

Abstract
Schizophrenia is a complex mental disorder that is still characterized by its symptoms rather than by biological markers because we have only a limited knowledge of its underlying molecular basis. In the past two decades, however, technical advances in genetics and brain imaging have provided new insights into the biology of the disease. Based on these advances we are now in a position to develop animal models that can be used to test specific hypotheses of the disease and explore mechanisms of pathogenesis. Here, we consider some of the insights that have emerged from studying in mice the relationship between defined genetic and molecular alterations and the cognitive endophenotypes of schizophrenia.
Introduction
The past year has brought a transformation in studying the genetics of schizophrenia. Like for most mental disorders, schizophrenia has a strong genetic component, as evidenced by family and twin studies. A monozygotic twin of a patient with schizophrenia has a 50% likelihood to also develop the disorder, whereas the prevalence in the general population is only 0.7%. However, despite a plethora of genetic association studies, even the most interesting and promising candidate genes for schizophrenia risk have failed to be replicated in multiple populations [1]. Most of the candidate genes that have been studied involve common allelic variants that individually confer only a slight increase in risk (odd ratios <2). Individually they cannot be responsible for the disease. Based on these findings, a ‘common disease–common allele’ model has been proposed according to which many different common allelic variants act in combination to confer predisposition for the disease. In addition, it is thought that some gene variants, or combinations of variants, might only become pathogenic under certain environmental conditions, adding another level of complexity. It is, therefore, not surprising that individual, often statistically underpowered, studies lead to conflicting results. To circumvent the problem of statistical power, a meta-analysis has recently been performed that analyzed >1000 published genetic association studies confirming that some common genetic risk factors are indeed consistently associated with schizophrenia [2]. This meta-analysis is regularly updated with newly published information and is freely accessible at www.szgene.org.
The common disease–common allele model is based on the idea that most disease-related mutations in the human genome are single nucleotide polymorphisms (SNPs). However, recent systematic studies of the human genome have revealed a completely new mechanism of mutations consisting not of SNPs but of variations (duplications and deletions) of large segments of DNA ranging in size from thousands to millions of nucleotides and often involving many genes. These copy number variations (CNVs) have now been mapped systematically and contribute to nucleotide diversity to a larger extent than SNPs.
As a result, this past year several papers have been published that directly challenge the common disease–common allele model and suggest that schizophrenia results from a common disease–rare allele model, according to which a large diversity of rare genetic variants individually account for a high risk for schizophrenia [3– 6]. The new studies find that patients with schizophrenia carry more CNVs than healthy controls and that some CNVs are highly enriched in patients and could be causative. In the extreme, this model implies that one family, or even one individual patient, could be the only existing carrier of one causative mutation, given that some cases might result from de novo mutations [6]. The common and rare allele models are not necessarily exclusive and it is likely that rare and common alleles interact in the generation of schizophrenia.
Irrespective of the mechanism, genetic association studies involving either SNPs or CNVs do not establish causative relationships. Neither do they reveal anything about the mechanism by which the affected molecule might be involved in the disease. To study the biological function of the genetic variants implicated in association studies, and to test their influence on biological processes affected in schizophrenia, animal models are crucial. Mice have emerged here, as in the study of other brain diseases, as the best mammalian genetic system in which to test targeted molecular alterations. Despite all obvious limitations, studying genetically modified mice is proving extremely valuable in the understanding of the cognitive deficits of schizophrenia. In this article we discuss the advantages and disadvantages of using genetically modified mice in the study of schizophrenia. We give examples of mouse models that have given important insights into the function of rare structural mutations and common allelic variants. In addition, because mouse models are helpful in studying the behavioral consequences of specific molecular alterations, they can be used to study specific hypotheses of the disease and mechanisms of pathogenesis. In this context we describe one recent model that is based on the dopamine hypothesis of schizophrenia in which a molecular alteration observed in patients has been modeled in the mouse.
