

Abstract
Surgical parabiosis of two animals of different genetic backgrounds creates a unique scenario to study cell-intrinsic versus cell-extrinsic roles for candidate genes of interest, migratory behaviors of cells, and secreted signals in distinct genetic settings. Because parabiotic animals share a common circulation, any blood or blood-borne factor from one animal will be exchanged with its partner and vice versa. Thus, cells and molecular factors derived from one genetic background can be studied in the context of a second genetic background. Parabiosis of adult mice has been used extensively to research aging, cancer, diabetes, obesity, and brain development. More recently, parabiosis of zebrafish embryos has been used to study the developmental biology of hematopoiesis. In contrast to mice, the transparent nature of zebrafish embryos permits the direct visualization of cells in the parabiotic context, making it a uniquely powerful method for investigating fundamental cellular and molecular mechanisms. The utility of this technique, however, is limited by a steep learning curve for generating the parabiotic zebrafish embryos. This protocol provides a step-by-step method on how to surgically fuse the blastulae of two zebrafish embryos of different genetic backgrounds to investigate the role of candidate genes of interest. In addition, the parabiotic zebrafish embryos are tolerant to heat shock, making temporal control of gene expression possible. This method does not require a sophisticated set-up and has broad applications for studying cell migration, fate specification, and differentiation in vivo during embryonic development.
Keywords: Developmental Biology, Issue 112, Parabiosis, Conjoined Embryos, Zebrafish, Hematopoietic Stem Cell, Zebrafish Chimera, Cell-Intrinsic and Cell-Extrinsic Functions
Introduction
Creation of genetic mosaics (chimeras) between wild-type and genetically modified animals is a well-established and classical strategy for investigating cell-intrinsic versus cell-extrinsic functions of candidate genes1-6. Blastula transplantation in zebrafish has been widely utilized to generate chimeric embryos for studies of cell-autonomy7-9. Depending on the tissue of interest, however, it can be challenging to predictably target donor cells to the desired tissue (e.g., blood) 1-3, 7-9. Mouse geneticists have long utilized parabiotic surgical methods to generate conjoined organisms with a shared circulation 10-14. Because the parabiotic animals share a common bloodstream, interactions between cells that originated from one animal with cells of the other animal of a different genetic background can be studied 10-16. Recently, Karima Kissa's group elegantly demonstrated the ability to create conjoined zebrafish embryos and then use this system to study hematopoietic stem and progenitor cell (HSPC) migration15. In addition, zebrafish parabiosis was recently used to investigate the role of endothelial cadherin 5 in hematopoietic stem cell (HSC) emergence and migration,16 and to study the role of stromal cells in the HSPC niche during zebrafish development 17.
Unlike mice, zebrafish embryos are transparent and allow for direct visualization of cells during parabiotic development, making the system uniquely powerful. The utility of parabiosis in zebrafish, however, is limited by a steep learning curve, and parabiotic surgery on the delicate embryos can be technically challenging without detailed instruction and visual demonstration. The goal of this protocol is to provide a set of clear, step-by-step instructions to accompany video-based tutorials on how to generate parabiotic zebrafish embryos for studying temporal, cell-intrinsic, or cell-extrinsic functions of a candidate gene(s) by surgical fusion of developing blastulae. Key modifications and additional recommendations for increasing parabiont survival and new experimental applications are included.
Protocol
This protocol was approved by Boston Children's Hospital Animal Care and Use Committee. This protocol is modified from a previously published method 15.
1. Preparation of Reagents (Days or Weeks in Advance)
Prepare 1.5% agarose-coated dishes.Add 1.5 g agarose to 100 ml Zebrafish E3 medium in an Erlenmeyer flask. Heat, dissolve the agarose, and then pour approximately 5 ml into 100 mm diameter x 20 mm deep petri dishes. Note: These are used for dechorionation of embryos (Step 3.1) and for the parabiotic surgery (Step 3.3). Store agarose-coated dishes at 4 °C for a few weeks; take them out of the fridge and warm to RT the morning of the day of parabiotic surgery.
- Prepare 4% methyl cellulose.Dissolve 4 g of methyl cellulose powder in 100 ml of E3 in an Erlenmeyer flask with a stir bar. Place the flask on a stir plate at 4 °C, set to the lowest speed, and let it mix for up to 2 days.
- Intermittently use a spatula to break up white clumps of methyl cellulose. The methyl cellulose crystals do not fully dissolve at this concentration. When the solution becomes clear and appears homogenous with no white clumps remaining, aliquot the solution into 1.5 ml microfuge tubes and freeze at -20 °C for long term storage. Note: Lower concentrations of methyl cellulose can be used; however, this higher percentage, more viscous methyl cellulose has yielded the best results.
Prepare High Calcium Ringer (HCR) solution.To make 400 ml of HCR solution, mix 8 ml of 5M NaCl (116 mM final), 400 µl of 3M KCl (2.9 mM final), 800 µl of 5M CaCl2 (10 mM final), and 2ml of 1M HEPES (5 mM final) with 390 ml of E3 media. Note: Store HCR solution at 4 °C for a few weeks; warm to RT the morning of the day of parabiotic surgery.
