

Abstract

The synthesis of functional polymers encoded with biomolecules has been an extensive area of research for decades. As such, a diverse toolbox of polymerization techniques and bioconjugation methods has been developed. The greatest impact of this work has been in biomedicine and biotechnology, where fully synthetic and naturally derived biomolecules are used cooperatively. Despite significant improvements in biocompatible and functionally diverse polymers, our success in the field is constrained by recognized limitations in polymer architecture control, structural dynamics, and biostabilization. This Perspective discusses the current status of functional biosynthetic polymers and highlights innovative strategies reported within the past five years that have made great strides in overcoming the aforementioned barriers.
Introduction
The structure and function of biopolymers found in nature has evolved over the past few billion years to form the underpinnings of life. Biosynthetic polymeric materials exemplify the diversity accessible through synthesis and semisynthesis that is inspired by and utilizes biopolymers (Figure 1a). Hancock and Ludersdorf prepared the first artificial polymer in 1840, through treatment of natural rubber with sulfur to create a tough and elastic material.1 It was another century before significant advances in polymer chemistry would enable the production of totally synthetic and complex polymeric materials. Within the past few decades, biologically compatible synthetic materials have emerged as one of the most exciting and prolific fields in polymer chemistry due to the widespread adoption of living and controlled polymerization methods (Figure 1b). These materials, herein referred to as biosynthetic polymers, are now used for a multitude of applications such as novel biomolecule stabilizers, drug-delivery vehicles, therapeutics, biosensors, biomedical adhesives, antifouling materials, and biomimetic scaffolds.2−5
Figure 1.

Various architectures of functional biosynthetic polymers via the conjugation of natural and synthetic moieties. (a) Biopolymers (polysaccharides, polynucleic acids, oligopeptides, and proteins) and their building blocks (nucleotides, monosaccharides, and amino acids) may be combined with (b) synthetic polymers (black) via a variety of polymerization methods. Representative controlled chain growth polymerization methods are depicted above uncontrolled versions. (c) The resulting functional biosynthetic polymers may act as an unstructured conjugate with various architectures or conjugate assemblies.
Biosynthetic polymers are materials that combine synthetic components with biopolymers or moieties prepared as mimics of those found in nature (Figure 1c).6 These materials consist of (a) synthetically modified biopolymers, such as functionalized hyaluronic acid derivatives7 or labeled proteins via cell-instruction.8 In the prior case concerning biopolymers such as polysaccharides or proteins, wherein reactive sites (amine, hydroxyl, thiol, carboxylic acid) are conventionally present as multiple copies, site-specific conjugation (graft-to) and subsequent purification are typically difficult. Other categories of biosynthetic polymers that enable more precise control over advanced architectures, functionalization, and subsequently dynamic function are (b) biomolecules conjugated to synthetic polymers produced by various grafting strategies9,10 or (c) bioinspired or fully synthetic polymers that act as biopolymer surrogates, which execute similar functions and occasionally exceed the performance of biopolymers.11 Considerable effort has been directed toward increasing the precision by which biomolecules are incorporated into polymers—in other words, expanding the so-called “bioconjugate toolbox”.12−14 With the advent of “click”-type chemistries, i.e. oxime,15,16 Staudinger ligation,17,18 thiol–ene,19 copper-catalyzed azide–alkyne cycloaddition (CuAAC),20 and strain-promoted azide/alkyne click (SPAAC),21 among others,22,23 biomolecule–polymer conjugates are not only readily attainable but achieve higher fidelity. Though many reports document interesting advancements, the focus of this paper is not to catalogue conjugation strategies but to describe paradigm shifts in the development of functional biosynthetic polymers.
As such, this Perspective discusses exciting and recently published works that address some of the most significant problems which still hinder progress in the field of functional biosynthetic polymers: (1) architecture control of synthetic components, (2) structural dynamics of polymer assemblies, and (3) biostabilization (storage, release, and bioresistance) of therapeutic cargos. Most of the examples discussed herein utilize controlled polymerization methods for addressing previously unmet challenges in architecture control and functional complexity, with some exceptions in which uncontrolled polymerization methods are necessary for simplicity and expense mitigation. The first section will discuss fundamental advances in polymer chemistry toward controlling primary sequence, tacticity, and functionality via grafting, which are paramount for the execution of complex biological functions, as demonstrated by the precise stereoregularity of biopolymers (i.e., proteins and DNA). The next two sections highlight significant progress made in advanced bulk functionality of unstructured and/or assembled biosynthetic polymers. Provided the number of articles that detail incremental advances in functionalization methods using various stimuli-responsive moieties, the authors abstain from a comprehensive discussion and encourage the reader to refer to published reviews on these topics.12,24,25 Throughout this Perspective, the merits of simple formulations for designing highly functional biosynthetic polymers are discussed. The conclusion gives a projected outlook on further progress in the field that hinges on the ability to overcome recurring limitations. In this context, our creative efforts to surpass what evolution has perfected are just beginning. In the foreseeable future, further advancements will no longer rely on copying nature for solutions but rather emerge from the limits of our own imagination.
Architecture Control
The basic informational biopolymers from which all life on earth is built are carbohydrates, nucleic acids, and proteins,26 with noninformational biogenic polymers including melanins making tremendous functional contributions.27 Biological organisms are capable of producing biopolymers with extreme complexity and high fidelity and accuracy, while using robust machinery and only a handful of simple monomers including saccharides, nucleotides, amino acids (and their derivatives), other metabolites, and fatty acids.26 Naturally, our current knowledge and abilities in the field of polymer chemistry pale in comparison to that achieved by billions of years of evolution. Nonetheless, researchers have devoted substantial efforts to synthesize polymers using libraries of novel monomers possessing different physical and chemical properties with diversities far beyond those prevalent in biological systems. Furthermore, strategic organization of these polymeric monomers can enhance the polymer complexity and overall mode of action.28,29 In this way, semisynthetic or fully synthetic materials may be tailored to mimic the highly versatile and functional properties of biopolymers. The following discussion focuses on current efforts to increase the control of polymer architecture as well as microstructure such as the specific arrangement of monomer sequence and stereoisomers.
Primary Sequence Control
Controlled polymerizations may be iterative, step growth, or chain growth in mechanism. In contrast to the others, chain growth strategies generally lack control over primary sequence despite controlled polymerization techniques. The discovery of living polymerizations by Michael Szwarc in 1956 was the first breakthrough in chain growth methods, whereby growth of a polymer chain proceeds at a constant rate, affording polymers with narrow molecular weight distributions or low dispersity.30 For the synthesis of precisely controlled polymers, the majority of suitable methods encompass reversible-deactivation radical polymerization (RDRP), including (a) atom transfer radical polymerization (ATRP), Single-Electron Transfer Living Radical Polymerization (SET LRP)31,32 or Supplemental Activator and Reducing Agent (SARA),33,34 Activators Regenerated by Electron Transfer (ARGET),35−37 Electrochemically mediated ATRP (eATRP),38,39 photoinduced ATRP (Photo-ATRP),40,41 and Metal-free Photoinduced Electron Transfer ATRP (PETATRP),42 and (b) reversible addition–fragmentation chain transfer (RAFT) polymerization, with alternatives such as PET-RAFT,43,44 (c) iniferter polymerization,45 and (d) nitroxide-mediated polymerization (NMP).46−49 Other well-characterized methods include ring-opening polymerization (ROP), with common variants including organocatalyzed,50 anionic,51−53 coordination–insertion,54 enzymatic,55,56N-carboxyanhydride polymerization,57,58 and ring-opening metathesis polymerization (ROMP),59 with alternatives such as alternating ROMP (AROMP)60−62 and metal-free ROMP.63 With uncontrolled methods, one cannot achieve significant control over primary sequence or advanced architectures. Despite the expansive inventory of controlled polymerization methods available, there is still no equivalent to the kind of sequence control afforded by solid phase synthesis first pioneered by R. B. Merrifield, even with the foremost controlled polymerization methods.64 Manual or automated iterative strategies have been used to synthesize sequence-controlled polymers;65 however, these approaches often incur the expenses of unsustainable practices and time. Regardless, stepwise approaches are still advantageous for developing sequence-controlled polymers for tuning properties such as single-chain morphologies.66 Meanwhile, efforts to develop streamlined chemistries are imperative; the following section describes some recent achievements toward the ability to control primary sequence.
Hawker and co-workers reported a new strategy for ROMP of sequence-controlled polymers using a macrocyclic monomer containing distinct ABCDE-type moieties.67 Prior to this work, efforts to synthesize sequence-controlled polymers via multisubstituted cyclooctadienes using ROMP have been limited in number and types of incorporated functionalities.68,69 During chain extension, the growing polymer sequence obtained ordered repeats of ABCDE units along a polyester backbone. To achieve this, the authors used a small molecule polymerization trigger derived from saccharin to synthesize an unstrained macrocycle. Close proximity of the macrocycle olefin to a terminal alkyne enabled fast intramolecular cyclization and subsequent rapid ROMP with Grubbs third generation catalyst (G3) (Figure 2). This work demonstrates a general synthetic strategy for ROMP of diverse repeat units such as ester, sulfonamide, heterocyclic, etc., incorporated within the polymer backbone. The strategy also provides one of the few known methods for synthesizing fully biodegradable ROMP polymers,70 a recurring challenge with highly functionalized biosynthetic polymers. Improvements in the AROMP method were also reported recently. Several examples describe iterative monomer addition by Ru-promoted isomerization of bicyclo[4.2.0]oct-7-ene-7-carboxamides62 or via living copolymerization of 1,1-disubstituted cyclopropenes with low-strain cyclic olefins.61 Though there is a great need for further improvement, especially in the context of tailoring these methods toward increased chemical diversity and demonstrated biofunctionality, these approaches chronicle an exciting movement toward efforts to control the primary sequence of copolymers.
Figure 2.

Strategy for the polymerization of unstrained macrocycles enabling primary sequence control. Monomers are composed of a ROMP polymerization trigger attached to a series of glycolate (Gly), (S)-lactate (Lact), (S)-phenyllactate (PhLact), and β-alanine (βAla). Reproduced with permission from ref (67).
A fascinating example of the use of biological machinery to control sequence selection during copper-mediated ATRP was recently published by Alexander and co-workers.71 In this example, GFP-labeled E. coli 539 served as a physical substrate for surface-promoted polymerization of acrylic quaternary amine-containing and sulphobetaine monomers in solution. The reductive environment of bacterial suspensions was utilized to generate catalytically active Cu(I) species, thereby initiating polymerization. This so-called “bacteria-instructed synthesis” produced templated polymers that specifically bound to the cell surface on which they were formed. In contrast, nontemplated polymers that formed in bulk solution had little affinity for the bacterial surface and were readily washed away. To demonstrate these polymers as a diagnostic tool, a presynthesized polymer bearing terminal alkyne groups was incubated with bacteria along with a “clickable” pro-fluorophore. Templated polymers sustained rapid fluorescence in the presence of the metabolically active environment due to copper-mediated cycloaddition. This work presents an innovative semisynthetic method for self-selective polymer sequence control entirely manipulated by bacteria. Its utility in detection and sequestration of matched pathogens is an exciting potential avenue in the field of adaptable diagnostics and antimicrobials.
Tacticity Control
Biopolymers containing tertiary and quaternary structures mediate a wide array of complex biological processes due to the preservation of their stereochemistry. It is known that the stereochemistry. It is known that the stereochemistry of polymers, or tacticity, can impose significant changes in physical and chemical properties of synthetic polymers.86,87 Even so, stereoregulation remains a barrier for precise control of polymer structure. In a recent example by the Johnson group, an iterative exponential growth (IEG)-inspired approach was demonstrated for the economically scalable synthesis of sequence- and stereocontrolled unimolecular polymers.88 In this IEG plus side chain functionality strategy, 1R and 1S epoxy alkynes were either subjected to azide substitution followed by functionalization or to deprotection in order to afford species that were coupled efficiently by CuAAC “click” chemistry, generating four different epoxy–alkyne diastereomers (Figure 3). These “dimers” were then matched appropriately to synthesize macromolecules with the desired tacticity through multiple cycles of azide-instructed epoxide opening, alkyne deprotection, and subsequent CuAAC click conjugation. Comparison of thermogravimetric analysis data for isotactic and syndiotactic hexadecamers revealed subtle differences in thermal properties, namely the glass transition temperatures Tg, suggesting that differences in intermolecular polymer interactions were at play. This method demonstrated the scalable synthesis of a 6300 Da syndiotactic polymer; however, the final product was recovered in approximately 1 week. In order to minimize the length of time required, semiautomated synthesis by Flow-IEG offers a favorable application of this method toward primary sequence and architecture control.89 With the increasing interest in tacticity control in mind, polymer chemists are actively pursuing advances in conventional chain growth methods. By improving control over primary structure and tacticity, more detailed analyses can be made to understand the correlation between these parameters and macromolecular assembly and function, thus bringing synthetic capabilities closer to the complexity afforded by nature.