Historically, schizophrenia is characterized by positive, negative and cognitive symptoms
Historically, schizophrenia has been characterized by three sets of symptoms: the positive, negative and cognitive. The positive or psychotic symptoms refer to false perceptions (hallucinations), abnormal believes (delusions) and disordered thought processes. The negative symptoms refer to social withdrawal, lack of motivation and abnormalities in social interaction. The cognitive symptoms refer to deficits in attention and working memory that lead to an inability to organize one's life and to work effectively. The reason why schizophrenia is characterized by its symptoms is because we do not know enough about the biological basis of the disease to develop biological markers that can be used for its diagnosis. However, recent technological advances in genetics and brain imaging have identified biological abnormalities that can help us to understand the etiology of the disease. Table 1 summarizes some of the most reliable biological alterations in schizophrenia.
Table 1. Beyond symptomatology: the biology of schizophrenia.
| General observations | Specific observations | Refs |
|---|---|---|
|
Genetic contributing factors Concordance in monozygotic twins is ∼50%, in dizygotic twins it is 15% |
Top candidate genes for common alleles with low risk Vesicular monoamine transporter 1 (SLC18A1) NR2B subunit of NMDA receptor (GRIN2B) β2 subunit of the GABAA receptor (GABRB2) Dopamine D2 receptor (DRD2) Semaphoring receptor (PLXNA2) (www.szgene.org on September 25th 2008) Rare structural mutations with high risk 1q21.1, 15q13.3 and 22q11 deletions t(1;11) translocation |
[4–6,105] |
|
Environmental contributing factors Concordance in monozygotic twins is not 100% |
Risk factors during pregnancy Maternal infections Malnutrition Risk factors during adolescence Marijuana use during early adolescence Stress |
[104,106,107,122,123] |
|
Developmental abnormalities Early symptoms including motor disturbances and speech difficulties during infancy |
MRI studies: Exaggerated cortical gray matter loss during adolescence in early-onset patients with schizophrenia |
[108,109,124] |
|
Molecular alterations All neurotransmitter systems might be affected in schizophrenia |
Alterations in the dopaminergic system Most antipsychotic drugs block dopamine D2 receptors Postmortem and PET imaging studies: Increased amphetamine induced dopamine release (striatum) Increased dopamine precursor uptake (striatum) Increased density and occupancy of D2 receptors (striatum) Altered density of D1 receptors (cortex) Decreased tyrosine hydroxylase immunoreactivity (cortex) Alterations in the GABAergic system Postmortem studies: Decreased GAD67 and GAT1 immunoreactivity (cortex) Increased GABAA immunoreactivity (cortex) Alterations in the glutamatergic system Chronic blockage of NMDA receptors leads to positive, negative and cognitive symptoms of schizophrenia mGluR2/3 agonists are the first non-D2 blockers that alleviate positive symptoms |
[92,94,95,110–113] |
|
Anatomical changes Several brain areas show structural alterations |
Structural MRI studies: Enlarged ventricles Decreased volumes of hippocampus, striatum and thalamus Structural alteration in prefrontal cortex Postmortem studies: Decreases in prefrontal neuropil Decreases in thalamic neurons |
[114–118] |
| Functional changes | Functional brain imaging: Altered activation of the prefrontal cortex during behavioral tasks that address the cognitive symptoms of schizophrenia |
[119] |
Modeling cognitive endophenotypes of schizophrenia in mice
Obviously it is impossible to model schizophrenia in its entirety including the positive, negative and cognitive symptoms in the mouse. The positive symptoms are particularly difficult to model because we cannot measure hallucinations or delusions in mice. However, mice can be used to study specific endophenotypes of the disease. Endophenotypes are individually measured traits or markers of a disease that alone represent only a component of the disease and, therefore, are often subclinical [7]. The use of endophenotypes was pioneered by Brown and Goldstein [8] who measured low-density lipoprotein (LDL) receptor levels as a subclinical endophenotype of atherosclerosis. They determined that the lack of adequate LDL receptors is the cause of familial hypercholesterolemia and heavily predisposes to atherosclerosis. An example of an endophenotype in schizophrenia is working memory as tested and scored in the N-back test. The idea underlying the endophenotype approach is that the genotype that is responsible for one specific endophenotype might be less complex than the genotype underlying the whole disease. In consequence, there might be a direct link between an individual gene and a specific endophenotype. There are both strengths and weaknesses to using the endophenotype approach in psychiatric genetics, which are discussed by Walters and Owen [9].