- Prepare 5 - 6 modified Pasteur pipettes for transferring blastula pairs. Briefly heat the end of the pipette over a Bunsen burner. Then, using large forceps, bend the end of the pipette to approximately 45 °C while it is still hot.
- To ensure there are no sharp edges that might damage embryos, hold the tip of the pipette briefly in the flame for around 3 sec. This will smooth/polish the end of the pipette. This modified glass Pasteur pipette is used in conjunction with a 10 ml pipette pump (Figure 1) in Step 3.5 below.
Prepare glass needle tools for surgical stitching of blastula pairs. Pull 2 - 3 glass needles as done for microinjection of zebrafish embryos. Use lab-film to fix each glass needle to the end of a wood- or plastic-handled teasing needle (Figure 1A). Using forceps, break off the end of the needle at a diameter of approximately 20 - 30 µm. Use a stage micrometer to guide this step (Figure 1B). Note: The size of the needle tip is important. If the tip is too large it becomes difficult to control precisely and can cause excessive damage. If the tip is too small it is difficult to sufficiently wound the embryos.
2. Setting up Breeding Pairs of Zebrafish and Collection of Embryos (Day -1 to Day 0)
Set up breeding pairs of adult zebrafish. Set up fish the night before the parabiosis surgery is to be conducted. Use dividers to separate males and females. To increase the odds that the desired embryos will be obtained, set up several pairs of each line. We find that typically 20 - 50% of the pairs will spawn.
The next morning, pull the dividers and allow mating. Collect any embryos using a fine-mesh tea strainer, then transfer them to a Petri dish containing E3 embryo medium18.
Micro-inject embryos as desired (e.g., with 25 pg plasmid DNA, 200 pg mRNA, or with 1 - 6 ng morpholino) within 1 hr of collection. Allow them to grow until 256-cell stage. If the DNA expression construct is placed under the control of a heat shock promoter (e.g., hsp70, Figure 4B), control the timing of gene expression by heat shocking the parabionts at 37 °C at a later stage (e.g., at 36 hpf). Note: As an alternative to a fluorescent transgene, fluorescent dextran can be added to the DNA, RNA, or morpholino injection mix (e.g., dextran, cascade blue at 2 mg/ml). The dextran dye will not interfere with normal development and will allow for the injected embryo to be identified under the appropriate fluorescent light after parabiotic fusion. Alternatively, the parabiosis of a pigmented strain (e.g., AB) with a non-pigmented strain (e.g., Casper) could be used to identify the injected embryo at later stages.
Incubate the embryos at 28.5 °C.Use a stereoscope to monitor their development periodically as they near the 256-cell developmental stage (approximately 2.5 hpf). Note: If needed, shift embryos to different temperatures to ensure stage matching between parabiotic partners (e.g., morpholino-injected embryos often develop slower than their un-injected counterparts. Shifting the un-injected embryos to RT while the morpholino-injected embryos are placed at 28.5 °C will result in their stages aligning after a couple hours of development).
3. Generation of Parabiotic Zebrafish Embryos by Surgical Fusion of Developing Blastulae
As the embryos approach the 256-cell stage (approximately 2.5 hpf), transfer the embryos from each genetic background (i.e., genetic mutant, transgenic lines and/or genetic manipulation) into separate agarose-coated dishes (e.g., one dish for morpholino-injected embryos and a second dish for control embryos). Alternatively, transfer the embryos to clean, scratch-free glass beakers or glass Petri dishes. Note: Avoid using uncoated plastic dishes as the embryos will stick to the plastic and be damaged once out of their chorions.
- Decant the E3 media until just enough liquid remains to cover the embryos (approximately 7 - 10 ml). Add 200 µl of 50 mg/ml mixture of proteases isolated from the extracellular fluid of Streptomyces griseus (proteases). Swirl the embryos gently every couple of minutes.
- When 50% of the embryos have come out of their chorions, which typically takes 5 - 10 min, rinse them three times with a generous volume (approximately 15 - 20 ml) of fresh E3 embryo medium.
- Dechorionate any embryos remaining in their chorions by gently drawing them up into the modified glass Pasteur pipette and then gently dispelling them. Once the embryos have been dechorionated and washed, replace the E3 media with HCR solution. As an alternative to proteases, remove the chorions manually using a fine forceps. Aim to have 60 - 100 dechorionated embryos available for each experimental condition. Note: This is several fold more than will ultimately be used, but many embryos are likely to be damaged during handling or the surgery for parabiosis. Once the embryos are dechorionated, keep them submerged and do not allow them to touch the surface of the water, as the surface tension will cause them to be destroyed. Keep in mind that using a higher dose of proteases allows the embryos to come out of their chorions sooner and in a more synchronized fashion. When using a lower dose of proteases, the embryos take longer to emerge from their chorions and do so in a less synchronous fashion, which can result in a portion of the embryos being damaged by prolonged exposure to proteases.
