

Abstract
Multi isolate whole genome sequencing (WGS) and typing for outbreak investigations has become a reality in the post-genomics era. We applied this technology to strains from Escherichia coli O157:H7 outbreaks. These include isolates from seven North America outbreaks, as well as multiple isolates from the same patient and from different infected individuals in the same household. Customized high-resolution bioinformatics sequence typing strategies were developed to assess the core genome and mobilome plasticity. Sequence typing was performed using an in-house single nucleotide polymorphism (SNP) discovery and validation pipeline. Discriminatory power becomes of particular importance for the investigation of isolates from outbreaks in which macrogenomic techniques such as pulse-field gel electrophoresis or multiple locus variable number tandem repeat analysis do not differentiate closely related organisms. We also characterized differences in the phage inventory, allowing us to identify plasticity among outbreak strains that is not detectable at the core genome level. Our comprehensive analysis of the mobilome identified multiple plasmids that have not previously been associated with this lineage. Applied phylogenomics approaches provide strong molecular evidence for exceptionally little heterogeneity of strains within outbreaks and demonstrate the value of intra-cluster comparisons, rather than basing the analysis on archetypal reference strains. Next generation sequencing and whole genome typing strategies provide the technological foundation for genomic epidemiology outbreak investigation utilizing its significantly higher sample throughput, cost efficiency, and phylogenetic relatedness accuracy. These phylogenomics approaches have major public health relevance in translating information from the sequence-based survey to support timely and informed countermeasures. Polymorphisms identified in this work offer robust phylogenetic signals that index both short- and long-term evolution and can complement currently employed typing schemes for outbreak ex- and inclusion, diagnostics, surveillance, and forensic studies.
Keywords: Escherichia coli, O157:H7, EHEC, phylogenomics, outbreaks, single nucleotide polymorphism, genomic epidemiology, whole genome sequence typing
Introduction
Microbial pathogens with a foodborne etiology present major challenges to public health. Escherichia coli has been divided into different pathovars based on key virulence factors that define their pathogenicity (Sadiq et al., 2014). One particularly daunting pathovar among the Shiga toxin producing E. coli (STEC) are strains of the enterohemorrhagic O157:H7 serotype, which can be transmitted by a variety of vehicles, and causes serious human disease (Tarr et al., 2005). Currently, there is no effective treatment or prophylaxis for hemolytic uremic syndrome (HUS) (Goldwater and Bettelheim, 2012), and use of antibiotics is not indicated (Freedman et al., 2016). Since its discovery in 1982, this lineage has rapidly evolved from a rare serotype into the now globally dominant enterohemorrhagic E. coli (EHEC) serotype. A remarkable feature is its low infectious dose; it is estimated that 10–100 colony-forming units (CFUs) are sufficient to cause disease (Tilden et al., 1996; Tuttle et al., 1999) For the above reasons, prevention of human infection is critical, and early identification of outbreaks is highly worthwhile. However, only rudimentary information exists regarding the genomic heterogeneity that can be expected within outbreaks (STEC Outbreaks). Moreover, current typing schemes, such as pulse field gel electrophoresis (PFGE) and multiple locus variable number of tandem repeats analysis (MLVA), often lack the resolution to differentiate organisms that form tightly clonal phylogenetic clusters within the O157:H7 clade (Eppinger et al., 2011b; Turabelidze et al., 2013; Underwood et al., 2013; Rusconi and Eppinger, 2016). Additionally, PFGE is subject to technological and interpretation challenges (Davis et al., 2003).
Increasing technologic economies offer new opportunities for sequence-based typing of microbial pathogens for public health purposes (den Bakker et al., 2014; Joensen et al., 2014; Leekitcharoenphon et al., 2014; Holmes et al., 2015). While it would be ideal to refer a clinical strain's sequence to a reference, of the 445 publicly available genomes of E. coli O157:H7 and its close relative O55:H7 (O157:H7 Genomes) (Kulasekara et al., 2009; Zhou et al., 2010; Eppinger et al., 2011a, 2013; Sanjar et al., 2014, 2015), to date only 11 have been closed (Hayashi et al., 2001; Perna et al., 2001; Kulasekara et al., 2009; Zhou et al., 2010; Eppinger et al., 2011b, 2013; Kyle et al., 2012; Xiong et al., 2012; Latif et al., 2014; Sanjar et al., 2014, 2015; Cote et al., 2015). Whole genome sequencing (WGS) can provide the necessary resolution power to investigate apparent single source outbreaks (Eppinger et al., 2011b; Hasan et al., 2012; Turabelidze et al., 2013) because the granularity of WGS data provides considerable confidence in assigning like vs. not-like status to two potentially linked pathogens (Gilchrist et al., 2015). Such data can also link pathogens to vehicles or environmental isolates most precisely (Bentley and Parkhill, 2015). WGS can offer additional advantages: serotypes and virulence loci within pathogens can be identified (Scheutz et al., 2012; Leekitcharoenphon et al., 2014; Lambert et al., 2015; Klemm and Dougan, 2016), and case management might theoretically be risk-optimized.
Optimization of E. coli O157:H7 sequence analysis methodologies depend on the scientific and epidemiologic inquiries and the data being analyzed. Pettengill et al. evaluated a number of single nucleotide polymorphism (SNP) predicting tools and phylogenetic methodologies in prokaryotes and concluded that a reference-based approach, which accommodates missing data as well as infers phylogenetic reconstruction, is the most appropriate (Pettengill et al., 2014). Such a reference-based approach was recently used by the Alberta Provincial Laboratory for Public Health to study E. coli O157:H7 outbreaks together with virulence profiling and other molecular methods (Berenger et al., 2015). No specific virulence pattern distinguished the outbreak strains from sporadic strains (Berenger et al., 2015). Recent studies have expanded WGS typing to globally distributed strains and identified geographical genomic structuring based on distribution of stx-converting phage integration sites and SNPs (Mellor et al., 2015; Strachan et al., 2015) and provided a more detailed subtyping of E. coli O157:H7 (Griffing et al., 2015). However, clarity can also be gained by comparing closely related isolates to each other, rather than to reference strains (Leopold et al., 2009; Turabelidze et al., 2013).
Here we adapt WGS to a specifically developed SNP-based pipeline for the high resolution typing of E. coli O157:H7 by identifying SNPs within the core genome. In addition to SNP analysis in the core genome we assessed plasticity in the mobilome by LS-BSR and plasmid comparison (phages and plasmids) (Eppinger et al., 2011a,b, 2014; Hasan et al., 2012; Jenkins et al., 2015). We tested this pipeline on isolates from seven retrospectively analyzed EHEC O157:H7 outbreaks, six intra-household cases, and five clinical “plate-mate” pairs, i.e., colonies from the same primary isolation plate from the clinical laboratory.
Materials and methods
Strains in study
We compared human isolates (Supplemental Table 1) of nine phylogenetic clades (Manning et al., 2008), so as to place the strains in the overall E. coli O157:H7 phylogenetic context. Strain-associated metadata of analyzed E. coli O157:H7 are provided in Supplemental Table 1. Outbreak strains were defined as a set of isolates from different cases of infection arising from a single point source, as determined by local health jurisdictions and/or the Centers for Disease Control and Prevention. Intra-household cluster strains were recovered from siblings within a household whose infections were not linked to a recognized outbreak. Because intra-household clusters could reflect co-primary infections rather than secondary transmission, we selected such pairings from among our strain set collection (Cornick et al., 2002; Besser et al., 2007) on the basis of prolonged intervals (4–6 days) between cases, so as to increase the likelihood that genomic diversity might emerge secondary to inter-host transmission. Plate mates are pairs of isolates from the same sorbitol-MacConkey agar plate used in clinical laboratories to diagnose the infection.
Bioinformatic analyses for polymorphisms discovery in core genome and mobilome
Developed bioinformatics workflows, methods and principles for SNP discovery and core and accessory genome analyses performed in this study are described in Figure 1 with external tools referenced in the legend. Multinucleotide insertions and deletions of polymorphic bases were not considered SNPs. To classify SNPs we mapped the annotation from the de novo annotated references with PROKKA and Prodigal ORF prediction (Hyatt et al., 2010), or the deposited annotation for EC4115 (Eppinger et al., 2011b). The core genome was defined as the set of genic and intragenic regions that were not repeated, did not contain phages, IS elements, or plasmid regions. Briefly for SNP discovery, reads were aligned with Bowtie2 (Langmead and Salzberg, 2012) to designated reference genomes. Resulting alignments were processed with Freebayes (Garrison and Marth, 2012) with the following threshold settings: mapping quality 30, base quality 20, coverage 30, and allelic frequency 0.9. To account for false positive calls we used several SNP curation strategies: (i) Reference reads were mapped against the reference genome and false positives were identified by Freebayes with the settings described above; (ii) If reads were not available, the post-assembly workflow created a reference-based NUCmer alignment and extracted SNPs with delta-filter and show-snps distributed with the MUMmer package (Delcher et al., 2003). SNP occurring in the excluded regions were removed. Cataloged SNPs from each genome were merged into a single SNP panel, and allelic status and chromosomal position were recorded. Curated SNPs were further processed by extracting the surrounding nucleotides (40 nt) and blastn against the query genomes (Altschul et al., 1990). Resulting alignments were parsed to remove SNP locations derived from ambiguous hits (≥2), non-uniformly distributed regions, and insertion or deletion events, as previously described (Myers et al., 2009; Morelli et al., 2010; Eppinger et al., 2011b, 2014; Vogler et al., 2011; Hasan et al., 2012).
Figure 1.

Bioinformatics analyses of the E. coli O157:H7 core and mobile genome. The majority of the analyses were performed on Galaxy (Goecks et al., 2010), an open-source web-based bioinformatics platform (gray box). Illumina reads, draft or closed genomes were retrieved from the NCBI SRA or WGS sequence repositories (Supplemental Table 1). Reads were assembled with SPAdes v 3.5.0 (Bankevich et al., 2012), and annotated with PROKKA v 1.11.0 (Seemann, 2014). A reference genome was selected for each outbreak analysis and distinct regions were excluded from SNP validation as follows: Repeats with NUCmer (Delcher et al., 2003), phages with PHAST (Zhou et al., 2011) plasmids by querying against a custom E. coli plasmid database and IS elements from ISFinder (Siguier et al., 2006). Optical maps were generated to facilitate accurate prophage and mobilome profiling (Eppinger et al., 2011b). Read based SNP discovery was based on Bowtie2 alignment (Langmead and Salzberg, 2012) and subsequent Freebayes SNP calling (Garrison and Marth, 2012) and for closed or draft genomes on NUCmer (Delcher et al., 2003), followed by blast- and PCR-based SNPs validations to curate for false-positives with strategies detailed in (Eppinger et al., 2011b, 2014). The identified SNPs were genotyped and used for phylogenetic reconstruction by maximum parsimony of the outbreaks with PAUP v4.0a146 (Wilgenbusch and Swofford, 2003). Consistency index and majority consensus trees were built with Mesquite (Maddison and Maddison, 2015). The mobilome was analyzed in regards to phage content with large scale-blast score ratio (LS-BSR) (Sahl et al., 2014) and computed matrices visualized with MeV (Saeed et al., 2003). Shiga toxins were identified by discontiguous megablast (Buhler, 2001; Ma et al., 2002) against the VirulenceFinder (Joensen et al., 2014) database. Plasmids were identified after alignment of draft genomes to the closest closed reference plasmid in Geneious (vR9) (Kearse et al., 2012). Unmapped remaining contigs were queried against NCBI nr plasmids by discontiguous megablast (Buhler, 2001; Ma et al., 2002). Identified plasmid homologs were compared and visualized with EasyFig (Sullivan et al., 2011) by tblastx (Altschul et al., 1990) or progressiveMauve (Darling et al., 2010). Plasmids and chromosomes were further surveyed for presence of resistance loci using ResFinder (Kleinheinz et al., 2014).
Optical maps
Optical mapping facilitated accurate phage profiling (Kotewicz et al., 2008). In total 12 maps were generated (Supplemental Table 1), either prepared by OpGen or contributed by FDA (Eppinger et al., 2011b, 2014). After gentle lysis and dilution, the extracted genomic DNA molecules from each strain were spread and immobilized onto derivatized glass slides. The genomic DNA was then digested with BamH1 restriction enzyme maintaining the DNA fragment order. Using the Argus™ Instrument, the DNA fragments were stained with YOYO-1 fluorescent dye and photographed using a fluorescent microscope interfaced with a digital camera. The optical data was converted to digital data, which defines single molecule restriction maps. Physical maps were complemented with in silico maps of other outbreak strains, and comparatively analyzed in MapSolver™ Optical Map Analysis software (Latreille et al., 2007; Zhou et al., 2007).
SNP PCR validation
SNPs in four isolates from two outbreaks for which we possessed archived cultures were subjected to PCR confirmation using primer pair (89750-F 5′- ACA ACG ATA TGA TCG ACC AGC, 89750-R 5′- TTG TAC AGA AGA CCA TGC TCG) and (27005-F 5′- AGA GTA CGG ATT CAC CTT GCC, 27005-R 5′- AGT CAG GCA ATT CCT CGT GG, 78298-F 5′- AGT CAT TAC CAG GAA CAG CAG 78298-R 5′- TGT TCG AGA TTC TGG TGA GTG) for strains from the Battle Ground Lake and Finley School District outbreak, respectively. Resulting amplicons were Sanger sequenced.
