

Abstract
A detailed protocol is provided here to identify amyloid Aβ plaques in brain sections from Alzheimer's disease mouse models before pre-embedding immunostaining (specifically for ionized calcium-binding adapter molecule 1 (IBA1), a calcium binding protein expressed by microglia) and tissue processing for electron microscopy (EM). Methoxy-X04 is a fluorescent dye that crosses the blood-brain barrier and selectively binds to β-pleated sheets found in dense core Aβ plaques. Injection of the animals with methoxy-X04 prior to sacrifice and brain fixation allows pre-screening and selection of the plaque-containing brain sections for further processing with time-consuming manipulations. This is particularly helpful when studying early AD pathology within specific brain regions or layers that may contain very few plaques, present in only a small fraction of the sections. Post-mortem processing of tissue sections with Congo Red, Thioflavin S, and Thioflavin T (or even with methoxy-X04) can label β-pleated sheets, but requires extensive clearing with ethanol to remove excess dye and these procedures are incompatible with ultrastructural preservation. It would also be inefficient to perform labeling for Aβ (and other cellular markers such as IBA1) on all brain sections from the regions of interest, only to yield a small fraction containing Aβ plaques at the right location. Importantly, Aβ plaques are still visible after tissue processing for EM, allowing for a precise identification of the areas (generally down to a few square millimeters) to examine with the electron microscope.
Keywords: Neuroscience, Issue 112, Alzheimer's disease, mouse model, amyloid β, microglia, immunohistochemistry, fluorescence microscopy, electron microscopy
Introduction
Amyloid Aβ plaque formation is the main neuropathological hallmark of Alzheimer's disease (AD). However increasing evidence suggests important roles of the immune system in disease progression1,2. In particular, new data from preclinical and clinical studies established immune dysfunction as a main driver and contributor to AD pathology. With these findings, central and peripheral immune cells have emerged as promising therapeutic targets for AD3. The following protocol combines light and electron microscopy (EM) to generate new insights into the relationship between Aβ plaque deposition and microglial phenotypic alterations in AD. This protocol allows the labeling of Aβ plaques in mouse models of AD using in vivo injection of the fluorescent dye methoxy-X04. Methoxy-X04 is a Congo Red derivative that can easily cross the blood-brain barrier to enter the brain parenchyma and bind β-pleated sheets with high affinity. Since the dye is fluorescent, it can be used for in vivo detection of Aβ plaque deposition with two-photon microscopy4. Once bound to Aβ, methoxy-X04 does not dissociate or redistribute away from plaques, and it retains its fluorescence over time. It is generally administered peripherally to allow for non-invasive imaging of brain dynamics5. The fluorescence also remains following aldehyde fixation, allowing for correlative post-mortem analyses, including investigation of neuronal death in the vicinity of Aβ plaques6.
This protocol takes advantage of the properties of methoxy-X04 to select brain sections from APPSWE/PS1A246E mice (APP-PS1; coexpressing a double mutation at APP gene Lys670Asn/Met671Leu, and human presenilin PS1-A264E variant)7 that exhibit Aβ plaques in specific regions of interest (hippocampus CA1, strata radiatum, and lacunosum-moleculare) prior to pre-embedding immunostaining against the microglial marker ionized calcium-binding adapter molecule 1 (IBA1) to visualize microglial cell bodies and processes with EM. The mice are given intraperitoneal injection of methoxy-X04 solution, 24 hr prior to brain fixation through transcardial perfusion. Coronal brain sections are obtained using a vibratome. Sections containing the hippocampus CA1 are screened under a fluorescent microscope for the presence of Aβ plaques in strata radiatum and lacunosum-moleculare. Immunostaining for IBA1, osmium tetroxide post-fixation, and plastic resin embedding are then performed on the selected brain sections. At the end of this protocol, the sections can be archived without further ultrastructural degradation, ready for ultrathin sectioning and ultrastructural examination. Importantly, the plaques are still fluorescent after immunostaining with different antibodies, for instance IBA1 as in the present protocol. They become darker than their surrounding neuropil following osmium tetroxide post-fixation, independently of methoxy-X04 staining, which helps to accurately identify the regions of interest, generally down to a few square millimeters, to be examined with the transmission electron microscope.
