

Abstract
This paper describes how to use a custom manufactured, commercially available enclosed cell culture system for basic and preclinical research. Biosafety cabinets (BSCs) and incubators have long been the standard for culturing and expanding cell lines for basic and preclinical research. However, as the focus of many stem cell laboratories shifts from basic research to clinical translation, additional requirements are needed of the cell culturing system. All processes must be well documented and have exceptional requirements for sterility and reproducibility. In traditional incubators, gas concentrations and temperatures widely fluctuate anytime the cells are removed for feeding, passaging, or other manipulations. Such interruptions contribute to an environment that is not the standard for cGMP and GLP guidelines. These interruptions must be minimized especially when cells are utilized for therapeutic purposes. The motivation to move from the standard BSC and incubator system to a closed system is that such interruptions can be made negligible. Closed systems provide a work space to feed and manipulate cell cultures and maintain them in a controlled environment where temperature and gas concentrations are consistent. This way, pluripotent and multipotent stem cells can be maintained at optimum health from the moment of their derivation all the way to their eventual use in therapy.
Keywords: Developmental Biology, Issue 112, Transduction, stem cells, sendai virus, hypoxic cell culture, sterility, closed-hood cell culture incubation, clinical cell production facility, good manufacturing practices
Introduction
Standard stem cell culture techniques suffer from several environmental constraints that place undue stresses on the cells and expose the cells to unacceptable risks of contamination. Among the stresses that cells may endure under standard cell culture conditions are precipitous changes in the levels of carbon dioxide and oxygen concentrations3,4. This occurs when the cells are moved from the incubator to the biosafety cabinet and/or microscope which may not be optimal for the cells. Previous studies have confirmed the advantages of culturing both pluripotent and neural stem cells in hypoxic conditions4,11, and for best results, these conditions need to be continuous. Moreover, risks of cellular contamination are higher as the laboratory environment and personnel impinge upon the cells at almost every step of their culture and manipulation. Traditional clean rooms comprise one effective method to greatly decrease contamination risks but they are expensive, have a large footprint and fail to address stressors related to carbon dioxide and oxygen concentrations.
A cell production facility that can address both contamination risks and gas concentrations and that can be qualified to meet cGMP criteria9 provides high quality cells for basic science research as well as clinical applications1,6,7. Such a cell production facility consists, at a minimum, of the following components: a process chamber, which acts as a heated workspace for the feeding and manipulation of cell cultures; a laminar flow hood, for the initial sterilization of reagents, tubes, and tools; two buffering airlock chambers in between the hood and the process chamber; two cell culture incubators accessible from the process chamber; a microscope chamber adjacent to the process chamber; and finally, computer software to set and monitor the conditions within these modules. Using this basic infrastructure, a wide variety of tasks can be performed, such as standard feeding and passaging of pluripotent stem cells and multipotent neural stem cells, as well as more specialized methods like Sendai virus-based reprogramming, in vitro migration studies, and differentiation of neural stem cells for electrophysiological characterization.
Protocol
1. Initial Setup
- Setting Gas and Temperature Concentrations
- Using the software, click the "Guest" tab located on the upper left corner of the graphical interface. Login to the system using a designated username and password. Make sure each user has their own user name and password.
- Click on a module to adjust (Figure 1). Within the new window displaying the current settings, click on the existing O2 set point value below "Set point" and enter the required O2 concentration level for this module. Enter 5% O2 if pluripotent stem cells will be grown or manipulated in this module, and 9% O2 if neural stem cells will be grown or manipulated. Click on the green check mark to confirm the set point. Note: Two modules are not gas-adjustable: the laminar hood, and the microscope chamber. The gas concentrations in the laminar flow hood are atmospheric while those in the microscope chamber are passively maintained by the gas concentrations in the process chamber.
- Repeat this step for CO2. Enter a set point of 5% CO2 for all modules except for the buffer chambers, which are only adjustable for O2.
- Monitor the current gas concentrations, which are labeled as "process values", to make sure that they reach the new set points.
- Adjust the gas set points of all modules to the appropriate values. Match the CO2 and O2 levels of chambers that will be exposed to one another. For example, adjust the process chamber to 5% O2 before opening an incubator that grows cells at 5% O2. Also adjust to 5% O2 any buffer chamber that is used to add items to the process chamber during this time.
- Set the temperature of the process chamber to 37 °C using the same screen for gas adjustments. Also set the process chamber floor temperature to 37 °C.
- Click on the incubation module banks underneath each incubator. Adjust the temperature of the banks to 37 °C.
- Operation of Buffer Chambers
- Gather all the supplies required for the given task (feeding, splitting, staining, etc.) in the beginning to prevent a delay in the work flow. Liberally spray all materials with 70% ethanol and allow them to dry in the laminar hood. Do not spray flasks or plates of cells - wipe them gently with a sterile gauze sponge saturated with 70% ethanol.
- Make sure both the outer and inner doors of the buffer chamber are securely closed. Then open the outside door, and place the items inside.