Some endophenotypes, especially those related to the cognitive symptoms, are tractable in the mouse. Kraepelin already recognized that patients with dementia praecox (i.e. schizophrenia) share many of the behavioral abnormalities observed in patients with lesions of the frontal cortex [10]. Both patients with prefrontal lesions and with dementia praecox are impaired in working memory, attention and other prefrontal dependent cognitive processes [11–14]. Based on imaging and lesion studies, working memory and behavioral flexibility have been associated specifically with the dorso-lateral prefrontal cortex (PFC) in humans and non-human primates [12,15]. Anatomical and lesion studies suggest that the homolog of the dorso-lateral PFC in primates is the medial PFC in the rat [15– 17]. Because of the utility of mouse genetic models, lesion studies have more recently been completed that extend this functional homology from rats to mice. To date, the mouse medial PFC has been found to be required for working memory tasks, conditioned associative learning, attentional set shifting and reversal learning [18–22]. This suggests that cognitive processes that are dependent on the PFC can be studied in the mouse as it has been done in the rat. Table 2 provides examples of mouse tasks that we and others designed to capture the cognitive processes underlying the successful performance of homologous tasks in humans. Although these homologous tasks use different sensory modalities, temporal schedules, rewards and motor demands in the two species, they each require PFC function and it is likely that they are solved by similar mental and physiological strategies.
Table 2. Behavioral tasks that address prefrontal cortical function in humans and in mice.
| Cognitive domains | Test administered to humans | Test administered to mice |
|---|---|---|
| Attentional set shifting Cognitive flexibility |
|

|
| Spatial working memory |

|

|
| Conditional associative learning |

|

|
| Sustained attention Selective attention |

|

|
Because the cognitive symptoms of schizophrenia present early in the disease and their severity is highly predictive of the long-term prognosis, understanding these symptoms is central for understanding schizophrenia. Moreover, unlike the positive symptoms, the cognitive symptoms are largely resistant to current treatment and persist throughout the lifetime of the individual, greatly compromising the patient's ability to function effectively in society. The great advantage of using mouse genetics to study schizophrenia is that it enables the establishment of a causal relationship between genotype and cognitive endophenotypes, a relationship that cannot be addressed in humans. In turn, this knowledge could lead to the development of more effective therapy in humans. Here, we describe three different categories of mouse models that have been used to explore the cognitive endophenotypes of schizophrenia.
Mouse models based on copy number variants
22q11 deletion syndrome
1:4000 newborns carry heterozygous deletions on chromosome 22 in the q11 region [23]. This deletion has a highly variable clinical presentation. Before the associated chromosomal deletion was identified, the variability of clinical features led to the description of a variety of syndromes including velocardiofacial syndrome, DiGeorge syndrome and others [24]. In ∼90% of cases the deletion spans 3 megabases, whereas in 8% of cases the deletion is only 1.5 megabases long. Almost all patients suffer from craniofacial abnormalities and over half suffer from cardiovascular disorders [25,26]. Patients also display cognitive deficits that range from impairments in working memory, executive function and mild learning disabilities to mental retardation [27,28]. About 30% of adult patients with 22q11 deletion syndromes are diagnosed with psychiatric disorders. These include schizophrenia, schizoaffective disorder, bipolar disorder, anxiety and affective disorders with schizophrenia by far the most prevalent [29–31]. The relative risk for schizophrenia in a patient with 22q11DS is ∼20–25 times the lifetime general population risk of ∼1%. Because the deletion confers such a high risk for schizophrenia, the genes that are rendered hemizygous by the deletion are promising candidate genes for study.