- Thaw the methylcellulose (made in Step 1.2 above) and centrifuge in a tabletop centrifuge on max speed for 10 min to pellet undissolved crystals which will damage the embryos or rupture their yolks.
- After centrifugation, use the clear upper fraction (approximately 75% of the volume). Transfer 1 - 1.5 cm diameter drops of methyl cellulose into a 1.5% agarose-coated Petri dish. Take care to avoid drawing up any of the pelleted crystals from the bottom of the tube.
- Use a marker to predetermine the position of each droplet on the bottom side of the dish. Place 12 - 15 drops (2 - 3 aliquots' worth) of methyl cellulose per 100 mm diameter petri dish. Prepare as many plates as needed per experiment. Note: Use each droplet for the fusion of a single blastula pair. The mark will designate the location of each droplet once the media is added-at which point the drops become nearly invisible.
- Prepare one 40 ml aliquot of antibiotic-supplemented HCR solution per agarose-coated dish prepared above in Step 3.3. To each 40 ml aliquot of HCR add penicillin-streptomycin at 50 U/ml, ampicillin at 50 U/ml, and kanamycin at 0.5 µg/ml.
- When ready to perform blastula fusions, carefully fill each of the dishes containing methyl cellulose droplets with 40 ml of the antibiotic-supplemented HCR solution. If the HCR solution is added too quickly, it can disrupt the droplets.
- Attach the modified glass Pasteur pipette (made in Step 1.4 above) to a 10 ml pipette pump. Under a stereoscope collect one dechorionated embryo from each background (e.g., one morpholino-injected embryo and one wild-type embryo).
- Before dispensing the two embryos, use the Pasteur pipette to make a small depression in the middle of the methyl cellulose drop, then gently deposit both embryos into the depression. Note: During the transfer process, turn the Pasteur pipette slightly upwards so that the embryos settle into the bend of the Pasteur pipette so as to avoid their making contact with the surface of the liquid at the top or bottom of the pipette (which can lead to their rupture).
- Immediately after depositing the embryos into the methyl cellulose, use a wood- or plastic-handled teasing needle with a gel-loading pipet tip on the end (Figure 1A) to carefully reposition the embryos within the methyl cellulose such that their animal poles are directly touching one another.
- Utilize gentle sweeping motions through the methyl cellulose close to the embryos to reposition them without directly touching them. Let the embryos sit for 5 min so the methyl cellulose will settle in around them. During this time, load the next pair into another drop. Note: Avoid making direct contact with the embryos and take special care not to touch any part of the yolk sac, which will easily rupture.
Using the glass needle tool, carefully wound each embryo at their point of contact. With a gentle sewing motion, cells from the first embryo can be pulled into the second embryo and then back again. At the end of this motion, hold the needle in place for 2 - 3 sec and then draw it away very slowly. Note: There is a period of about 2 - 3 sec after the embryos are wounded where the two tissues seem to be more adhesive and more likely to attach to one another. Thus, we believe that holding the needle in place briefly after wounding helps to keep the wounded tissues of each embryo in contact for the initial period of time immediately after wounding.
Allow these embryos to sit for 10 - 15 min at RT. In the meantime, continue to stitch new pairs in other drops of methyl cellulose. After 10 - 15 min, assess whether the first pair has remained attached (Figure 2A). Note: If the embryos are longer attached, repeat the stitching process for as long as the embryos are younger than around the 30% epiboly stage. Typically this allows for up to three attempts to stitch the embryos together. If the embryos appear to have maintained an attachment but the connection is not substantial, perform additional stitching at the edge of the connection to reinforce it. Avoid shaking or moving dishes during the parabiotic surgery and incubation periods. Note: Despite their early developmental stage and perceived fragility, the cell masses of the embryos can tolerate a substantial amount of manipulation (i.e., wounding and stitching), and still develop normally. The yolk, however, can rupture with only the slightest touch, killing the embryo. When stitching the two blastulae with the glass needle, do not to make contact with the interface between the cell mass and yolk of each embryo.
Incubate the embryos O/N at 28.5 °C in the same dish and media (do not change the media). By the following morning, the embryos will have moved out of the mostly dissolved methyl cellulose droplets. Remove any embryos that were not successfully fused. Decant the media and replace with a fresh 25 ml E3. Note: If working with a pigmented strain, add 500 µl 0.15% (50x) phenylthiourea (PTU) to a final concentration of 0.003% to block pigmentation. Embryos will need to remain continuously in PTU to maintain suppression of pigmentation.
4. Microscope Setup, Image Acquisition, Processing and Analysis
- Prepare 0.8% low melting point (LMP) agarose: Add 0.8 g LMP agarose to 100 ml E3 and heat until fully dissolved. While hot, aliquot 1 ml of LMP agarose into 1.5 ml microfuge tubes in a 37 °C heating block. The LMP agarose will remain molten at 37 °C. Add 40 µl of 4 mg/ml (25x), pH 7.0 tricaine to each 1 ml aliquot of LMP agarose at 37 °C. Note: If working with a pigmented strain, add 20 µl of 0.15% (50x) PTU to each 1ml aliquot.