- Close the outer door. Within the software, select the buffer module, and click on the dilution factor tab. Enter 1 into the log factor box. Click start. Note: A "dilution factor" is defined to mean that the buffer module's atmosphere will be evacuated and replaced 1 time with clean, HEPA-filtered gas.
- Once the graphical interface has stopped flashing "dilution factor", open the buffer chamber's inner door and bring the materials inside the process chamber. Caution: Do not allow the inner and outer doors to ever be open at the same time and do not open the inner door if the buffer chamber has not undergone a dilution factor.
- In order to remove items from the process chamber, first ensure that the buffer chamber has undergone a dilution factor. Then open the inner door, place the items to be removed inside, and close the inner door. Then open the outer door and remove the items. Note: A dilution factor does not need to be run before opening the outer door.
2. Introducing and Feeding Cells
- Incubator Setup
- Place 3 Petri dishes in the water pan at the base of each incubator. Fill these dishes with sterile water. Do not directly fill the water pan. Maintain the water level in these dishes, as they are critical for maintaining the relative humidity (RH) set point in the incubators.
- Within the graphical interface, click on the incubator. Click on the existing value for relative humidity (RH) under set point and enter 85%. Click on the green check mark to accept this value.
- Preparation of the Cell Production Facility for Cells
- If not already done so, adjust the buffer chambers, the process chamber, and an incubator to the gas set points required for the type of cell being introduced.
- Clean the surface of the process chamber with sterile gauze and a non-flammable disinfectant. Do not use any peracetic acid-based disinfectant, as in a closed system the strong, lingering vapors are toxic to mammalian cells. Instead, use benzalkonium chloride-based products.
- Continue disinfecting. Clean the gloves and surfaces that are commonly touched, such as door handles. Use the disinfectant thoroughly but sparingly, as excess liquid in the process chamber will contribute to humidity, condensation and possible microbial growth.
- Clean the surface of the laminar flow hood and buffer chamber with 70% ethanol, and allow it to dry. Place the flask or plate of cells (in our case, PSCs or NSCs) in the hood, and briefly wipe its exterior surface with a sterile gauze sponge saturated with ethanol.
- Put the flask or plate in the buffer chamber, and run a dilution factor (step 1.2.3).
- Upon completion of the dilution factor, immediately move the flask or plate to the appropriate incubator. As the incubators are at the back of the process chamber, pull a shelf out into the process chamber for cell placement. Avoid opening the incubator doors unnecessarily or for extended periods of time, as the process chamber's air is very dry compared to that of the incubators, and will result in a significant loss of humidity in the incubator.
- Feeding Cell Cultures
- Gather medium components, serological pipettes, an electronic pipettor, gauze and disinfectant. Also gather a waste container for spent cell culture medium, but do not fill it with bleach. Bring the materials into the process chamber through a buffer chamber as described in Section 1.2. Prepare cell culture medium in the process chamber, as previously described9,10 (also see Materials Table)
- Arrange materials in such a manner as to enable an optimal working space. Avoid placing items not being used in the center of the process chamber.
- Allow for the medium to equilibrate with the CO2 and O2 levels present within the system by leaving the media container slightly uncapped. Some PSC medium manufacturers indicate not to warm media at 37 ºC; if this is the case, use a buffer chamber to equilibrate the medium. Allow the medium to equilibrate for 20 min.
- Remove the old medium from wells using an appropriate stereological pipette and an electronic pipettor. For PSCs, remove all but a small remaining amount of the old medium, enough to prevent desiccation during the feeding process. For NSCs, remove half of the old medium. Pipet the waste into the waste container.
- Place the used pipette back into its original wrapper and move it to the side of the process chamber, or into a buffer chamber designated for waste removal. With a fresh sterile serological pipette, add fresh medium to cell culture plates and place the plates back into their designated incubator.
- At the end of feeding, spray gauze with disinfectant and clean the floor and door handles of the process chamber thoroughly. If there are multiple cell lines being grown in the system, sterilize in between feeding each cell line. This helps minimize the risk of cross contamination.
- Upon completion, remove waste or other unneeded items through one of the buffer chambers.
3. Splitting Cell Cultures
- Enzymatic Passaging of PSCs or NSCs
- Gather all items needed for passaging. This includes media, enzyme or non-enzymatic cell dissociation buffer (the latter is for NSCs only), DPBS, plates or flasks, extracellular matrix (ECM), conical tubes, and serological pipettes. Bring them into the system using a buffer chamber.
- Coat fresh plates or flasks with ECM, as previously described9,10. Some ECMs -such as those derived from mouse sarcoma cell lines - are only liquid between 4 and 15 °C, so use a sterilized cold block or gel ice pack to keep the ECM cold while working in the heated process chamber.
- Pipet off the spent medium from the culture and dispose of it in the waste container.
- Rinse the well or flask surface using 1 ml of DPBS/ well and dispose of the DPBS.