To understand which genes in the 22q11 deletion might be responsible for the different physical and behavioral phenotypes, mouse models of the deletion have been developed [32,33]. In the mouse the syntenic region is located on chromosome 16 and it carries almost all of the genes of the human 22q11 region although the organization of the locus is altered (Figure 1). In an elegant collection of studies performed by several laboratories, the transcription factor Tbx-1 has been identified as the mediator for some of the physical alterations observed in the 22q11 deletion syndrome [34–37] (Figure 1). First, the 1.5 megabase deletion was introduced into the mouse genome resulting in cardiovascular abnormalities comparable to those observed in patients. Second, several sub-deletions were generated that enabled narrowing down the number of potential candidate genes, and a small genomic fragment carrying these genes could rescue the deletion phenotype when expressed from an artificial chromosome. Third, targeted inactivation of one copy of the Tbx-1 gene revealed that haploinsufficiency of this gene alone led to the cardiovascular phenotype.
Figure 1.

Studying the 22q11 deletion in mice. The top shows the 1.5 megabase region of chromosome (chr.) 22 that is deleted in 22q11 carriers in humans. The syntenic region in the mouse is located on chromosome 16. The names of the genes in both regions are colored to emphasize the difference in the organization of the loci. Chromosomal deletions: seven chromosomal 16 deletions of different lengths that have been generated in the mouse and analyzed for behavior are shown [38–40]. Individual gene knockouts: eight knockout mice have been analyzed for behavior [41,77,120,121]. Behavioral analysis: comparative summary of the behavioral analysis of the 15 different mouse models. Because only one copy is deleted in the 22q11 deletion, the behavioral analysis of heterozygous mice are shown unless only homozygous mice (—/—) have been evaluated. * Denotes that a deficit in pre-pulse inhibition was observed in Tbx1 heterozygous mutant mice when backcrossed to C57b6 but not on a mixed genetic background.
The same strategy has been used to identify the genes responsible for the behavioral abnormalities. So far, the behavioral analysis has largely focused on testing pre-pulse inhibition (PPI), a measure for sensory motor gating that is impaired in patients with certain mental illnesses including schizophrenia and patients with the 22q11 deletion syndrome [38–40] (Figure 1). In these studies mice with heterozygous deletion of individual genes from the deletion region were tested for PPI and they showed PPI deficits if they carried individual mutations in three genes: Dgcr8, Gnb1l and Tbx-1. This indicates that the sensorimotor gating defect in patients with deletions is not due to haploinsufficiency of any one individual gene. Dgcr8 by itself might also contribute to the deficit in working memory observed in carriers of the deletion [40]. The Dgcr8 gene is interesting because it encodes a double-stranded RNA-binding protein that is involved in the processing of immature micro RNAs (miRNAs) to mature miRNAs. In consequence, Dgcr8 heterozygous mice show reduced mature miRNAs [40]. Because miRNAs are regulators of protein expression, the altered Dgcr8 expression might have strong pleiotropic effects. More studies are needed to see whether additional genes in the deletion might be responsible for the behavioral deficits of the deletion carriers. Conceivably different genes interact in their hemizygous state in the modulation of cognitive processes. Indeed, a functional interaction between two 22q11 genes, the Prodh gene (which encodes proline dehydrogenase) and the Comt gene (which encodes catechol-O-methyltransferase, COMT), has already been demonstrated [41].