Multi drug resistance (MDR) profiling
Susceptibility to amikacin, ampicillin, amoxicillin-clavulanic acid, cefoxitin, ceftiofur, ceftriaxone, chloramphenicol, ciprofloxacin, gentamicin, kanamycin, nalidixic acid, streptomycin, sulfisoxazole, tetracycline, and trimethoprim-sulfamethoxazole was assessed at FDA according to the NARMS methodology and manufacturer's instructions with the Sensititre automated system (Trek Diagnostic Systems, Westlake, OH) (Zhao et al., 2008). Resistance was determined by comparing MICs to Clinical and Laboratory Standards Institute (CLSI) values (Institute, 2013).
Results and discussion
Epidemiology of investigated strains
We analyzed 36 strains from seven US outbreaks as recognized by the CDC that occurred between 1998 and 2009 (Supplemental Table 1): (1) 11 children were infected after consumption of contaminated ground beef tacos in the Finley School District (FS) in 1998; (2) 28 swimmers at Battle Ground (BL) Lake State Park, WA, and eight secondary cases were infected in 1999; (3) 81 cases were attributed to lettuce served at multiple outlets of a taco chain (Taco John) in 2006; (4) 71 people were infected in a multistate outbreak after eating at Taco Bell (TB) in 2006; though the vehicle was not identified (Taco Bell); (5) 21 infections were attributed to a prolonged multi-state outbreak linked to the consumption of Totino's or Jeno's contaminated pepperoni pizza (Totino's pizza) (TP) in 2007; (6) 76 cases were attributed to a nationwide outbreak of contaminated cookie dough (CD) (Cookie dough) in 2009; (7) 26 patients from eight states were infected by beef traced to Fairbank Farms (FF) in 2009 (Fairbank Farms).
We further studied (#12) strains from six intrahousehold illnesses (IH), in which the pathogen probably spread between patients based on the long intervals between onset in the individual family members (Supplemental Table 1). Though we cannot exclude the possibility of infection from the same source (co-primary). The median incubation period of E. coli O157:H7 infections is 3 days (Bell et al., 1994), and onsets ranged between 4 and 6 days. We also studied pairs of isolates from the same primary plate in the clinical laboratory (plate-mates, PM) from six patients (Figure 2). The clinical strains were compared to strains representing the nine phylogenetic clades reported by Manning et al. (2008) (Figure 2, Supplemental Table 1).
Figure 2.
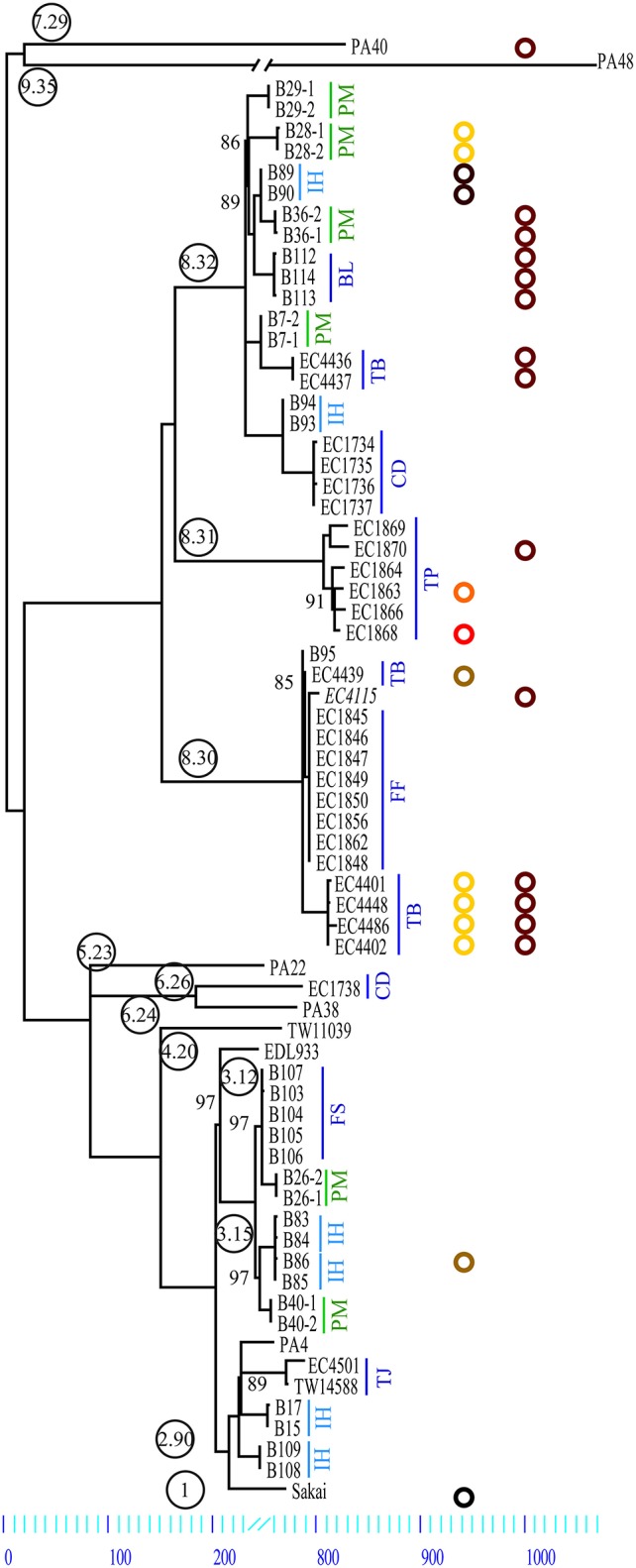
Maximum parsimony tree of E. coli O157:H7. Comparison of 70 genomes yielded a total of 3313 SNPs of which 1266 were parsimony informative. The tree shown is a majority-consensus tree of 28 equally parsimonious trees with a consistency index of 0.998. Trees were recovered using a heuristic search in Paup 4.0b10 (Wilgenbusch and Swofford, 2003). Only nodes with bootstrap values below 100 are listed. Phylogenetic clade association is provided in circled numbers (Manning et al., 2006). Strains investigated are comprised of PM, Plate mates; IH, intrahousehold infections; BL, Battle Ground Lake; CD, Cookie Dough; FS, Finley School District; FF, Fairbank Farms; TB, Taco Bell; TJ, Taco John; and TP, Totino's Pizza. Our plasmid survey confirmed that all strains carry the lineage-specific plasmid pO157. Further we identified other plasmids with a size range from 34 to 78 kb. Plasmid type prevalence is represented in colored circles: p78 (yellow), p63 (orange), p55 (light brown), p39 (red), p36 (brown), p34 (dark brown), and a small 3.3 kb plasmid (black) (Makino et al., 1998). Plasmid p36 is homologous to pEC4115 (Eppinger et al., 2011b).
Core genome phylogeny
We applied WGS typing strategies to determine the phylogenetic relatedness of the individual outbreak strains in the context of outbreak etiology, and to place them into the larger phylogenomic framework of the E. coli O157:H7 lineage (Leopold et al., 2009; Eppinger et al., 2011b; Dallman et al., 2015; Holmes et al., 2015; Jenkins et al., 2015). Of the 3313 SNPs identified in these 70 genomes, 2797 were intragenic and 516 were intergenic (Table 1, Supplemental Dataset 1). We observed significantly more SNPs in intergenic regions (chi square test, p < 10−14) than would be expected when considering the average intergenic frequency in EC4115 of 11.1% when compared to the percentage (15.5%) delineated from the cataloged SNPs. We note that, even though we excluded repeated regions and phages/mobilome during SNP discovery, thereby reducing the genome content by 20%, the coding to non-coding ratio of the remaining core genome remained stable. Homoplasy was negligible: only seven homoplastic SNPs were found dispersed throughout the chromosome (Supplemental Table 2) evidenced by a consistency index of 0.998. Of the seven homoplastic SNPs four are in rpoS, which is known to be highly polymorphic in E. coli O157:H7 (Uhlich et al., 2013). In line with our previous findings (Leopold et al., 2009; Eppinger et al., 2011a,b), SNPs were evenly distributed throughout the chromosome (Figure 2) without any mutational hot spots as found in other enteric pathogens (Hasan et al., 2012; Eppinger et al., 2014). From the cataloged SNP panel we delineated a total of 77 individual SNP genotypes. These genotypes represent only two-thirds of the 115 nodes in the tree (Figure 2, Supplemental Table 2), which can be attributed to the lack of terminal strain-specific SNPs (Figure 2). Among the cataloged 3313 SNPs, approximately one-third (#1266) is parsimony-informative. The SNPs in PA40 and PA48 are not strain-specific, but indicate the relative phylogenetic distance that separates these clade 7 and 9 strains from the other clades (Manning et al., 2008). As evidenced in the tree topology, approximately half of the parsimony non-informative SNPs (#1046) is introduced by reference strain PA48 from clade 9 (Figure 2, Supplemental Table 2). Among investigated strains PA48 is phylogenetically closest to the progenitor O55:H7 serotype (Feng et al., 1998; Manning et al., 2008; Zhou et al., 2010). This clade is also within the most ancient cluster of E. coli O157:H7 (Leopold et al., 2009) and higher SNP counts are indicative of more time to accrue mutations than in other phylogenetic groups that have emerged more recently.
Table 1.
SNPs characteristics for 70 E. coli O157:H7.
| Total | NSYN | NSYN/ NSYN | SYN | SYN/SYN | Intergenic | Genic | |
|---|---|---|---|---|---|---|---|
| SNPs | 3313 | 1712 | 1 | 1083 | 1 | 516 | 2797 |
| NI | 2048 | 1054 | 1 | 656 | 1 | 335 | 1712 |
| PI | 1266 | 658 | 0 | 427 | 0 | 181 | 1085 |
| Genes | 1821 | 1264 | 1 | 891 | 1 | 0 | 2157 |
| Stop gain | 50 | 57 | 0 | 0 | 0 | 0 | 57 |
| Stop loss | 16 | 17 | 0 | 0 | 0 | 0 | 17 |
| Hypothetical proteins | 245 | 269 | 1 | 103 | 0 | 0 | 373 |
| Transition | 2254 | 1037 | 1 | 852 | 1 | 363 | 1891 |
| Transversion | 1064 | 676 | 1 | 231 | 1 | 155 | 909 |
| Multiallelic | 5 | 1 | 1 | 2 | 1 | 2 | 5 |
Genomic epidemiology of north american outbreaks
Guided by the established phylogenomic framework (Figure 2), we analyzed outbreak specific “genome” characteristics and polymorphic heterogeneity in seven different North American outbreaks using a common (EC4115), as well as outbreak-specific references. We specifically applied this dual reference genome approach to improve resolution power by enabling polymorphism discovery in parts of the core genome integral to the outbreak-associated strains, but not necessarily present in a more phylogenetic distant reference like EC4115 (Figure 2). We note here that our investigation of the 2006 Spinach (SP) outbreak revealed a number of subtle polymorphisms distinguishing all the recovered Maine isolates from the remainder of SP strains. Such subtle polymorphisms would have clearly evaded detection by using a reference from outside the SP outbreak (Eppinger et al., 2011b). In general we observed limited plasticity among related outbreak strains when compared to the closed reference genome EC4115 (Tables 2–5) (Eppinger et al., 2011b). Strains with increased SNP numbers were either from cases that were epidemiologically predicted to be outliers, or that could not be read-corrected. For example, the TB outbreak associated strains included four strains classified by CDC as temporal outliers (Supplemental Table 1), two (EC4436, EC4437) separated by 285 SNPs and one (EC4439) by 27 SNPs from the core outbreak cluster (Figure 2, Tables 2, 3). According to our SNP analysis, the remaining strain EC4448 should be considered to be derived from a single point source, even if this isolate is separated by a single homoplastic nonsynonymous (nsyn) SNP (Figures 2, 3, Tables 2, 3, Supplemental Table 2). Notably, rpoS carries this homoplastic stop codon mutation, which is known to be highly polymorphic in E. coli O157:H7; particularly in regards to premature stop codons that affect curli expression and biofilm formation (Uhlich et al., 2013). The same SNP was identified with an outbreak specific reference EC4401 in addition to multiple (#31) reference specific alleles (Figure 4A, Tables 2, 3, Supplemental Dataset 1). These SNPs were mainly located in intergenic (#30) regions and probably caused by over-predictions because of a lack of reads in the genome repository, and consequently inability to perform quality control. We observed the same phenomenon of over-prediction for the TJ outbreak strains, separated by 24 SNPs (Tables 2, 3, Supplemental Dataset 1); again no read data were available to us. We found the majority of predicted SNPs clustered mainly in close proximity either in intergenic regions or within the boundaries of the same gene, indicative of low quality sequence regions. Intragenic SNPs were identical to those found in EC4115, except for two additional SNPs in the lac repressor and in rpoS (Supplemental Dataset 1). The CD outbreak set underwent both contig and read-based discovery, which again over-predicted SNPs for EC1734 (no reads) due to a lack of reads for quality control (Figures 2, 4B, Tables 2, 3). Moreover, the production lot isolate EC1738 was placed on a distant branch (clade 6.26), separated from all human isolates tightly clustered in clade 8.30 (Figure 1, Table 2, Supplemental Table 1). Hence, we consider this strain as an outlier, which is phylogenetically unrelated to the case isolates. Among the outbreak-specific SNPs we detected one synonymous (syn) and two nsyn SNPs in EC1736, but the syn was also detected using EC4115 as a reference (Table 2, Supplemental Dataset 1). Archived strains were not available for this outbreak and we could therefore not confirm if EC1736 truly carries these 3 SNPs, which would question its inclusion into the outbreak.