- Store the remaining LMP agarose for several months in sealed 50 ml tubes at 4 °C.
Anesthetize parabiotic embryos to be imaged by adding 1 ml of 4 mg/ml (25x), pH 7.0 tricaine to the 25 ml of E3 that the embryos are already in.
- Gently swirl the dish so that the embryos will pool in the middle. Then, using a wide-tipped plastic transfer pipette draw up the embryos in as little liquid as possible. Turn the pipette upright and gently bounce the embryos so that they settle to the very bottom of the pipette.
- With the embryos at the bottom of the pipette, transfer them to a 1 ml aliquot of LMP agarose by lightly touching the pipette tip to the surface of the agarose. Avoid transferring excess liquid as this will dilute the agarose.
After expelling the excess liquid from the pipette, use the pipette to gently mix the embryos within the agarose, then use the pipette to transfer all of the agarose and embryos to the well of a glass-bottom (No. 1.5 coverslip), 6-well plate. Note: Be sure to discard the pipette after transferring the embryos to the imaging plate. Residual agarose will set inside the pipette, rendering it ineffective for further use. Rather, use a new pipette each time.
Under a stereoscope, use a gel-loading pipette tip fixed on the end of a wood-handled teasing needle to position the embryos down towards the cover glass (to ensure they are within the working distance of the microscope objective) and in the desired orientation for imaging. Note: Reposition continuously until the agarose has fully set. Take care to mount the parabiotic embryos according to which embryo of the pair is to be imaged. Given the random orientation of the conjoined embryos, for some pairs it may be difficult to image both embryos. In this scenario, recover the embryos from the agarose using a forceps and a wide-tipped plastic transfer pipette and then remount for imaging from a different perspective.
Once the agarose has set, cover with 2 - 3 ml of E3 supplemented with tricaine (and PTU if necessary).
Image the embryos using an inverted wide-field epi-fluorescence, laser-scanning confocal, or spinning disk confocal microscope. Select the objective most appropriate for the desired imaging. For a whole-embryo field of view, use a 4x objective. Select a higher magnification, such as a 20x objective, to image a specific tissue16, 19. Note: If using an upright microscope, mount in LMP agarose (similar to above) on a glass cover slip. Invert the glass coverslip onto a glass microscope slide that has a ring of vacuum grease filled with the same E3-tricaine-PTU16, 19.
Process acquired images using free image analysis software packages such as NIH Image J/FIJI16, 19. Note: Count cell numbers and quantify dynamics such as cell migration and cell division using segmentation and tracking algorithms16, 19.
Representative Results
Consistent with previously published studies15, successful parabiotic fusion of zebrafish embryos depends on the staging and orientation of the two embryos and the concentration of methylcellulose. With just a few simple, inexpensive tools, surgically fused developing blastulae were generated that grew into parabiotic embryos with shared circulation. These tools included a modified Pasteur pipette, a 10 ml pipette pump, and wood handled teasing needles which were used either alone or with a gel-loading tip or glass microinjection needle fixed to the end by lab-film (Figure 1).
After placing the two blastulae in 4% methyl cellulose and using the gel-loading tip to orient them with their animal poles facing one another (Figure 2A), the embryos were carefully wounded at their point of contact with the glass microinjection needle (Figure 2B). A greater degree of wounding (compare Figure 2B to Figure 2C), and revisiting embryo pairs to bolster an initial connection with additional wounding, increased the likelihood of the embryos maintaining a connection that resulted in successful fusion (Figure 3A-C, Movie 1), and without additional morphological defects or delayed development. When done carefully, nearly 100% of the embryo pairs were fused, although a fraction of these embryos (around 25%) never established healthy circulation in both halves. By orienting the two blastulae with their animal poles directly aligned, reliable head-to-head or yolk sac-to-yolk sac fusions were generated with shared circulation. In most instances, the embryos had two hearts pumping a shared common circulation (Figure 3D).
To confirm that the embryos indeed shared their circulation, fluorescent dextran was injected into circulation, which could be seen subsequently circulating through both embryos (Movie 2). Additionally, transgenic embryos that had GFP+ erythrocytes (lcr:eGFP) were fused to embryos that had mCherry+ vascular endothelial cells (flk1:HRAS-mCherry). By 48 hpf, GFP+ erythrocytes were observed circulating through the flk1:HRAS-mCherry partner embryo (Figure 4A and Movie 3). To expand the utility of the parabiotic system, temporal control of gene expression was added. Prior to fusion, one of the two embryos was injected with an hsp70:eGFP DNA construct 20. While the fused embryos were growing at 28.5 °C, no fluorescence was observed. In contrast, after a brief 30 min heat shock at 37 °C, a clear GFP signal was visible in one of the two embryos (Figure 4B). In some instances GFP+ cells were observed circulating in the un-injected partner embryo. Thus, placing a gene of interest under the control of a heat shock promoter provides additional temporal control to studies investigating cell-intrinsic or cell-extrinsic functions of candidate genes.