This correlative approach offers an efficient way to identify specific brain sections to examine at the ultrastructural level. This is particularly helpful when studying early AD pathology, within specific brain regions or layers that may only contain a few Aβ plaques, present in only a small fraction of tissue sections. During these times especially, it would be inefficient to use immunostaining for Aβ (and dual labeling for other cellular markers such as IBA1) on several brain sections simply to yield a small fraction containing Aβ plaques at the right location. In addition, injection of live mice with methoxy-X04 prior to sacrifice and tissue processing does not compromise the ultrastructural preservation. Alternative methods such as post-mortem staining with Congo Red, Thioflavin S, Thioflavin T or methoxy-X04 on fixed tissue sections require staining differentiation in ethanol,8-11 which causes osmotic stress and disrupts the ultrastructure. Congo Red is also a known human carcinogen12.
Protocol
Note: All experiments were approved and performed under the guidelines of the Institutional animal ethics committee, in conformity with the Canadian Council on Animal Care guidelines as administered by the Animal Care Committee of Université Laval. APP-PS1 male mice between 4 and 21 months of age were used. These animals were housed under a 12 hr light-dark cycle at 22 - 25 °C with free access to food and water.
1. Methoxy-X04 Solution Preparation
- Prepare a 5 mg/ml solution of methoxy-X049 by dissolving methoxy-X04 into a solution containing 10% dimethyl sulfoxide (DMSO), 45% propylene glycol, and 45% sodium phosphate buffered saline (0.9% NaCl in 100 mM phosphate buffer, pH 7.4).
- Using a microbalance, weigh 5 mg of the methoxy-X04 compound. Under a fume hood, dissolve the methoxy-X04 in DMSO and stir until a clear greenish solution is obtained. Successively add propylene glycol and phosphate buffer saline while stirring with each addition.
- Stir the solution at 4 °C on a rotator O/N. Obtain a yellowish green emulsion. The solution may be stored at 4 °C for up to two months without degradation.
2. Methoxy-X04 Solution Injection
24 hr prior to perfusion, weigh the mice and give each mouse an intraperitoneal injection of methoxy-X04 at a dose of 10 mg/kg of body weight using a 27 ½ G needle.
3. Transcardial Perfusion of the Injected Mice
On the day before the perfusion, prepare appropriate volumes of phosphate buffered saline (PBS), 3.5% acrolein, and 4% paraformaldehyde (PFA) solutions13 and store them at 4 °C O/N. Note: These solutions will be used for both perfusion and pre-embedding immunohistochemistry. Take special care when working with acrolein, as it is corrosive and toxic to both researchers and the environment. In addition, since acrolein is corrosive to plastic, always use suitable glassware to prepare acrolein solutions.
On the day of perfusion, filter the PFA and acrolein by using coarse filter paper of 25 µm particle retention.
- During the perfusion, use a peristaltic pump to deliver 15 ml of PBS, 75 ml of acrolein, and 150 ml of PFA successively into the mouse circulation, at a flow rate of 25 ml/min. Use caution. Perform perfusion strictly inside a fume hood to avoid harmful fumes of PFA and acrolein.
- To set up the pump, insert one end into the PBS solution, fill the tubing (holding approximately 15 ml) with PBS, and fix a 25 G winged blood collection needle to the other end. At all times, ensure that the tubing is free from any trapped air bubbles.
- Place the tube directly in the acrolein bottle after setting the perfusion pump with the tubing filled with PBS.
Anaesthetize one mouse at a time with a 90 mg/kg of body weight dose of sodium pentobarbital injected intraperitoneally using a 27 ½ G needle. Assess responses to tail/toe pinches. Proceed only if the mouse is unresponsive to such aversive, painful stimuli.
- Secure the mouse in the supine position (lying on the back with face upward) by taping the forepaws and hindpaws to the work surface and carefully expose the heart without causing damage to other organs. Be sure to work quickly after puncturing the diaphragm.
- Perform an incision through the skin with surgical scissors along the thoracic midline beginning caudal to the ribcage and proceed rostral to the clavicle.
- Make two additional lateral incisions along the base of the ventral ribcage. Gently move and pin the two flaps of skin, making sure to expose the entire thoracic cavity.
- Hold the xiphoid process with blunt forceps to expose the thoracic cavity and steady the pointed scissors. Cut through the diaphragm and ribcage, being careful to avoid puncturing the lungs, and continue the incision rostral to the clavicles. Be sure to work quickly after puncturing the diaphragm.
Gently tear the pericardial sac with blunt forceps. Hold the heart steady with blunt forceps, cut the right atrium, and start the infusion of PBS. Immediately insert the blood collection needle into the left ventricle.
Perfuse the mouse with PBS (in the tubing) followed by acrolein for 3 min (corresponding to 75 ml) then switch to PFA for 6 min (corresponding to 150 ml). Be sure to pause the pump flow before switching solution to prevent bubbles from entering the tube.
- Decapitate the mouse and extract the fixed brain and place it directly into a glass vial containing 4% PFA for at least 2 hr at 4 °C.