Figure 3.

IEG-inspired iterative synthesis of sequence and stereocontrolled polymers. (a) Example of a 32-mer prepared by (b) orthogonal azidification, functionalization and silyl deprotection of two chiral monomers (1S, 1R) followed by CuAAC “click” of key stereoisomeric intermediates to generate polymers with precise sequences and stereochemistry. Adapted with permission from ref (88).
Grafting Control
While researchers are investigating novel methods for finely tuning polymer primary sequence, controlled polymerization strategies enable the incorporation of complex biomolecules that in themselves possess absolute sequence control. Therefore, by gaining excellent control over biomolecule graft polymers, materials are generated with 2-dimenional architectural control including the polymer backbone and side chains as a biologically interactive system. In this subsection, we highlight the impact of 2-dimensional architectures on biological mode of action.
Efforts have been directed at the expansion of graft-through methodologies with ROMP in order to avoid the large kinetic barrier implicit in postpolymerization conjugation of macromolecules. Successful conjugation of macromolecules is limited by steric hindrance, which often results in variable degrees of grafting, difficult purification and low reproducibility of polymer bioconjugates. As such, considerable effort has been devoted toward the direct polymerization of complex peptides,72 nucleobases,73 bioderived polyesters,74 imaging agents,75 and therapeutic drugs.76 These biosynthetic polymers possess extreme complexity with rigorous control over polymer assembly and in some cases biofunctionality as tumor targeting77 and protease resistant materials.78 In particular, protected and/or deprotected peptide-based monomers, which range in size from 5 to 30 amino acids, can be polymerized into dense brushes as homopolymers79 or amphiphilic block copolymers that self-assemble into micellar nanoparticles.80 This modular approach allows very large peptides of any given sequence to be polymerized in the presence of Grubbs modified second generation catalyst. Attachment of 6-aminohexanoyl spacers to separate the polymerizable norbornene subunit from the peptide sequence further enhances polymerization rates, maintains low dispersity, and enables higher degrees of polymerization of biomacromonomers. Other strategies that incorporate complex functionality may rely on a grafting from approach, such as the preparation of high-chain density cylindrical copolypeptide brushes, via two rounds of N-carboxyanhydride polymerization in a one-pot procedure, with controlled segment lengths.81 Accompanying the unmistakable advantage that graft-through and some graft from strategies present for generating highly dense peptide polymers,82 unique modes of action may be accessed, such as restricted proteolytic degradation.78
Regardless of which strategy is utilized, varying polymer architecture via grafting has the potential to modulate biological function. For instance, Sumerlin and co-workers used graft-to polymer bioconjugates in order to improve the therapeutic function of osteoprotegerin (OPG), which is a protein that restricts osteoclast formation and subsequently bone resorption in accelerated bone loss disorders.83 Specifically, the role of side-chain grafting density was examined for OPG-polymer bioconjugates using linear, loosely branched, and densely branched poly(ethylene glycol) (PEG) architectures. Modest restoration of bone mineral density was achieved for the loosely branched conjugate/analogue in comparison to the other architectures. In another example, Tew and co-workers synthesized bioinspired protein transduction domain mimics with varying degrees of hydrophilic guanidine and hydrophobic phenyl group segregation.84 Three types of polymers were analyzed for their membrane affinity and cellular internalization characteristics: nonsegregated homopolymers, intermediately segregated gradient copolymers, and strongly segregated block copolymers. Gradient copolymers with intermediate segregation displayed the highest activity and solubility with low cytotoxicity. Insight from this structure–activity survey was used for efficient siRNA delivery and gene knockdown in human T cells.85 Thus, architecture control via selective grafting strategies can potentially improve efficacy of biosynthetic polymers, depending on the function required.
Conclusions for Architecture Control
Researchers are becoming increasingly aware of the importance of architecture90 and tacticity91 in self-assembly. Precise control over both stereo- and regiochemistry may accelerate opportunities for regulating geometries such as polymer tertiary and quaternary structures. Just as nature employs DNA and RNA sequences to encode biological information, and harnesses protein tertiary and quaternary structures to confer specialized activities, so too can researchers aspire to use semisynthetic or fully synthetic polymers for the preparation of artificial viruses or enzymes, or cofactors in cascade pathways. Further, the ability to directly polymerize large biomolecules has the advantage of loading dense arrays of information. This contrasts with previous synthetic efforts of postpolymerization modification, which have little sequence control, lower grafting densities, and are often difficult to characterize. Achieving complex sequences using simple synthetic methods is paramount; continued efforts in this regard will undoubtedly surpass practically tenuous methods in the movement toward synthetic biological mimics.
Structural Dynamics of Polymer Assemblies
Complexing biosynthetic polymers into assemblies can increase their versatility and function. For example, nano- and micron-scale particles, vesicles, films, and hydrogels have been developed using self-assembled polymers.92−94 Polymeric assemblies encompassing these architectures have been developed for various functions including stabilizing internal cargos, slowing clearance within biological systems, performing as supportive scaffolds, and/or serving as vehicles for signaling and detection. Many research groups have sought to develop assemblies with environmentally adaptive characteristics and therefore have devoted efforts to develop stimuli-responsive biomaterials for triggered signaling,95 drug release,5 and/or degradation.22 Motivations for this aim arise from the desire to mimic natural behaviors like blood clotting and wound healing. However, controlling structurally dynamic behaviors within these assemblies is still difficult. Success along this avenue can advance previously unrealized opportunities for functionally diverse materials that surpass the limited utility of structurally inert designs. A notable example in this regard is the enhanced therapeutic efficacy of assemblies with active targeting capabilities compared to ones with passive abilities at low doses.96 Nonetheless, in the interest of translating these systems for in vivo use, increasing functional complexity without forfeiting synthetic simplicity is necessary. The following discussion highlights examples that evaluate these two considerations.
Dynamic Particle Assemblies
The clinical relevance of shape-changing biosynthetic polymers has been demonstrated in both a myocardial infarction (MI) and a fibrosarcoma tumor model.76,77,80,97 In post-MI treatment specifically, developing systems capable of noninvasive delivery and heart retention for periods longer than 1 week is currently a significant challenge. By utilizing discrete fluorescent nanoparticles that can enzymatically assemble into aggregated scaffolds, researchers observed a signal enhancement for the targeted diseased tissues relative to healthy tissue (Figure 4). Matrix metalloproteinases (MMPs), which are overexpressed in areas of severe inflammation, cleave the peptides displayed on the nanoparticle surface. A shift in the polymer amphiphilicity, a consequence of proteolytic cleavage of hydrophilic peptide fragments, then causes nanoparticle reassembly into micron-scale aggregates in the infarct tissue. These enlarged aggregates are slow to clear, which prolongs their tissue retention and enhances diagnostic ability by way of the colocalization of fluorogenic material with the site of damage. Retention of these materials within infarcted tissue up to 28 days was observed, which greatly exceeds insufficient retention times of hours to days observed with other active-targeting nanoparticle formulations.300,200 A similar shapechanging system demonstrated active cargo release of bound drugs at the site of scaffold assembly in a fibrosarcoma tumor model.76 These proof-of-concept works demonstrate exceptional utility achieved by the dynamic morphological response of enzyme-responsive micellar assemblies in two ways: first as a discrete vehicle for noninvasive intravenous delivery and second as a stationary scaffold for diagnostics and localized drug release.
Figure 4.

Enzyme-responsive peptide–polymer amphiphiles change shape in response to biological stimulus. (a) Diagram of a dye-labeled brush peptide–polymer amphiphile (PPA) bearing an MMP-9 specific recognition sequence, shown underlined. PPAs self-assemble into nanoparticles through hydrophobic–hydrophilic interactions when dialyzed into aqueous buffer. (b) Responsive nanoparticles aggregate in response to enzymatic cleavage by TEM. (c) Injection of particles into an infarcted heart (left) result in infarct-specific aggregation and retention over healthy tissue by fluorescence (middle and right). Scale bar: 100 μm. Adapted with permission from ref (97).
Alternative stimuli, such as temperature or pH, have been shown to provoke changes in peptide-based particle assemblies. In one example, researchers developed a “nanopeptifier” system, which relies on thermally triggered self-assembly of elastin-like polypeptide amphiphiles containing a cell-penetrating peptide (CPP) domain on the hydrophilic block.98 Once assembled into micelles, the high density CPP surface array enhances cellular uptake. To assess therapeutic payload delivery, a proapoptotic peptide was attached to the hydrophobic domain; the resulting nanopeptifier acts as a dynamic switch, inducing apoptosis only in micellar form above the lower critical solution temperature (LCST). In another example by Savin and co-workers, poly(l-lysine)-b-poly(propylene oxide)-b-poly(l-lysine) triblock copolymers containing different lysine fractions were found to adopt distinct morphological transitions, either spherical micelle to vesicle or spherical micelle to disk micelle structures, as a function of pH.99 Dynamic morphologies like this may have the capacity to alter in vivo biodistribution and shape-change induced drug release.
Despite increasing efforts for instilling stimuli-responsiveness in polymeric assemblies, some basic questions governing the spatial organization of these assemblies still remain. Specifically, how does conformational fluidity of a biofunctional polymer assembly impact its interaction at biological interfaces, in contrast to inert analogues? For example, amphiphilic block copolymers are promising in their use as artificial biological membranes, which are known to stabilize membrane proteins.100 An interesting observation emerged from this study by Meier and co-workers, which determined that high flexibility of poly(dimethylsiloxane)-containing block copolymers may be responsible for successful integration of model membrane proteins despite mismatches in their hydrophobic domain sizes. As such, the identity of block copolymers within an assembly must be carefully considered when designing biomimetic membranes and analogous systems.
Self-Healing Hydrogels via Noncovalent Interactions
Polymer hydrogels represent a class of materials well-suited for a range of biological applications such as tissue engineering, regenerative medicines, controlled drug delivery, diagnostics, biological sensors, and microarrays due to their high water content and tunable mechanical properties.101−103 These viscoelastic materials are achieved via permanent covalent chemical cross-links, through reactions between functional groups or free radicals, or noncovalent physical cross-links, such as hydrogen bonding, electrostatic interactions, metal–ligand coordination, and host–guest recognition. Covalent cross-links form robust and elastic hydrogels for load-bearing support, whereas noncovalently cross-linked hydrogels have the ability to flow under shear thinning conditions, an important quality for in vivo delivery. However, when covalent hydrogels are damaged by breaks or when noncovalent hydrogels resist reassembly after shearing from injection, they inevitably lose their function in biological systems. Thus, many therapeutic hydrogel strategies are not currently viable for noninvasive delivery or prolonged clinical application.104 In one approach to combat this problem, researchers imparted a permanently cross-linked hydrogel with pH-dependent self-healing by integrating noncovalent physical cross-linking capability, specifically side chains capable of hydrogen bonding.105 Nevertheless, this combinatorial strategy only enabled healed hydrogels to retain up to 70% of their pristine fracture stress after a single break. Recent accounts using polymer–nanoparticle and host–guest interactions provide simple and adaptable methods for uniform and complete healing via concentrated pockets of strong noncovalent interactions following shear thinning damage; in essence, these interactions act as transient covalent cross-link mimics which provide rapid healing on the order of seconds. These types of composite materials may be a plausible alternative for soft hydrogel-based therapeutic delivery.
Inspired by work with clay particle in polymer dispersions that improve stiffness and elasticity of resulting gels,106,107 Marcellan, Leibler, and co-workers used TM-50 silicon nanoparticle solutions as adhesives for severed noncovalent gels and biological tissues.108 This strategy relies on the absorption of nanoparticles onto polymer gels, which then bridge polymer chains. By simply pressing them together in the nanoparticle solutions, separate pieces of poly(dimethylacrylamide) adhered on the order of tens of seconds. Interestingly, glued gels were tougher by lap-shear adhesion tests and resisted further damage at the bonding junction compared to unsevered regions. Within this bonding junction, polymer chains adhered to the nanoparticles reorganized and dissipated energy under stress, thus maintaining flexibility and structural support at the newly repaired tear site. This facile method of noncovalent hydrogel repair contrasts with those that rely on harsher conditions such as chemical, thermal, pH, UV irradiation, or an electric field.
Langer and co-workers tackled a complementary issue with shear-thinning, noncovalently cross-linked hydrogels.109 Hydroxypropylmethylcellulose derivatives (HPMC-x) were combined with core–shell PEG-b-PLA nanoparticles to create shear thinning hydrogels that fully recovered their equilibrium viscoelasticity within 10 s (Figure 5). In contrast, the absence of nanoparticles yielded low-viscosity liquids. Transient and reversible hydrophobic forces between the polymer and nanoparticle chains were responsible for rapid disassembly when traversing the needle under force followed by rapid recovery after exiting. Given this efficient healing process, an encapsulated model therapeutic protein, fluorescein isothiocyanate-labeled bovine serum albumin, remained within the matrix for localized and gradual release over several days following subcutaneous injection into C57BL/6 mice. Another study utilized shear-thinning nanocomposite hydrogels composed of synthetic silicate nanoplatelets and gelatin for the treatment of hermorrhage in a mouse liver bleeding model.110 Rapid mechanical recovery of the hydrogels and promotion of coagulation enabled sealing of severe incompressible wounds. The results of these studies demonstrate a simple method for minimally invasive delivery of polymeric hydrogels with the capacity for encoding biomolecules.