- Add 1 - 2 ml of warm enzyme to each well. Only very dense cultures require 2 ml.
- Immediately move the culture dish or flask to the microscope chamber, and observe the culture carefully. Watch for signs of individual cells beginning to loosen off the dish. Do not wait until the cells float into suspension before proceeding to the next step.
- Return the cells to the main process chamber. Using a 5 ml serological pipette, add 4 ml of DPBS for each 1 ml of enzyme, and then forcefully pipet up and down to dislodge the cells from the well surface. If passaging multiple wells within a multiwall plate, add the DPBS to each well before dislodging the cells from the individual wells.
- Transfer the enzyme and PBS cell suspension to an appropriately-sized conical tube.
- If a centrifuge is not available in the cell production facility, tightly seal the conical tube, place it in a buffer chamber and seal the door closed.
- Remove the conical tube from the buffer chamber from the laminar flow side and spin the cells at 100 x g for 5 min at RT.
- Spray the conical tube with 70% ethanol, and bring it into the process chamber using a buffer chamber and dilution factor.
- Pipet off the supernatant, and resuspend the cells in 2 ml of PSC or NSC medium. If passaging PSCs, make sure to include ROCK inhibitor in the medium at a concentration of 10 µM, as previously described9,10.
- Count the cells using a hemocytometer, and determine the number of wells or plates required. Plate PSCs at 5 x 104 - 1 x 105 cells/cm2. Split the NSCs at a 1:2 ratio. Note: NSCs often clump and resist accurate counting in a hemocytometer.
- If cryopreservation of the cells is required, follow the appropriate protocol9,10, but ensure that all supplies and freezing media are prepared in advance and placed inside the process chamber. DMSO is very toxic to cells at 37 ºC, so work very quickly. Keep the isopropanol-jacketed freezing container at RT in the laminar flow hood. Once the cryopreservation vials are filled and sealed, immediately remove the vials from the process chamber by passing them through a buffer chamber.
- Manual Passaging of PSCs
- Confirm that the culture needs to be passaged, as previously described8. If so, prepare fresh plates or flasks coated with extracellular matrix. Then change the medium entirely.
- Clean the microscope chamber with sterile gauze moistened with a sparing amount non-flammable disinfectant - too much will result in excess fogging in the chamber. Only clean the gloves, floor area, and stage. Bring several 200 µl or 1,000 µl individually wrapped pipet tips into the chamber.
- Bring the plate of PSCs into the microscope chamber, remove the lid, and place it face down on the freshly cleaned floor. Leaving the lid up exposes the underside directly to air currents, and to potential contamination.
- Unwrap a pipet tip, and using the microscope to visualize the culture, pick apart colonies that have good morphology using the tip of the pipet. Note: The pipet tip may be attached to a pipet if the individual user finds this more comfortable.
- Replace the lid on the plate, and move the plate back to the process chamber.
- Pipet off excess ECM from the new plate, and transfer the media from the old plate (containing the colony pieces in suspension) to the new plate. If the old plate still contains colonies that are not ready to be picked, feed it as well.
4. Specialized Culture Techniques
- Sendai Transduction of Fibroblasts into PSCs
- Within the cell production facility, expand human skin fibroblasts1 in fibroblast media containing DMEM/F12, 10% FBS, and 20 ng/ml FGF2, at 5% O2. Use 6-well culture dishes coated with 0.1% gelatin.
- Prepare the cells to be transduced by dissociating them with 1 ml of 0.25% trypsin per well. Inactivate the trypsin with 1 ml fibroblast medium. Spin at 200 x g, resuspend the cells in 1 ml of fibroblast medium, and count them using a hemocytometer.
- Resuspend 2.0 x 105 cells in 2 ml fibroblast medium, and add the cell suspension to 1 well of a gelatin-coated 6 well plate (2.1 x 104 cells/cm2). Put the cells back in the incubator.
- Allow 24 hr for the cells to attach to the bottom of the plate.
- Place fresh fibroblast medium inside the process chamber (1 ml of medium per well of cells to be transduced), and allow it to equilibrate to the gases.
- Sterilize a pre-cooled cold block with 70% ethanol and place it in a buffer chamber. Use a gel ice pack with holes cut in it if a commercial cold block is not available. Note: There are 3 - 4 separate Sendai viruses (depending on the version of the transduction kit), which must be thawed quickly, but kept in the cold block once thawed.
- Dip the bottom half of the tubes in a 37 °C water bath for 15 sec (do not submerge the tubes), then immediately clean the tube exteriors with 70% ethanol. Place the tubes on the sterilized cold block in the buffer chamber and run a dilution factor.
- Working quickly, add the necessary volume of each Sendai reprogramming virus to the equilibrated medium2. This volume is determined by three variables - the number of cells, the titer of each virus, and the desired multiplicity of infection for each virus. Consult the kit manual for recommended MOI. The formula is: [(number of cells being transfected) (multiplicity of infection) (1,000)] / (viral titer) = required µl of virus
- Remove the supernatant from the well(s) of cells to be reprogrammed. For each well, add 1 ml of the medium containing the viruses. Gently rock the plate, being careful not to spill media, and place the plate back in the 5% O2 incubator.