- To extract the brain, use scissors and tweezers to cut through the skin to expose the skull. Carefully break the skull open in between the eyes, and chip off small pieces of it to expose the underlying brain.
- Carefully remove the exposed brain and post fix it with 4% PFA for an additional 2 hr before proceeding for vibratome sectioning.
4. Brain Sectioning Using a Vibratome
Wash the fixed brain 3 times with chilled PBS. Using a sharp razor blade, remove the olfactory bulb (unless this region is under investigation) and cut the brain transversally into 2 - 3 pieces of approximately equal height, all of which can be sectioned simultaneously in order to accelerate the procedure.
Glue the pieces of brain tissue vertically onto the specimen plate secured into the tray. Make sure that the smooth cut surface sticks firmly to the specimen plate and does not get dislodged during the sectioning procedure. Add enough PBS solution into the tray until the entire brain surface is completely submerged. It is important to keep the brain pieces and subsequent sections fully immersed in PBS throughout this step.
Place the tray in the vibratome, adjust the sectioning speed to 0.5 mm/sec, the sectioning frequency to 90 Hz, and the feed thickness to 50 µm in order to yield 50 µm thick sections. Then transfer the sections into 20 ml glass vials containing cryoprotectant solution (40% PBS, 30% ethylene glycol, and 30% glycerol) using a fine paintbrush. Store the vials at -20 °C until further use. Alternatively, keep the sections in PBS for immediate screening as described in the following steps.
5. Section Screening for the Presence of Methoxy-X04-stained Plaques
With the aid of a stereotaxic mouse brain atlas14, select brain sections containing the region of interest, for instance, the hippocampus CA1 as used in the present example. Place each section into a well within a 24-well culture plate containing enough cryoprotectant solution so as to prevent the sections from drying out.
- Examine each section successively, to avoid drying out the sections, using the following procedure:
- Add a droplet of PBS to a microscope slide with a disposable pipette, and place the section on the droplet.
- Examine the section under a fluorescent microscope to identify regions containing methoxy-X04 labeled Aβ plaques. Note: methoxy-X04 can be easily visualized using a fluorescence ultraviolet (UV) filter (excitation 340 - 380 nm).
Capture images of regions of interest (ROI) in bright field as well as in fluorescence mode without moving the microscope stage, since each image must show the same region in both fields in order to correlate the presence of the plaques directly to the structural region of the tissue section.
Save and name the images taken according to the animal number. Also record the well number in the plate and the field of the pictures taken. For example, Ex: 9978A1B; Animal number: 9978; Well number: A1; Field: Bright. Place the section back in its designated well once the imaging is completed. Note: In the end, for each section examined, two pictures are obtained, one in bright field and another in UV field.
Open the two images of the same ROI (one in bright field and the other in UV field) in Image J, and using the MosaicJ plugin, align the edges of the two images and save the aligned and combined image according to the well number it came from.
Save the picture in a separate folder with the number of the particular animal. Combine the images to identify and localize the plaques in the tissue section, and have a full view of the whole section in the two different fields (bright and UV).
After the screening process is completed, store the examined sections at -20 °C in a 24-well culture plate containing cryoprotectant until immunostaining or further processing is carried out.
6. Pre-embedding Immunostaining for IBA1
Note: Perform the immunostaining for IBA1 on selected freely-floating sections by placing the plate on a slowly moving rocker at RT.
Thoroughly wash the sections 3 times with approximately 1 ml PBS, each wash lasting for 10 min.
Quench endogenous peroxidases with 0.3% hydrogen peroxide in PBS solution for 10 min. This step prevents non-specific peroxidase activity, which is important to avoid background staining.
Wash the tissue in PBS 3 times for 10 min each.
Incubate the sections in 0.1% sodium borohydride in PBS solution for 30 min. This step is important to reduce any remaining aldehydes from the fixation step, and is especially important if acrolein is used in tissue fixation.
Wash the tissue in PBS 3 times for 10 min each time and make sure to remove all the bubbles.
Block the sections for 1 hr. Different antibodies may require different blocking times and solutions. For IBA1, prepare blocking buffer containing 10% fetal bovine serum, 3% bovine serum albumin, and 0.01% Triton X-100 in 50 mM Tris-buffered saline (TBS; pH 7.4). Note: The blocking step is to prevent nonspecific binding of the primary antibody. The low concentration of Triton X-100 allows slight permeabilization of membranes for better staining, and is low enough to preserve most ultrastructural features of tissue under EM.
Remove the blocking buffer from the tissue, and incubate in primary antibody solution (rabbit anti-IBA1, diluted [1:1,000] in blocking buffer) at 4 °C O/N.