Figure 5.
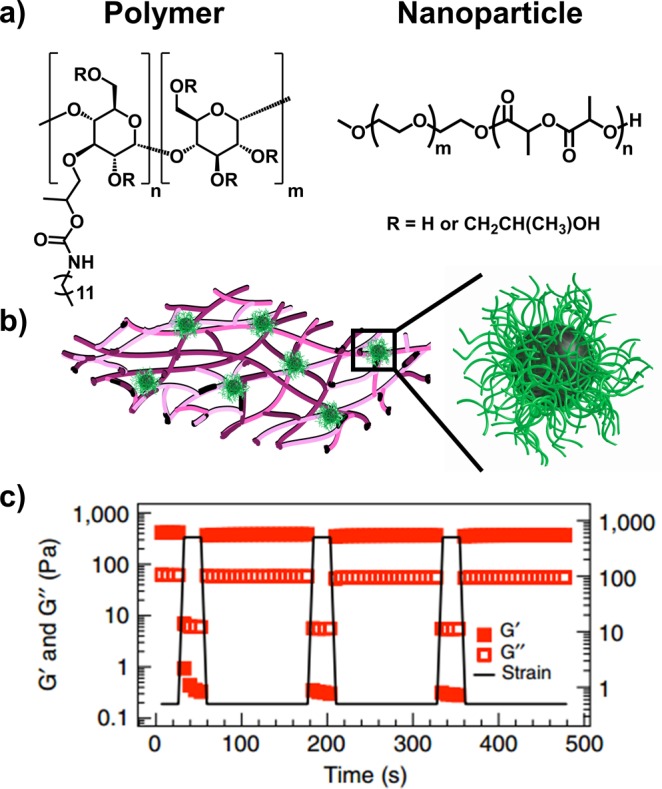
Self-assembled hydrogels utilizing polymer–nanoparticle (PNP) interactions. (a) Dodecyl-derivatized hydroxypropylmethylcellulose (HPMC-C12) polymer and core–shell PEG-b-PLA based NPs are (b) mixed to form hydrogels where polymer chains adsorb to NPs to create transient noncovalent interactions. (c) These hydrogels shear-thin and self-heal repeatedly as shown by rheology. Adapted with permission from ref (109).
Noncovalent hydrogels utilizing host–guest recognition were first demonstrated in 1994 with cyclodextrins (CDs),111 which can act as a host for a variety of molecules such as PEG, ferrocene, and adamantine.94 These interactions have the advantage of providing specificity for selective recognition and stimuli-responsive healing. Yin and co-workers utilized a host–guest complex between β-CD-modified poly(l-glutamic acid) (PLGA-g-β-CD) and cholesterol-modified triblock (PLGA-b-PEG-b-PLGA)-g-Chol to create self-healing and degradable supramolecular hydrogels.112 A complementary system developed by Scherman and co-workers utilized polysaccharides functionalized with phenylalanine–cysteine dipeptides as a host-responsive biosynthetic progelator.10 Upon addition of cucurbit[8]uril, strong 1:2 “homoternary” complexes with the pendant phenylalanine residues caused a sol-to-gel transition. Yet another system by Ravoo and co-workers used amphiphilic β-CD host-bound vesicles and adamantane guest-bound hydroxyethyl cellulose in the formation of supramolecular hydrogels.113 In contrast to self-healing hydrogels, which rely on nonspecific ionic interactions and hydrogen bonding, these dynamic host–guest interactions mitigated unwanted side reactions leading to passivation of the exposed surface areas;114 therefore, long-term stability was maintained.
Many polymeric hydrogels possess viscoelastic properties similar to those of soft biological tissues;115 these materials subsequently make excellent tissue scaffolds for wound repair or delivery vehicles for biomolecules. To ensure durability and/or practical delivery of these polymeric scaffolds in a biological system, however, they must possess dynamic assembly characteristics that enable self-healing despite excess shear stress. New studies on polymer–NP and host–guest interactions in hydrogels can bridge the gap between the discussed disparate advantages of covalently and noncovalently cross-linked polymer hydrogels in biomedical applications.
Self-Healing Hydrogels via Reversible Covalent Cross-Links
Considering applications in which stiffer hydrogels are necessary, developing systems containing covalent cross-links may be required. To compensate for structural damage through depreciation as a biological scaffold, creative methods to increase intrinsic dynamic properties are necessary. Self-healing polymers, for example, have been conceived in the past decade in response to this problem;116 self-healing polymeric hydrogels that rely on reversible cross-links are an even newer development.
Chen and co-workers, for instance, designed a polysaccharide-based self-healing hydrogel cross-linked via two types of dynamic covalent bonds.117 These hydrogels were composed of biocompatible oxidized sodium alginate (OSA) cross-linked through dynamic acylhydrazone bonds, by way of condensation between OSA and adipic acid dihydrazide (ADH), and further cross-linked with carboxyethyl chitosan (CEC) through more reactive and neutral condition-tolerant imine bonds via Schiff base reaction between the two polysaccharides. The resulting CEC-I-OSA-I-ADH hydrogel underwent complete viscoelastic recovery within a matter of seconds following excess surface strain. Upon severing a prepared hydrogel disk, reconnected pieces exhibited a healing efficiency, defined as the ratio of the healed and pristine sample breaking point strengths, of up to 95 ± 2.2% after immersion in PBS for 12 h at 37 °C. Given the combination of dynamic reversible covalent bonds, broken hydrogel pieces from a syringe injection immediately self-assembled and self-healed into a single mold.
In one strategy, the hydrolytic instability of oxime linkages was leveraged for the development of reversible oxime-cross-linked hydrogels that autonomously healed following mechanical damage.16 Complementary polymeric hydrogels containing reversible boronate ester linkages is another example of dynamic self-healing materials, wherein pH tolerance of the intramolecular cross-links may provide stability in areas of tissue inflammation.118 Consequently, a distinct advantage is that the healed regions of these materials are chemically identical to that of the bulk hydrogel. Hydrogels that heal through reversible covalent cross-links or noncovalent polymer–nanoparticle interactions, as discussed earlier, can address existing issues with injection strategies and prolonged integrity of soft tissue mimics.
Adhesive and Shape-Memory Polymers for Tissue Repair and Replacement
Fast annealing, adhesive bioinspired polymers are interesting materials for on-site repair of existing tissues. Despite ongoing progress in the design of self-healing cohesive polymers, maintaining tissue adhesion in aqueous environments is another issue. The general stipulation that biosynthetic polymer glues adhere to aqueous biological tissues yet maintain resistance to biological fluids seems paradoxical. Likewise, a biodegradable material that resists structural damage during tissue healing is another seemingly irreconcilable scenario. Creative solutions to these problems over the past couple years have drawn upon synthetic modification, biological mimicry, or a combination of both.
Work from the del Nido and Karp groups have resulted in blood-resistant surgical glues for repair of blood vessels and heart defects.119 Rapid adhesion took place when a gelatinous solution of poly(glycerol sebacate acrylate) mixed with a photoinitiator was chemically cross-linked via UV light. Researchers demonstrated that direct application to the heart wall or carotid artery converted the dynamic polymer into a water-tight yet flexible sealant to act as a tailored suture or patch, respectively. In comparison to currently available sealants, such as fibrin, this hydrophobic light-activated adhesive exhibited superior pull-off adhesion strength.
Mussels, which can affix to a wide variety of surfaces, such as metals, rocks, wood, and polymers, have been a source of inspiration for adhesive polymers.120−123 Adhesive plaque formation is achieved through strong bidentate hydrogen bonding of the catechol moieties in 3,4-dihydroxyphenylalinine rich proteins secreted under reducing conditions. Waite and co-workers utilized semi-rigid polyacrylate and more rigid polymethacrylate polymers functionalized with silyl-protected catechol moieties to afford wetting-induced, bioinspired glues.124 Deprotection under mildly acidic conditions exposes hydrogen-bonding moieties on the implant surface for wetting-induced stickiness (Figure 6). One could envision a similar biosynthetic polymer to prevent implant loosening, which is common with knee replacements.125 This surface activation strategy could be further advantageous for long-term implant durability. In regards to stress fracturing or slow surface erosion, newly exposed protected catechols could reinforce adhesion near sites of inflammation in vivo, where the local environment is acidic. Complementary adhesive materials utilize the downstream mechanism of mussel plaque curing through metal chelation and covalent cross-linking of catechol moieties.126
Figure 6.

Mussel-inspired polycatechols as healable gels. (a) UV radical polymerization of silyl protected catechol acrylate monomer using a photoinitiator. (b) Adhesive interactions measured between semi-rigid polymer films functionalized with silyl protected catechols (catechol blocked) and deprotected catechols (catechol exposed). Dashed lines with open circles indicate delamination of the polymer from the substrate surface. (c) Scheme depicting the self-healing property of semi-rigid polycatechol acrylate rods after an initial incision, immersion in pH 3 buffer for silyl deprotection, and subsequent rejoining. Inset diagram shows hydrogen bonding interactions between deprotected catechols. Adapted with permission from ref (124).
So far, the discussions for tissue repair have focused on soft and elastic materials. In situations where load-bearing tissue such as bone requires mending or partial replacement, shape-memory polymers (SMP) may be useful. Grunland and Hahn recently designed a thermoresponsive SMP that were able to self-fit into irregular cranio-maxillo facial bone defects and exhibited interconnected porous morphology similar to that of bone.127 Poly(ε-caprolactone) scaffolds were softened by heating to above their melting transition temperature (Tm ∼ 55 °C) for manual compression and subsequent expansion into irregular boundaries upon release of pressure. Cooling locked the final scaffold conformation for tailored bone implants. Furthermore, adhesion to adjacent bone tissue and enhanced bone regeneration was achieved when scaffolds were precoated with poly(dopamine). Other materials such as amino acid-based poly(ester urea)s have also been developed as premolded osteogenic scaffolds.128 Poly(leucine)-based versions had a reported Tg of 57 °C,129 which enabled these materials to also act as self-fitting implants.
The shift in structural dynamics from free-flowing or moldable polymers to locked sealants exemplifies a new class of dynamic biosynthetic polymers for minimally invasive structural repair in a clinical setting. These advanced materials have the potential to replace static implants, such as invasive autografts and premolded scaffolds, or traditional wound closing devices, such as sutures and staples, during surgery.
Conclusions for Structural Dynamics of Polymer Assemblies
Within the past several decades, interest in dynamic biosynthetic polymers has expanded and with it new approaches to develop materials that encompass both the complex functions intrinsic to natural biomolecules and tunable capabilities like stimuli responsiveness constructed in synthetic polymers. By introducing dynamic complexity into polymeric assemblies such as nanoparticles, hydrogels, elastomers, adhesives, and foams, we may build a better understanding of biological processes that govern tissue assembly and preservation; ideally, these mimetic polymeric scaffolds could emulate natural soft and hard tissues in complexity to afford wound healing, tissue regeneration, and load-bearing support.
Biotherapeutic Stabilization: Storage, Release, and Bioresistance
The widespread availability of biotherapeutics such as therapeutic peptides and proteins, antibodies, engineered fusion proteins, and conjugates has been used to treat conditions including cardiovascular disease, cancer, inflammation, infectious diseases, and genetic disorders.130,131 They are advantageous over small molecule drugs due to higher specificity and potency, lower systemic toxicity, and reduced off-target biodistribution. However, the nature of their large sizes and complex structures introduces challenges in the production, storage, and administration of these therapeutics. Functional biosynthetic polymers have been used as vehicles or supports for storing and safely delivering biotherapeutics in formulations such as polymer scaffolds, polymer conjugates, and polymeric and liposomal particles.24,93,132 In some instances, these approaches are prohibitively expensive or unstable for industrial and agricultural applications.133 In the following section, efforts to stabilize biotherapeutics are discussed.