- After 2 hr gently rock the plate again and repeat again after another 2 hr.
- After 24 hr (day 1 post-transduction), feed the well with 1 ml of pluripotent stem cell medium. Repeat on day 2, but do not remove medium from the well.
- On days 3 and 4, change half of the medium.
- On day 5 and beyond, change the entire medium.
- When colonies appear, stain the culture using sterile antibodies to confirm that the colonies are indeed pluripotent, as described previously2. As with feeding cells, ensure that the PSC media containing the antibodies has been equilibrated to the gases in the cell production facility. Note: In a successful reprogramming, iPSC colonies should be present approximately two weeks after the transduction1.
- When the colonies have grown large enough, use the microscope to mark and mechanically passage them onto an ECM-coated plate, as described in section 3.2.
- Expand the colonies either manually or enzymatically, as previously described9,10.
5. Cleaning Up After Daily Use
Place all waste materials, including the liquid waste flask, into a sterile buffer chamber. Wipe down the surface of the processing chamber and all the areas that have come in contact by supplies with sterile gauze and non-flammable disinfectant.
Remove all solid waste materials from the buffer chamber and discard in the appropriate waste container.
Discard liquid waste in an appropriate waste container, or simply mix with bleach and pour down the drain. Clean the waste container with soap and a brush. Sterilize the container with 70% ethanol and move it back to the process chamber via a buffer chamber. Note: It is not recommended to use bleach in the waste container while it is inside the system, as bleach fumes may recirculate and harm cell cultures.
For hygiene considerations, wipe down the front plastic surface of the system's processing chamber with 70% ethanol. This helps to clean off sweat and skin oils from the user's forehead.
If condensation has built up inside the process chamber, run a dilution factor to clear the condensation. Allow at least 1 hr for this to complete.
6. Routine Non-daily Tasks
Replenish the water dishes in the cell culture incubators with sterile, distilled water as needed. Top off the dishes at least once a week. However, the evaporation rate will fluctuate based on how often the incubator doors are opened, the volume of liquid in cultures, and the incubator settings.
Once a month, recalibrate the O2 and CO2 settings in all chambers. This requires the use of SPAN (calibration) gas, which contains known concentrations levels of O2 and CO2 as a standard of reference. Make sure that there is adequate SPAN gas supply before starting calibration. The system uses specific gas sensors, located in each module, to detect gas concentrations; thus, the use of high quality SPAN gases ensures that the sensors reliably yield accurate gas concentrations.
Replace the gloves in the process chamber and microscope chamber on a regular basis. Replace the gloves every 2 months, regardless of how often the system is utilized. Before installation, sterilize the new gloves with disinfectant. Run a dilution factor on the process chamber after the gloves have been replaced. Note: When stretched over the cuffs of the system, nitrile gloves have a tendency to develop holes in areas exposed to physical stress and heat. This is normal and unavoidable wear and tear.
Change out the syringe filters in all chambers every 6 months.
Replace or wash the prefilter on the laminar flow hood as soon as it begins to appear dirty.
Refer to the manufacturer's documents regularly for information on the replacement of major parts.
7. Cleaning of the System
In the event of a major contamination event affecting the majority of cell cultures, move any surviving cell cultures out of the system, or (if possible) into an unaffected incubator.
Turn off the gas control for all chambers except that of unaffected incubators.
Remove the flexible front panel of the apparatus, and remove the stainless steel shelves from the affected cell culture incubator. Also remove the water pan.
Autoclave the incubator shelves and water pan, and put them back into the incubator. Wipe the interior surfaces of the incubator with 70% ethanol. Allow the ethanol to evaporate, and close the incubator door.
Wipe the surfaces of the process and microscope chamber with 70% ethanol. Replace the front panel of the apparatus.
Wipe the interior surfaces of the apparatus, including that of the affected incubator, with non-flammable disinfectant. Leave the door of the affected incubator open, and run a dilution factor on the process chamber. Make sure the microscope chamber doors are also open.
For routine cleaning of the cell culture incubators, ensure that the incubator and process chamber are at the same atmospheric settings. Open the incubator door, and place all cell culture dishes on the surface of the process chamber.
Wipe down the incubator shelves and interior surfaces with non-flammable disinfectant. Allow the disinfectant to evaporate, and move the cell cultures back to the incubator. Work as quickly as possible to avoid dessication of the cell cultures.