 Figure 1. Tools for Generating Parabiotic Zebrafish Embryos.(A) Annotated image shows tools used for surgical fusion of developing blastulae. Tools include a modified Pasteur pipette (top), which is used in conjunction with a 10 ml pipette pump (green). Wood handled teasing needles are used either alone or with a plastic gel-loading pipette tip or pulled glass microinjection needled fixed to the end with lab-film. (B) Image shows the ends of three glass microinjection needles that have been broken with a forceps at different diameters. The needles are lying on top of a micrometer; the smallest lines are spaced 10 µm apart. Please click here to view a larger version of this figure.
Figure 1. Tools for Generating Parabiotic Zebrafish Embryos.(A) Annotated image shows tools used for surgical fusion of developing blastulae. Tools include a modified Pasteur pipette (top), which is used in conjunction with a 10 ml pipette pump (green). Wood handled teasing needles are used either alone or with a plastic gel-loading pipette tip or pulled glass microinjection needled fixed to the end with lab-film. (B) Image shows the ends of three glass microinjection needles that have been broken with a forceps at different diameters. The needles are lying on top of a micrometer; the smallest lines are spaced 10 µm apart. Please click here to view a larger version of this figure.
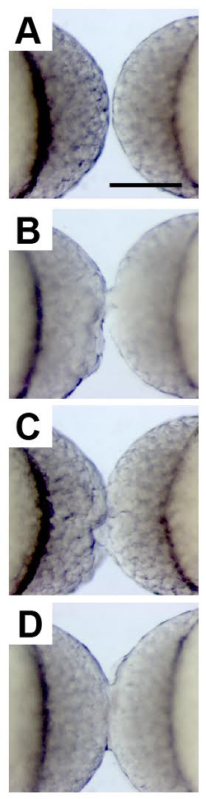 Figure 2. Surgical Fusion of Developing Blastulae. High-magnification images show two zebrafish embryos at the high stage of development, oriented with their animal poles facing one another, prior to surgical stitching (A), immediately after minimal wounding (B), and after more substantial wounding (C). 20 min later it is apparent that the embryos have been successfully fused based on the uninterrupted bridge of cells between the two blastulae (D). Scale bar represents 250 µm. Please click here to view a larger version of this figure.
Figure 2. Surgical Fusion of Developing Blastulae. High-magnification images show two zebrafish embryos at the high stage of development, oriented with their animal poles facing one another, prior to surgical stitching (A), immediately after minimal wounding (B), and after more substantial wounding (C). 20 min later it is apparent that the embryos have been successfully fused based on the uninterrupted bridge of cells between the two blastulae (D). Scale bar represents 250 µm. Please click here to view a larger version of this figure.
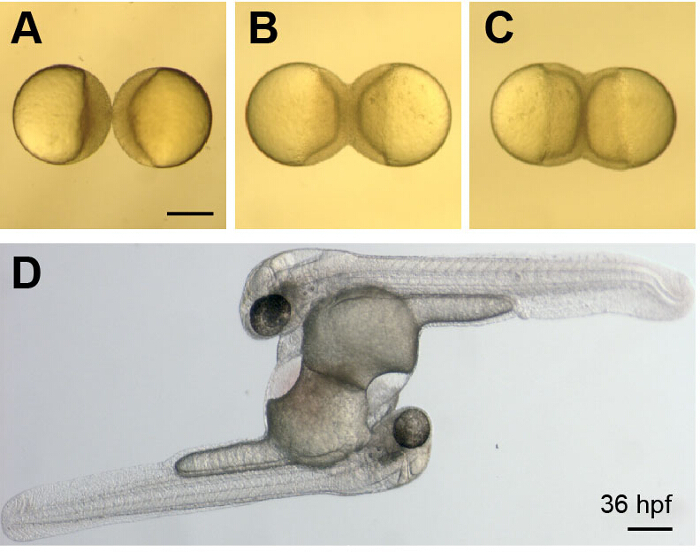 Figure 3. Development of Parabiotic Zebrafish Embryos.(A-C) Images show a whole-parabiont view of early development just after wounding (A) and as the embryos continue to develop and undergo epiboly (B and C) These images correspond to Movie 1. (D) Image shows a parabiotic embryo pair at 36 hpf. The embryos are connected at their yolk sacs, have two hearts and share a common circulation. Scale bars represent 250 µm. Please click here to view a larger version of this figure.
Figure 3. Development of Parabiotic Zebrafish Embryos.(A-C) Images show a whole-parabiont view of early development just after wounding (A) and as the embryos continue to develop and undergo epiboly (B and C) These images correspond to Movie 1. (D) Image shows a parabiotic embryo pair at 36 hpf. The embryos are connected at their yolk sacs, have two hearts and share a common circulation. Scale bars represent 250 µm. Please click here to view a larger version of this figure.
