X magnified view of region indicated in middle panels. Middle panels: 10X magnified view of region indicated in lower panels. (A, middle panel) High magnification view of neuropil showing synapse in (A, lower panel), synaptic vesicles (SVs) and postsynaptic density (PD). (B, Middle panel) High magnification view of amoeboid microglia or possibly a macrophage (middle panel, M) showing typical amoeboid microglial features (lower panel) including prominent Golgi apparatus (G), inclusions including lysosomal vacuoles (L). Scale bars: 50 µm (top), 5 µm (middle) and 0.5 µm (bottom). This figure has been modified from5. Please click here to view a larger version of this figure, or visit the full data online (nanotomy.org).
Discussion
EM allows analysis of cellular context with high resolution imaging of macromolecules in biological context. However, this typically limits the field of view. Larger scale 3D EM is particularly suitable for mapping neuronal connectivity by creating nanoscale resolution 3 dimensional reconstructions, requiring complex data processing10. In contrast, 2D large scale EM requires only a single section and stitching of the imaging data, and assessment of the data is possible by anyone with access to an Internet browser. We and others previously used large scale EM to analyze tissues and whole animals. As for most approaches, TEM and SEM-based stitching do have their own advantages. Here, scanning transmission EM (STEM) was used that allows generation of a large field of view at high resolution. Typically, one STEM image is equivalent to the fields of view of approximately 100 TEM images, significantly reducing the amount of stitching when imaging large fields of view at high resolution. If higher resolution is needed, TEM may be advantageous. STEM has the advantage over TEM that non-contrasted samples can be used with good ultrastructural contrast6 .
Additionally, the method described here can be simply adjusted for use with back scattered electron detection (BSD) on sections mounted on silica wafers, broadening the use to multiple microscope systems. HTML-zoomable tissue EM files are very useful for quantification, sharing data that may not have been analyzed to its full content, combining LM and EM data (CLEM)8, presentation purposes in scientific research, to analyze patient data, and for education. Alternatively, in large-scale EM in a SEM, but not in a TEM, silica wafers can be used, which has two main advantages: BSD can be used, which is more generally available on scanning electron microscopes than STEM detection. Second, mounting of large sections (> 1 mm2 areas of interest) is straightforward. Mounting on single slotted grids is labor intensive and technically challenging. Detailed comparison between TEM, SEM and STEM is detailed elsewhere6. A disadvantage of BSD imaging is that, compared to STEM, images have increased noise. This can partly be compensated by increasing the pixel dwell time, resulting in much longer acquisition times.
Although for sample preparation relatively standard EM processing (fixation, embedding and sectioning) is needed5-7 , it is technically challenging to cut large ultrathin sections completely devoid of artifacts. Sections are very fragile, easily break, fold or are destroyed during imaging, which typically takes multiple hours per dataset. However, because of the online sharing of the raw unbiased data, it should become possible to compare more easily to published data, and use published dataset in the open domain as controls. Currently, few EM devices capable of large-scale analysis are in use, and therefore accessibility to this technique is somewhat limited, though most imaging centers welcome collaborative efforts.
For large-scale sections the tissue has to be properly fixed throughout the sample. That is why a mixture of the fast but moderate fixative PFA is used in combination with the slow but strong fixative GA. Cutting and picking up large sections without artifacts is difficult. Working with wafers in combination with BSD is easier compared to collecting sections on single hole grids. Heavy metal post staining is more critical compared to classical EM. Since the whole section is imaged every artifact will be visible. TEM users typically find it difficult to switch to a SEM, because of the difference in microscope operation.
Quantification and data sharing - Quantifying subcellular features is difficult in single EM images. The possibility to zoom in and out of large-scale images readily allows identification of cells of interest, which can be followed by nanoscale measurements inside the cells. These datasets indicate that specific cell types can be rapidly identified and quantified in an unbiased manner in these large tissue sections, based on features detectable at different scales. For example microglia can be identified based on their morphology and dense cytoplasm. Subsequently, upon zooming in on the individual cells, nanoscale subcellular and molecular features of these cells can be measured within the same dataset, as we previously showed for ER width within a large scale EM tissue culture dataset2. An additional advantage of nanotomy is that hosting of large scale datasets online will allow others to inspect the data, maybe for other features, and draw their conclusions on new hypothesis.
CLEM - In addition to facilitating quantitative EM, large scale EM makes it easier to correlate light microscopic labeling to EM level8. In the present example the presence of phagocytic microglia in a zebrafish ablation model is shown. A major question in neuroscience is what the individual functions and contributions are of microglia and potential other sources of phagocytic and immune cells. Early EM studies have shown distinctive subcellular features of microglia in disease18. Unfortunately, it is difficult to selectively label microglia in particular in a pathological setting, as they exhibit large overlap with other immune cells in gene expression, morphology and function. Therefore, it is unclear whether and when microglia at the ultrastructural level differ from immune cells from other sources including monocyte derived infiltrating macrophages. Understanding whether there are ultrastructural differences between these cells will provide a starting point for analysis of functional differences. Combining selective transgenic or expression markers and CLEM allows detection of ultrastructural features selective to specific populations.