Representative Results
This manuscript describes in some detail a cell production facility, and the unique aspects of culturing cells inside a closed system. The bulk of the cell production facility comprises a custom-made, enclosed, cell culture apparatus which has a small footprint and can be easily housed within a 6 x 7.5 m2 room (Figure 2). At no time are the cells within the apparatus exposed to laboratory personnel or environment. The apparatus consists of several modules: a process chamber, a laminar flow hood, 2 buffering airlock chambers in between the hood and the process chamber, two cell culture incubators, and a microscope chamber adjacent to the process chamber (Figure 3). The system is controlled by software allowing environmental variables to be controlled and monitored (Figure 1). The computer running this software is on an uninterruptible power supply. Gases are fed into the system by a set of manifolds for oxygen, nitrogen and carbon dioxide (Figure 4). Gases for the device are supplied by manifold systems that have two sets of tanks each - an active set and a reserve set. When the active set is drawn down, the manifold automatically switches to the reserve set and staff order replacement tanks. The power is on a backup generator system.
Pluripotent and neural stem cells can be grown in hypoxic conditions in this facility using previously developed protocols9, with the added complications inherent to the novel equipment. Pluripotent stem cells can be derived using Sendai virus, using pluripotency-specific antibodies to identify fully transfected colonies, which are then isolated and expanded. (Figure 5A and B), From these iPSCs, neural stem cells and neurons can be derived using dual SMAD inhibition, and their phenotype verified using NSC-specific antibodies (Figure 5C and D).
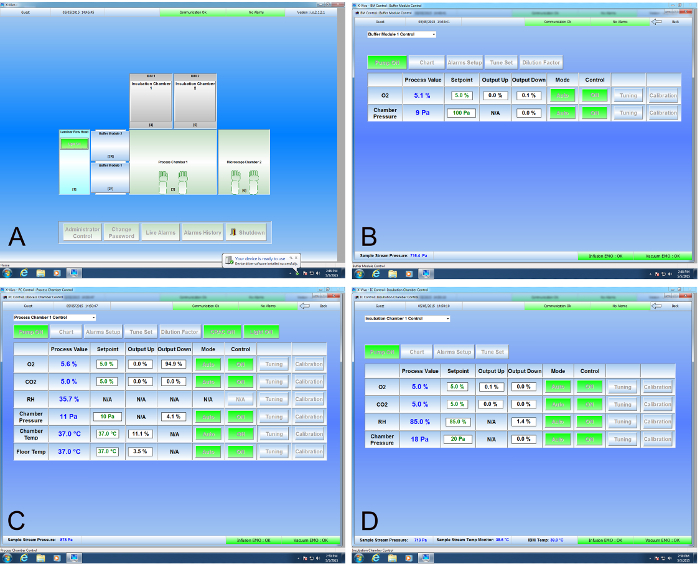 Figure 1. Graphical Interface for the Cell Production Facility. (A) The graphical representation of the CPF is the default screen and matches the CAD drawing shown in Figure 3. A click of the mouse over the Buffer Module 1 (2F in Figure 1) opens its control screen (B) where O2 values are set to match the processing chamber (3 in Figure 3). Similarly, clicking on the Process Chamber or Incubator 1 brings up their respective control screens (C and D, respectively), where values may be changed or monitored as appropriate. Please click here to view a larger version of this figure.
Figure 1. Graphical Interface for the Cell Production Facility. (A) The graphical representation of the CPF is the default screen and matches the CAD drawing shown in Figure 3. A click of the mouse over the Buffer Module 1 (2F in Figure 1) opens its control screen (B) where O2 values are set to match the processing chamber (3 in Figure 3). Similarly, clicking on the Process Chamber or Incubator 1 brings up their respective control screens (C and D, respectively), where values may be changed or monitored as appropriate. Please click here to view a larger version of this figure.
 Figure 2. The Cell Production Facility. (A) The system as seen from a front right view. Note the power and gas connections in the ceiling and the cart with the microscope accessories and computer to the right. (B) The laminar flow hood with the access doors to the buffer modules seen at the right. (C) The process chamber with the two incubators (black doors) seen at the rear. (D) A view through the right side of the system showing the microscope and monitor with the doors to the buffer chambers seen in the distance. Please click here to view a larger version of this figure.
Figure 2. The Cell Production Facility. (A) The system as seen from a front right view. Note the power and gas connections in the ceiling and the cart with the microscope accessories and computer to the right. (B) The laminar flow hood with the access doors to the buffer modules seen at the right. (C) The process chamber with the two incubators (black doors) seen at the rear. (D) A view through the right side of the system showing the microscope and monitor with the doors to the buffer chambers seen in the distance. Please click here to view a larger version of this figure.