Table 2.
Comparison of common vs. outbreak-specific reference genic SNPs.
| Isolate | Sequences analyzeda | Synonymous SNPs in isolates, among backbone ORFs | Nonsynonymous SNPs in isolates, among backbone ORFs | Comments | ||
|---|---|---|---|---|---|---|
| Compared to reference EC4115 | Compared to outbreak strain | Compared to reference EC4115 | Compared to outbreak strain | |||
| FINLEY SCHOOL OUTBREAK, ALL ISOLATES CLADE 3.15 | ||||||
| B103 | A | 0 | 0 | 0 | 1 | All co-primary cases |
| B104 | A | 0 | 0 | 0 | 1 | |
| B105 | A | 0 | Reference | 0 | Reference | |
| B106 | A | 0 | 0 | 0 | 0 | |
| B107 | A | 0 | 0 | 0 | 1 | |
| TACO BELL OUTBREAK, ALL CLADE 8.30 EXCEPT EC4436-7 CLADE 8.32 | ||||||
| EC4401 | B | 1 | Reference | 0 | Reference | Eppinger et al., 2011b |
| EC4402 | A | 0 | 0 | 0 | 0 | Case isolate |
| EC4436 | A | 90 | Excluded | 155 | Excluded | Temporal outliers |
| EC4437 | A | 90 | Excluded | 155 | Excluded | |
| EC4439 | A | 5 | Excluded | 17 | Excluded | |
| EC4448 | A | 0 | 0 | 1 | 2 | |
| EC4486 | B | 0 | 1 | 4 | 4 | Eppinger et al., 2011b |
| TACO JOHN, ALL ISOLATES CLADE 2.90 | ||||||
| EC4501 | B | 1 | 0 | 6 | 8 | Eppinger et al., 2011b |
| TW14588 | C | 0 | Reference | 0 | Reference | |
| FAIRBANK FARMS OUTBREAK, ALL ISOLATES CLADE 8.30 | ||||||
| EC1845 | A | 0 | 0 | 0 | 2 | Case isolate |
| EC1846 | A | 0 | 0 | 0 | 2 | |
| EC1847 | A | 0 | 0 | 0 | 2 | |
| EC1848 | A | 0 | 0 | 0 | 2 | |
| BATTLEGROUND LAKE OUTBREAK, ALL ISOLATES CLADE 8.32 | ||||||
| B112 | A | 1 | 1 | 0 | 0 | Case isolates |
| B113 | A | 0 | 0 | 0 | 0 | |
| B114 | A | 0 | Reference | 0 | Reference | |
| COOKIE DOUGH OUTBREAK, EC1738 CLADE 6.26, EC1734-7 CLADE 8.32 | ||||||
| EC1738 | B | 168 | excluded | 298 | excluded | Product isolate |
| EC1734 | B | 0 | 5 | 2 | 5 | Case isolates |
| EC1735 | A | 0 | 1 | 0 | 2 | |
| EC1736 | A | 1 | Reference | 1 | Reference | |
| EC1737 | A | 0 | 1 | 0 | 2 | |
| TOTINO's, PIZZA OUTBREAK ALL ISOLATES CLADE 8.31 | ||||||
| EC1863 | A | 6 | Reference | 11 | Reference | Case isolates |
| EC1864 | A | 9 | 10 | 10 | 8 | |
| EC1866 | A | 6 | 7 | 12 | 8 | |
| EC1868 | A | 6 | 3 | 6 | 6 | |
| EC1869 | A | 7 | 15 | 9 | 21 | |
| EC1870 | A | 10 | 20 | 9 | 21 | |
| FAIRBANK FARMS OUTBREAK, ALL ISOLATES CLADE 8.30 | ||||||
| EC1849 | A | 0 | 0 | 0 | 2 | Case isolate |
| EC1850 | A | 0 | 0 | 0 | 2 | |
| EC1856 | A | 0 | Reference | 0 | Reference | |
| EC1862 | A | 0 | 0 | 0 | 3 | |
This study/short reads in NCBI = A, WGS = B, assembled genomes in NCBI = C.
Table 3.
Comparison of common vs. outbreak-specific reference intergenic SNPs.
| Isolate | Sequences analyzeda | SNPs in isolates, in intergenic regions | Commentsb | ||
|---|---|---|---|---|---|
| Compared to reference EC4115 | Compared to outbreak strain | ||||
| FS | B103 | A | 1 | 1 | Reference specific allele |
| B104 | A | 0 | 1 | ||
| B105 | A | 0 | Reference | ||
| B106 | A | 0 | 1 | ||
| B107 | A | 0 | 1 | ||
| BG | B112 | A | 0 | 0 | |
| B113 | A | 0 | 0 | ||
| B114 | A | 0 | Reference | ||
| TB | EC4401 | B | 1 | Reference | F |
| EC4402 | A | 0 | 27 | ||
| EC4436 | A | 40 | Excluded | ||
| EC4437 | A | 40 | Excluded | ||
| EC4439 | A | 5 | Excluded | ||
| EC4448 | A | 0 | 27 | ||
| EC4486 | B | 3 | 31 | ||
| TJ | EC4501 | B | 8 | 16 | F |
| TW14588 | C | 1 | Reference | ||
| CD | EC1738 | B | 67 | Excluded | F |
| EC1734 | B | 0 | 4 | ||
| EC1735 | A | 0 | 0 | ||
| EC1736 | A | 0 | Reference | ||
| EC1737 | A | 0 | 0 | ||
| TP | EC1863 | A | 2 | Reference | High diversity |
| EC1864 | A | 1 | 5 | ||
| EC1866 | A | 2 | 6 | ||
| EC1868 | A | 2 | 5 | ||
| EC1869 | A | 10 | 14 | ||
| EC1870 | A | 6 | 11 | ||
| FF | EC1845 | A | 0 | 1 | Reference specific allele |
| EC1846 | A | 0 | 1 | ||
| EC1847 | A | 0 | 1 | ||
| EC1848 | A | 0 | 1 | ||
| EC1849 | A | 0 | 1 | ||
| EC1850 | A | 0 | 1 | ||
| EC1856 | A | 0 | Reference | ||
| EC1862 | A | 0 | 1 | ||
This study/short reads in NCBI = A, WGS = B, assembled genomes in NCBI = C.
F = mixed analysis with reads missing for some strains.
Figure 3.

Genome-wide distribution of SNPs. Chromosomal positions of the 3313 identified SNPs were plotted along the EC4115 chromosome using a 1000 bp sliding window. We found SNPs dispersed throughout the chromosome providing no indication for mutational hotspots. Deserted regions lacking SNP calls correspond to excluded mobile regions, such as stx-prophages.
Figure 4.
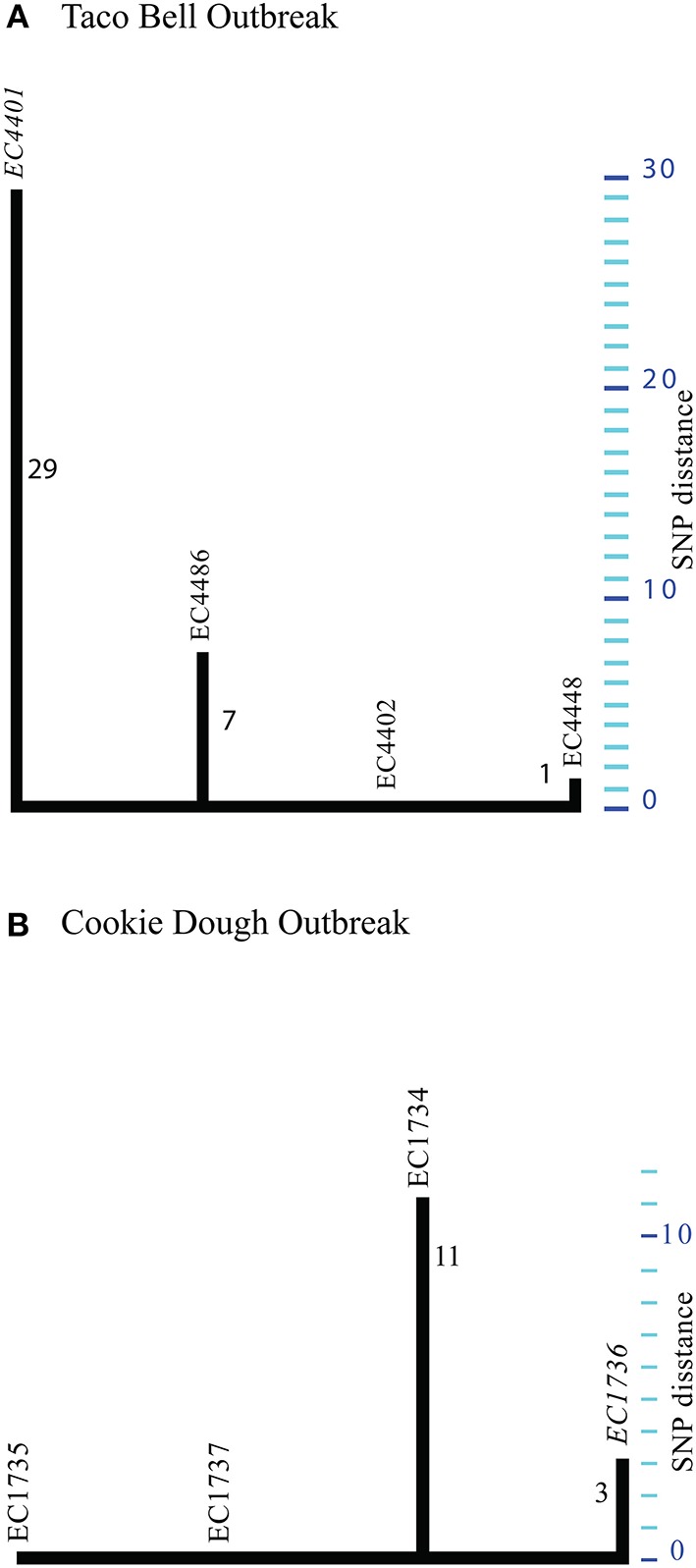
Phylogenetic tree for Taco Bell (A) and Cookie dough (B) outbreak strains. Maximum parsimony phylogeny computed from 37 TB and 14 CD SNPs identified in the respective outbreak associated strains. (A) By using a TB outbreak-specific reference (EC4401) we increased the phylogenetic resolution for these highly clonal strains. (B) Maximum parsimony phylogeny delineated from 14 SNPs discovered in the four CD outbreak associated strains. Strain EC1736 served as reference for the read based discovery. Majority of SNPs (#11) were specific to EC1734 and probably false positives from through assembly based discovery.
For three outbreaks (FF, FS, and BL) we identified only a single or no SNP when referenced to EC4115 (Figure 2, Tables 2, 3). A single intergenic and three nsyn mutations were identified when using an FF outbreak-specific reference strain EC1856 (Tables 2, 3). The three nsyn SNPs did not affect domain prediction in Pfam (Finn et al., 2016). B112 of the BL outbreak had a syn SNP in a tRNA-histidine ligase (#3460738) not found in any other E. coli O157:H7 genome deposited (nr or WGS). This SNP was identified in both instances when using EC4115 or an outbreak-specific reference (Supplemental Dataset 1). This SNP was confirmed using PCR amplicon sequencing. Using the alternative FS outbreak-specific reference one intergenic SNP in B105 and one nsyn SNP affecting the Nitrogen regulation protein NR(I) (ECH74115_RS26390) (B107 and B105) were identified (Tables 2, 3). SNP discovery predicted an outbreak-specific allele in three strains. However, these SNPs are false positives, as they could not be confirmed by PCR sequencing. The SNP (#693920) in FS strain B103 was identified as false-positive homoplastic SNP also observed in plate mate and intrahousehold strains with an allelic frequency below 0.9 (Supplemental Table 2). During SNP prediction we identified 52 SNPs in strain B103 that were not found in the other FS outbreak strains. These 52 SNPs were all located in a phage region that corresponds to the tandem integrated SP1/2 phages (Hayashi et al., 2001). The SNPs were all false-positives due to the presence of an additional phage in B103 related to a prophage from organism pro483 (NC_028943) (Supplemental Figure 3). The tail fiber proteins of these two phages were sufficiently similar to misalign reads for B103. This exemplifies the importance of SNP curation and assessment according to the genomic region in which they originate, as independent horizontal acquisition of segments can introduce epidemiologically misleading SNPs (Pettengill et al., 2014), also known as epidemiological type 2 errors of attribution.