The balanced t(1;11) translocation affecting DISC1
In 1990 St Clair et al. [42] characterized a Scottish family that carried a balanced autosomal translocation and suffered a high prevalence of mental illness. From the 29 carriers of the translocation, seven were later diagnosed with schizophrenia, one with bipolar disorder and ten with depression [43]. Positional cloning identified two genes DISC1 and DISC2 that are disrupted by the translocation [44]. Although this translocation has only been identified in one family, the high incidence of a mental disorder point to DISC1, DISC2 and other genes close to the translocation break point as interesting candidate genes for psychosis and affective disorders. In addition, two large independent association studies have identified haplotypes within the DISC1 gene that seem to contribute to genetic risk for schizophrenia [45,46]. So far, molecular studies have been focused on DISC1 because DISC2 does not code for a protein. However, DISC2 might code for a regulatory RNA that could have biological importance.
DISC1 seems to serve as a scaffolding protein interacting with many proteins ranging from transcription factors, phosphodiesterases, cytoskeletal proteins to centrosomal proteins [47–53]. As a result, the function of DISC1 might be important for a variety of cell biological processes. Consistent with this idea, studies in cell culture and in vivo studies in Drosophila melanogaster and mouse found that disruption of DISC1 affects intracellular signaling, gene expression and cytoskeletal function, in addition to neuronal migration, positioning, differentiation and neurite extension [49,53–55]. Expression of DISC1 is widespread in the brain and is strong during embryonic development. In the adult brain, expression is largely restricted to the granule cells of the hippocampus and to the neurons in the olfactory bulb, two neuronal populations that are regenerated in the adult mouse [56]. The expression studies and the functional studies, therefore, suggest that altered DISC1 function owing to the translocation might affect brain development.
To study the consequences of altered Disc1 function on endophenotypes involved in depression and schizophrenia, seven different mouse models have been generated (Figure 2). Four of these overexpress a transdominant-negative peptide in the brain. Three of the four overexpress the truncated protein or N-terminal peptide that could potentially be made in human t(1;11) carriers but the presence of which has not yet been validated [49,57–60]. Because the translocation disrupts the coding sequence, the transcribed mRNA might not be polyadenylated and might, therefore, be unstable and not translated. The fourth mouse model expresses a peptide from the C-terminal part of DISC1 that binds two interacting partners of DISC1, NUDEL and LIS1. One possible limitation of this transgenic approach is that transgenic DISC1 is overexpressed constantly throughout development and into adulthood. However, there are very dynamic changes in DISC1 expression during development. In mice there is one sharp peak of expression around embryonic day 13.5 and a second dramatic increase in expression level at puberty [61]. One model circumvents this problem by using the endogenous Disc1 promoter to express the peptide [60]. Moreover, because DISC1 interacts with many proteins, overexpression might unspecifically interfere with the function of these proteins.
Figure 2.
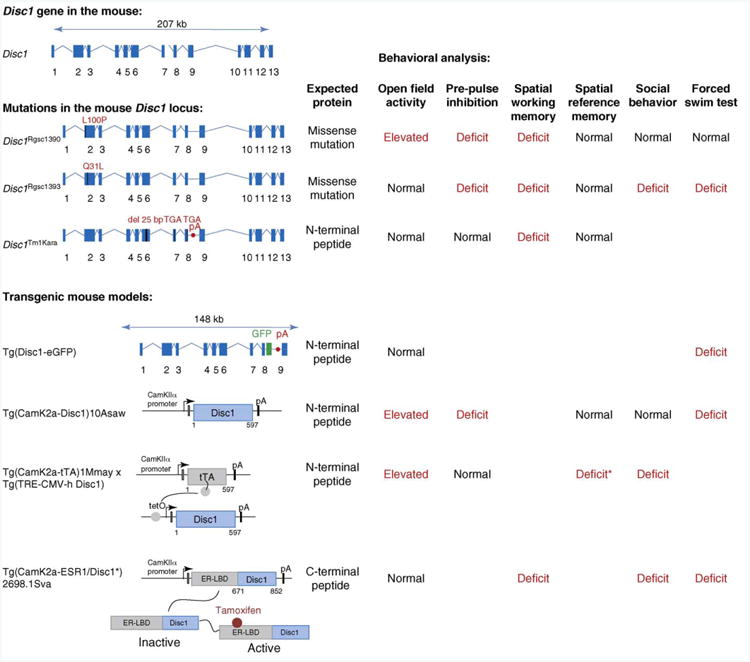