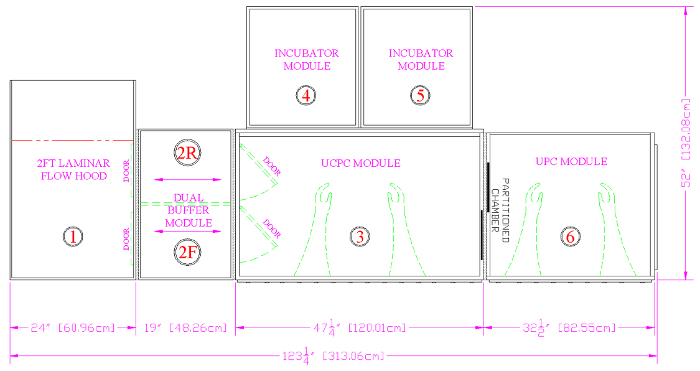 Figure 3. A CAD Drawing of the Cell Production Facility. All items entering the device are first wiped down with alcohol and air dried in the laminar flow hood (1). Items are then transitioned to the appropriate atmosphere in the front buffer module (2F) before being passed into the UCPC module, or Process Chamber (3). Cells are cultured in the two incubators (4, 5). Live cell staining, colony picking, assessment of general cellular morphology and migration analysis takes place in the UPC module, or Microscope Chamber (6). Finally, waste exits the system through the rear buffer module (2R). Please click here to view a larger version of this figure.
Figure 3. A CAD Drawing of the Cell Production Facility. All items entering the device are first wiped down with alcohol and air dried in the laminar flow hood (1). Items are then transitioned to the appropriate atmosphere in the front buffer module (2F) before being passed into the UCPC module, or Process Chamber (3). Cells are cultured in the two incubators (4, 5). Live cell staining, colony picking, assessment of general cellular morphology and migration analysis takes place in the UPC module, or Microscope Chamber (6). Finally, waste exits the system through the rear buffer module (2R). Please click here to view a larger version of this figure.
 Figure 4. Sources of Gases and Power. (A) The CO2 manifold is a 4 x 4 setup as it supplies all the incubators of the laboratory. (B) The O2 manifold is a 2 x 2 setup and supplies only the cell production facility. Nitrogen for the system is generated in a vaporizer (C, just below the Exit sign) attached to a liquid nitrogen manifold (to the right) that also automatically fills cryofreezers (bottom left). The manifold is a 2 x 2 setup being supplied by two pairs of 160 L liquid nitrogen tanks. CO2, O2, and N2 are supplied from the ceiling through shutoffs (D). House vacuum is also supplied at this location. Seen just behind the supply gas shutoffs are a pair of power cables that supply power to the system. Please click here to view a larger version of this figure.
Figure 4. Sources of Gases and Power. (A) The CO2 manifold is a 4 x 4 setup as it supplies all the incubators of the laboratory. (B) The O2 manifold is a 2 x 2 setup and supplies only the cell production facility. Nitrogen for the system is generated in a vaporizer (C, just below the Exit sign) attached to a liquid nitrogen manifold (to the right) that also automatically fills cryofreezers (bottom left). The manifold is a 2 x 2 setup being supplied by two pairs of 160 L liquid nitrogen tanks. CO2, O2, and N2 are supplied from the ceiling through shutoffs (D). House vacuum is also supplied at this location. Seen just behind the supply gas shutoffs are a pair of power cables that supply power to the system. Please click here to view a larger version of this figure.
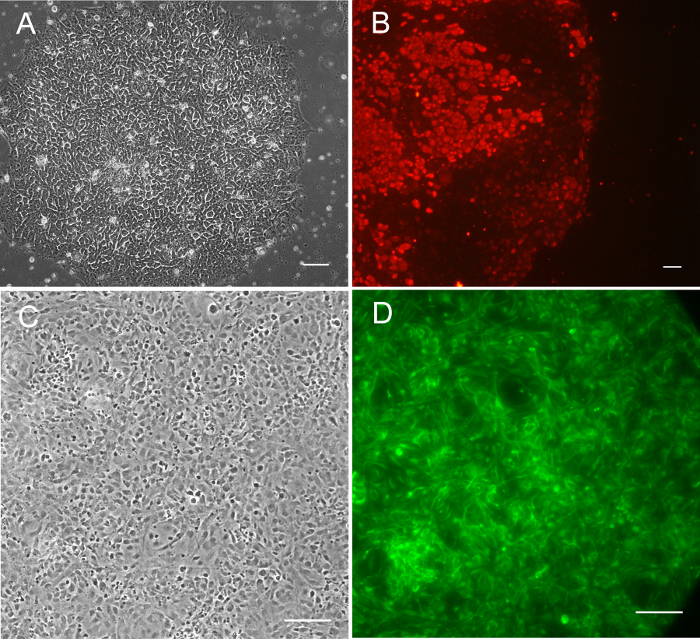 Figure 5. Pluripotent Stem Cells Derived in Hypoxia with Sendai virus. (A) SC187-SF4-2I0-E3 iPSCs iPSCs (nomenclature found in Stover et al.9), phase contrast 10X. (B) SC88.1-UH1-2I0 iPSCs live-stained with AF-594-labeled Tra-1-60, 1:100 dilution in media. All iPSCs were transduced in the cell production facility in 5% O2 with Sendai virus. (C) Phase contrast of SC68.1-UH0-2I0-M0S13-N2G6 iPSC-derived NSCs, grown at 5% O2 in chamber slides. Cells were then fixed in 4% paraformaldehyde and stained with anti-human Nestin primary antibody, and Alex-488 secondary antibody. All scale bars are at 100 µm. Please click here to view a larger version of this figure.