Matrix-Based Assemblies for Biopolymer Storage
Hydrogels are one of the most heavily investigated biomaterials for enzyme stabilization and controlled release of small molecule drugs, nanoparticles, and biotherapeutics, due to simple syntheses.134 However, current hydrogel formulations still remain unsuitable for maintaining prolonged biological activity of many fragile biomacromolecules such as monoclonal antibodies and therapeutic enzymes.135 Hydrogels formed through covalent cross-linking or noncovalent interactions can cause denaturation and aggregation, respectively. Recently, however, Maynard and Langer demonstrated the utility of trehalose-based polymeric hydrogels for enhancing long-term functional stability and heat resistance of unstable biotherapeutics.133,136
Trehalose is a nonreducing disaccharide which is extremely effective as a protein- and poly(nucleic acid)-stabilizing excipient.137 Maynard and co-workers analyzed the stabilizing effects of trehalose-based polymeric hydrogels on phytase, a thermally unstable enzyme heavily used in the animal feed industry.133 Radical polymerization of vinyl-substituted trehalose and simple purification procedures presented a scalable and economical two-step method for preparing trehalose hydrogels for phytase stabilization (Figure 7). Investigators observed 100% phytase activity retention in the presence of trehalose hydrogels even when heated to 90 °C. Interestingly, a noncontrolled polymerization technique still allowed investigators to access highly functional biosynthetic polymers without rigorous control over polymer architecture. In examples such as this, one asks the question, “how vital is discrete polymer architecture for some applications?”
Figure 7.

Trehalose and ethoxylated polyol (EP) hydrogel components for stabilizing model proteins. (a) Thiol–ene cross-linking of diacrylate functionalized trehalose (TDA) with EP, showing hydrogen-bonding interactions between trehalose and horseradish peroxidase (HRP) within the hydrogel network. (b) Increasing trehalose content in the hydrogel correlates with greater percent recovery of active protein during release. (c) Measured stability of HRP within the trehalose gel 1, 72, or 48 h of lyophilization and subsequent rehydration for 24 h. Adapted with permission from ref (136).
In a related study, Langer and co-workers fabricated synthetic trehalose hydrogels through thiol–ene cross-linking of diacrylate-functionalized trehalose (TDA) within a known ethoxylated polyol (EP) hydrogel platform.136 Horseradish peroxidase (HRP) isoform C, glucose oxidase, and α-chymotrypsin were used as model unstable proteins and into preformed hydrogel disks. Protein stability was directly proportional to trehalose content within EP hydrogel networks, with high levels of biological activity recovery in high trehalose-containing hydrogels. Enhanced stabilization under heat and lyophilization is thought to be attributed in part to uniquely strong hydrogen-bonding character within the trehalose hydrogel network, which is hydration dependent. Furthermore, modular release kinetics was achieved through variable hydrolyzable ester content from trehalose incorporation.
These studies represent new strides in the field of biomolecule-functionalized synthetic polymers for broad utility as biopolymer protectants. Notable are biomimetic polymeric hydrogels containing heparin, hyaluronic acid, and collagen; other native moieties have been used as biotherapeutic storage and release matrices with less impressive stabilization results.138,139
Stable and Controlled Release of Biomaterials from Layered Surfaces and Networks
A popular tool introduced in 1992 for functionalization of surfaces involves the layer-by-layer (LbL) deposition of polyelectrolyte films,140 polysaccharides,141−144 poly(amino acids),145 poly(acrylic acid),146 and various other polyelectrolytes.147,148 The Hammond Lab has made considerable progress with these materials for temporary protection and variable-timed release of therapeutic cargos via diffusion, stimuli-response, and/or network degradation.149−153 For instance, naturally derived LbL films were assembled through electrostatic complexation between anionic poly(β-l-malic acid) and cationic chitosan in aqueous conditions.154 By increasing chitosan composition, which stabilized film growth and robustness, protein release kinetics were slowed from total enzyme release in tens of minutes to multiple days to promote burst release or sustain degradative release, respectively. The investigators also noted that release kinetics were unaffected by increasing the total number of surface layers, enabling short-term or prolonged dosing. Application of bFGF-loaded multilayer films in NIH3T3 fibroblast cells demonstrated dose-responsive cell proliferation. Comparison with equal quantities of as-received bFGF showed that film-released bFGF significantly increased proliferative activity. As such, corelease with chitosan was believed to stabilize this liberated bFGF from heat-inactivation and proteolysis.
Rigorous control over biotherapeutic release from a protected state is key for creating personalized medicine. Given the variety of known diseases that affect different physiological pathways and to variable degrees, appropriate dosing is salient for proper treatment. To be seriously considered as a viable system for general application in clinical treatment, biosynthetic polymer assemblies such as those discussed above need to provide rigorously controlled and versatile cargo release profiles.
Bioresistance with Biopolymer Conjugates
Polymer conjugation has been a successful method for prolonging the circulation half-life and activity of unstable biopolymers. Since the late 1970s, protein–polymer conjugates have been widely explored as therapeutics, even gaining FDA approval as PEG conjugates.155 PEGylation has been widely used because of its solubilizing effect, high biocompatibility, and ability to minimize interactions with blood components. However, recent studies have reported controversial accounts of PEGylation including immunogenicity, degradation under stress, and accumulation in the body above excretion limits. As such, charged zwitterionic polymers,156 polyglycidols,157 and biodegradable alternatives158 like poly(amino acids)s, chitosan mimics, and heparin mimics are currently being explored. A noncytotoxic heparin-mimic polymer PDS-b-P(SS-co-PEGMA) bearing styrenesulfonate (SS) moieties and a pyridyl disulfide (PDS) functionalizable end group was synthesized via ATRP.159 Conjugation of this polymer to basic fibroblast growth factor (bFGF), a notoriously unstable protein involved in bone regeneration and wound healing, resulted in superior resistance to extreme temperatures used in storage and delivery, low pH, and proteolysis (Figure 8). Bioactivity of the bFGF-p(SS-co-PEGMA) heparin-mimic conjugate was significantly higher than the bFGF-pPEGMA control in which the styrenesulfonate units were omitted. Close proximity of the heparin-mimic polymer to bFGF was shown to protect it from denaturation more so than a simple mixture of heparin and bFGF, the conventional method of stabilizing bFGF for drug delivery. This is the first example of a stabilized bFGF conjugate and may provide an avenue for increasing the scope of biostabilization through polymer conjugates.
Figure 8.

A heparin-mimicking polymer conjugate stabilizes bFGF. (a) Conjugation of bFGF to a styrenesulfonate and poly(ethylene glycol) bearing methyl methacrylate copolymer (bFGF-p(SS-co-PEGMA)) (b) stabilizes the protein to prolonged storage, heat, acidic conditions, and enzymatic degradation. Adapted with permission from ref (159).
In some formulations, polymer conjugates self-assemble into particles.160 Nanoparticle and liposomal formulations with unconjugated cargos have been used as secondary carriers to load and shield biomolecules, but their formulation stability and/or loading capacities often remain low.161 This issue is addressed in a study where nucleic acid delivery potency was enhanced through conjugation of DNA with functional synthetic polymers that self-assemble. Inspired by early work from Mirkin and co-workers with spherical nucleic acids on gold nanoparticles162 and their templated metal-free counterparts,163 Gianneschi and co-workers directly conjugated DNA to a hydrophobic homopolymer to generate informational amphiphiles.164 These nucleic acid–polymer conjugates self-assembled into micellar nanoparticles displaying a dense shell of nucleic acids on the surface. The high surface curvature and packing density of the micellar nanoparticle promoted nuclease resistance and rapid cellular uptake for prolonged knockdown of survivin mRNA in HeLa cancer cells.165 This work shows the utility of simple nucleic acid–polymer conjugates, which protect themselves through spontaneous self-assembly, for targeted biotherapeutic delivery.
Conclusion for Biostabilization
Expensive biomolecules and biotherapeutics often suffer from instability and rapid degradation or clearance. Thus, the push for stabilization in biotechnology and biomedicine is high. The discussed packaging strategies have the advantage of reduced effective dosages, prolonged treatment, and/or enhanced targeting. Because of the nature of these expensive cargos, however, equally expensive biostabilizing polymers are impractical, as the combined cost would be prohibitive. Therefore, in this field, the necessity for economic viability in the form of adaptability of the technology, scalability, and reproducibility is paramount.
Conclusions and Outlook
The divide between pure synthetic and natural macromolecules continues to narrow in the pursuit of biologically relevant materials with increasingly complex functionality. Indeed, recent developments in biosynthetic polymer chemistry are allowing scientists to prepare, analyze, and use polymeric systems in unprecedented ways. Synthetic and especially multivalent polymers can partially mimic biopolymers. Conversely, biopolymers can be engineered to generate unnatural materials. Refinements in both controlled polymerization techniques, such as RDRP methods and olefin metathesis reactions, and bioconjugation strategies have enabled chemists to prepare bespoke multifunctional materials with designed architectures. Bioinspired linear polymers with properties similar to natural materials are also now accessible, while new functional materials with biological activity beyond those of natural polymers are in the first stages of development. Perhaps the greatest obstacle still in place is achieving functional complexity through simple design and manufacturing. Several examples presented here are still synthetically challenging and prompt initiatives for modifying existing techniques in order to develop simple and streamlined protocols. Just as personal or hand-held computers were, at worst, not even imagined and, at best, thought to be a near impossibility in the era of large scale computers in the 1960s, functional biosynthetic polymers may in the next few decades become commonplace in numerous industries. Despite great strides being made in this field, a pioneering perspective from 1984 persists as an exciting yet astounding reality in 2016.166 Macromolecular chemistry as we know is far from capturing the processes so prevalent and active in living systems. However, we remain inspired by the possibilities that present themselves in the attempt to achieve these lofty heights. The key is an attitude that multidisciplinary approaches and collaborations with neighboring fields will open doors and opportunities for the field itself and for biomedicine alike.
Acknowledgments
We acknowledge generous support from the AFOSR through a PECASE (FA9550-11-1-0105) and a BRI (FA9550-12-1-0414). We also acknowledge support from the NIH through a Director’s New Innovator Award (1DP2OD008724), through the NIBIB (1R01EB011633), and for a Transformative Award (NHLBI R01HL117326). Further support comes from the ARO (W911NF-14-1-0169 and a MURI: W911NF-15-1-0568). A.S.C. thanks the NSF for a Graduate Research Fellowship (DGE-1144086).
Glossary
Abbreviations
- DNA
deoxyribonucleic acid;
- RNA
ribonucleic acid
- PBS
phosphate buffered saline
- TEM
transmission electron microscopy
- PEG
poly(ethylene glycol)
Biographies

Andrea S. Carlini attended Virginia Tech, completing her B.S. degrees in Chemistry and Biological Sciences with honors in 2012. She is currently a Ph.D. candidate in Chemistry and Biochemistry at the University of California, San Diego, in the laboratory of Prof. Nathan C. Gianneschi as a National Science Foundation Graduate Research Fellow. Her research focuses on responsive polymeric biomaterials for the treatment of postmyocardial infarction.

Lisa Adamiak attended the University of California, Santa Cruz, completing a B.S. degree in Chemistry with honors in 2010. After graduating, she earned an ORISE research fellowship and worked under the guidance of Dr. Daron Freedberg at the FDA in Bethesda, MD, until 2011. Lisa is currently pursuing a Ph.D. under the supervision of Professor Nathan C. Gianneschi at the University of California, San Diego. Her research involves directing self-assembly processes of responsive polymers using enzymatic triggers, specifically used towards the development of active-targeting diagnostics in vivo and responsive liquid crystal–polymer composites.

Nathan C. Gianneschi received his B.Sc(Hons) at the University of Adelaide, Australia, in 1999. In 2005 he completed his Ph.D at Northwestern University. Following a Dow Chemical postdoctoral fellowship at The Scripps Research Institute, in 2008 he began his independent career at the University of California, San Diego, where he is currently Associate Professor. The Gianneschi group takes an interdisciplinary approach to nanomaterials research with a focus on multifunctional materials with interests that include biomedical applications, programmed interactions with biomolecules and cells, and basic research into nanoscale materials design, synthesis, and characterization. For this work he has been awarded the NIH Director’s New Innovator Award, the NIH Director’s Transformative Research Award, and the White House’s highest honor for young scientists and engineers with a Presidential Early Career Award for Scientists and Engineers. Prof. Gianneschi was awarded a Dreyfus Foundation Fellowship, is a Kavli Fellow of the National Academy of Sciences, and is an Alfred P. Sloan Foundation Fellow.
The authors declare no competing financial interest.