The TP outbreak strains revealed a highly distinct SNP pattern compared to the genomic plasticity reported for other outbreaks (Figures 1, 5, Tables 2, 3). Two distinct phylogenetic clusters separated by 16 SNPs were observed. Additionally, each strain carried at least 4–17 strain-specific SNPs. Comparison to outbreak-specific reference EC1863 confirmed the relative high number of strain-specific SNPs (Tables 2, 3, Figure 5). In contrary to our observations for strain-specific SNPs in the above discussed outbreaks, these SNPs are neither concentrated in specific regions nor more frequent in intergenic than in genic regions (Tables 2, 3). The EC1869/EC1870 branch contributes roughly 60% of all SNPs (Supplemental Dataset 1). Based on the established phylogenetic topology we hypothesize that two closely related but different E. coli O157:H7 contaminated a common vehicle, if, indeed, all cases had the same exposure. Two-thirds of the SNPs were strain-specific, denoting a particular high diversity within this outbreak (Figures 1, 5, Tables 2, 3). Such a degree of genomic plasticity among epidemiologically linked strains has rarely been observed in E. coli O157:H7. Several scenarios could have led to this radial expansion: (i) the epidemiology linked cases together that actually were from different simultaneous outbreaks, (ii) the SNPs identified in silico are false positives and only PCR-confirmation could really confirm the true distance among the strains, (iii) the high rate of accumulated SNPs could be caused by a mutator genotype resulting in the accumulation of mutations in a short time span, (iv) the heterogeneity could be related to the protracted duration of the outbreak (3 months), vs. single, brief, single source-exposures as in the FS outbreak, or (v) heterogeneity caused by increased strain mutation rates during outbreaks as have been discussed for other enterics (Morelli et al., 2010). In support of our findings, Dallman et al. noted correlations between the length of the strain collection intervals and respective numbers of SNPs observed (Dallman et al., 2015).
Figure 5.

Phylogenetic tree for Totino's Pizza outbreak strains. Maximum parsimony phylogeny of the Totino's Pizza outbreak using reference EC1863. The tree topology shows the genotypic heterogeneity among the outbreak associated strains, forming “two” distinct phylogenetic groups.
The clonal nature of E. coli O157:H7 outbreaks was confirmed in the majority of the outbreak strains analyzed here, consistent with prior findings from SNP typing in other O157:H7 outbreaks (Turabelidze et al., 2013; Dallman et al., 2015; Holmes et al., 2015; Jenkins et al., 2015; Munns et al., 2016). We found the number of SNPs to be inversely proportional to the availability of reads. This highlights the critical importance of quality control for accurate SNP discovery by accounting for both underlying sequence quality and evolutionary context of the SNP carrying loci to curate for false-positives. In this regard, the relevance of excluding mobile regions when inferring outbreak relatedness is evidenced in the loss of at least two thirds of predicted SNPs that if considered would impair phylogenetic accuracy.
WGS typing of plate mates recovered from human infections
In the medical praxis typically a single colony is retrieved from a primary isolation plate and sent for further molecular analysis. It is therefore not clear how much genotypic diversity exists among infecting isolates of E. coli O157:H7 as shed from the same individual in a single stool. To answer this question, plate-mates (pairs of colonies) were separately saved from five patients (Figure 2, Supplemental Table 1) enrolled in a multi-state study of E. coli O157:H7 infections (Wong et al., 2012). In the EC4115 reference-based discovery, two PM possessed the same homoplastic intergenic SNP (Figure 2, Tables 4, 5), which was not confirmed after allelic verification. When using an internal reference these strains were undistinguishable. The results are in accordance with those of Dallman et al., who reported 0–2 SNPs among same patient isolates, with most (70%) having no SNP differences at all (Dallman et al., 2015). Our results from this limited study, therefore, point toward infection with a single E. coli O157:H7 clone as the underlying cause for the majority of infections. We previously reported that a single laboratory passage can produce SNPs in E. coli O157:H7, but SNPs arise only rarely (Eppinger et al., 2011b). In the course of naturally acquired human infections, our data endorse that E. coli O157:H7 SNPs are exceptionally rare events.
Table 4.
Comparison of common vs. PM/IH-specific genic SNPs.
| Isolate | Sequences analyzeda | Synonymous SNPs in isolates, among backbone ORFs | Nonsynonymous SNPs in isolates, among backbone ORFs | |||
|---|---|---|---|---|---|---|
| Compared to reference EC4115 | Compared to outbreak strain | Compared to reference EC4115 | Compared to outbreak strain | |||
| PM | B26-1 | A | 0 | 0 | 0 | 0 |
| B26-2 | A | 0 | Reference | 0 | Reference | |
| PM | B28-1 | A | 0 | Reference | 0 | Reference |
| B28-2 | A | 0 | 0 | 0 | 0 | |
| PM | B29-1 | A | 0 | 0 | 0 | 0 |
| B29-2 | A | 0 | Reference | 0 | Reference | |
| PM | B36-1 | A | 0 | 0 | 0 | 0 |
| B36-2 | A | 0 | Reference | 0 | Reference | |
| PM | B40-1 | A | 0 | Reference | 0 | Reference |
| B40-2 | A | 0 | 0 | 0 | 0 | |
| PM | B7-1 | A | 0 | 0 | 0 | 0 |
| B7-2 | A | 0 | Reference | 0 | Reference | |
| IH | B83 | A | 0 | 0 | 0 | 0 |
| B84 | A | 0 | Reference | 0 | Reference | |
| IH | B85 | A | 0 | Reference | 0 | Reference |
| B86 | A | 0 | 0 | 0 | 0 | |
| IH | B89 | A | 0 | 0 | 0 | 0 |
| B90 | A | 0 | Reference | 0 | Reference | |
| IH | B93 | A | 0 | Reference | 0 | Reference |
| B94 | A | 0 | 0 | 0 | 0 | |
| IH | B15 | A | 0 | 0 | 0 | 0 |
| B17 | A | 0 | Reference | 0 | Reference | |
| IH | B108 | A | 0 | 0 | 0 | 0 |
| B109 | A | 0 | Reference | 0 | Reference | |
This study/short reads in NCBI = A, WGS = B, assembled genomes in NCBI = C.
Table 5.
Comparison of common vs. PM/IH-specific intergenic SNPs.
| Isolate | Sequences analyzeda | SNPs in isolates, in intergenic regions | Commentsb | ||
|---|---|---|---|---|---|
| Compared to reference EC4115 | Compared to outbreak strain | ||||
| PM | B26-1 | A | 0 | 0 | |
| B26-2 | A | 0 | Reference | ||
| PM | B28-1 | A | 1 | Reference | Homoplastic FP |
| B28-2 | A | 0 | 0 | ||
| PM | B29-1 | A | 0 | 0 | |
| B29-2 | A | 0 | Reference | ||
| PM | B36-1 | A | 1 | 0 | Homoplastic FP |
| B36-2 | A | 0 | Reference | ||
| PM | B40-1 | A | 0 | Reference | |
| B40-2 | A | 0 | 0 | ||
| PM | B7-1 | A | 0 | 0 | |
| B7-2 | A | 0 | Reference | ||
| IH | B83 | A | 1 | 0 | Homoplastic FP |
| B84 | A | 0 | Reference | ||
| IH | B85 | A | 0 | Reference | Homoplastic FP |
| B86 | A | 1 | 0 | ||
| IH | B89 | A | 0 | 0 | |
| B90 | A | 0 | Reference | ||
| IH | B93 | A | 0 | Reference | |
| B94 | A | 0 | 0 | ||
| IH | B15 | A | 0 | 0 | Homoplastic FP |
| B17 | A | 1 | Reference | ||
| IH | B108 | A | 0 | 0 | |
| B109 | A | 0 | Reference | ||
This study/short reads in NCBI = A, WGS = B, assembled genomes in NCBI = C.
False positive = FP.
WGS typing of strains from intrahousehold infections
To determine if genomic changes in infecting E. coli O157:H7 occur during probable intrahousehold (IH) transmission, we analyzed a cohort of six pair isolates from IH infections where onset was quite delayed between cases (Figure 2). As with the PM pairs, EC4115 based SNP discovery resulted only in false positive homoplastic intergenic SNPs (Figure 2, Tables 3, 4) that were absent in the pair-wise analysis. Dallman et al. observed similar SNP distributions in household transmission cases in the UK, with 40% having no such differences in the core genome (Dallman et al., 2015). Interestingly, two IH cases of clade type 3.15 clustered together (Figure 2). A single syn SNP was specific to the B83/B84 cluster. These cases were all from the same state and occurred in the same year, but epidemiological investigations suggest they are separate cases of IH transmissions with over 6 weeks between occurrence and 80 miles distance between the zip codes in which the cases resided. This application of WGS typing analysis can genomically link clusters that were not previously identified epidemiologically (Dallman et al., 2015).
In general the frequency of SNPs in intergenic and genic regions were similar, highlighting the random nature of SNPs identified. While there is clearly no applicable universal gold standard or criteria for outbreak ex- or inclusion in regards to SNP matrix distances, we note that a number of outbreak investigations have found between four to seven SNPs among strains with putative epidemiological links (Underwood et al., 2013; Joensen et al., 2014; Dallman et al., 2015; Holmes et al., 2015). However, these analyses are limited by only including the genic portions of the genomes and/or did not use an outbreak-specific reference for SNP discovery. This prevents identification of variations in parts of the core genome that are unique to outbreak-associated strains and not necessarily present in a distantly related closed reference strain. Moreover, only few studies use confirmatory PCR or other resequencing to validate in silico delineated SNPs (Eppinger et al., 2011b; Underwood et al., 2013).
Phage profiles of clinical U.S. strains
The abundance of lambdoid phages in the EHEC O157:H7 genome hinders assembly of phage regions based on short reads alone (Eppinger et al., 2011b). Contig breaks often occur within the phage borders due to the conserved nature of structural and replication proteins and hinder individual phage-level comparisons in the fragmented phage assemblies. Therefore, we applied an alternative genome-scale strategy to comprehensively analyze stx allele status and losses or gains in the strain's phage ORF-omes.
Major virulence traits of E. coli O157:H7 are encoded on members of the mobilome that are usually stably integrated into the chromosome, such as the locus of enterocyte effacement (LEE) and stx-converting phages (Nataro and Kaper, 1998). Phages are key components of pathogenome evolution and their acquisitions are important events in the emergence of E. coli O157:H7 from an ancestral cell closely related to E. coli O55:H7 (Feng et al., 1998, 2007; Zhou et al., 2010). Moreover, analyses such as SNP typing that are limited to the core genome cannot provide information about the conferred pathogenic potential anchored in the mobilome. Our analysis of the 2006 SP outbreak exemplifies genomic heterogeneity that can be found in a single outbreak of O157:H7 in regards to mobilome (Eppinger et al., 2011b). Within the prophage pool (Hayashi et al., 2001) the stx-converting bacteriophages are of particular interest, as they encode a potent cytotoxin, Shiga toxin or Stx (Karmali et al., 2010) as direct mediator of EHEC O157:H7 disease (Krüger and Lucchesi, 2015). In E. coli O157:H7 the chromosomal backbone is highly conserved and genomic alterations chiefly relate to phage complement, plasticity, and respective integration sites (Shaikh and Tarr, 2003; Abu-Ali et al., 2009; Eppinger et al., 2011b, 2013; Smith et al., 2012; Yin et al., 2015). Three stx alleles, stx1a, stx2a, and stx2c, are found predominantly in this lineage (Scheutz et al., 2012). We used discontiguous megablast against the VirulenceFinder database to determine the toxin subtypes present in each outbreak (Joensen et al., 2014). All IH, PM, and outbreak strains carry the more potent allelic variant stx2a (Supplemental Table 1). In addition, all FS and TJ outbreak strains, PMs B40-1/2 and B26-1/2 and two separate IH transmission cases (B83/B84, B85/B86) carry an stx1-converting phage. Co-carriage of Stx2 and Stx1 can reduce Stx2a production (Serra-Moreno et al., 2008) and also attenuates end-organ toxicity of Stx2a (Donohue-Rolfe et al., 2000; Russo et al., 2016). Noteworthy, the 2006 SP outbreak associated with hypervirulence (Kulasekara et al., 2009; Abu-Ali et al., 2010) features the Stx2a/2c toxin type, with an almost complete stx1-converting phage occupies yehV. However, this atypical phage lacks stx1genes (Eppinger et al., 2011b). We also note that the TJ lettuce isolate TW14588 harbors two stx2a-converting phages integrated at argW and wrbA (Supplemental Table 1). We speculate that double stx2a-converting phage occupancy might also increase pathogenic potential, such as through phage dosage effects, also considering that stx2a is the most potent allelic subtype (Tesh et al., 1993; Tesh, 2010; Fogg et al., 2012). We note that this information cannot be gathered by PCR-based Stx-subtyping (Scheutz et al., 2012), as this approach does not determine copy number, highlighting the increased resolution obtained by WGS in regards to the pathogenic potential of the outbreak (Holmes et al., 2015). All other outbreaks except BL possess stx2c-converting bacteriophages. The interplay between these two stx2-converting phage types is not known, although both variants have been linked to HUS (Friedrich et al., 2002; Persson et al., 2007). We observed only two variations in the stx2 allelic profiles in the CD and TP outbreaks (Supplemental Table 1). The non-clinical CD outlier EC1738 collected in the production plant is distinguished by lack of the stx2a allele. TP strain EC1870 lacks the stx2c allele that is present in all other TP outbreak strains (Supplemental Table 1).
On average, the phage complement in the strains represents 14% of predicted coding regions in the genomes, in accordance with other studies (Asadulghani et al., 2009; Smith et al., 2012). As expected when considering the close relation between outbreak strains (Figure 2), the variability in phage-borne ORFs was low (5%) (Supplemental Figure 1; Table 3). This small variability represents the noise caused by clustering of related proteins into centroids in the LS-BSR analysis rather than differences in phage regions. The TB associated strains had more variability (11.2%) (Supplemental Table 3), due to inclusion of the temporal outliers EC4436, EC4437, and EC4439 (Figure 6). Variome analysis highlighted phage regions that were unique to each temporal outlier group, resulting in the same clustering as in the SNP-based analysis (Figures 2, 6). The CD outbreak also had greater variability (16.5%) despite the exclusion of outlier EC1738 (Supplemental Table 3), likely attributable to differences in fragmentation of the analyzed genomes. We observed a correlation of quality of PHAST prediction with size of contigs and reduced genome fragmentation. Closed genomes and genomes with larger contigs had up to 20% more predicted phage regions that also served to increase the noise, compared to more fragmented genomes (Supplemental Table 1).