Figure 5. Pluripotent Stem Cells Derived in Hypoxia with Sendai virus. (A) SC187-SF4-2I0-E3 iPSCs iPSCs (nomenclature found in Stover et al.9), phase contrast 10X. (B) SC88.1-UH1-2I0 iPSCs live-stained with AF-594-labeled Tra-1-60, 1:100 dilution in media. All iPSCs were transduced in the cell production facility in 5% O2 with Sendai virus. (C) Phase contrast of SC68.1-UH0-2I0-M0S13-N2G6 iPSC-derived NSCs, grown at 5% O2 in chamber slides. Cells were then fixed in 4% paraformaldehyde and stained with anti-human Nestin primary antibody, and Alex-488 secondary antibody. All scale bars are at 100 µm. Please click here to view a larger version of this figure.
Discussion
Cells grown within the CPF see no changes in oxygen or carbon dioxide concentrations as they move from incubator to processing chamber to microscope chamber and back. It is critical that conditions in each chamber are matched to the particular incubator in which the cells are kept before the cells are removed from the incubator. The atmosphere within the apparatus is continuously HEPA-filtered and is customizable with regard to oxygen and carbon dioxide concentrations. Cells can be grown at standard concentrations for PSCs or NSCs, 5% and 9%, respectively; or alternative concentrations can be chosen for different cell types or for specific experiments. Thus, the apparatus is supplied with constant sources of medical grade oxygen, carbon dioxide, and nitrogen (Figure 4). All three of these gases are supplied by gas-specific manifold systems that ensure constant supplies. The apparatus is also supplied with a calibration gas mixture consisting of 10% (± 0.01%) carbon dioxide in oxygen. The manifold systems are housed outside the cell production facility and the gases are piped into the facility through the ceiling. The calibration gas is housed within the facility. The apparatus is additionally supplied with house vacuum, also through the ceiling. Using an electronic monitoring system and wireless sending units, the output pressures of all manifolds are constantly monitored. In the event that any pressure falls out of range, the cell production facility operators are automatically telephoned and notified such that appropriate action may be taken.
The power requirements of the apparatus are met by six dedicated 120 V circuits descending from the ceiling and connected to the hospital's back-up generators to ensure a constant supply. Operation of the apparatus is controlled via software on a PC-based computer powered through an uninterruptible power supply. These power and computer arrangements ensure that the system functions continuously even in the event of a public power system failure. The software controlling the apparatus has a user-friendly graphical interface (Figure 1) which allows for the control of oxygen and carbon dioxide concentrations as well as temperature, humidity, and chamber pressures. The values of all these parameters are continuously recorded to provide a running record of all apparatus parameters. This data is backed up onto a remote server every night to protect their integrity. The computer and software can be accessed remotely by administrative users to assess and/or change any parameter. Additionally, the computer and software can be accessed remotely, allowing interactive assessment of apparatus parameters and troubleshooting with local users. An additional alarm sending unit is connected to the apparatus such that cell production facility operators are notified of any out-of-range condition of the apparatus. The remote access capabilities allow log in and assessment of the specifics of the out-of-range condition.
The apparatus is designed as a modular system both in a macro and a micro sense. Individual cell culture modules, such as incubators and processing chambers, can be customized in regard to their dimensions and requirements as well as in their layout with respect to each other. Additionally, most of the control functions of the individual modules are themselves modular such that individual atmospheric gas controllers, for example, may be easily replaced without significant disruption to the system.
Specialized processing chambers, such as one for microscopic visualization and manipulation of cell cultures, are easily adapted to the system. Both phase-contrast and fluorescence microscope are inside the system (Figure 6) so that cells can be live stained, and colonies can be dissected in the same atmospheric conditions as inside the incubators. Routing of cables through sealed grommets in the side walls of the processing chamber allows equipment such as power supplies and computers to be kept outside of the apparatus, usually on a cart (Figure 6).
The processing chambers in the cell production facility have a different airflow pattern than conventional BSCs. In conventional BSCs, airflow flows down from a central exhaust vent and splits into two separate streams, which are then taken up by two different intake vents in the forward and aft portion of the cabinet's floor. By contrast, the CPF has a single vent in the forward portion of the ceiling. Air flows downwards and towards the back of the chamber, where it is then drawn upwards into an intake vent. Although the CPF is inherently very clean, this unique airflow pattern means that technicians have to slightly adjust their technique to reduce the risk of contamination. As with a conventional BSC, a lab worker should avoid placing their hands upstream of open cell culture plates and media bottles. However, the direction which is upstream has been altered in the CPF
The cell production facility laboratory itself is fairly standard and comes equipped with a -20 °C freezer, a -80 °C freezer, a 4 °C refrigerator, a centrifuge, and a water bath. The laboratory also has a sink with foot controls for convenient hands-free operation. In order for this laboratory to become a functional clinical cell production facility, however, several additional modifications must still be made. Firstly, the apparatus itself must be upgraded to have the capacity to monitor volatile organic compounds, particulates, and concentrations of chlorine dioxide which is used for decontamination. Secondly, a processing chamber containing a FACS machine may be housed and connected to the rest of the apparatus via a buffer module. This will allow for cell sorting and purification of transplantable cell populations under the appropriate environmental conditions. Lastly, the entire apparatus must be housed within a soft wall clean room. This provides an International Organization for Standardization (ISO) class 8 environment for the apparatus5.