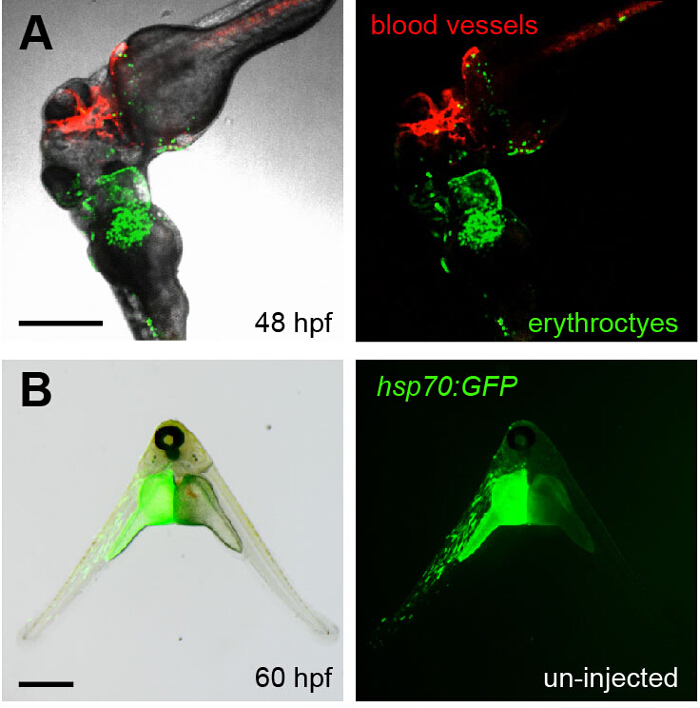 Figure 4. Visualizing Parabiosis with Genetically Encoded Fluorescent Proteins.(A) Images (fluorescence alone, right; overlay with transmitted light, left) show the head region of conjoined embryos at 48 hpf. The bottom embryo of this pair is transgenic for lcr:eGFP (erythroctyes, green) while its partner (top) is transgenic for flk1:HRAS-mCherry, which labels vascular endothelial cells (red). Green erythrocytes can be seen circulating through the partner embryo. This image corresponds to Movie 3. (B) The left embryo in this pair was injected with an hsp70:eGFP construct at the one-cell stage and then fused to an un-injected partner (right). When the animals were heat shocked at 37 °C for 30 min at 36 hpf, the green fluorescence of GFP was observed in the left embryo at 60 hpf. Scale bars represent 500 µm. Please click here to view a larger version of this figure.
Figure 4. Visualizing Parabiosis with Genetically Encoded Fluorescent Proteins.(A) Images (fluorescence alone, right; overlay with transmitted light, left) show the head region of conjoined embryos at 48 hpf. The bottom embryo of this pair is transgenic for lcr:eGFP (erythroctyes, green) while its partner (top) is transgenic for flk1:HRAS-mCherry, which labels vascular endothelial cells (red). Green erythrocytes can be seen circulating through the partner embryo. This image corresponds to Movie 3. (B) The left embryo in this pair was injected with an hsp70:eGFP construct at the one-cell stage and then fused to an un-injected partner (right). When the animals were heat shocked at 37 °C for 30 min at 36 hpf, the green fluorescence of GFP was observed in the left embryo at 60 hpf. Scale bars represent 500 µm. Please click here to view a larger version of this figure.
 Movie 1. Early development of surgically fused zebrafish blastulae.(Right click to download). Movie shows the early development of a pair of surgically fused zebrafish blastulae. Epiboly can be seen occurring simultaneously in each embryo. This movie is related to Figure 3A-C.
Movie 1. Early development of surgically fused zebrafish blastulae.(Right click to download). Movie shows the early development of a pair of surgically fused zebrafish blastulae. Epiboly can be seen occurring simultaneously in each embryo. This movie is related to Figure 3A-C.
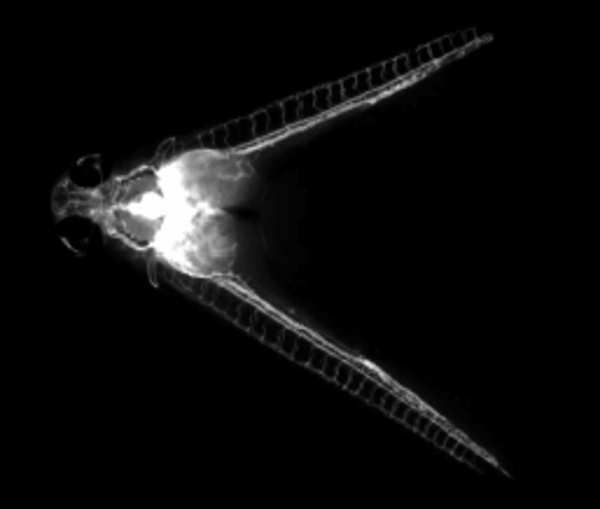 Movie 2. Fluorescent dextran injection illuminates a shared circulation.(Right click to download). Movie shows fluorescent dextran being injected into the common cardinal vein of a parabiotic embryo pair at 60 hpf. The fluorescent dye can be seen subsequently circulating through both embryos.