Figure 6.

Hierarchical clustering of the computed phage variome for Cookie dough (A) and Taco Bell (B) outbreaks. Phage inventories within the respective outbreak clusters compared by LS-BSR and variome differences are presented in this heatmap. BSR values range from 0 (blue, absent) to 1 (yellow, identical). (A) Strain EC1734 contributed the majority of varying regions within the CD phage complement. (B) The tree topology derived from SNP typing for the TB outbreak (Figure 2) is in accordance with the variome clustering. Most of the variation was introduced by phages prevalent in the three temporal outlier strains EC4436, EC4437, and EC4439.
The identified phage complements of the FS isolates were highly similar. However, we found phage sequences unique to strain B103 (Supplemental Figure 2A). Discontiguous megablast of the phage region (Buhler, 2001; Ma et al., 2002) against closed bacteriophages identified Escherichia phage pro483 (KR073661), originally isolated from an avian pathogenic E. coli DE048. This prophage was previously described in SP strains (Eppinger et al., 2011b) and a supershedder strain SS17 (Cote et al., 2015). Unlike the yegQ insertion in SP outbreak strains (Eppinger et al., 2011b), this phage disrupts the colicin immunity protein (WP_001303895) in strain B103. Using the phage pro483 (KR073661) as a genomic anchor for the B103 draft contigs we recovered 86% of the phage genome with 97% identity. We further identified a 12 bp (ACCAATAACTGA) repeat at both ends of the phage borders, indicative of the phage integration mechanism (Campbell, 1992). The SP outbreak strain EC4115 however features an 18 bp repeat (Eppinger et al., 2011b). The genomic architecture is syntenic and largely conserved throughout the phage genome, except for insertion or deletion introduced by an exonuclease (ECH74115_RS15445) and a hypothetical protein (ECH74115_RS15450) only present in EC4115 (Supplemental Figure 3).
Acquisition or loss of phages secondary to recombination events during the course of an outbreak creates interstrain plasticity. Thus, analysis of a single archetypical outbreak strain might underestimate the mobilome and core chromosome plasticity (Eppinger and Cebula, 2015). Comprehensive analyses did not reveal significant differences in phage content of the BL and FF outbreak clusters (Figure 2) to further distinguish these clonal outbreaks featuring only one and four SNPs per outbreak cluster, respectively (Supplemental Figures 2B,C). In contrast, the TP outbreak strains displayed a higher degree of mobilome plasticity (Supplemental Figure 2D), in line with the higher number of predicted SNPs (#98) for this outbreak (Figure 2, Supplemental Dataset 1).
Plasmid prevalence in clinical E. coli O157:H7 strains
The E. coli O157:H7 lineage is distinguished from other serotypes by the presence of the large virulence plasmid pO157 (Burland et al., 1998). For this serotype, additional plasmids have been occasionally characterized at sequence level (Makino et al., 1998; Eppinger et al., 2011b) or by plasmid profiling (Ostroff et al., 1989; Meng et al., 1995). To facilitate plasmid discovery and survey we reassembled the genomes with SPAdes (Bankevich et al., 2012). Even though deposited genomes from 454 and Illumina Celera hybrid assemblies (Denisov et al., 2008) had fewer contigs compared to SPAdes assemblies from Illumina reads only (Supplemental Table 1), reassembly typically produced longer contigs, in particular for plasmid-originating regions. If Illumina reads only were processed, the SPAdes assemblies clearly outperformed NCBI deposited Velvet assemblies in regards to sensitivity for plasmid prediction (Supplemental Table 1). We queried plasmid sequences against the NCBI nr plasmid database using discontiguous megablast (Buhler, 2001; Ma et al., 2002).
Using this approach we discovered five plasmids at the sequence level (p78, p34, p55, p63, and p39) that have not been previously described in deposited E. coli O157:H7 genomes. Among these is a homolog of a 37 kb conjugal transfer pEC4115, referred to as p36, originally described in the SP outbreak strains (Eppinger et al., 2011b). We found the TB and TP outbreaks to be most diverse in regards to plasmid carriage (Figure 2). The TB associated strains contained three distinct plasmid profiles, which correlated with the clustering from the core genome SNP discovery (Figure 2).
Plasmid p78, the largest plasmid, shows homology to the conjugative IncI1 group E. coli plasmid pC49-108 and Salmonella enterica plasmids (Fricke et al., 2011; Kröger et al., 2012; Wang et al., 2014a). p78 varied in length, from 78 to 88 kb in clade 8 strains (Figure 7). The related plasmid pC49-108 carries multiple antibiotic resistance genes (Wang et al., 2014a), including a beta-lactamase (blaCTX−M−1) (Wang et al., 2014a), dihydrofolate reductase (dfrA17) and aminoglycoside adenylyltransferase (aadA5) found both adjacent to a class 1 integron (Wang et al., 2014a). In similarity to the blaCTX−M−1 located next to a mobile element (ISECp1), we found another class C beta-lactamase gene in S. enterica CVM 22462, again found next to a mobile transposase locus. We speculate that colocalization to mobile elements might affect locus stability and explains the scattered prevalence of these resistances in the plasmid homologs (Wang et al., 2014b) (Figure 7).
Figure 7.

Alignment of plasmid p78 and homologs. Plasmid architecture and gene inventories were compared by tblastx, and respective annotations were mapped in Geneious vR9 (CDS, green). The plasmid architecture was highly conserved with a high identity level throughout the entire length of the plasmid [100% identity yellow (inverted fragment, orange), 36% identity blue (inverted fragment light blue)]. Plasmid p78 homologs were widespread, such as in TB outbreak associated strains, B28 plate mates, other E. coli O157:H7, and S. enterica. We found a locus for a RelB/E tox/antitoxin system present in all plasmids, with the exception of PM strains B28 (blue box). Resistance loci are highlighted in red boxes.
Resistance to antibiotics has been observed in E. coli O157:H7, but the genetic basis remains largely unknown (Meng et al., 1998). We previously linked multi drug resistance (MDR), a rare occurrence in E. coli O157:H7, to phage-borne antibiotic resistance loci (Eppinger et al., 2011a). Strain EC4402, part of the core TB outbreak cluster, was identified as a MDR isolate (Figure 2). This strain displays elevated MICs for several cephalosporins and aminoglycosides, sulfisoxazole and nalidixic acid (quinolone). However, our in silico analysis with ResFinder (Kleinheinz et al., 2014) did not reveal any potential underlying resistance loci. Here we note that resistance phenotypes can be conferred by loci not previously linked to antibiotic resistance (Gibson et al., 2016). We speculate that the resistance loci might have been either lost from the original p78, or were an integral part of other MDR plasmids lost during laboratory cultivation prior to the sequencing of EC4402. Alternatively, the antibiotic resistance might be conferred by yet unknown loci not represented in queried resistance databases.
Plasmid p36 was highly homologous to other conjugal transfer plasmids, such as S. enterica plasmid pCFSAN000111_01 (NZ_CP007599) (Timme et al., 2012) and pEC4115 (Eppinger et al., 2011b) (Supplemental Figure 4). While p78 was found solely in clade 8, p36 seems to be more widespread (Cote et al., 2015), and present in non-O157:H7 E. coli serotypes. Co-carriage of a p78-p36 combination was also found in clade 8 strains K4405 and K4406 (Figure 7, Supplemental Figure 4). The TB outlier strain EC4439 lacks both p78 and p36, but carries a p55 plasmid with high homology to Klebsiella pneumoniae pDMC1097-77.775 kb (87% coverage, 99% identity) (Wright et al., 2014) (Supplemental Figure 5). This IncI2 group plasmid carries multiple resistances, which are absent in the E. coli plasmid homologs (Supplemental Figure 5). Interestingly, this plasmid is also present in IH strain B86, but absent from strain B85, either because of independent acquisition or secondary loss in B85, respectively (Figure 2). Our findings on plasmid prevalence are in accordance with those of Dallman et al. who showed that epidemiologically linked strains can vary largely in their plasmid inventory (Holmes et al., 2015).
The IH strains B89 and B90 harbor p34 (Supplemental Figure 6), which is related to E. coli pVR50, an F-like conjugative MDR plasmid (Beatson et al., 2015). While the overall plasmid backbone is conserved, p34 lacks any resistance loci (Supplemental Figure 6). The TP strains also possess strain-specific plasmids: p36 in EC1870, p63 in EC1863, and p38 in EC1868 (Figure 2). Plasmid p63 has partial homology to pO26-Vir, an IncK group plasmid, a mosaic of multiple plasmids (Fratamico et al., 2011). In 1863 (p63) we found homologous loci for conjugal transfer and type IV pili (Supplemental Figure 7), which have been implicated in cell adherence and biofilm formation (Dudley et al., 2006), and notably, these phenotypes are strain-dependent in E. coli O157:H7 (Vogeleer et al., 2014). A 39 kb plasmid fragment in EC1868 (p39) was found to be homologous to a 87 kb INcFII plasmid from E. coli (pGUE-NDM) (Bonnin et al., 2012) (Figure 2).
The observed variability in plasmid type and prevalence in the individual strains clearly highlights genomic plasticity that exists even among closely related isolates of the same origin and can be utilized for strain attribution (Eppinger et al., 2011b). The identified heterogeneity between the mobilome of outbreak strains stresses the importance of studying a number of isolates from the same outbreak instead of using archetypal outbreak strains, which as shown might not fully reflect the plasticity in the outbreak population (Eppinger and Cebula, 2015). Interestingly, all the above described E. coli O157:H7 plasmids lack antibiotic resistance loci, even though our plasmid survey found widespread resistances among homologous plasmids in other serotypes and species.
Conclusion
While some of these clinical isolates have been studied previously using molecular epidemiology techniques (Samadpour et al., 2002), we have for the first time applied whole genomics epidemiology approaches (Eppinger et al., 2011b). Through these high resolution methods we established a detailed understanding of the genomic heterogeneity found among the studied E. coli O157:H7 outbreak populations from the U.S. The gathered phylogenomic data were critical to define the genetic relatedness of individual strains in the context of outbreak etiology and phylogenetic positions in the broader model of E. coli O157:H7 evolution and epidemiology. In this study, we detected previously unnoted polymorphic genome features in the core and mobile genome, such as an array of new plasmids not previously associated with this lineage. The cataloged polymorphic signatures aided in strain attribution and allowed us to precisely define the outbreak boundaries. This allowed us to discern the distinct phylogenetic boundaries of the studied EHEC strains when placed into a larger phylogenomic framework of E. coli O157:H7 from North America (Figure 2) assessing both core and mobilome (Figures 6, 7) (Eppinger and Cebula, 2015). The developed WGS typing approach provided us with the necessary resolution power to study the individual dynamics in highly clonal outbreaks (Morelli et al., 2010; Eppinger et al., 2011b, 2014; Hasan et al., 2012; Berenger et al., 2015; Holmes et al., 2015; Jenkins et al., 2015).
While the majority of outbreaks were caused by pathogens that form tight clonal clusters, one outbreak (“Totino's Pizza”) was associated with isolates showing considerable genomic heterogeneity (Figures 2, 5). Apparent SNPs in other outbreaks are associated with a paucity of reads for quality control, falsely increasing the diversity among the outbreak isolates. Since outbreaks can have high economic impacts, such as nationwide recalls of contaminated product, multiple samples from the same outbreak should be concomitantly sequenced instead of using archetypal outbreak strains to provide strong evidence for inclusion or exclusion, strain and source attribution (Eppinger et al., 2010, 2014; Morelli et al., 2010; Hasan et al., 2012). Additionally, these high resolution approaches allow for the discovery of emerging pathotypes, and, potentially, to better assess the pathogenic potential of individual bacterial clones (Berenger et al., 2015; Klemm and Dougan, 2016). Expanding these sequence-based analyses to the publicly available EHEC sequence pool will improve public health response in the event of an outbreak allowing timely and informed countermeasures. Canonical SNPs can be implemented in efficient typing assays offering robust phylogenetic signals for outbreak exclusion/inclusion that surpass classical technologies (Riordan et al., 2008; Elhadidy et al., 2015; Rusconi and Eppinger, 2016).