Wash in TBS 3 times for 10 min each time.
Incubate in secondary antibody (goat anti-rabbit conjugated to biotin) [1:200] in 0.05 M TBS for 90 min.
Wash in TBS 5 times for 5 min each time.
Incubate in Avidin-Biotin complex solution (Avidin [1:100], Biotin [1:100]) in TBS solution for 1 hr.
Wash in TBS 5 times for 5 min each time.
Reveal the IBA1 staining with 0.05% diaminobenzidine (DAB) and 0.015% hydrogen peroxide in TBS for 8 min. Note: The timing of DAB development may vary from protocol to protocol and should be determined by quickly examining the stained sections under a light microscope. Note: For IBA1, one should be able to clearly visualize the cell bodies and processes of the IBA1-stained microglia at 20X (see Figure 2 for representative example). Be cautious using DAB, as it is a known carcinogen.
Avoid over-developing by carefully monitoring the sections (as described above) during this step. Stop the reaction by washing the sections in chilled PB 5 times for 5 min each time.
7. Processing for Electron Microscopy
Prepare 1% osmium tetroxide solution in PBS in a glass vial. Osmium tetroxide is photosensitive; cover the glass vial with aluminum foil to protect the solution from light. Be cautious using osmium tetroxide, as it is a very hazardous chemical. Perform this and the following steps inside a fume hood.
Remove the PBS from the sections and spread them flat using a fine paintbrush. Perform this step one well at a time to keep the sections from drying out. Be sure to flatten the sections immediately before adding osmium tetroxide, as any folds in the sections will become permanent and attempting to flatten tissue post osmium fixation will only break the sections.
Immerse the sections in osmium tetroxide for 30 min at RT, adding one drop of osmium tetroxide at a time with a transfer pipette, to prevent the sections from folding. Cover the well with aluminum foil to protect the sections from light. Note: This step fixes the lipids within the sections. The tissue will appear very dark after osmium post-fixation.
While sections are in osmium tetroxide, prepare plastic resin in a disposable beaker by mixing 20 g component A, 20 g component B, 0.6 g component C, and 0.4 g component D. Combine the components together in the above order and mix them well using a 10 ml serological pipette until a uniform color is obtained.
Transfer the prepared mixture to aluminum weighing dishes. They will receive the tissue sections once they have been dehydrated.
Dehydrate the sections in increasing concentrations of ethanol for 2 min in the following order: 35%, 35%, 50%, 70%, 80%, 90%, 100%, 100%, 100%.
To remove residual ethanol, immerse the sections in propylene oxide 3 times for 2 min each time. Remove the dehydrated-sections from the 24-well culture plate into 20 ml glass vials. Always use glass vials when working with propylene oxide as it dissolves plastic, and maintain caution, as it is hazardous.
Use a bent glass pipette tip or a fine paintbrush to transfer the sections from propylene oxide solution into the resin and leave O/N for infiltration at RT. Be careful not to dilute the resin with propylene oxide.
- Embed the section with the resin on poly-chloro-tri-fluoro-ethylene (PCTFE) filmsheets:
- Place the aluminum weighing dishes containing the specimens into a 50 - 60 °C oven for 10 - 15 min prior to embedding the sections on PCTFE filmsheets.
- When embedding sections, work with one aluminum weighing dish at a time. Using a fine paintbrush, paint a thin layer of resin onto one PCTFE film sheet.
- Move one section of tissue at a time from the aluminum weighing dish to the PCTFE film sheet. Remove excess resin from around the tissue, being careful not to disturb it.
- After moving all the sections from one weighing dish to the PCTFE film sheet, place a second PCTFE sheet over the first, creating a sandwich of tissue and resin in between 2 sheets.
Polymerize the resin in an incubator for 3 days at 55 - 60 °C.
The material is now ready for ultrathin sectioning and ultrastructural examination, specialized techniques which are often performed by EM core facilities. Store the samples between PCTFE film sheets safely at RT without ultrastructural degradation. Note: The subsequent steps for ultrathin sectioning and transmission electron microscopy examination are explained in a separate protocol13.