References
- Günay K. A.; Theato P.; Klok H.-A.. History of Post-Polymerization Modification. In Functional Polymers by Post-Polymerization Modification; Wiley-VCH Verlag GmbH & Co. KGaA: 2012; pp 1–44. [Google Scholar]
- Khan F.; Tanaka M.; Ahmad S. R. Fabrication of polymeric biomaterials: a strategy for tissue engineering and medical devices. J. Mater. Chem. B 2015, 3 (42), 8224–8249. 10.1039/C5TB01370D. [DOI] [PubMed] [Google Scholar]
- Theato P.; Sumerlin B. S.; O’Reilly R. K.; Epps T. H. III Stimuli responsive materials. Chem. Soc. Rev. 2013, 42 (17), 7055–7056. 10.1039/c3cs90057f. [DOI] [PubMed] [Google Scholar]
- Muskovich M.; Bettinger C. J. Biomaterials-Based Electronics: Polymers and Interfaces for Biology and Medicine. Adv. Healthcare Mater. 2012, 1 (3), 248–266. 10.1002/adhm.201200071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A. P.; Kammeyer J. K.; Rush A. M.; Callmann C. E.; Hahn M. E.; Gianneschi N. C. Stimuli-Responsive Nanomaterials for Biomedical Applications. J. Am. Chem. Soc. 2015, 137 (6), 2140–2154. 10.1021/ja510147n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole-Warren L. A.; Patton A. J.. 1 - Introduction to biomedical polymers and biocompatibility. In Biosynthetic Polymers for Medical Applications; Poole-Warren L., Martens P., Green R., Eds.; Woodhead Publishing: 2016; pp 3–31. [Google Scholar]
- Vasi A.-M.; Popa M. I.; Butnaru M.; Dodi G.; Verestiuc L. Chemical functionalization of hyaluronic acid for drug delivery applications. Mater. Sci. Eng., C 2014, 38, 177–185. 10.1016/j.msec.2014.01.052. [DOI] [PubMed] [Google Scholar]
- Ngo J. T.; Schuman E. M.; Tirrell D. A. Mutant methionyl-tRNA synthetase from bacteria enables site-selective N-terminal labeling of proteins expressed in mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (13), 4992–4997. 10.1073/pnas.1216375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Zheng L.; Liu Q.; de Vries J. W.; Gerasimov J. Y.; Herrmann A. Nucleic Acid Chemistry in the Organic Phase: From Functionalized Oligonucleotides to DNA Side Chain Polymers. J. Am. Chem. Soc. 2014, 136 (40), 14255–14262. 10.1021/ja5080486. [DOI] [PubMed] [Google Scholar]
- Rowland M. J.; Atgie M.; Hoogland D.; Scherman O. A. Preparation and Supramolecular Recognition of Multivalent Peptide–Polysaccharide Conjugates by Cucurbit[8]uril in Hydrogel Formation. Biomacromolecules 2015, 16 (8), 2436–2443. 10.1021/acs.biomac.5b00680. [DOI] [PubMed] [Google Scholar]
- Bapat A. P.; Roy D.; Ray J. G.; Savin D. A.; Sumerlin B. S. Dynamic-Covalent Macromolecular Stars with Boronic Ester Linkages. J. Am. Chem. Soc. 2011, 133 (49), 19832–19838. 10.1021/ja207005z. [DOI] [PubMed] [Google Scholar]
- Cobo I.; Li M.; Sumerlin B. S.; Perrier S. Smart hybrid materials by conjugation of responsive polymers to biomacromolecules. Nat. Mater. 2015, 14 (2), 143–159. 10.1038/nmat4106. [DOI] [PubMed] [Google Scholar]
- Espeel P.; Du Prez F. E. Click”-Inspired Chemistry in Macromolecular Science: Matching Recent Progress and User Expectations. Macromolecules 2015, 48 (1), 2–14. 10.1021/ma501386v. [DOI] [Google Scholar]
- Patterson D. M.; Nazarova L. A.; Prescher J. A. Finding the Right (Bioorthogonal) Chemistry. ACS Chem. Biol. 2014, 9 (3), 592–605. 10.1021/cb400828a. [DOI] [PubMed] [Google Scholar]
- Alconcel S. N. S.; Kim S. H.; Tao L.; Maynard H. D. Synthesis of Biotinylated Aldehyde Polymers for Biomolecule Conjugation. Macromol. Rapid Commun. 2013, 34 (12), 983–989. 10.1002/marc.201300205. [DOI] [PubMed] [Google Scholar]
- Mukherjee S.; Hill M. R.; Sumerlin B. S. Self-healing hydrogels containing reversible oxime crosslinks. Soft Matter 2015, 11 (30), 6152–6161. 10.1039/C5SM00865D. [DOI] [PubMed] [Google Scholar]
- Sletten E. M.; Bertozzi C. R. From Mechanism to Mouse: A Tale of Two Bioorthogonal Reactions. Acc. Chem. Res. 2011, 44 (9), 666–676. 10.1021/ar200148z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S.; Edgar K. J. Staudinger Reactions for Selective Functionalization of Polysaccharides: A Review. Biomacromolecules 2015, 16 (9), 2556–2571. 10.1021/acs.biomac.5b00855. [DOI] [PubMed] [Google Scholar]
- Stamenović M. M.; Espeel P.; Camp W. V.; Du Prez F. E. Norbornenyl-Based RAFT Agents for the Preparation of Functional Polymers via Thiol–Ene Chemistry. Macromolecules 2011, 44 (14), 5619–5630. 10.1021/ma200799b. [DOI] [Google Scholar]
- Canalle L. A.; van der Knaap M.; Overhand M.; van Hest J. C. M. A Comparison of Triazole-forming Bioconjugation Techniques for Constructing Comb-Shaped Peptide–Polymer Bioconjugates. Macromol. Rapid Commun. 2011, 32 (2), 203–208. 10.1002/marc.201000507. [DOI] [PubMed] [Google Scholar]
- van Oers M. C. M.; Rutjes F. P. J. T.; van Hest J. C. M. Tubular Polymersomes: A Cross-Linker-Induced Shape Transformation. J. Am. Chem. Soc. 2013, 135 (44), 16308–16311. 10.1021/ja408754z. [DOI] [PubMed] [Google Scholar]
- Kharkar P. M.; Kloxin A. M.; Kiick K. L. Dually degradable click hydrogels for controlled degradation and protein release. J. Mater. Chem. B 2014, 2 (34), 5511–5521. 10.1039/C4TB00496E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih H.-W.; Prescher J. A. A Bioorthogonal Ligation of Cyclopropenones Mediated by Triarylphosphines. J. Am. Chem. Soc. 2015, 137 (32), 10036–10039. 10.1021/jacs.5b06969. [DOI] [PubMed] [Google Scholar]
- Klok H.-A. Peptide/Protein–Synthetic Polymer Conjugates: Quo Vadis. Macromolecules 2009, 42 (21), 7990–8000. 10.1021/ma901561t. [DOI] [Google Scholar]
- Romulus J.; Henssler J. T.; Weck M. Postpolymerization Modification of Block Copolymers. Macromolecules 2014, 47 (16), 5437–5449. 10.1021/ma5009918. [DOI] [Google Scholar]
- Reece J. B.; Urry L. A.; Cain M. L.; Wasserman S. A.; Minorsky P. V.; Jackson R.; Campbell N. A.. Campbell Biology; Pearson: 2014. [Google Scholar]
- García-Borrón J. C.; Olivares Sánchez M. C.. Biosynthesis of Melanins. In Melanins and Melanosomes; Wiley-VCH Verlag GmbH & Co. KGaA: 2011; pp 87–116. [Google Scholar]
- Lutz J.-F.; Sumerlin B.; Matyjaszewski K. Some More Insights on Precisely Controlled Polymer Architectures. Macromol. Rapid Commun. 2014, 35 (4), 377–377. 10.1002/marc.201400072. [DOI] [PubMed] [Google Scholar]
- Noel A.; Borguet Y. P.; Raymond J. E.; Wooley K. L. Poly(ferulic acid-co-tyrosine): Effect of the Regiochemistry on the Photophysical and Physical Properties en Route to Biomedical Applications. Macromolecules 2014, 47 (20), 7109–7117. 10.1021/ma5015534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szwarc M. `Living’ Polymers. Nature 1956, 178 (4543), 1168–1169. 10.1038/1781168a0. [DOI] [Google Scholar]
- Zhang Q.; Collins J.; Anastasaki A.; Wallis R.; Mitchell D. A.; Becer C. R.; Haddleton D. M. Sequence-Controlled Multi-Block Glycopolymers to Inhibit DC-SIGN-gp120 Binding. Angew. Chem., Int. Ed. 2013, 52 (16), 4435–4439. 10.1002/anie.201300068. [DOI] [PubMed] [Google Scholar]
- Samanta S. R.; Anastasaki A.; Waldron C.; Haddleton D. M.; Percec V. SET-LRP of hydrophobic and hydrophilic acrylates in tetrafluoropropanol. Polym. Chem. 2013, 4 (22), 5555–5562. 10.1039/c3py00901g. [DOI] [Google Scholar]
- Costa J. R. C.; Mendonça P. V.; Maximiano P.; Serra A. C.; Guliashvili T.; Coelho J. F. J. Ambient Temperature “Flash” SARA ATRP of Methyl Acrylate in Water/Ionic Liquid/Glycol Mixtures. Macromolecules 2015, 48 (19), 6810–6815. 10.1021/acs.macromol.5b01795. [DOI] [Google Scholar]
- Konkolewicz D.; Krys P.; Góis J. R.; Mendonça P. V.; Zhong M.; Wang Y.; Gennaro A.; Isse A. A.; Fantin M.; Matyjaszewski K. Aqueous RDRP in the Presence of Cu0: The Exceptional Activity of CuI Confirms the SARA ATRP Mechanism. Macromolecules 2014, 47 (2), 560–570. 10.1021/ma4022983. [DOI] [Google Scholar]
- Mackenzie M. C.; Shrivats A. R.; Konkolewicz D.; Averick S. E.; McDermott M. C.; Hollinger J. O.; Matyjaszewski K. Synthesis of Poly(meth)acrylates with Thioether and Tertiary Sulfonium Groups by ARGET ATRP and Their Use as siRNA Delivery Agents. Biomacromolecules 2015, 16 (1), 236–245. 10.1021/bm501449a. [DOI] [PubMed] [Google Scholar]
- Mendonça P. V.; Averick S. E.; Konkolewicz D.; Serra A. C.; Popov A. V.; Guliashvili T.; Matyjaszewski K.; Coelho J. F. J. Straightforward ARGET ATRP for the Synthesis of Primary Amine Polymethacrylate with Improved Chain-End Functionality under Mild Reaction Conditions. Macromolecules 2014, 47 (14), 4615–4621. 10.1021/ma501007j. [DOI] [Google Scholar]
- Simakova A.; Averick S. E.; Konkolewicz D.; Matyjaszewski K. Aqueous ARGET ATRP. Macromolecules 2012, 45 (16), 6371–6379. 10.1021/ma301303b. [DOI] [Google Scholar]
- Shida N.; Koizumi Y.; Nishiyama H.; Tomita I.; Inagi S. Electrochemically Mediated Atom Transfer Radical Polymerization from a Substrate Surface Manipulated by Bipolar Electrolysis: Fabrication of Gradient and Patterned Polymer Brushes. Angew. Chem., Int. Ed. 2015, 54 (13), 3922–3926. 10.1002/anie.201412391. [DOI] [PubMed] [Google Scholar]
- Magenau A. J. D.; Bortolamei N.; Frick E.; Park S.; Gennaro A.; Matyjaszewski K. Investigation of Electrochemically Mediated Atom Transfer Radical Polymerization. Macromolecules 2013, 46 (11), 4346–4353. 10.1021/ma400869e. [DOI] [Google Scholar]
- Ribelli T. G.; Konkolewicz D.; Pan X.; Matyjaszewski K. Contribution of Photochemistry to Activator Regeneration in ATRP. Macromolecules 2014, 47 (18), 6316–6321. 10.1021/ma501384q. [DOI] [Google Scholar]
- Ribelli T. G.; Konkolewicz D.; Bernhard S.; Matyjaszewski K. How are Radicals (Re)Generated in Photochemical ATRP?. J. Am. Chem. Soc. 2014, 136 (38), 13303–13312. 10.1021/ja506379s. [DOI] [PubMed] [Google Scholar]
- Liu X.; Zhang L.; Cheng Z.; Zhu X. Metal-free photoinduced electron transfer-atom transfer radical polymerization (PET-ATRP) via a visible light organic photocatalyst. Polym. Chem. 2016, 7, 689. 10.1039/C5PY01765C. [DOI] [Google Scholar]
- Shanmugam S.; Boyer C. Stereo-, Temporal and Chemical Control through Photoactivation of Living Radical Polymerization: Synthesis of Block and Gradient Copolymers. J. Am. Chem. Soc. 2015, 137 (31), 9988–9999. 10.1021/jacs.5b05903. [DOI] [PubMed] [Google Scholar]
- Xu J.; Shanmugam S.; Boyer C. Organic Electron Donor–Acceptor Photoredox Catalysts: Enhanced Catalytic Efficiency toward Controlled Radical Polymerization. ACS Macro Lett. 2015, 4 (9), 926–932. 10.1021/acsmacrolett.5b00460. [DOI] [PubMed] [Google Scholar]
- Otsu T. Iniferter concept and living radical polymerization. J. Polym. Sci., Part A: Polym. Chem. 2000, 38 (12), 2121–2136. . [DOI] [Google Scholar]
- Tesch M.; Hepperle J. A. M.; Klaasen H.; Letzel M.; Studer A. Alternating Copolymerization by Nitroxide-Mediated Polymerization and Subsequent Orthogonal Functionalization. Angew. Chem., Int. Ed. 2015, 54 (17), 5054–5059. 10.1002/anie.201412206. [DOI] [PubMed] [Google Scholar]