Our study strongly endorses that quality of SNPs and choice of an appropriate reference strain in WGST approaches are equally critical to achieve phylogenetic resolution and accuracy (Table 6). Here we also demonstrate that in order to avoid type 2 error of attribution, the quality of SNP data obtained from WGS approaches is crucial (Table 6). For read-based discovery approaches we would like to emphasize the importance of SRA data availability, which is not only foundational to determine coverage and quality of detected SNP positions, but also to optimize assembly quality should assemblers with improved algorithms become available (Supplemental Table 1). SNP discovery with an appropriate outbreak-specific reference strain is critical for reference based WGS typing. To fully assess the genomic plasticity, the reference should be phylogenetically related and not too distant to the strains of interest, as evidenced by the resolution power gained using a within outbreak reference (Figures 2, 4, 5). By extending our analysis to the mobilome, we detected plasticity among clonal strains in phage and plasmid content describing novel plasmids not previously associated with E. coli O157:H7. We also would like to stress the importance of publicly available strain associated clinical, environmental, and epidemiological metadata concomitantly to the genomic data as prerequisite for informed source attribution (Table 6) (Eppinger and Cebula, 2015). We anticipate that NGS long-read technology, such as contemporary SMRT technology (English et al., 2012), or other platforms under development (Feng et al., 2015; Rhoads and Au, 2015) will tremendously benefit WGS typing strategies as it pertains to the highly homogenous E. coli O157:H7 lineage (Zhang et al., 2006, 2007; Eppinger et al., 2011b). In particular, long-read technologies will produce (near) closed genomes and thus allow to accurately determine the stx-virulence status by defining not only stx allele type, but also stx-converting phage combination, plasticity, and location, all factors that have been associated with alterations in Stx-production as direct mediator of EHEC disease (Ogura et al., 2015; Toro et al., 2015; Yin et al., 2015).
Table 6.
Proposed criteria and practices for SNP-based epidemiological outbreak inclusion or exclusion.
| Ideal case | |
|---|---|
| PRIMARY CRITERIA | |
| Highly credible exposure | Point source (clustered in time and space), pathogen isolated from incriminated vehicle |
| High quality sequencing | Contig numbers should be comparable to previous assemblies performed with the same assembly method, minimum coverage required will depend on technology used |
| Mobilome exclusion | Data focus on the most immutable part of the genome, most commonly the genome backbone |
| SNP validation | Second method, PCR confirmation or re-sequencing to validate mutations in discriminatory SNPs |
| SECONDARY CRITERIA | |
| Collection of multiple isolates from cases for accurate attribution | The larger the sample that demonstrates homogeneity, the greater the likelihood of a common source |
| If a common PFGE/MLVA type is present in the same region, confirmation that allelic differences exist between outbreak strain and background non-outbreak strains | |
| SNP calling | Reference and outbreak based SNP comparisons |
| Allelic frequency >=0.9 | |
| Base and mapping quality control | |
| Novel SNP | If a tolerance is set at >0 for SNPs as a cut point for assigning isolates as being from the same source, a SNP that is not in the database (i.e., apparently de novo) would be given more credence than one that has been described previously, and which probably represents a different lineage or located in a known polymorphic gene, such as rpoS |
| QUESTIONABLE CRITERIA | |
| Variances in the complementary mobilome, such as presence of plasmids and phages | Loci of likely mobile origin are not reliable as a differentiating trait for epidemiologic purposes |
| BEST PRACTICES IN REPORTING RESULTS | |
| Report exclusion criteria | A list of loci and regions that were excluded from SNP discovery |
| Provide list of SNP loci and alleles | Provide information on location of SNP, product, resulting codon change |
| Provide reads | Deposition of sequences and strain associated metadata in public repositories |
Our data provide insight into the maximum number of permissible SNPs two strains can have and still designate them of the same origin. In prior work, we found no SNPs between 24 isolates of the same point-source cluster, focusing on backbone ORFs (Turabelidze et al., 2013). Dallman et al. and others tolerated up to 4 SNPs in the core genome before assigning two isolates to different sources (Underwood et al., 2013; Joensen et al., 2014; Dallman et al., 2015; Holmes et al., 2015). We found one bona fide SNP in the course of a single point-source, short-term outbreak. Since no gold standards have yet been accepted for E. coli O157:H7 WGS typing we propose the following criteria (Table 6) for inclusion (presumably of same source) vs. exclusion (presumably of different source) of investigated isolates: (i) High-quality whole genome sequence fortified with extensive epidemiological outbreak data, (ii) genome-scale SNP discovery based on high quality sequencing with reference, (iii) exclusion of mobilome and repeats (to reduce epidemiological noise), followed by (iv) PCR-confirmation of eventual SNPs for definitive in-/exclusion, and (v) mobilome discovery which can significantly contribute to the genomic plasticity. Moreover, for cases that are quite dispersed in time and space, there should be greater stringency in assigning “like” status to two strains that are even differentiated by a single SNP. When outbreaks occur, there are often large product liability issues at stake, and considerable obligation on disease control authorities to identify such clusters and molecular typing serves an increasingly important role. Therefore, diligence should be exercised in choice of sequence-based typing protocols, and in their analysis.
Finally, while we eagerly anticipate the introduction of sequence-based pathogen typing as a public health and disease prevention tool (Sadiq et al., 2014; Eppinger and Cebula, 2015), we share the concern of Osterholm (2015), who stresses that this powerful technology be employed as an adjunct to, and not a replacement for, case interviewing (descriptive epidemiology) and environmental investigations. Also, we are entering an era of non-culture diagnosis of enteric infections, including those caused by E. coli O157:H7 (Schatz and Phillippy, 2012; Klemm and Dougan, 2016). The high resolution data presented in this article would not have been possible without classic diagnostic microbiology laboratory recovery of the pathogen of interest. We hope that resources will be devoted to recovering these agents from submitted specimens, so as to complement case investigation by local healthy jurisdictions.
Accession number
The sequence data sets analyzed in this study have been retrieved from the short read archive (SRA) and whole genome shotgun repository at NCBI. Accession numbers for the genomes are provided in Supplemental Table 1.
Author contributions
Conceived and designed the experiments: ME. Analyzed the data: BR, FS, SK, MM, PT, ME. Contributed reagents/materials/analysis tools: MM, PT, ME. Wrote the paper: BR, SK, MM, PT, ME.
Funding
The study was supported by the National Institute of Allergy and Infectious Diseases, National Institute of Health, Department of Health and Human Services under contract SC2AI120941, the US Department of Homeland Security under contract 2014-ST-062-000058, the Department of Biology, the South Texas Center for Emerging Infectious Diseases (STCEID) at the University of Texas at San Antonio, and the High Performance Computing Center (HPC) under contract 2G12RR013646-12. Strain collection and archiving was funded by R01DK52081 and P30DK052574 for the Biobank Core. BR was supported in part by the Swiss National Science Foundation Early Postdoc Mobility Fellowship (P2LAP3-151770). FS was supported in part by the South Texas Center for Emerging Infectious Diseases (STCEID) and through an UTSA Teaching Fellowship (UTF).
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work received computational support from Computational System Biology Core, funded by the National Institute on Minority Health and Health Disparities (G12MD007591) from the National Institutes of Health. We would like to thank Armando L. Rodriguez for technical support and maintenance of the Galaxy platform and Dr. Anna Allue-Guardia and Heidi Gildersleeve for critically reading the manuscript. Further we are grateful to Dr. Nurmohammad Shaikh for assistance with SNP PCR confirmation. We are also grateful to the diagnostic microbiologists, whose diligence in identifying the isolates on the original agar plates were necessary to the subsequent sequences that underlie our analysis. We are further indebted to the various outbreak investigation teams that identified the outbreaks described in this manuscript.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2016.00985
References
- Abu-Ali G. S., Lacher D. W., Wick L. M., Qi W., Whittam T. S. (2009). Genomic diversity of pathogenic Escherichia coli of the EHEC 2 clonal complex. BMC Genomics 10:296. 10.1186/1471-2164-10-296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Ali G. S., Ouellette L. M., Henderson S. T., Lacher D. W., Riordan J. T., Whittam T. S., et al. (2010). Increased adherence and expression of virulence genes in a lineage of Escherichia coli O157:H7 commonly associated with human infections. PLoS ONE 5:e10167. 10.1371/journal.pone.0010167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- Asadulghani M., Ogura Y., Ooka T., Itoh T., Sawaguchi A., Iguchi A., et al. (2009). The defective prophage pool of Escherichia coli O157: prophage-prophage interactions potentiate horizontal transfer of virulence determinants. PLoS Pathog. 5:e1000408. 10.1371/journal.ppat.1000408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A., Nurk S., Antipov D., Gurevich A. A., Dvorkin M., Kulikov A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatson S. A., Ben Zakour N. L., Totsika M., Forde B. M., Watts R. E., Mabbett A. N., et al. (2015). Molecular analysis of asymptomatic bacteriuria Escherichia coli strain VR50 reveals adaptation to the urinary tract by gene acquisition. Infect. Immun. 83, 1749–1764. 10.1128/IAI.02810-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell B. P., Goldoft M., Griffin P. M., Davis M. A., Gordon D. C., Tarr P. I., et al. (1994). A multistate outbreak of Escherichia coli O157:H7-associated bloody diarrhea and hemolytic uremic syndrome from hamburgers. The Washington experience. JAMA 272, 1349–1353. 10.1001/jama.1994.03520170059036 [DOI] [PubMed] [Google Scholar]
- Bentley S. D., Parkhill J. (2015). Genomic perspectives on the evolution and spread of bacterial pathogens. Proc. Biol. Sci. 282:20150488. 10.1098/rspb.2015.0488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenger B. M., Berry C., Peterson T., Fach P., Delannoy S., Li V., et al. (2015). The utility of multiple molecular methods including whole genome sequencing as tools to differentiate Escherichia coli O157:H7 outbreaks. Euro Surveill. 20:30073. 10.2807/1560-7917.ES.2015.20.47.30073 [DOI] [PubMed] [Google Scholar]
- Besser T. E., Shaikh N., Holt N. J., Tarr P. I., Konkel M. E., Malik-Kale P., et al. (2007). Greater diversity of Shiga toxin-encoding bacteriophage insertion sites among Escherichia coli O157:H7 isolates from cattle than in those from humans. Appl. Environ. Microbiol. 73, 671–679. 10.1128/AEM.01035-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnin R. A., Poirel L., Carattoli A., Nordmann P. (2012). Characterization of an IncFII plasmid encoding NDM-1 from Escherichia coli ST131. PLoS ONE 7:e34752. 10.1371/journal.pone.0034752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler J. (2001). Efficient large-scale sequence comparison by locality-sensitive hashing. Bioinformatics 17, 419–428. 10.1093/bioinformatics/17.5.419 [DOI] [PubMed] [Google Scholar]
- Burland V., Shao Y., Perna N. T., Plunkett G., Sofia H. J., Blattner F. R. (1998). The complete DNA sequence and analysis of the large virulence plasmid of Escherichia coli O157:H7. Nucleic Acids Res. 26, 4196–4204. 10.1093/nar/26.18.4196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell A. M. (1992). Chromosomal insertion sites for phages and plasmids. J. Bacteriol. 174, 7495–7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornick N. A., Jelacic S., Ciol M. A., Tarr P. I. (2002). Escherichia coli O157:H7 infections: discordance between filterable fecal shiga toxin and disease outcome. J. Infect. Dis. 186, 57–63. 10.1086/341295 [DOI] [PubMed] [Google Scholar]
- Cote R., Katani R., Moreau M. R., Kudva I. T., Arthur T. M., Debroy C., et al. (2015). Comparative analysis of super-shedder strains of Escherichia coli O157:H7 reveals distinctive genomic features and a strongly aggregative adherent phenotype on bovine rectoanal junction squamous epithelial cells. PLoS ONE 10:e0116743. 10.1371/journal.pone.0116743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallman T. J., Byrne L., Ashton P. M., Cowley L. A., Perry N. T., Adak G., et al. (2015). Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin. Infect. Dis. 61, 305–312. 10.1093/cid/civ318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling A. E., Mau B., Perna N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 5:e11147. 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. A., Hancock D. D., Besser T. E., Call D. R. (2003). Evaluation of pulsed-field gel electrophoresis as a tool for determining the degree of genetic relatedness between strains of Escherichia coli O157:H7. J. Clin. Microbiol. 41, 1843–1849. 10.1128/JCM.41.5.1843-1849.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher A. L., Salzberg S. L., Phillippy A. M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics Chapter 10:Unit 10.13. 10.1002/0471250953.bi1003s00 [DOI] [PubMed] [Google Scholar]