Representative Results
This section illustrates the results that can be obtained at different critical steps of the protocol. In particular the results show examples of brain sections containing methoxy-X04 stained plaques in specific region and layers of interest: the hippocampus CA1, strata radiatum, and lacunosum-moleculare. The plaques and regional/lamellar organization of the hippocampus are successively visualized using a combination of UV and bright field filters (Figure 1). The selected brain sections are subsequently immunostained and processed for EM while keeping track of their Aβ plaques, considering that they are still fluorescent following immunostaining, and become darker than the surrounding neuropil following treatment with osmium tetroxide and embedding (Figure 1). This allows one to identify the areas (generally a few millimeters squared) to examine with the electron microscope based on the location of plaques. Furthermore, an improved protocol for pre-embedding immunostaining of microglia with IBA1 is described here. This protocol yields an exceptional visualization of microglial cell bodies, large and small processes (Figure 2) as well as penetration of the antibodies within the brain sections. It thus facilitates the identification of microglial cell bodies and processes at the ultrastructural level, and the study of their interactions with Aβ plaques (Figures 3 and 4).
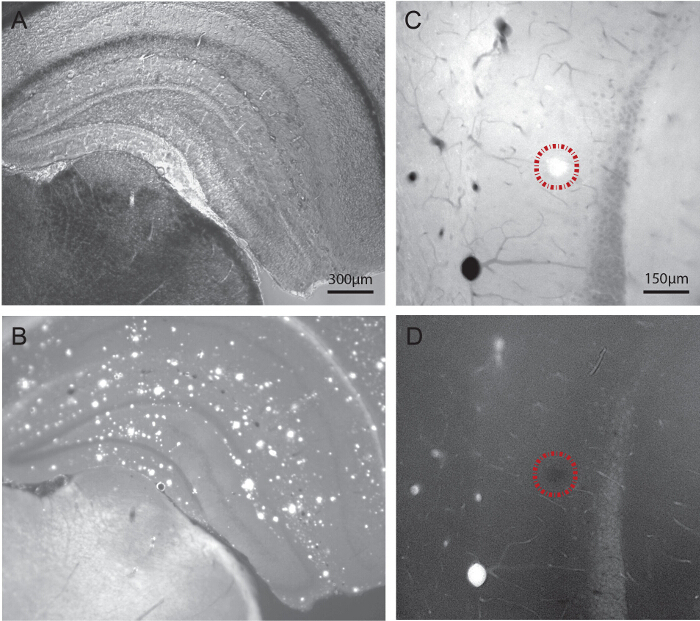 Figure 1. Visualization of Aβ Plaques in 21-month-old APP-PS1 Mice Using Light Microscopy, Following Systemic Injection of the Fluorescent Dye Methoxy-X04. A-B) Dual imaging of one hippocampal section using bright field and fluorescence modes. The regions and layers of interest are visualized under bright field (A), and the Aβ plaques successively localized using an UV filter at a range of 340 - 380 nm (B). C-D) Dual imaging of another hippocampal section before (C) and after (D) pre-embedding immunostaining for IBA1, followed by post-fixation in osmium tetroxide, dehydration in ethanol, and embedding in a plastic resin as required for transmission electron microscopy. To be noted, the plaque (encircled by a dotted line) is still visible upon tissue processing for electron microscopy. Visualizing the plaques when trimming the tissue blocks allows for a precise selection of the areas to image at high spatial resolution. Scale bars = 300 μm for A and B, 150 μm for C and D. Please click here to view a larger version of this figure.
Figure 1. Visualization of Aβ Plaques in 21-month-old APP-PS1 Mice Using Light Microscopy, Following Systemic Injection of the Fluorescent Dye Methoxy-X04. A-B) Dual imaging of one hippocampal section using bright field and fluorescence modes. The regions and layers of interest are visualized under bright field (A), and the Aβ plaques successively localized using an UV filter at a range of 340 - 380 nm (B). C-D) Dual imaging of another hippocampal section before (C) and after (D) pre-embedding immunostaining for IBA1, followed by post-fixation in osmium tetroxide, dehydration in ethanol, and embedding in a plastic resin as required for transmission electron microscopy. To be noted, the plaque (encircled by a dotted line) is still visible upon tissue processing for electron microscopy. Visualizing the plaques when trimming the tissue blocks allows for a precise selection of the areas to image at high spatial resolution. Scale bars = 300 μm for A and B, 150 μm for C and D. Please click here to view a larger version of this figure.