The high sterility and computer-controlled nature of the CPF makes it an ideal system for future applications with cell-based therapy and good manufacturing processes. The risk of contamination is greatly mitigated, but more importantly, the conditions of cell expansion are automatically recorded and archived by the computer system. Deviations in gas concentrations, temperature, humidity, and all events of access into the system are rigorously documented. This can greatly help when investigating product quality problems. However, there are still limitations. The usage of any and all reagents and supplies (e.g., media components, pipettes, plates) must be documented separately. Additionally, there are a multitude of potential problems (including many forms of human error) that can arise which are completely unrelated to the variables documented by the CPF's monitoring system. Thus, the need for highly trained personnel and detailed manual documentation of tasks remains in place.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge the staff at Biospherix for their help in learning to use the Xvivo enclosed cell culture system, especially Matt Freeman; the staff of Miles & Kelley Construction Company, Inc. for their work in setting up the laboratory infrastructure, especially Russ Hughes; the staff of Children's Hospital of Orange County department of Facilities and Support Services for their work in coordinating the laboratory remodel, especially Adam Lukhard and Devin Hugie; the staff of Children's Hospital of Orange County department of Information Systems for their help in setting up the data management infrastructure and remote access, especially Viet Tran; the Children's Hospital of Orange County Executive Management Team for their longstanding support of the project, especially Dr. Maria Minon and Brent Dethlefs. This work was funded by Children's Hospital of Orange County and the California Institute for Regenerative Medicine through grant TR3-05476 to PHS. All authors contributed equally to this work.
References
- Brick DJ, et al. The Autism Spectrum Disorders Stem Cell Resource at Children's Hospital of Orange County: Implications for Disease Modeling and Drug Discovery. Stem Cells Transl.Med. 2014;3(11):1275–1286. doi: 10.5966/sctm.2014-0073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nethercott HE, Brick DJ, Schwartz PH. Derivation of induced pluripotent stem cells by lentiviral transduction. Methods Mol.Biol. 2011;767:67–85. doi: 10.1007/978-1-61779-201-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pistollato F, et al. Hypoxia and HIF1alpha repress the differentiative effects of BMPs in high-grade glioma. Stem Cells. 2009;27(1):7–17. doi: 10.1634/stemcells.2008-0402. [DOI] [PubMed] [Google Scholar]
- Pistollato F, Chen HL, Schwartz PH, Basso G, Panchision DM. Oxygen tension controls the expansion of human CNS precursors and the generation of astrocytes and oligodendrocytes. Mol.Cell Neurosci. 2007;35:424–435. doi: 10.1016/j.mcn.2007.04.003. [DOI] [PubMed] [Google Scholar]
- Raval JS, Koch E, Donnenberg AD. Real-time monitoring of non-viable airborne particles correlates with airborne colonies and represents an acceptable surrogate for daily assessment of cell-processing cleanroom performance. Cytotherapy. 2012;14(9):1144–1150. doi: 10.3109/14653249.2012.698728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PH. The potential of stem cell therapies for neurological diseases. Expert.Rev.Neurother. 2006;6:153–161. doi: 10.1586/14737175.6.2.153. [DOI] [PubMed] [Google Scholar]
- Schwartz PH, Brick DJ. Stem cell therapies for the lysosomal storage diseases - the quintessential neurodegenerative diseases. Curr.Stem Cell Res.Ther. 2008;3(2):88–98. doi: 10.2174/157488808784223041. [DOI] [PubMed] [Google Scholar]
- Schwartz PH, Brick DJ, Nethercott HE, Stover AE. Traditional human embryonic stem cell culture. Methods Mol.Biol. 2011;767:107–123. doi: 10.1007/978-1-61779-201-4_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover AE, et al. Process-based expansion and neural differentiation of human pluripotent stem cells for transplantation and disease modeling. J Neurosci.Res. 2013;91(10):1247–1262. doi: 10.1002/jnr.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover AE, Schwartz PH. Adaptation of human pluripotent stem cells to feeder-free conditions in chemically defined medium with enzymatic single-cell passaging. Methods Mol.Biol. 2011;767:137–146. doi: 10.1007/978-1-61779-201-4_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida Y, Takahashi K, Okita K, Ichisaka T, Yamanaka S. Hypoxia enhances the generation of induced pluripotent stem cells. Cell Stem Cell. 2009;5(3):237–241. doi: 10.1016/j.stem.2009.08.001. [DOI] [PubMed] [Google Scholar]