- Guégain E.; Guillaneuf Y.; Nicolas J. Nitroxide-Mediated Polymerization of Methacrylic Esters: Insights and Solutions to a Long-Standing Problem. Macromol. Rapid Commun. 2015, 36 (13), 1227–1247. 10.1002/marc.201500042. [DOI] [PubMed] [Google Scholar]
- Garcia-Valdez O.; Champagne-Hartley R.; Saldivar-Guerra E.; Champagne P.; Cunningham M. F. Modification of chitosan with polystyrene and poly(n-butyl acrylate) via nitroxide-mediated polymerization and grafting from approach in homogeneous media. Polym. Chem. 2015, 6 (15), 2827–2836. 10.1039/C5PY00028A. [DOI] [Google Scholar]
- Delplace V.; Harrisson S.; Ho H. T.; Tardy A.; Guillaneuf Y.; Pascual S.; Fontaine L.; Nicolas J. One-Step Synthesis of Azlactone-Functionalized SG1-Based Alkoxyamine for Nitroxide-Mediated Polymerization and Bioconjugation. Macromolecules 2015, 48 (7), 2087–2097. 10.1021/acs.macromol.5b00178. [DOI] [Google Scholar]
- Kamber N. E.; Jeong W.; Waymouth R. M.; Pratt R. C.; Lohmeijer B. G. G.; Hedrick J. L. Organocatalytic Ring-Opening Polymerization. Chem. Rev. 2007, 107 (12), 5813–5840. 10.1021/cr068415b. [DOI] [PubMed] [Google Scholar]
- McGrath A. J.; Shi W.; Rodriguez C. G.; Kramer E. J.; Hawker C. J.; Lynd N. A. Synthetic strategy for preparing chiral double-semicrystalline polyether block copolymers. Polym. Chem. 2015, 6 (9), 1465–1473. 10.1039/C4PY01503G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S.; Ishizone T.; Hirao A. Precise Synthesis of New Exactly Defined Graft Copolymers Made up of Poly(alkyl methacrylate)s by Iterative Methodology Using Living Anionic Polymerization. Macromolecules 2015, 48, 8307. 10.1021/acs.macromol.5b01908. [DOI] [Google Scholar]
- Hirao A.; Goseki R.; Ishizone T. Advances in Living Anionic Polymerization: From Functional Monomers, Polymerization Systems, to Macromolecular Architectures. Macromolecules 2014, 47 (6), 1883–1905. 10.1021/ma401175m. [DOI] [Google Scholar]
- Crawford K. E.; Sita L. R. Stereoengineering of Poly(1,3-methylenecyclohexane) via Two-State Living Coordination Polymerization of 1,6-Heptadiene. J. Am. Chem. Soc. 2013, 135 (24), 8778–8781. 10.1021/ja402262x. [DOI] [PubMed] [Google Scholar]
- Kundu S.; Bhangale A. S.; Wallace W. E.; Flynn K. M.; Guttman C. M.; Gross R. A.; Beers K. L. Continuous Flow Enzyme-Catalyzed Polymerization in a Microreactor. J. Am. Chem. Soc. 2011, 133 (15), 6006–6011. 10.1021/ja111346c. [DOI] [PubMed] [Google Scholar]
- Castano M.; Zheng J.; Puskas J. E.; Becker M. L. Enzyme-catalyzed ring-opening polymerization of ε-caprolactone using alkyne functionalized initiators. Polym. Chem. 2014, 5 (6), 1891–1896. 10.1039/C3PY01536J. [DOI] [Google Scholar]
- Deming T. J. Synthesis of Side-Chain Modified Polypeptides. Chem. Rev. 2016, 116 (3), 786–808. 10.1021/acs.chemrev.5b00292. [DOI] [PubMed] [Google Scholar]
- Cao J.; Hu P.; Lu L.; Chan B. A.; Luo B.-H.; Zhang D. Non-ionic water-soluble “clickable” α-helical polypeptides: synthesis, characterization and side chain modification. Polym. Chem. 2015, 6 (8), 1226–1229. 10.1039/C4PY01560F. [DOI] [Google Scholar]
- Chatterjee A. K.; Toste F. D.; Choi T.-L.; Grubbs R. H. Ruthenium-Catalyzed Olefin Cross Metathesis of Styrenes as an Alternative to the Heck and Cross-Coupling Reactions. Adv. Synth. Catal. 2002, 344 (6–7), 634–637. . [DOI] [Google Scholar]
- Romulus J.; Tan L.; Weck M.; Sampson N. S. Alternating ROMP copolymers containing charge-transfer units. ACS Macro Lett. 2013, 2 (8), 749–752. 10.1021/mz4002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling B. R.; Xia Y. Living Alternating Ring-Opening Metathesis Polymerization Based on Single Monomer Additions. J. Am. Chem. Soc. 2015, 137 (31), 9922–9926. 10.1021/jacs.5b05497. [DOI] [PubMed] [Google Scholar]
- Tan L.; Li G.; Parker K. A.; Sampson N. S. Ru-Catalyzed Isomerization Provides Access to Alternating Copolymers via Ring-Opening Metathesis Polymerization. Macromolecules 2015, 48 (14), 4793–4800. 10.1021/acs.macromol.5b01058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K. A.; Goetz A. E.; Boydston A. J. Metal-Free Ring-Opening Metathesis Polymerization. J. Am. Chem. Soc. 2015, 137 (4), 1400–1403. 10.1021/ja512073m. [DOI] [PubMed] [Google Scholar]
- Merrifield R. B. Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. J. Am. Chem. Soc. 1963, 85 (14), 2149–2154. 10.1021/ja00897a025. [DOI] [Google Scholar]
- Ouahabi A. A.; Kotera M.; Charles L.; Lutz J.-F. Synthesis of Monodisperse Sequence-Coded Polymers with Chain Lengths above DP100. ACS Macro Lett. 2015, 4 (10), 1077–1080. 10.1021/acsmacrolett.5b00606. [DOI] [PubMed] [Google Scholar]
- Roy R. K.; Lutz J.-F. Compartmentalization of Single Polymer Chains by Stepwise Intramolecular Cross-Linking of Sequence-Controlled Macromolecules. J. Am. Chem. Soc. 2014, 136 (37), 12888–12891. 10.1021/ja507889x. [DOI] [PubMed] [Google Scholar]
- Gutekunst W. R.; Hawker C. J. A General Approach to Sequence-Controlled Polymers Using Macrocyclic Ring Opening Metathesis Polymerization. J. Am. Chem. Soc. 2015, 137 (25), 8038–8041. 10.1021/jacs.5b04940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Matta M. E.; Hillmyer M. A. Synthesis of Sequence-Specific Vinyl Copolymers by Regioselective ROMP of Multiply Substituted Cyclooctenes. ACS Macro Lett. 2012, 1 (12), 1383–1387. 10.1021/mz300535r. [DOI] [PubMed] [Google Scholar]
- Thomas R. M.; Grubbs R. H. Synthesis of Telechelic Polyisoprene via Ring-Opening Metathesis Polymerization in the Presence of Chain Transfer Agent. Macromolecules 2010, 43 (8), 3705–3709. 10.1021/ma902749q. [DOI] [Google Scholar]
- Fishman J. M.; Kiessling L. L. Synthesis of Functionalizable and Degradable Polymers by ROMP. Angew. Chem., Int. Ed. 2013, 52 (19), 5061. 10.1002/anie.201300293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magennis E. P.; Fernandez-Trillo F.; Sui C.; Spain S. G.; Bradshaw D. J.; Churchley D.; Mantovani G.; Winzer K.; Alexander C. Bacteria-instructed synthesis of polymers for self-selective microbial binding and labelling. Nat. Mater. 2014, 13 (7), 748–755. 10.1038/nmat3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M. E.; Randolph L. M.; Adamiak L.; Thompson M. P.; Gianneschi N. C. Polymerization of a peptide-based enzyme substrate. Chem. Commun. 2013, 49 (28), 2873–2875. 10.1039/c3cc40472b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James C. R.; Rush A. M.; Insley T.; Vuković L.; Adamiak L.; Král P.; Gianneschi N. C. Poly(oligonucleotide). J. Am. Chem. Soc. 2014, 136 (32), 11216–11219. 10.1021/ja503142s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veccharelli K. M.; Tong V. K.; Young J. L.; Yang J.; Gianneschi N. C. Dual responsive polymeric nanoparticles prepared by direct functionalization of polylactic acid-based polymers via graft-from ring opening metathesis polymerization. Chem. Commun. 2016, 52, 567. 10.1039/C5CC07882B. [DOI] [PubMed] [Google Scholar]
- Thompson M. P.; Randolph L. M.; James C. R.; Davalos A. N.; Hahn M. E.; Gianneschi N. C. Labelling polymers and micellar nanoparticles via initiation, propagation and termination with ROMP. Polym. Chem. 2014, 5 (6), 1954–1964. 10.1039/C3PY01338C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callmann C. E.; Barback C. V.; Thompson M. P.; Hall D. J.; Mattrey R. F.; Gianneschi N. C. Therapeutic Enzyme-Responsive Nanoparticles for Targeted Delivery and Accumulation in Tumors. Adv. Mater. 2015, 27 (31), 4611–4615. 10.1002/adma.201501803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien M.-P.; Thompson M. P.; Barback C. V.; Ku T.-H.; Hall D. J.; Gianneschi N. C. Enzyme-Directed Assembly of a Nanoparticle Probe in Tumor Tissue. Adv. Mater. 2013, 25 (26), 3599–3604. 10.1002/adma.201300823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blum A. P.; Kammeyer J. K.; Yin J.; Crystal D. T.; Rush A. M.; Gilson M. K.; Gianneschi N. C. Peptides Displayed as High Density Brush Polymers Resist Proteolysis and Retain Bioactivity. J. Am. Chem. Soc. 2014, 136 (43), 15422–15437. 10.1021/ja5088216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammeyer J. K.; Blum A. P.; Adamiak L.; Hahn M. E.; Gianneschi N. C. Polymerization of protecting-group-free peptides via ROMP. Polym. Chem. 2013, 4 (14), 3929–3933. 10.1039/c3py00526g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien M.-P.; Carlini A. S.; Hu D.; Barback C. V.; Rush A. M.; Hall D. J.; Orr G.; Gianneschi N. C. Enzyme-Directed Assembly of Nanoparticles in Tumors Monitored by in Vivo Whole Animal Imaging and ex Vivo Super-Resolution Fluorescence Imaging. J. Am. Chem. Soc. 2013, 135 (50), 18710–18713. 10.1021/ja408182p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes A. J.; Deming T. J. Tandem Catalysis for the Preparation of Cylindrical Polypeptide Brushes. J. Am. Chem. Soc. 2012, 134 (47), 19463–19467. 10.1021/ja308620h. [DOI] [PubMed] [Google Scholar]
- Lin E.-W.; Maynard H. D. Grafting from Small Interfering Ribonucleic Acid (siRNA) as an Alternative Synthesis Route to siRNA–Polymer Conjugates. Macromolecules 2015, 48 (16), 5640–5647. 10.1021/acs.macromol.5b00846. [DOI] [Google Scholar]
- Tucker B. S.; Stewart J. D.; Aguirre J. I.; Holliday L. S.; Figg C. A.; Messer J. G.; Sumerlin B. S. Role of Polymer Architecture on the Activity of Polymer–Protein Conjugates for the Treatment of Accelerated Bone Loss Disorders. Biomacromolecules 2015, 16 (8), 2374–2381. 10.1021/acs.biomac.5b00623. [DOI] [PubMed] [Google Scholar]
- Sgolastra F.; Minter L. M.; Osborne B. A.; Tew G. N. Importance of Sequence Specific Hydrophobicity in Synthetic Protein Transduction Domain Mimics. Biomacromolecules 2014, 15 (3), 812–820. 10.1021/bm401634r. [DOI] [PubMed] [Google Scholar]
- deRonde B. M.; Torres J. A.; Minter L. M.; Tew G. N. Development of Guanidinium-Rich Protein Mimics for Efficient siRNA Delivery into Human T Cells. Biomacromolecules 2015, 16 (10), 3172–3179. 10.1021/acs.biomac.5b00795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas C.; Mitra K.; Singh S.; Ramesh K.; Misra N.; Maiti B.; Panda A.; Maiti P.; Kamigaito M.; Okamoto Y.; Ray B. Study of the effect of isotacticity on some physical properties of poly(N-isopropylacrylamide). Colloid Polym. Sci. 2015, 293 (6), 1749–1757. 10.1007/s00396-015-3562-3. [DOI] [Google Scholar]
- Cameron D. J. A.; Shaver M. P. Control of thermal properties and hydrolytic degradation in poly(lactic acid) polymer stars through control of isospecificity of polymer arms. J. Polym. Sci., Part A: Polym. Chem. 2012, 50 (8), 1477–1484. 10.1002/pola.25927. [DOI] [Google Scholar]