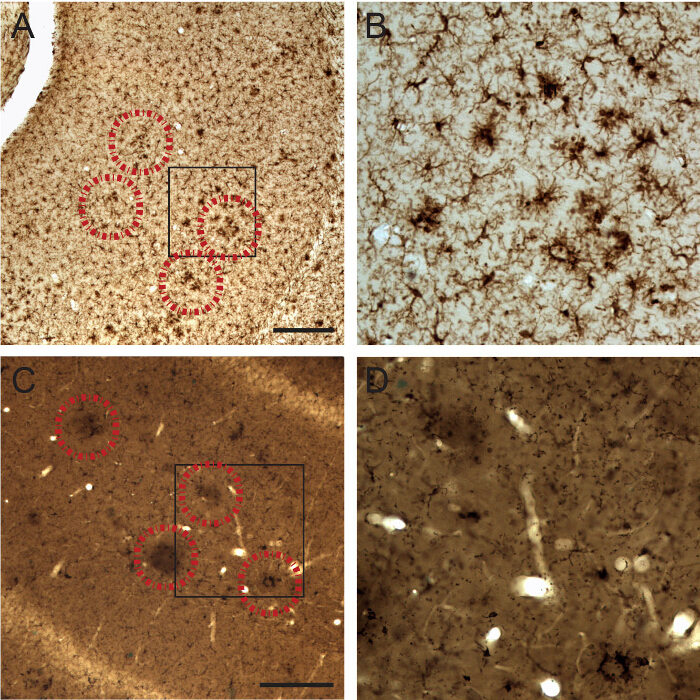 Figure 2. Visualization of Microglial Cell Body and Fine Processes in 21-month-old APP-PS1 Mice by IBA1 Staining at the Light Microscopic Level. A-B) Examples of IBA1-stained microglia showing a clustered distribution when they associate with Aβ plaques (identified by correlative fluorescence imaging of methoxy-X04; encircled by a dotted line) as observed before osmium tetroxide post-fixation and plastic resin embedding. C-D) Examples of IBA1-stained microglia associated with Aβ plaques after osmium tetroxide post-fixation and plastic resin embedding. Note that the plaques become darker than their surrounding neuropil following osmium post-fixation, thus allowing tracking of their location. Scale bar = 250 μm. Please click here to view a larger version of this figure.
Figure 2. Visualization of Microglial Cell Body and Fine Processes in 21-month-old APP-PS1 Mice by IBA1 Staining at the Light Microscopic Level. A-B) Examples of IBA1-stained microglia showing a clustered distribution when they associate with Aβ plaques (identified by correlative fluorescence imaging of methoxy-X04; encircled by a dotted line) as observed before osmium tetroxide post-fixation and plastic resin embedding. C-D) Examples of IBA1-stained microglia associated with Aβ plaques after osmium tetroxide post-fixation and plastic resin embedding. Note that the plaques become darker than their surrounding neuropil following osmium post-fixation, thus allowing tracking of their location. Scale bar = 250 μm. Please click here to view a larger version of this figure.
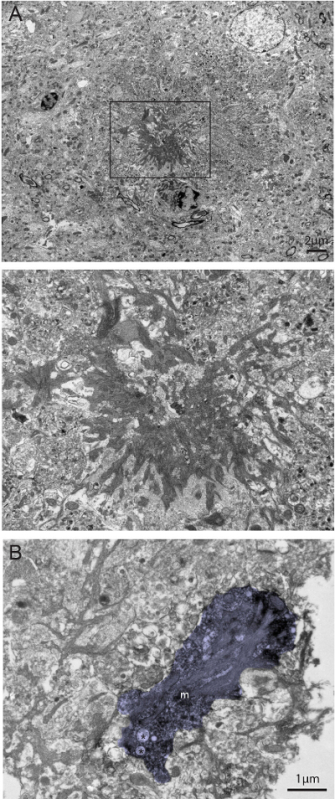 Figure 3. Dual Visualization of Aβ plaques and IBA1-immunostaining in 6-month-old APP-PS1 Mice at the Ultrastructural Level. A) Example of dense core Aβ plaque recognized by its compact fibrillary structure (see inset) and association with dystrophic neurites with ultrastructural features of autophagy. B) Example of IBA1-stained microglial process (m; colored in violet) found nearby the Aβ plaque that contains amyloid deposits. Mitochondrial alterations (shown by asterisks) can also be seen within the microglial process. To be noted, this picture was acquired at the tissue-resin border (white, at the right of the picture) where the penetration of antibodies and staining intensity is maximal. Scale bars = 2 μm for A, 1 μm for B. Please click here to view a larger version of this figure.
Figure 3. Dual Visualization of Aβ plaques and IBA1-immunostaining in 6-month-old APP-PS1 Mice at the Ultrastructural Level. A) Example of dense core Aβ plaque recognized by its compact fibrillary structure (see inset) and association with dystrophic neurites with ultrastructural features of autophagy. B) Example of IBA1-stained microglial process (m; colored in violet) found nearby the Aβ plaque that contains amyloid deposits. Mitochondrial alterations (shown by asterisks) can also be seen within the microglial process. To be noted, this picture was acquired at the tissue-resin border (white, at the right of the picture) where the penetration of antibodies and staining intensity is maximal. Scale bars = 2 μm for A, 1 μm for B. Please click here to view a larger version of this figure.