- den Bakker H. C., Allard M. W., Bopp D., Brown E. W., Fontana J., Iqbal Z., et al. (2014). Rapid whole-genome sequencing for surveillance of Salmonella enterica serovar enteritidis. Emerg. Infect. Dis. 20, 1306–1314. 10.3201/eid2008.131399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov G., Walenz B., Halpern A. L., Miller J., Axelrod N., Levy S., et al. (2008). Consensus generation and variant detection by Celera assembler. Bioinformatics 24, 1035–1040. 10.1093/bioinformatics/btn074 [DOI] [PubMed] [Google Scholar]
- Donohue-Rolfe A., Kondova I., Oswald S., Hutto D., Tzipori S. (2000). Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J. Infect. Dis. 181, 1825–1829. 10.1086/315421 [DOI] [PubMed] [Google Scholar]
- Dudley E. G., Abe C., Ghigo J. M., Latour-Lambert P., Hormazabal J. C., Nataro J. P. (2006). An IncI1 plasmid contributes to the adherence of the atypical enteroaggregative Escherichia coli strain C1096 to cultured cells and abiotic surfaces. Infect. Immun. 74, 2102–2114. 10.1128/IAI.74.4.2102-2114.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elhadidy M., Elkhatib W. F., Piérard D., De Reu K., Heyndrickx M. (2015). Model-based clustering of Escherichia coli O157:H7 genotypes and their potential association with clinical outcome in human infections. Diagn. Microbiol. Infect. Dis. 83, 198–202. 10.1016/j.diagmicrobio.2015.06.016 [DOI] [PubMed] [Google Scholar]
- English A. C., Richards S., Han Y., Wang M., Vee V., Qu J., et al. (2012). Mind the gap: upgrading genomes with Pacific biosciences RS long-read sequencing technology. PLoS ONE 7:e47768. 10.1371/journal.pone.0047768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger M., Cebula T. A. (2015). Future perspectives, applications and challenges of genomic epidemiology studies for food-borne pathogens: a case study of Enterohemorrhagic Escherichia coli (EHEC) of the O157:H7 serotype. Gut Microbes 6, 194–201. 10.4161/19490976.2014.969979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger M., Daugherty S., Agrawal S., Galens K., Sengamalay N., Sadzewicz L., et al. (2013). Whole-genome draft sequences of 26 enterohemorrhagic Escherichia coli O157:H7 strains. Genome Announc. 1:e00134-00112. 10.1128/genomeA.00134-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger M., Mammel M. K., Leclerc J. E., Ravel J., Cebula T. A. (2011a). Genome Signatures of Escherichia coli O157:H7 Isolates from the bovine host reservoir. Appl. Environ. Microbiol. 77, 2916–2925. 10.1128/AEM.02554-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger M., Mammel M. K., Leclerc J. E., Ravel J., Cebula T. A. (2011b). Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. U.S.A. 108, 20142–20147. 10.1073/pnas.1107176108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger M., Pearson T., Koenig S. S., Pearson O., Hicks N., Agrawal S., et al. (2014). Genomic epidemiology of the Haitian cholera outbreak: a single introduction followed by rapid, extensive, and continued spread characterized the onset of the epidemic. MBio 5:e01721. 10.1128/mBio.01721-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eppinger M., Worsham P. L., Nikolich M. P., Riley D. R., Sebastian Y., Mou S., et al. (2010). Genome sequence of the deep-rooted yersinia pestis strain angola reveals new insights into the evolution and pangenome of the plague bacterium. J. Bacteriol. 192, 1685–1699. 10.1128/JB.01518-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng P. C., Monday S. R., Lacher D. W., Allison L., Siitonen A., Keys C., et al. (2007). Genetic diversity among clonal lineages within Escherichia coli O157:H7 stepwise evolutionary model. Emerg. Infect. Dis. 13, 1701–1706. 10.3201/eid1311.070381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng P., Lampel K. A., Karch H., Whittam T. S. (1998). Genotypic and phenotypic changes in the emergence of Escherichia coli O157:H7. J. Infect. Dis. 177, 1750–1753. 10.1086/517438 [DOI] [PubMed] [Google Scholar]
- Feng Y., Zhang Y., Ying C., Wang D., Du C. (2015). Nanopore-based fourth-generation DNA sequencing technology. Genomics Proteomics Bioinformatics 13, 4–16. 10.1016/j.gpb.2015.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn R. D., Coggill P., Eberhardt R. Y., Eddy S. R., Mistry J., Mitchell A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. 10.1093/nar/gkv1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogg P. C., Saunders J. R., McCarthy A. J., Allison H. E. (2012). Cumulative effect of prophage burden on Shiga toxin production in Escherichia coli. Microbiology 158, 488–497. 10.1099/mic.0.054981-0 [DOI] [PubMed] [Google Scholar]
- Fratamico P. M., Yan X., Caprioli A., Esposito G., Needleman D. S., Pepe T., et al. (2011). The complete DNA sequence and analysis of the virulence plasmid and of five additional plasmids carried by Shiga toxin-producing Escherichia coli O26:H11 strain H30. Int. J. Med. Microbiol. 301, 192–203. 10.1016/j.ijmm.2010.09.002 [DOI] [PubMed] [Google Scholar]
- Freedman S. B., Xie J., Neufeld M. S., Hamilton W. L., Hartling L., Tarr P. I., et al. (2016). Shiga toxin-producing Escherichia coli Infection, antibiotics, and risk of developing hemolytic uremic syndrome: a meta-analysis. Clin. Infect. Dis. 62, 1251–1258. 10.1093/cid/ciw099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke W. F., Mammel M. K., McDermott P. F., Tartera C., White D. G., Leclerc J. E., et al. (2011). Comparative genomics of 28 Salmonella enterica isolates: evidence for CRISPR-mediated adaptive sublineage evolution. J. Bacteriol. 193, 3556–3568. 10.1128/JB.00297-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedrich A. W., Bielaszewska M., Zhang W. L., Pulz M., Kuczius T., Ammon A., et al. (2002). Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 185, 74–84. 10.1086/338115 [DOI] [PubMed] [Google Scholar]
- Garrison E., Marth G. (2012). Haplotype-Based Variant Detection from Short-Read Sequencing. ArXiv e-prints [Online], 1207. Available online at: http://adsabs.harvard.edu/abs/2012arXiv1207.3907G (Accessed July 1, 2012).
- Gibson M. K., Wang B., Ahmadi S., Burnham C.-A. D., Tarr P. I., Dantas G., et al. (2016). Developmental dynamics of the preterm infant gut microbiota and antibiotic resistome. Nat. Microbiol. 1:16024 10.1038/nmicrobiol.2016.24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist C. A., Turner S. D., Riley M. F., Petri W. A., Jr., Hewlett E. L. (2015). Whole-genome sequencing in outbreak analysis. Clin. Microbiol. Rev. 28, 541–563. 10.1128/CMR.00075-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goecks J., Nekrutenko A., Taylor J., Galaxy T. (2010). Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 11:R86. 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldwater P. N., Bettelheim K. A. (2012). Treatment of enterohemorrhagic Escherichia coli (EHEC) infection and hemolytic uremic syndrome (HUS). BMC Med. 10:12. 10.1186/1741-7015-10-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffing S. M., MacCannell D. R., Schmidtke A. J., Freeman M. M., Hyytia-Trees E., Gerner-Smidt P., et al. (2015). Canonical single nucleotide polymorphisms (SNPs) for high-resolution subtyping of shiga-toxin producing Escherichia coli (STEC) O157:H7. PLoS ONE 10:e0131967. 10.1371/journal.pone.0131967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasan N. A., Choi S. Y., Eppinger M., Clark P. W., Chen A., Alam M., et al. (2012). Genomic diversity of 2010 Haitian cholera outbreak strains. Proc. Natl. Acad. Sci. U.S.A. 109, E2010–E2017. 10.1073/pnas.1207359109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T., Makino K., Ohnishi M., Kurokawa K., Ishii K., Yokoyama K., et al. (2001). Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 8, 11–22. 10.1093/dnares/8.1.11 [DOI] [PubMed] [Google Scholar]
- Holmes A., Allison L., Ward M., Dallman T. J., Clark R., Fawkes A., et al. (2015). Utility of whole-genome sequencing of escherichia coli o157 for outbreak detection and epidemiological surveillance. J. Clin. Microbiol. 53, 3565–3573. 10.1128/JCM.01066-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt D., Chen G. L., Locascio P. F., Land M. L., Larimer F. W., Hauser L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Institute C. A. L. S. (2013). Performance Standards for Antimicrobial Susceptibility Testing: 23rd Informational Supplement. Wayne, PA: CLSI. [Google Scholar]
- Jenkins C., Dallman T. J., Launders N., Willis C., Byrne L., Jorgensen F., et al. (2015). Public health investigation of two outbreaks of shiga toxin-producing Escherichia coli O157 associated with consumption of watercress. Appl. Environ. Microbiol. 81, 3946–3952. 10.1128/AEM.04188-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joensen K. G., Scheutz F., Lund O., Hasman H., Kaas R. S., Nielsen E. M., et al. (2014). Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 52, 1501–1510. 10.1128/JCM.03617-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karmali M. A., Gannon V., Sargeant J. M. (2010). Verocytotoxin-producing Escherichia coli (VTEC). Vet. Microbiol. 140, 360–370. 10.1016/j.vetmic.2009.04.011 [DOI] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinheinz K. A., Joensen K. G., Larsen M. V. (2014). Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 4:e27943. 10.4161/bact.27943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm E., Dougan G. (2016). Advances in understanding bacterial pathogenesis gained from whole-genome sequencing and phylogenetics. Cell Host Microbe 19, 599–610. 10.1016/j.chom.2016.04.015 [DOI] [PubMed] [Google Scholar]
- Kotewicz M. L., Mammel M. K., LeClerc J. E., Cebula T. A. (2008). Optical mapping and 454 sequencing of Escherichia coli O157:H7 isolates linked to the US 2006 spinach-associated outbreak. Microbiology 154, 3518–3528. 10.1099/mic.0.2008/019026-0 [DOI] [PubMed] [Google Scholar]
- Kröger C., Dillon S. C., Cameron A. D., Papenfort K., Sivasankaran S. K., Hokamp K., et al. (2012). The transcriptional landscape and small RNAs of Salmonella enterica serovar Typhimurium. Proc. Natl. Acad. Sci. U.S.A. 109, E1277–E1286. 10.1073/pnas.1201061109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger A., Lucchesi P. M. (2015). Shiga toxins and stx phages: highly diverse entities. Microbiology 161, 451–462. 10.1099/mic.0.000003 [DOI] [PubMed] [Google Scholar]
- Kulasekara B. R., Jacobs M., Zhou Y., Wu Z., Sims E., Saenphimmachak C., et al. (2009). Analysis of the genome of the Escherichia coli O157:H7 2006 spinach-associated outbreak isolate indicates candidate genes that may enhance virulence. Infect. Immun. 77, 3713–3721. 10.1128/IAI.00198-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyle J. L., Cummings C. A., Parker C. T., Quiñones B., Vatta P., Newton E., et al. (2012). Escherichia coli serotype O55:H7 diversity supports parallel acquisition of bacteriophage at Shiga toxin phage insertion sites during evolution of the O157:H7 lineage. J. Bacteriol. 194, 1885–1896. 10.1128/JB.00120-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert D., Carrillo C. D., Koziol A. G., Manninger P., Blais B. W. (2015). GeneSippr: a rapid whole-genome approach for the identification and characterization of foodborne pathogens such as priority Shiga toxigenic Escherichia coli. PLoS ONE 10:e0122928. 10.1371/journal.pone.0122928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B., Salzberg S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latif H., Li H. J., Charusanti P., Palsson B. Ø., Aziz R. K. (2014). A Gapless, unambiguous genome sequence of the enterohemorrhagic Escherichia coli O157:H7 strain EDL933. Genome Announc. 2:e00821-14. 10.1128/genomeA.00821-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latreille P., Norton S., Goldman B. S., Henkhaus J., Miller N., Barbazuk B., et al. (2007). Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics 8:321. 10.1186/1471-2164-8-321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leekitcharoenphon P., Nielsen E. M., Kaas R. S., Lund O., Aarestrup F. M. (2014). Evaluation of whole genome sequencing for outbreak detection of Salmonella enterica. PLoS ONE 9:e87991. 10.1371/journal.pone.0087991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leopold S. R., Magrini V., Holt N. J., Shaikh N., Mardis E. R., Cagno J., et al. (2009). A precise reconstruction of the emergence and constrained radiations of Escherichia coli O157 portrayed by backbone concatenomic analysis. Proc. Natl. Acad. Sci. U.S.A. 106, 8713–8718. 10.1073/pnas.0812949106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B., Tromp J., Li M. (2002). PatternHunter: faster and more sensitive homology search. Bioinformatics 18, 440–445. 10.1093/bioinformatics/18.3.440 [DOI] [PubMed] [Google Scholar]
- Maddison W. P., Maddison D. R. (2015). Mesquite: a Modular System for Evolutionary Analysis. Version 3.04. [Google Scholar]
- Makino K., Ishii K., Yasunaga T., Hattori M., Yokoyama K., Yutsudo C. H., et al. (1998). Complete nucleotide sequences of 93-kb and 3.3-kb plasmids of an enterohemorrhagic Escherichia coli O157:H7 derived from Sakai outbreak. DNA Res. 5, 1–9. 10.1093/dnares/5.1.1 [DOI] [PubMed] [Google Scholar]
- Manning S. D., Motiwala A. S., Springman A. C., Qi W., Lacher D. W., Ouellette L. M., et al. (2008). Variation in virulence among clades of Escherichia coli O157:H7 associated with disease outbreaks. Proc. Natl. Acad. Sci. U.S.A. 105, 4868–4873. 10.1073/pnas.0710834105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning S. D., Whittam T. S., Steinsland H. (2006). EcMLST [Online]. Available online at: http://www.shigatox.net (Accessed September 25 2014).