- Barnes J. C.; Ehrlich D. J. C.; Gao A. X.; Leibfarth F. A.; Jiang Y.; Zhou E.; Jamison T. F.; Johnson J. A. Iterative exponential growth of stereo- and sequence-controlled polymers. Nat. Chem. 2015, 7 (10), 810–815. 10.1038/nchem.2346. [DOI] [PubMed] [Google Scholar]
- Leibfarth F. A.; Johnson J. A.; Jamison T. F. Scalable synthesis of sequence-defined, unimolecular macromolecules by Flow-IEG. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (34), 10617–10622. 10.1073/pnas.1508599112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright D. B.; Patterson J. P.; Pitto-Barry A.; Lu A.; Kirby N.; Gianneschi N. C.; Chassenieux C.; Colombani O.; O’Reilly R. K. The Copolymer Blending Method: A New Approach for Targeted Assembly of Micellar Nanoparticles. Macromolecules 2015, 48 (18), 6516–6522. 10.1021/acs.macromol.5b01426. [DOI] [Google Scholar]
- Ren J. M.; Satoh K.; Goh T. K.; Blencowe A.; Nagai K.; Ishitake K.; Christofferson A. J.; Yiapanis G.; Yarovsky I.; Kamigaito M.; Qiao G. G. Stereospecific Cyclic Poly(methyl methacrylate) and Its Topology-Guided Hierarchically Controlled Supramolecular Assemblies. Angew. Chem., Int. Ed. 2014, 53 (2), 459–464. 10.1002/anie.201308366. [DOI] [PubMed] [Google Scholar]
- Mai Y.; Eisenberg A. Selective Localization of Preformed Nanoparticles in Morphologically Controllable Block Copolymer Aggregates in Solution. Acc. Chem. Res. 2012, 45 (10), 1657–1666. 10.1021/ar2003144. [DOI] [PubMed] [Google Scholar]
- De Koker S.; De Cock L. J.; Rivera-Gil P.; Parak W. J.; Auzély Velty R.; Vervaet C.; Remon J. P.; Grooten J.; De Geest B. G. Polymeric multilayer capsules delivering biotherapeutics. Adv. Drug Delivery Rev. 2011, 63 (9), 748–761. 10.1016/j.addr.2011.03.014. [DOI] [PubMed] [Google Scholar]
- Buwalda S. J.; Boere K. W. M.; Dijkstra P. J.; Feijen J.; Vermonden T.; Hennink W. E. Hydrogels in a historical perspective: From simple networks to smart materials. J. Controlled Release 2014, 190, 254–273. 10.1016/j.jconrel.2014.03.052. [DOI] [PubMed] [Google Scholar]
- Kramer J. R.; Deming T. J. Multimodal Switching of Conformation and Solubility in Homocysteine Derived Polypeptides. J. Am. Chem. Soc. 2014, 136 (15), 5547–5550. 10.1021/ja500372u. [DOI] [PubMed] [Google Scholar]
- Singh R.; Norret M.; House M. J.; Galabura Y.; Bradshaw M.; Ho D.; Woodward R. C.; Pierre T. G. S.; Luzinov I.; Smith N. M.; Lim L. Y.; Iyer K. S. Dose-Dependent Therapeutic Distinction between Active and Passive Targeting Revealed Using Transferrin-Coated PGMA Nanoparticles. Small 2016, 12 (3), 351–359. 10.1002/smll.201502730. [DOI] [PubMed] [Google Scholar]
- Nguyen M. M.; Carlini A. S.; Chien M.-P.; Sonnenberg S.; Luo C.; Braden R. L.; Osborn K. G.; Li Y.; Gianneschi N. C.; Christman K. L. Enzyme-Responsive Nanoparticles for Targeted Accumulation and Prolonged Retention in Heart Tissue after Myocardial Infarction. Adv. Mater. 2015, 27 (37), 5547–5552. 10.1002/adma.201502003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvir T.; Bauer M.; Schroeder A.; Tsui J. H.; Anderson D. G.; Langer R.; Liao R.; Kohane D. S. Nanoparticles Targeting the Infarcted Heart. Nano Letters 2011, 11 (10), 4411–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Lin T.; Zhang W.; Jiang Y.; Jin H.; He H.; Yang V. C.; Chen Y.; Huang Y. A Prodrug-type, MMP-2-targeting Nanoprobe for Tumor Detection and Imaging. Theranostics 2015, 5 (8), 787–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacEwan S. R.; Chilkoti A. Controlled Apoptosis by a Thermally Toggled Nanoscale Amplifier of Cellular Uptake. Nano Lett. 2014, 14 (4), 2058–2064. 10.1021/nl5002313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray J. G.; Naik S. S.; Hoff E. A.; Johnson A. J.; Ly J. T.; Easterling C. P.; Patton D. L.; Savin D. A. Stimuli-Responsive Peptide-Based ABA-Triblock Copolymers: Unique Morphology Transitions With pH. Macromol. Rapid Commun. 2012, 33 (9), 819–826. 10.1002/marc.201100881. [DOI] [PubMed] [Google Scholar]
- Itel F.; Najer A.; Palivan C. G.; Meier W. Dynamics of Membrane Proteins within Synthetic Polymer Membranes with Large Hydrophobic Mismatch. Nano Lett. 2015, 15 (6), 3871–3878. 10.1021/acs.nanolett.5b00699. [DOI] [PubMed] [Google Scholar]
- Hunt J. A.; Chen R.; van Veen T.; Bryan N. Hydrogels for tissue engineering and regenerative medicine. J. Mater. Chem. B 2014, 2 (33), 5319–5338. 10.1039/C4TB00775A. [DOI] [PubMed] [Google Scholar]
- Peppas N. A.; Hilt J. Z.; Khademhosseini A.; Langer R. Hydrogels in Biology and Medicine: From Molecular Principles to Bionanotechnology. Adv. Mater. 2006, 18 (11), 1345–1360. 10.1002/adma.200501612. [DOI] [Google Scholar]
- Ahmed E. M. Hydrogel: Preparation, characterization, and applications: A review. Journal of Advanced Research 2015, 6 (2), 105–121. 10.1016/j.jare.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungerleider J. L.; Christman K. L. Concise Review: Injectable Biomaterials for the Treatment of Myocardial Infarction and Peripheral Artery Disease: Translational Challenges and Progress. Stem Cells Transl. Med. 2014, 3 (9), 1090–1099. 10.5966/sctm.2014-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phadke A.; Zhang C.; Arman B.; Hsu C.-C.; Mashelkar R. A.; Lele A. K.; Tauber M. J.; Arya G.; Varghese S. Rapid self-healing hydrogels. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (12), 4383–4388. 10.1073/pnas.1201122109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Q.; Schexnailder P.; Gaharwar A. K.; Schmidt G. Silicate Cross-Linked Bio-Nanocomposite Hydrogels from PEO and Chitosan. Macromol. Biosci. 2009, 9 (10), 1028–1035. 10.1002/mabi.200900080. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Mynar J. L.; Yoshida M.; Lee E.; Lee M.; Okuro K.; Kinbara K.; Aida T. High-water-content mouldable hydrogels by mixing clay and a dendritic molecular binder. Nature 2010, 463 (7279), 339–343. 10.1038/nature08693. [DOI] [PubMed] [Google Scholar]
- Rose S.; Prevoteau A.; Elziere P.; Hourdet D.; Marcellan A.; Leibler L. Nanoparticle solutions as adhesives for gels and biological tissues. Nature 2014, 505 (7483), 382–385. 10.1038/nature12806. [DOI] [PubMed] [Google Scholar]
- Appel E. A.; Tibbitt M. W.; Webber M. J.; Mattix B. A.; Veiseh O.; Langer R. Self-assembled hydrogels utilizing polymer–nanoparticle interactions. Nat. Commun. 2015, 6, 6295. 10.1038/ncomms7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaharwar A. K.; Avery R. K.; Assmann A.; Paul A.; McKinley G. H.; Khademhosseini A.; Olsen B. D. Shear-Thinning Nanocomposite Hydrogels for the Treatment of Hemorrhage. ACS Nano 2014, 8 (10), 9833–9842. 10.1021/nn503719n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J.; Harada A.; Kamachi M. Sol-Gel Transition During Inclusion Complex-Formation Between Alpha-Cyclodextrin and High-Molecular Weight Poly(ethylene glycol)s in Aqueous Solution. Polym. J. 1994, 26 (9), 1019–1026. 10.1295/polymj.26.1019. [DOI] [Google Scholar]
- Li G.; Wu J.; Wang B.; Yan S.; Zhang K.; Ding J.; Yin J. Self-Healing Supramolecular Self-Assembled Hydrogels Based on Poly(l-glutamic acid). Biomacromolecules 2015, 16 (11), 3508–3518. 10.1021/acs.biomac.5b01287. [DOI] [PubMed] [Google Scholar]
- Himmelein S.; Lewe V.; Stuart M. C. A.; Ravoo B. J. A carbohydrate-based hydrogel containing vesicles as responsive non-covalent cross-linkers. Chemical Science 2014, 5 (3), 1054–1058. 10.1039/c3sc52964a. [DOI] [Google Scholar]
- McKee J. R.; Appel E. A.; Seitsonen J.; Kontturi E.; Scherman O. A.; Ikkala O. Healable, Stable and Stiff Hydrogels: Combining Conflicting Properties Using Dynamic and Selective Three-Component Recognition with Reinforcing Cellulose Nanorods. Adv. Funct. Mater. 2014, 24 (18), 2706–2713. 10.1002/adfm.201303699. [DOI] [Google Scholar]
- Zhu J.; Marchant R. E. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev. Med. Devices 2011, 8 (5), 607–626. 10.1586/erd.11.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urquhart J.Underwater self-healing polymer mimics mussels. Chemistry World, 2014. [Google Scholar]
- Wei Z.; Yang J. H.; Liu Z. Q.; Xu F.; Zhou J. X.; Zrínyi M.; Osada Y.; Chen Y. M. Novel Biocompatible Polysaccharide-Based Self-Healing Hydrogel. Adv. Funct. Mater. 2015, 25 (9), 1352–1359. 10.1002/adfm.201401502. [DOI] [Google Scholar]
- Deng C. C.; Brooks W. L. A.; Abboud K. A.; Sumerlin B. S. Boronic Acid-Based Hydrogels Undergo Self-Healing at Neutral and Acidic pH. ACS Macro Lett. 2015, 4 (2), 220–224. 10.1021/acsmacrolett.5b00018. [DOI] [PubMed] [Google Scholar]
- Lang N.; Pereira M. J.; Lee Y.; Friehs I.; Vasilyev N. V.; Feins E. N.; Ablasser K.; O’Cearbhaill E. D.; Xu C.; Fabozzo A.; Padera R.; Wasserman S.; Freudenthal F.; Ferreira L. S.; Langer R.; Karp J. M.; del Nido P. J. A Blood-Resistant Surgical Glue for Minimally Invasive Repair of Vessels and Heart Defects. Sci. Transl. Med. 2014, 6 (218), 218ra6–218ra6. 10.1126/scitranslmed.3006557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo J.; Kang T.; Jang S. G.; Hwang D. S.; Spruell J. M.; Killops K. L.; Waite J. H.; Hawker C. J. Improved Performance of Protected Catecholic Polysiloxanes for Bioinspired Wet Adhesion to Surface Oxides. J. Am. Chem. Soc. 2012, 134 (49), 20139–20145. 10.1021/ja309044z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Defante A. P.; Lin F.; Xu Y.; Yu J.; Gao Y.; Childers E.; Dhinojwala A.; Becker M. L. Adhesion Properties of Catechol-Based Biodegradable Amino Acid-Based Poly(ester urea) Copolymers Inspired from Mussel Proteins. Biomacromolecules 2015, 16 (1), 266–274. 10.1021/bm501456g. [DOI] [PubMed] [Google Scholar]
- Krogsgaard M.; Behrens M. A.; Pedersen J. S.; Birkedal H. Self-Healing Mussel-Inspired Multi-pH-Responsive Hydrogels. Biomacromolecules 2013, 14 (2), 297–301. 10.1021/bm301844u. [DOI] [PubMed] [Google Scholar]
- Holten-Andersen N.; Harrington M. J.; Birkedal H.; Lee B. P.; Messersmith P. B.; Lee K. Y. C.; Waite J. H. pH-induced metal-ligand cross-links inspired by mussel yield self-healing polymer networks with near-covalent elastic moduli. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (7), 2651–2655. 10.1073/pnas.1015862108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn B. K.; Lee D. W.; Israelachvili J. N.; Waite J. H. Surface-initiated self-healing of polymers in aqueous media. Nat. Mater. 2014, 13 (9), 867–872. 10.1038/nmat4037. [DOI] [PubMed] [Google Scholar]
- Rousseau M.-A.; Lazennec J.-Y.; Catonné Y. Early mechanical failure in total knee arthroplasty. International Orthopaedics 2008, 32 (1), 53–56. 10.1007/s00264-006-0276-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedó J.; Saiz-Poseu J.; Busqué F.; Ruiz-Molina D. Catechol-Based Biomimetic Functional Materials. Adv. Mater. 2013, 25 (5), 653–701. 10.1002/adma.201202343. [DOI] [PubMed] [Google Scholar]