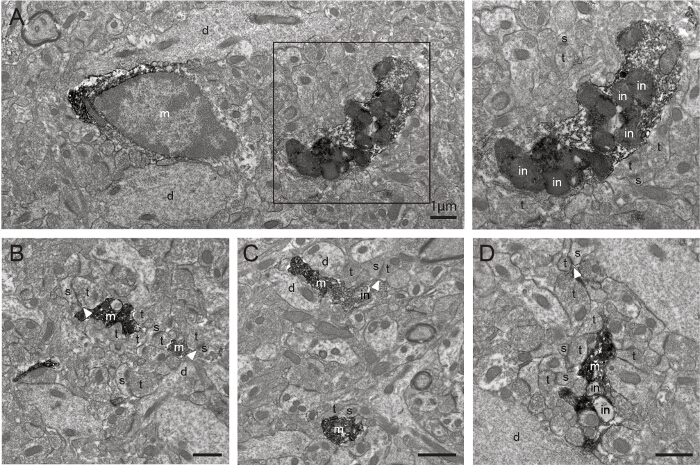 Figure 4. Additional Examples of IBA1-immunostained Microglia in 6-month-old APP-PS1 Mice as Observed with Transmission Electron Microscopy. A-D) Examples of IBA1-stained microglial cell body and processes (m) acquired farther from the tissue-resin border, showing an excellent penetration of the antibodies and staining intensity deeper within the section using this protocol. bv=blood vessel, d=dendrite, in=inclusion, s=dendritic spine, t=axon terminal. The arrowheads show synaptic clefts. Scale bars = 1 μm. Please click here to view a larger version of this figure.
Figure 4. Additional Examples of IBA1-immunostained Microglia in 6-month-old APP-PS1 Mice as Observed with Transmission Electron Microscopy. A-D) Examples of IBA1-stained microglial cell body and processes (m) acquired farther from the tissue-resin border, showing an excellent penetration of the antibodies and staining intensity deeper within the section using this protocol. bv=blood vessel, d=dendrite, in=inclusion, s=dendritic spine, t=axon terminal. The arrowheads show synaptic clefts. Scale bars = 1 μm. Please click here to view a larger version of this figure.
Discussion
This protocol explains a correlative approach for targeting dense core Aβ plaques with EM. Methoxy-X04 in vivo injection allows rapid selection of brain sections that contain Aβ plaques within particular regions and layers of interest, for instance the hippocampus CA1, strata radiatum, and lacunosum-moleculare. In the present example, methoxy-X04 pre-screening was combined with pre-embedding immunostaining for IBA1 to study how different microglial phenotypes interact with synapses at the ultrastructural level in the presence of dense core Aβ plaques.
Microglia are incredibly responsive to their surroundings and their inflammatory activity has been long studied in the context of undermining normal brain function and enhancing the advancement of AD. In particular, these cells were implicated in neurodegenerative processes due to their extensive activation and release of pro-inflammatory cytokines, leading to chronic neuroinflammation, and disruption of normal brain homeostasis15. In recent years, however, these resident immune cells were also shown to actively remodel neuronal circuits in the healthy brain, leading to identification of new pathogenic mechanisms16,17. Their physiological roles at synapses may become dysregulated during neuroinflammation in AD, resulting in exacerbated synaptic loss, currently the best pathological correlate of cognitive decline across various neurodegenerative conditions18,19.
The modifications made to the previous pre-embedding IBA1 staining method13 now allow better penetration of antibodies across the brain sections, as well as visualization of microglial cell bodies and fine processes at the ultrastructural level. Overall, the protocol needs to be performed rigorously from mouse perfusion until plastic resin embedding of the tissue sections, considering that ultrastructural degradation occurring at any in-between step could compromise the integrity of cellular membranes, organelles, cytoskeletal elements, etc.
This protocol could be performed without any immunostaining (by omitting section 6) to solely visualize dense core Aβ plaques, but it also provides a powerful tool to study the complex mechanisms of AD pathogenesis with relation to Aβ deposition, as different antibodies may be used to focus on different cell types, including peripherally-derived macrophages, endogenous microglia, astrocytes, oligodendrocytes and their progenitors, as well as many neuronal subtypes.
Considering the in vivo injection of methoxy-X04, this protocol cannot be performed on fixed brain sections, which precludes its use with post-mortem human brain samples. Also, contrarily to immunostaining against Aβ which labels soluble and insoluble forms of Aβ, the use of dyes binding to β-pleated sheet protein structures such as methoxy-X04 only allows visualization of dense core plaques, which constitutes another limitation.