Diagnosis & presentation and education - By visualizing nanoscale to microscale within a single dataset EM data is greatly facilitated to a broad audience. With the increased possibilities and tools for correlative and large-scale EM we anticipate a revival of EM in basic and medical research. Our method represented here is applied to a zebrafish brain injury model5, but has been used on human tissue7, rat brain3, in a rat model for diabetes4 and in cell culture2, and can also be used in combination with a TEM-based approach1 showing the versatility of this method. The microscope operator is no longer recording highly selected, and hence biased images, but all numerous ultrastructural features are recorded and open for worldwide analysis.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Majority of the work has been performed at the UMCG Microscopy and Imaging Center (UMIC), which is sponsored by NWO 175-010-2009-023 and ZonMW 91111006; STW "Microscopy Valley 12718" to B.N.G.G.. This work was sponsored by a ZonMW VENI grant, a Marie Curie career integration grant (saving dying neurons) and an Alzheimer Nederland fellowship to T.J.v.H.
References
- Faas FG, et al. Virtual nanoscopy: generation of ultra-large high resolution electron microscopy maps. J Cell Biol. 2012;198:457–469. doi: 10.1083/jcb.201201140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers J, et al. FLIPPER, a combinatorial probe for correlated live imaging and electron microscopy, allows identification and quantitative analysis of various cells and organelles. Cell Tissue Res. 2015;360:61–70. doi: 10.1007/s00441-015-2142-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima M, Mendenhall JM, Lindsey LF, Harris KM. Automated transmission-mode scanning electron microscopy (tSEM) for large volume analysis at nanoscale resolution. PLoS One. 2013;8:e59573. doi: 10.1371/journal.pone.0059573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravelli RB, et al. Destruction of tissue, cells and organelles in type 1 diabetic rats presented at macromolecular resolution. Sci Rep. 2013;3:1804. doi: 10.1038/srep01804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham TJ, et al. Intravital correlated microscopy reveals differential macrophage and microglial dynamics during resolution of neuroinflammation. Dis Model Mech. 2014;7:857–869. doi: 10.1242/dmm.014886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuipers J, de Boer P, Giepmans BN. Scanning EM of non-heavy metal stained biosamples: Large-field of view, high contrast and highly efficient immunolabeling. Exp Cell Res. 2015;15477:202–207. doi: 10.1016/j.yexcr.2015.07.012. [DOI] [PubMed] [Google Scholar]
- Sokol E, et al. Large-Scale Electron Microscopy Maps of Patient Skin and Mucosa Provide Insight into Pathogenesis of Blistering Diseases. J Invest Dermatol. 2015;135:1763–1770. doi: 10.1038/jid.2015.109. [DOI] [PubMed] [Google Scholar]
- de Boer P, Hoogenboom JP, Giepmans BN. Correlated light and electron microscopy: Ultrastructure lights up! Nat Methods. Nat Methods. 2015;12:503–513. doi: 10.1038/nmeth.3400. [DOI] [PubMed] [Google Scholar]
- Lam SS, et al. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat Methods. 2015;12:51–54. doi: 10.1038/nmeth.3179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter M, et al. Connectomic reconstruction of the inner plexiform layer in the mouse retina. Nature. 2013;500:168–174. doi: 10.1038/nature12346. [DOI] [PubMed] [Google Scholar]
- Oosterhof N, Boddeke E, van Ham TJ. Immune cell dynamics in the CNS: Learning from the zebrafish. Glia. 2015;63:719–735. doi: 10.1002/glia.22780. [DOI] [PubMed] [Google Scholar]
- van Ham TJ, Kokel D, Peterson RT. Apoptotic cells are cleared by directional migration and elmo1- dependent macrophage engulfment. Curr Biol. 2012;22:830–836. doi: 10.1016/j.cub.2012.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davison JM, et al. Transactivation from Gal4-VP16 transgenic insertions for tissue-specific cell labeling and ablation in zebrafish. Dev Biol. 2007;304:811–824. doi: 10.1016/j.ydbio.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distel M, Wullimann MF, Koster RW. Optimized Gal4 genetics for permanent gene expression mapping in zebrafish. Proc Natl Acad Sci U S A. 2009;106:13365–13370. doi: 10.1073/pnas.0903060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham TJ, Mapes J, Kokel D, Peterson RT. Live imaging of apoptotic cells in zebrafish. FASEB J. 2010;24:4336–4342. doi: 10.1096/fj.10-161018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds ES. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J Cell Biol. 1963;17:208–212. doi: 10.1083/jcb.17.1.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwajima M, Mendenhall JM, Harris KM. Large-volume reconstruction of brain tissue from high-resolution serial section images acquired by SEM-based scanning transmission electron microscopy. Methods Mol Biol. 2013;950:253–273. doi: 10.1007/978-1-62703-137-0_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stensaas LJ, Reichert WH. Round and amoeboid microglial cells in the neonatal rabbit brain. Z Zellforsch Mikrosk Anat. 1971;119:147–163. doi: 10.1007/BF00324517. [DOI] [PubMed] [Google Scholar]
- Mori S, Leblond CP. Identification of microglia in light and electron microscopy. J Comp Neurol. 1969;135:57–80. doi: 10.1002/cne.901350104. [DOI] [PubMed] [Google Scholar]