Movie 2. Fluorescent dextran injection illuminates a shared circulation.(Right click to download). Movie shows fluorescent dextran being injected into the common cardinal vein of a parabiotic embryo pair at 60 hpf. The fluorescent dye can be seen subsequently circulating through both embryos.
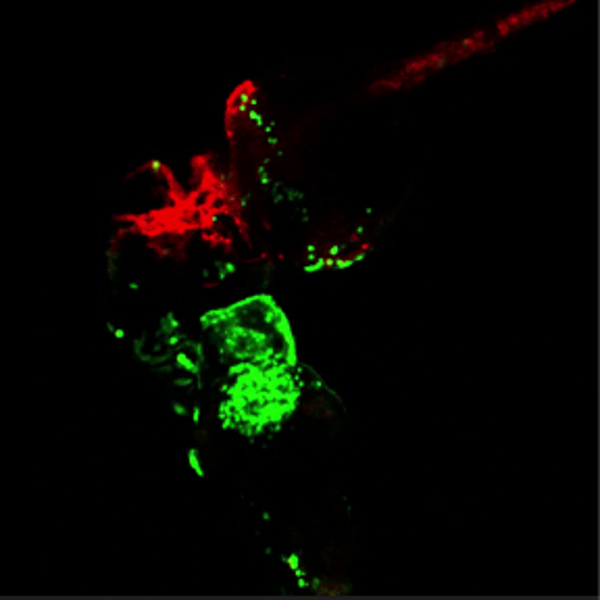 Movie 3. Green blood flowing through red vessels.(Right click to download). Movie shows the head region of conjoined embryos at 48 hpf. The bottom embryo of this pair is transgenic for lcr:eGFP (erythroctyes, green) while its partner (top) is transgenic for flk1:HRAS-mCherry, which labels vascular endothelial cells (red). Green erythrocytes are seen circulating through the partner embryo. This movie is related to Figure 4A.
Movie 3. Green blood flowing through red vessels.(Right click to download). Movie shows the head region of conjoined embryos at 48 hpf. The bottom embryo of this pair is transgenic for lcr:eGFP (erythroctyes, green) while its partner (top) is transgenic for flk1:HRAS-mCherry, which labels vascular endothelial cells (red). Green erythrocytes are seen circulating through the partner embryo. This movie is related to Figure 4A.
Discussion
Parabiotic fusion has been a powerful tool to investigate cellular functions of candidate genes in adult murine models and chick embryos10-14. More recently, a blastula fusion method has been described for generating conjoined zebrafish embryos15. In the present protocol, video-based tutorials are used to demonstrate and better describe the methodology for creating parabiotic zebrafish embryos of different genetic backgrounds in order to study temporal, cell-intrinsic, and cell-extrinsic roles of candidate genes of interest in hematopoietic development and beyond.
The method described in this paper was recently used to track HSC emergence in one embryo, migration via blood-circulation to an embryo of different genetic background, and subsequent engraftment and differentiation 16. In agreement with the Kissa group’s findings, staging and embryo positioning were found to determine the success rate and anatomical nature of the parabiotic fusion. With a few key modifications to the protocol, parabionts were generated with a significantly higher survival rate. These include wounding the blastulae to a greater degree, holding the needle in place for 2 - 3 sec after wounding, and then revisiting blastula pairs after 15 min to reinforce an initial connection with additional wounding, if necessary. With these additional recommendations, nearly 100% of the parabionts survived and remained attached, and 75% of them shared a common circulation.
To incorporate an additional element of experimental control to the method, parabiotic embryos are demonstrated here to be tolerant to heat shock, allowing for temporal control of gene expression within the parabiotic context 20. A limitation of this technique is that in some instances the parabiotic partner embryos are not in the same plane after parabiotic fusion. This can make it difficult to mount embryos in LMP agarose to track cells in both embryos simultaneously using time-lapse microscopy. To circumvent this potential issue, one should plan to generate multiple parabiotic embryos to ensure the desired orientation is achieved. Alternatively, embryos can be recovered from the LMP using a fine forceps and Pasteur pipette and re-mounted for imaging from a different perspective. The goal of this protocol was to provide a set of clear, concise instructions to accompany video-based tutorials on how to surgically fuse developing blastulae to generate parabiotic zebrafish embryos.
With the key modifications and additional recommendations for increasing parabiont survival, this protocol will hopefully empower investigators who wish to utilize conjoined embryos to investigate factors regulating cell migration, fate specification, and differentiation in a range of tissues and cellular contexts.
Disclosures
The authors declare that they have no conflict of interest.
Acknowledgments
We thank Julie R. Perlin for helpful comments on the manuscript. D.I.S. is supported by grants from the American Society of Hematology, the Cooley's Anemia Foundation, and the NIH (K01DK085217 and R03DK100672). E.J.H. is a Howard Hughes Medical Institute Fellow of the Helen Hay Whitney Foundation. B.L. is a Howard Hughes Medical Institute Medical Research Fellow. B.W.B. is supported by an Irvington Fellowship from the Cancer Research Institute and a Young Investigator Award from the Conquer Cancer Foundation of ASCO. L.I.Z. is supported by grants from the NIH (R01CA103846, P01HL032262, and R01HL04880), Taub Foundation for MDS Research, Harvard Stem Cell Institute, and is an investigator of the Howard Hughes Medical Institute.