- Mellor G. E., Fegan N., Gobius K. S., Smith H. V., Jennison A. V., D'astek B. A., et al. (2015). Geographically distinct Escherichia coli O157 isolates differ by lineage, Shiga toxin genotype, and total shiga toxin production. J. Clin. Microbiol. 53, 579–586. 10.1128/JCM.01532-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J., Zhao S., Doyle M. P., Joseph S. W. (1998). Antibiotic resistance of Escherichia coli O157:H7 and O157:NM isolated from animals, food, and humans. J. Food Prot. 61, 1511–1514. [DOI] [PubMed] [Google Scholar]
- Meng J., Zhao S., Zhao T., Doyle M. P. (1995). Molecular characterisation of Escherichia coli O157:H7 isolates by pulsed-field gel electrophoresis and plasmid DNA analysis. J. Med. Microbiol. 42, 258–263. 10.1099/00222615-42-4-258 [DOI] [PubMed] [Google Scholar]
- Morelli G., Song Y., Mazzoni C. J., Eppinger M., Roumagnac P., Wagner D. M., et al. (2010). Yersinia pestis genome sequencing identifies patterns of global phylogenetic diversity. Nat. Publish. Group 42, 1140–1143. 10.1038/ng.705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munns K. D., Zaheer R., Xu Y., Stanford K., Laing C. R., Gannon V. P., et al. (2016). Comparative Genomic Analysis of Escherichia coli O157:H7 isolated from super-shedder and low-shedder cattle. PLoS ONE 11:e0151673. 10.1371/journal.pone.0151673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers G. S., Mathews S. A., Eppinger M., Mitchell C., O'Brien K. K., White O. R., et al. (2009). Evidence that human Chlamydia pneumoniae was zoonotically acquired. J. Bacteriol. 191, 7225–7233. 10.1128/JB.00746-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataro J. P., Kaper J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y., Mondal S. I., Islam M. R., Mako T., Arisawa K., Katsura K., et al. (2015). The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 5:16663. 10.1038/srep16663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterholm M. T. (2015). Editorial commentary: the detection of and response to a foodborne disease outbreak: a cautionary tale. Clin. Infect. Dis. 61, 910–911. 10.1093/cid/civ434 [DOI] [PubMed] [Google Scholar]
- Ostroff S. M., Tarr P. I., Neill M. A., Lewis J. H., Hargrett-Bean N., Kobayashi J. M. (1989). Toxin genotypes and plasmid profiles as determinants of systemic sequelae in Escherichia coli O157:H7 infections. J. Infect. Dis. 160, 994–998. 10.1093/infdis/160.6.994 [DOI] [PubMed] [Google Scholar]
- Perna N. T., Plunkett G., III, Burland V., Mau B., Glasner J. D., Rose D. J., et al. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529–533. 10.1038/35054089 [DOI] [PubMed] [Google Scholar]
- Persson S., Olsen K. E., Ethelberg S., Scheutz F. (2007). Subtyping method for Escherichia coli shiga toxin (verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 45, 2020–2024. 10.1128/JCM.02591-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettengill J. B., Luo Y., Davis S., Chen Y., Gonzalez-Escalona N., Ottesen A., et al. (2014). An evaluation of alternative methods for constructing phylogenies from whole genome sequence data: a case study with Salmonella. PeerJ 2:e620. 10.7717/peerj.620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhoads A., Au K. F. (2015). PacBio Sequencing and its applications. Genomics Proteomics Bioinformatics 13, 278–289. 10.1016/j.gpb.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riordan J. T., Viswanath S. B., Manning S. D., Whittam T. S. (2008). Genetic differentiation of Escherichia coli O157:H7 clades associated with human disease by real-time PCR. J. Clin. Microbiol. 46, 2070–2073. 10.1128/JCM.00203-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusconi B., Eppinger M. (2016). Whole genome sequence typing for strategies for enterohemorrhagic Escherichia coli of the O157:H7 serotype, in The Handbook of Microbial Bioresources, eds Gupta V. K., Sharma G. D., Tuohy M. G., Gaur R. (Wallingford, CT: Commonwealth Agricultural Bureaux International (CABI)), 656. [Google Scholar]
- Russo L. M., Melton-Celsa A. R., O'Brien A. D. (2016). Shiga Toxin (Stx) Type 1a reduces the Oral Toxicity of Stx Type 2a. J. Infect. Dis. 213, 1271–1279. 10.1093/infdis/jiv557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadiq M. S., Hazen T. H., Rasko D. A., Eppinger M. (2014). EHEC genomics: Past, Present, and Future, in Enterohemorrhagic Escherichia coli and other shiga Toxin-Producing E. coli, eds Sperandio V., Hovde C. J. (Herndon, VA: ASM Press; ), 55–71. [Google Scholar]
- Saeed A. I., Sharov V., White J., Li J., Liang W., Bhagabati N., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. BioTechniques 34, 374–378. [DOI] [PubMed] [Google Scholar]
- Sahl J. W., Caporaso J. G., Rasko D. A., Keim P. (2014). The large-scale blast score ratio (LS-BSR) pipeline: a method to rapidly compare genetic content between bacterial genomes. Peer J. 2:e332. 10.7717/peerj.332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samadpour M., Stewart J., Steingart K., Addy C., Louderback J., Mcginn M., et al. (2002). Laboratory investigation of an E. coli O157:H7 outbreak associated with swimming in Battle Ground Lake, Vancouver, Washington. J. Environ. Health 64, 16–20. 26, 25. [PubMed] [Google Scholar]
- Sanjar F., Hazen T. H., Shah S. M., Koenig S. S. K., Agrawal S., Daugherty S., et al. (2014). Genome sequence of Escherichia coli O157:H7 Strain 2886-75, associated with the first reported case of human infection in the united states. Genome Announc. 2:e01120-13. 10.1128/genomeA.01120-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjar F., Rusconi B., Hazen T. H., Koenig S. S., Mammel M. K., Feng P. C., et al. (2015). Characterization of the pathogenome and phylogenomic classification of enteropathogenic Escherichia coli of the O157:non-H7 serotypes. Pathog. Dis. 73:ftv033. 10.1093/femspd/ftv033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz M. C., Phillippy A. M. (2012). The rise of a digital immune system. Gigascience 1:4. 10.1186/2047-217X-1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheutz F., Teel L. D., Beutin L., Piérard D., Buvens G., Karch H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. 10.1128/JCM.00860-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- Serra-Moreno R., Jofre J., Muniesa M. (2008). The CI repressors of Shiga toxin-converting prophages are involved in coinfection of Escherichia coli strains, which causes a down regulation in the production of Shiga toxin 2. J. Bacteriol. 190, 4722–4735. 10.1128/JB.00069-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh N., Tarr P. I. (2003). Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 185, 3596–3605. 10.1128/JB.185.12.3596-3605.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siguier P., Perochon J., Lestrade L., Mahillon J., Chandler M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. 10.1093/nar/gkj014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. L., Rooks D. J., Fogg P. C., Darby A. C., Thomson N. R., McCarthy A. J., et al. (2012). Comparative genomics of Shiga toxin encoding bacteriophages. BMC Genomics 13:311. 10.1186/1471-2164-13-311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strachan N. J., Rotariu O., Lopes B., MacRae M., Fairley S., Laing C., et al. (2015). Whole genome sequencing demonstrates that geographic variation of Escherichia coli O157 genotypes dominates host association. Sci. Rep. 5:14145. 10.1038/srep14145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan M. J., Petty N. K., Beatson S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. 10.1093/bioinformatics/btr039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarr P. I., Gordon C. A., Chandler W. L. (2005). Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet 365, 1073–1086. 10.1016/s0140-6736(05)71144-2 [DOI] [PubMed] [Google Scholar]
- Tesh V. L. (2010). Induction of apoptosis by Shiga toxins. Future Microbiol. 5, 431–453. 10.2217/fmb.10.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesh V. L., Burris J. A., Owens J. W., Gordon V. M., Wadolkowski E. A., O'Brien A. D., et al. (1993). Comparison of the relative toxicities of Shiga-like toxins type I and type II for mice. Infect. Immun. 61, 3392–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tilden J., Jr., Young W., Mcnamara A. M., Custer C., Boesel B., Lambert-Fair M. A., et al. (1996). A new route of transmission for Escherichia coli: infection from dry fermented salami. Am. J. Public Health 86, 1142–1145. 10.2105/AJPH.86.8_Pt_1.1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timme R. E., Allard M. W., Luo Y., Strain E., Pettengill J., Wang C., et al. (2012). Draft genome sequences of 21 Salmonella enterica serovar enteritidis strains. J. Bacteriol. 194, 5994–5995. 10.1128/JB.01289-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro M., Rump L. V., Cao G., Meng J., Brown E. W., Gonzalez-Escalona N. (2015). Simultaneous presence of insertion sequence-excision enhancer (IEE) and insertion sequence IS629 correlates with increased diversity and virulence in Shiga-toxin producing Escherichia coli (STEC). J. Clin. Microbiol. 53, 3466–3473. 10.1128/JCM.01349-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turabelidze G., Lawrence S. J., Gao H., Sodergren E., Weinstock G. M., Abubucker S., et al. (2013). Precise dissection of an Escherichia coli O157:H7 outbreak by single nucleotide polymorphism analysis. J. Clin. Microbiol. 51, 3950–3954. 10.1128/JCM.01930-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle J., Gomez T., Doyle M. P., Wells J. G., Zhao T., Tauxe R. V., et al. (1999). Lessons from a large outbreak of Escherichia coli O157:H7 infections: insights into the infectious dose and method of widespread contamination of hamburger patties. Epidemiol. Infect. 122, 185–192. 10.1017/S0950268898001976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlich G. A., Chen C. Y., Cottrell B. J., Hofmann C. S., Dudley E. G., Strobaugh T. P., Jr., et al. (2013). Phage insertion in mlrA and variations in rpoS limit curli expression and biofilm formation in Escherichia coli serotype O157:H7. Microbiology 159, 1586–1596. 10.1099/mic.0.066118-0 [DOI] [PubMed] [Google Scholar]
- Underwood A. P., Dallman T., Thomson N. R., Williams M., Harker K., Perry N., et al. (2013). Public health value of next-generation DNA sequencing of enterohemorrhagic Escherichia coli isolates from an outbreak. J. Clin. Microbiol. 51, 232–237. 10.1128/JCM.01696-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogeleer P., Tremblay Y. D., Mafu A. A., Jacques M., Harel J. (2014). Life on the outside: role of biofilms in environmental persistence of Shiga-toxin producing Escherichia coli. Front. Microbiol. 5:317. 10.3389/fmicb.2014.00317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogler A. J., Chan F., Wagner D. M., Roumagnac P., Lee J., Nera R., et al. (2011). Phylogeography and molecular epidemiology of Yersinia pestis in Madagascar. PLoS Negl. Trop. Dis. 5:e1319. 10.1371/journal.pntd.0001319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Stephan R., Power K., Yan Q., Hächler H., Fanning S. (2014a). Nucleotide sequences of 16 transmissible plasmids identified in nine multidrug-resistant Escherichia coli isolates expressing an ESBL phenotype isolated from food-producing animals and healthy humans. J. Antimicrob. Chemother. 69, 2658–2668. 10.1093/jac/dku206 [DOI] [PubMed] [Google Scholar]
- Wang J., Stephan R., Zurfluh K., Hächler H., Fanning S. (2014b). Characterization of the genetic environment of bla ESBL genes, integrons and toxin-antitoxin systems identified on large transferrable plasmids in multi-drug resistant Escherichia coli. Front. Microbiol. 5:716. 10.3389/fmicb.2014.00716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenbusch J. C., Swofford D. (2003). Inferring evolutionary trees with PAUP*. Curr. Protoc. Bioinformatics Chapter 6, Unit 6 4. 10.1002/0471250953.bi0604s00 [DOI] [PubMed] [Google Scholar]
- Wong C. S., Mooney J. C., Brandt J. R., Staples A. O., Jelacic S., Boster D. R., et al. (2012). Risk factors for the hemolytic uremic syndrome in children infected with Escherichia coli O157:H7: a multivariable analysis. Clin. Infect. Dis. 55, 33–41. 10.1093/cid/cis299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright M. S., Perez F., Brinkac L., Jacobs M. R., Kaye K., Cober E., et al. (2014). Population structure of KPC-producing Klebsiella pneumoniae isolates from midwestern U.S. hospitals. Antimicrob. Agents Chemother. 58, 4961–4965. 10.1128/AAC.00125-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y., Wang P., Lan R., Ye C., Wang H., Ren J., et al. (2012). A novel Escherichia coli O157:H7 clone causing a major hemolytic uremic syndrome outbreak in China. PLoS ONE 7:e36144. 10.1371/journal.pone.0036144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S., Rusconi B., Sanjar F., Goswami K., Xiaoli L. Z., Eppinger M., et al. (2015). Escherichia coli O157:H7 strains harbor at least three distinct sequence types of Shiga toxin 2a-converting phages. BMC Genomics 16:733. 10.1186/s12864-015-1934-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Qi W., Albert T. J., Motiwala A. S., Alland D., Hyytia-Trees E. K., et al. (2006). Probing genomic diversity and evolution of Escherichia coli O157 by single nucleotide polymorphisms. Genome Res. 16, 757–767. 10.1101/gr.4759706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Laing C., Steele M., Ziebell K., Johnson R., Benson A. K., et al. (2007). Genome evolution in major Escherichia coli O157:H7 lineages. BMC Genomics 8:121. 10.1186/1471-2164-8-121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S., White D. G., Friedman S. L., Glenn A., Blickenstaff K., Ayers S. L., et al. (2008). Antimicrobial resistance in Salmonella enterica serovar Heidelberg isolates from retail meats, including poultry, from 2002 to 2006. Appl. Environ. Microbiol. 74, 6656–6662. 10.1128/AEM.01249-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S., Bechner M. C., Place M., Churas C. P., Pape L., Leong S. A., et al. (2007). Validation of rice genome sequence by optical mapping. BMC Genomics 8:278. 10.1186/1471-2164-8-278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Liang Y., Lynch K. H., Dennis J. J., Wishart D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. 10.1093/nar/gkr485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Li X., Liu B., Beutin L., Xu J., Ren Y., et al. (2010). Derivation of Escherichia coli O157:H7 from its O55:H7 precursor. PLoS ONE 5:e8700. 10.1371/journal.pone.0008700 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.