Nevertheless, important applications could include studying the roles of microglia and other various cell types, which overlap and augment their activities, and could have direct effects on plaque homeostasis and neuronal function over the course of AD pathology.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are grateful to Dr. Sachiko Sato and Julie-Christine Lévesque at the Bioimaging Platform of the Centre de recherche du CHU de Québec for their technical assistance. Grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) RGPIN-2014-05308, The Banting Research Foundation, and The Scottish Rite Charitable Foundation of Canada to M.E.T supported this work.
H.E.H. is recipient of a scholarship from the Lebanese Ministry of Education and Higher Education, and K.B. from the Faculté de médecine of Université Laval.
References
- Heneka MT, Carson MJ, et al. Neuroinflammation in Alzheimer's disease. The Lancet Neurol. 2015;14(4):388–405. doi: 10.1016/S1474-4422(15)70016-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrup K. The case for rejecting the amyloid cascade hypothesis. Nat. Neurosci. 2015;18(6):794–799. doi: 10.1038/nn.4017. [DOI] [PubMed] [Google Scholar]
- Heppner FL, Ransohoff RM, Becher B. Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015;16(6):358–372. doi: 10.1038/nrn3880. [DOI] [PubMed] [Google Scholar]
- Klunk WE, Bacskai BJ, et al. Imaging Abeta plaques in living transgenic mice with multiphoton microscopy and methoxy-X04, a systemically administered Congo red derivative. J. Neuropathol. Exp. Neurol. 2002;61(9):797–805. doi: 10.1093/jnen/61.9.797. [DOI] [PubMed] [Google Scholar]
- Liebscher S, Meyer-Luehmann M. A peephole into the brain: Neuropathological features of Alzheimer's disease revealed by in vivo two-photon imaging. Front Psychiatry. 2012;3:26. doi: 10.3389/fpsyt.2012.00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, et al. Microglial Cx3cr1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat. Neurosci. 2010;13(4):411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, et al. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19(4):939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Ly PTT, Cai F, Song W. Detection of neuritic plaques in Alzheimer's disease mouse model. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Sadowski M, Pankiewicz J, et al. Targeting prion amyloid deposits in vivo. J. Neuropathol. Exp. Neurol. 2004;63(7):775–784. doi: 10.1093/jnen/63.7.775. [DOI] [PubMed] [Google Scholar]
- Bussière T, Bard F, et al. Morphological characterization of Thioflavin-S-positive amyloid plaques in transgenic Alzheimer mice and effect of passive Abeta immunotherapy on their clearance. Am. J. Pathol. 2004;165(3):987–995. doi: 10.1016/s0002-9440(10)63360-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamohamedsait HB, Sigurdsson EM. Histological staining of amyloid and pre-amyloid peptides and proteins in mouse tissue. Methods Mol. Biol. 2012;849:411–424. doi: 10.1007/978-1-61779-551-0_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afkhami A, Moosavi R. Adsorptive removal of Congo red, a carcinogenic textile dye, from aqueous solutions by maghemite nanoparticles. J. Hazard. Mater. 2010. pp. 398–403. [DOI] [PubMed]
- Tremblay M-E, Riad M, Majewska A. Preparation of mouse brain tissue for immunoelectron microscopy. J Vis Exp. 2010. [DOI] [PMC free article] [PubMed]
- Konsman J-P. Psychoneuroendocrinology. 6. Vol. 28. New York: Academic Press; 2003. The mouse brain in stereotaxic coordinates. [Google Scholar]
- Norden DM, Muccigrosso MM, Godbout JP. Microglial priming and enhanced reactivity to secondary insult in aging, and traumatic CNS injury, and neurodegenerative disease. Neuropharmacology. 2014;96((Pt A)):29–41. doi: 10.1016/j.neuropharm.2014.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay M-E, Stevens B, Sierra A, Wake H, Bessis A, Nimmerjahn A. The role of microglia in the healthy brain. J. Neurosci. 2011;31(45):16064–16069. doi: 10.1523/JNEUROSCI.4158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šišková Z, Tremblay Mè. Microglia and synapse: Interactions in health and neurodegeneration. Neural Plast. 2013;2013:425845. doi: 10.1155/2013/425845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeKosky ST, Scheff SW, Styren SD. Structural correlates of cognition in dementia: quantification and assessment of synapse change. Neurodegeneration. 1996;5(4):417–421. doi: 10.1006/neur.1996.0056. [DOI] [PubMed] [Google Scholar]
- Terry RD, Masliah E, et al. Physical basis of cognitive alterations in Alzheimer's disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991;30(4):572–580. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]