References
- Zon LI, Orkin SH. Hematopoiesis: An evolving paradigm for stem cell biology. Cell. 2008;132(4):631–644. doi: 10.1016/j.cell.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzierzak E, Speck NA. Of lineage and legacy: the development of mammalian hematopoietic stem cells. Nat Immunol. 2008;9(2):129–136. doi: 10.1038/ni1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, et al. Epoxyeicosatrienoic acids enhance embryonic haematopoiesis and adult marrow engraftment. Nature. 2015;523(7561):468–471. doi: 10.1038/nature14569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tam PP, Rossant J. Mouse embryonic chimeras: tools for studying mammalian development. Development. 2003;130(25):6155–6163. doi: 10.1242/dev.00893. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Gertsenstein M, Rossant J, Nagy A. Mash2 acts cell autonomously in mouse spongiotrophoblast development. Dev. Biol. 1997;190(1):55–65. doi: 10.1006/dbio.1997.8685. [DOI] [PubMed] [Google Scholar]
- Morin-Kensicki EM, Faust C, LaMantia C, Magnuson T. Cell and tissue requirements for the gene eed during mouse gastrulation and organogenesis. Genesis. 2001;31(4):142–146. doi: 10.1002/gene.10017. [DOI] [PubMed] [Google Scholar]
- Ho RK, Kane DA. Cell-autonomous action of zebrafish spt-1 mutation in specific mesodermal precursors. Nature. 1999;348(6303):728–730. doi: 10.1038/348728a0. [DOI] [PubMed] [Google Scholar]
- Parker L, Stainier DY. Cell-autonomous and non-autonomous requirements for the zebrafish gene cloche in hematopoiesis. Development. 1999;126(12):2643–2651. doi: 10.1242/dev.126.12.2643. [DOI] [PubMed] [Google Scholar]
- Liao EC, Trede NS, Ransom D, Zapata A, Kieran M, Zon LI. Non-cell autonomous requirement for the bloodless gene in primitive hematopoiesis of zebrafish. Development. 2002;3(3):649–659. doi: 10.1242/dev.129.3.649. [DOI] [PubMed] [Google Scholar]
- Kamran P, Sereti KI, Zhao P, Ali SR, Weissman IL, Ardehali R. Parabiosis in mice: a detailed protocol. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Wagers AJ, Sherwood RI, Christensen JL, Weissman IL. Little evidence for developmental plasticity of adult hematopoietic stem cells. Science. 2002;297(5590):2256–2259. doi: 10.1126/science.1074807. [DOI] [PubMed] [Google Scholar]
- Hervey GR. The effects of lesions in the hypothalamus in parabiotic rats. J. Physiol. 1959;145(2):336–352. doi: 10.1113/jphysiol.1959.sp006145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman DC, Bailey AS, Pfaffle DL, Al Masri A, Christian JL, Fleming WH. BMP4 regulates the hematopoietic stem cell niche. Blood. 2009;114(20):4393–4401. doi: 10.1182/blood-2009-02-206433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieterlen-Lièvre F, Martin C, Beaupain D. Quail-chick chimaeras and parabionts: several new models to investigate early developmental events in the haemopoietic system. Folia Biol (Praha) 1979;25(5):293–295. [PubMed] [Google Scholar]
- Demy DL, Ranta Z, Giorgi JM, Gonzalez M, Herbomel P, Kissa K. Generating parabiotic zebrafish embryos for cell migration and homing studies) Nat. Methods. 2013;10(3):256–258. doi: 10.1038/nmeth.2362. [DOI] [PubMed] [Google Scholar]
- Anderson H, et al. Hematopoietic stem cells develop the absence of endothelial cadherin 5 expression. Blood. 2015;126(26):2811–2820. doi: 10.1182/blood-2015-07-659276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama E, Sarris M, Redd M, Le Guyader D, Vivier C, Horsley W, Trede N, Herbomel P. NACA deficiency reveals the crucial role of somite-derived stromal cells in haematopoietic niche formation. Nat Commun. 2015;28(6):8375. doi: 10.1038/ncomms9375. [DOI] [PubMed] [Google Scholar]
- Shah DI, et al. Mitochondrial Atpif1 regulates haem synthesis in developing erythroblasts. Nature. 2012;491(7425):608–612. doi: 10.1038/nature11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamplin OJ, et al. Hematopoietic stem cell arrival triggers dynamic remodeling of the perivascular niche. Cell. 2015;160(1-2):241–252. doi: 10.1016/j.cell.2014.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adám A, Bártfai R, Lele Z, Krone PH, Orbán L. Heat-inducible expression of a reporter gene detected by transient assay in zebrafish. Exp. Cell Res. 2000;256(1):282–290. doi: 10.1006/excr.2000.4805. [DOI] [PubMed] [Google Scholar]