- Zhang D.; George O. J.; Petersen K. M.; Jimenez-Vergara A. C.; Hahn M. S.; Grunlan M. A. A bioactive “self-fitting” shape memory polymer scaffold with potential to treat cranio-maxillo facial bone defects. Acta Biomater. 2014, 10 (11), 4597–4605. 10.1016/j.actbio.2014.07.020. [DOI] [PubMed] [Google Scholar]
- Policastro G. M.; Lin F.; Smith Callahan L. A.; Esterle A.; Graham M.; Sloan Stakleff K.; Becker M. L. OGP Functionalized Phenylalanine-Based Poly(ester urea) for Enhancing Osteoinductive Potential of Human Mesenchymal Stem Cells. Biomacromolecules 2015, 16 (4), 1358–1371. 10.1021/acs.biomac.5b00153. [DOI] [PubMed] [Google Scholar]
- Stakleff K. S.; Lin F.; Smith Callahan L. A.; Wade M. B.; Esterle A.; Miller J.; Graham M.; Becker M. L. Resorbable, amino acid-based poly(ester urea)s crosslinked with osteogenic growth peptide with enhanced mechanical properties and bioactivity. Acta Biomater. 2013, 9 (2), 5132–5142. 10.1016/j.actbio.2012.08.035. [DOI] [PubMed] [Google Scholar]
- Walsh G. Biopharmaceutical benchmarks 2010. Nat. Biotechnol. 2010, 28 (9), 917–924. 10.1038/nbt0910-917. [DOI] [PubMed] [Google Scholar]
- Hall M. P. Biotransformation and In Vivo Stability of Protein Biotherapeutics: Impact on Candidate Selection and Pharmacokinetic Profiling. Drug Metab. Dispos. 2014, 42 (11), 1873–1880. 10.1124/dmd.114.058347. [DOI] [PubMed] [Google Scholar]
- van Eldijk M. B.; Smits F. C. M.; Vermue N.; Debets M. F.; Schoffelen S.; van Hest J. C. M. Synthesis and Self-Assembly of Well-Defined Elastin-Like Polypeptide–Poly(ethylene glycol) Conjugates. Biomacromolecules 2014, 15 (7), 2751–2759. 10.1021/bm5006195. [DOI] [PubMed] [Google Scholar]
- Lee J.; Ko J. H.; Lin E.-W.; Wallace P.; Ruch F.; Maynard H. D. Trehalose hydrogels for stabilization of enzymes to heat. Polym. Chem. 2015, 6 (18), 3443–3448. 10.1039/C5PY00121H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong R.; Pang Y.; Su Y.; Zhu X. Supramolecular hydrogels: synthesis, properties and their biomedical applications. Biomater. Sci. 2015, 3 (7), 937–954. 10.1039/C4BM00448E. [DOI] [PubMed] [Google Scholar]
- Harding S. E. Some observations on the effects of bioprocessing on biopolymer stability. J. Drug Targeting 2010, 18 (10), 732–740. 10.3109/1061186X.2010.512470. [DOI] [PubMed] [Google Scholar]
- O’Shea T. M.; Webber M. J.; Aimetti A. A.; Langer R. Covalent Incorporation of Trehalose within Hydrogels for Enhanced Long-Term Functional Stability and Controlled Release of Biomacromolecules. Adv. Healthcare Mater. 2015, 4 (12), 1802–1812. 10.1002/adhm.201500334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue L.; Kelkar S. S.; Wang X.; Ma J.; Madsen L. A.; Reineke T. M. A theranostic polycation containing trehalose and lanthanide chelate domains for siRNA delivery and monitoring. RSC Adv. 2015, 5 (90), 74102–74106. 10.1039/C5RA14325J. [DOI] [Google Scholar]
- Vermonden T.; Censi R.; Hennink W. E. Hydrogels for Protein Delivery. Chem. Rev. (Washington, DC, U. S.) 2012, 112 (5), 2853–2888. 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- Vaishya R.; Khurana V.; Patel S.; Mitra A. K. Long-term delivery of protein therapeutics. Expert Opin. Drug Delivery 2015, 12 (3), 415–40. 10.1517/17425247.2015.961420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decher G.; Hong J. D.; Schmitt J. Buildup of ultrathin multilayer films by a self-assembly process: III. Consecutively alternating adsorption of anionic and cationic polyelectrolytes on charged surfaces. Thin Solid Films 1992, 210–211 (Part 2), 831–835. 10.1016/0040-6090(92)90417-A. [DOI] [Google Scholar]
- Almodóvar J.; Bacon S.; Gogolski J.; Kisiday J. D.; Kipper M. J. Polysaccharide-Based Polyelectrolyte Multilayer Surface Coatings can Enhance Mesenchymal Stem Cell Response to Adsorbed Growth Factors. Biomacromolecules 2010, 11 (10), 2629–2639. 10.1021/bm1005799. [DOI] [PubMed] [Google Scholar]
- Almodóvar J.; Place L. W.; Gogolski J.; Erickson K.; Kipper M. J. Layer-by-Layer Assembly of Polysaccharide-Based Polyelectrolyte Multilayers: A Spectroscopic Study of Hydrophilicity, Composition, and Ion Pairing. Biomacromolecules 2011, 12 (7), 2755–2765. 10.1021/bm200519y. [DOI] [PubMed] [Google Scholar]
- Cado G.; Kerdjoudj H.; Chassepot A.; Lefort M.; Benmlih K.; Hemmerlé J.; Voegel J. C.; Jierry L.; Schaaf P.; Frère Y.; Boulmedais F. Polysaccharide Films Built by Simultaneous or Alternate Spray: A Rapid Way to Engineer Biomaterial Surfaces. Langmuir 2012, 28 (22), 8470–8478. 10.1021/la300563s. [DOI] [PubMed] [Google Scholar]
- Crouzier T.; Szarpak A.; Boudou T.; Auzély-Velty R.; Picart C. Polysaccharide-Blend Multilayers Containing Hyaluronan and Heparin as a Delivery System for rhBMP-2. Small 2010, 6 (5), 651–662. 10.1002/smll.200901728. [DOI] [PubMed] [Google Scholar]
- Porcel C. H.; Izquierdo A.; Ball V.; Decher G.; Voegel J. C.; Schaaf P. Ultrathin Coatings and (Poly(glutamic acid)/Polyallylamine) Films Deposited by Continuous and Simultaneous Spraying. Langmuir 2005, 21 (2), 800–802. 10.1021/la047570n. [DOI] [PubMed] [Google Scholar]
- Almquist B. D.; Castleberry S. A.; Sun J. B.; Lu A. Y.; Hammond P. T. Combination Growth Factor Therapy via Electrostatically Assembled Wound Dressings Improves Diabetic Ulcer Healing In Vivo. Adv. Healthcare Mater. 2015, 4 (14), 2090–2099. 10.1002/adhm.201500403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C. A.; Tabrizian M. Enhanced MC3T3 preosteoblast viability and adhesion on polyelectrolyte multilayer films composed of glycol-modified chitosan and hyaluronic acid. J. Biomed. Mater. Res., Part A 2012, 100A (2), 518–526. 10.1002/jbm.a.33305. [DOI] [PubMed] [Google Scholar]
- Hsu B. B.; Park M.-H.; Hagerman S. R.; Hammond P. T. Multimonth controlled small molecule release from biodegradable thin films. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (33), 12175–12180. 10.1073/pnas.1323829111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu B. B.; Hagerman S. R.; Jamieson K.; Castleberry S. A.; Wang W.; Holler E.; Ljubimova J. Y.; Hammond P. T. Multifunctional Self-Assembled Films for Rapid Hemostat and Sustained Anti-infective Delivery. ACS Biomater. Sci. Eng. 2015, 1 (3), 148–156. 10.1021/ab500050m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J.-H.; Kim S.-O.; Linardy E.; Dreaden E. C.; Zhdanov V. P.; Hammond P. T.; Cho N.-J. Adsorption of hyaluronic acid on solid supports: Role of pH and surface chemistry in thin film self-assembly. J. Colloid Interface Sci. 2015, 448, 197–207. 10.1016/j.jcis.2015.01.060. [DOI] [PubMed] [Google Scholar]
- Monteiro I. P.; Shukla A.; Marques A. P.; Reis R. L.; Hammond P. T. Spray-assisted layer-by-layer assembly on hyaluronic acid scaffolds for skin tissue engineering. J. Biomed. Mater. Res., Part A 2015, 103 (1), 330–340. 10.1002/jbm.a.35178. [DOI] [PubMed] [Google Scholar]
- Shah N. J.; Hyder M. N.; Quadir M. A.; Dorval Courchesne N.-M.; Seeherman H. J.; Nevins M.; Spector M.; Hammond P. T. Adaptive growth factor delivery from a polyelectrolyte coating promotes synergistic bone tissue repair and reconstruction. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (35), 12847–12852. 10.1073/pnas.1408035111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J.; Braatz R. D.; Hammond P. T. Tunable staged release of therapeutics from layer-by-layer coatings with clay interlayer barrier. Biomaterials 2014, 35 (8), 2507–2517. 10.1016/j.biomaterials.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu B. B.; Hagerman S. R.; Jamieson K.; Veselinovic J.; O’Neill N.; Holler E.; Ljubimova J. Y.; Hammond P. T. Multilayer Films Assembled from Naturally-Derived Materials for Controlled Protein Release. Biomacromolecules 2014, 15 (6), 2049–2057. 10.1021/bm5001839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knop K.; Hoogenboom R.; Fischer D.; Schubert U. S. Poly(ethylene glycol) in Drug Delivery: Pros and Cons as Well as Potential Alternatives. Angew. Chem., Int. Ed. 2010, 49 (36), 6288–6308. 10.1002/anie.200902672. [DOI] [PubMed] [Google Scholar]
- Shao Q.; Jiang S. Molecular Understanding and Design of Zwitterionic Materials. Adv. Mater. 2015, 27 (1), 15–26. 10.1002/adma.201404059. [DOI] [PubMed] [Google Scholar]
- Thomas A.; Müller S. S.; Frey H. Beyond Poly(ethylene glycol): Linear Polyglycerol as a Multifunctional Polyether for Biomedical and Pharmaceutical Applications. Biomacromolecules 2014, 15 (6), 1935–1954. 10.1021/bm5002608. [DOI] [PubMed] [Google Scholar]
- Kamaly N.; Yameen B.; Wu J.; Farokhzad O. C. Degradable Controlled-Release Polymers and Polymeric Nanoparticles: Mechanisms of Controlling Drug Release. Chem. Rev. 2016, 116 (4), 2602–2663. 10.1021/acs.chemrev.5b00346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T. H.; Kim S.-H.; Decker C. G.; Wong D. Y.; Loo J. A.; Maynard H. D. A heparin-mimicking polymer conjugate stabilizes basic fibroblast growth factor. Nat. Chem. 2013, 5 (3), 221–227. 10.1038/nchem.1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding K.; Alemdaroglu F. E.; Börsch M.; Berger R.; Herrmann A. Engineering the Structural Properties of DNA Block Copolymer Micelles by Molecular Recognition. Angew. Chem., Int. Ed. 2007, 46 (7), 1172–1175. 10.1002/anie.200603064. [DOI] [PubMed] [Google Scholar]
- Kim S.; Shi Y.; Kim J. Y.; Park K.; Cheng J.-X. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle–cell interaction. Expert Opin. Drug Delivery 2010, 7 (1), 49–62. 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 1996, 382 (6592), 607–609. 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- Cutler J. I.; Zhang K.; Zheng D.; Auyeung E.; Prigodich A. E.; Mirkin C. A. Polyvalent Nucleic Acid Nanostructures. J. Am. Chem. Soc. 2011, 133 (24), 9254–9257. 10.1021/ja203375n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush A. M.; Thompson M. P.; Tatro E. T.; Gianneschi N. C. Nuclease-Resistant DNA via High-Density Packing in Polymeric Micellar Nanoparticle Coronas. ACS Nano 2013, 7 (2), 1379–1387. 10.1021/nn305030g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush A. M.; Nelles D. A.; Blum A. P.; Barnhill S. A.; Tatro E. T.; Yeo G. W.; Gianneschi N. C. Intracellular mRNA Regulation with Self-Assembled Locked Nucleic Acid Polymer Nanoparticles. J. Am. Chem. Soc. 2014, 136 (21), 7615–7618. 10.1021/ja503598z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Büschl R.; Folda T.; Ringsdorf H. Polymeric monolayers and liposomes as models for biomembranes. How to bridge the gap between polymer science and membrane biology?. Makromol. Chem. 1984, 6 (S19841), 245–258. 10.1002/macp.1984.020061984119. [DOI] [Google Scholar]