Studying the function of Disc1 in the mouse. Top left shows the structure of the Disc1 gene on mouse chromosome 8. Below the genomic loci or the transgenic constructs of seven genetically modified mice are depicted. Mutations in the mouse Disc1 locus: three mouse models carry mutations in the mouse Disc1 locus. Two models carry missense mutations in exon 2 introduced by a chemical-induced mutagenesis screen (Disc1Rgsc1390 and Disc1Rgsc1393) [62]. The third model, Disc1Tm1Kara [63,64], features a stop codon in exon 8 and a polyadenylation signal (pA) after this exon to model the breakpoint of the human translocation. However, because this mutation was generated in a 129S6/SveV background it also carries an intrinsic stop codon in exon 7 resulting in an N-terminal peptide that ends at amino acid 542. This peptide is 50 amino acid smaller than the 597 amino acid long N-terminal peptide that could potentially be expressed in t(1;11) carriers. Transgenic mouse models: four transgenic mouse models have been generated that all overexpress truncated DISC1 proteins or peptides in neurons of the brain of the mouse. One expresses the truncated DISC1 protein suggested to be present in t(1;11) carriers from the endogenous Disc1 mouse locus using a bacterial artificial chromosome that spans 148 kb of the Disc1 gene [60]. This strategy prevents ectopic expression of the transgene. In this mouse DISC1 is fused to enhanced green fluorescent protein (GFP) to visualize protein expression. Three models use the CamKIIa promoter to restrict expression to the forebrain. Two models overexpress the N-terminal 597 amino acid peptide: Tg(CamK2a-Disc1)10Asaw [57] and Tg(CamK2a-tTA)1Mmay x Tg(TRE-CMV-hDisc1) [58]. Note that for the second mouse model the expression of transgenic DISC1 can be regulated at the level of transcription using the tetracycline transactivator (tTA) system. The tTA transcription factor drives expression from the tetO promoter in the absence of doxycycline. When animals are fed with a doxycycline-supplemented diet, tTA dissociates from the promoter and the transgene is switched off. The third mouse model expresses a C-terminal peptide designed to work as a transdominant negative protein by sequestering out the binding partners NUDEL or LIS1 [59]. The peptide is fused to a mutated ligand-binding domain of the estrogen receptor, permitting regulation of the activity of the fusion protein. Treating the animals with the synthetic steroid tamoxifen activates the fusion protein. Behavioral analysis: the right side gives a comparative summary of the behavioral analysis performed with the six mouse models. * Denotes that the deficit in spatial reference memory of Tg(CamK2a-tTA)1Mmay x Tg(TRE-CMV-hDisc1) mice was observed in females but not in males.
In addition to the four transgenic mouse lines, three models have been generated that are germ-line mutations. Two models carry missense mutations, of which the immediate functional impact is not known, and one model carries a Disc1 gene that encodes the truncated peptide [62–64]. Although these missense mutations importantly link mutations in the Disc1 gene to phenotypes related to schizophrenia, they might not be directly comparable to the human genetic variations involved in the disease because the amino acid sequences involved are not conserved between humans and rodents [65]. The germ-line mutation that expresses the truncated peptide is probably closest to the situation in human t(1;11) carriers, although it might still express DISC1 isoforms that are not present in translocation carriers [66].
Together, these models show quite clearly that altering DISC1 function leads to deficits in endophenotypes of schizophrenia and depression including deficits in sensory-motor gating, spatial working memory, social interaction and the forced swim test (Figure 2). In line with the role of DISC1 in regulating neuronal morphology, all mouse models show morphological alterations in the brain that are similar to the ones observed in patients with schizophrenia (Table 1). For example, some models were assayed by structural magnetic imaging (MRI) and showed increased lateral ventricles or decreased brain size reminiscent of MRI studies performed in patients with schizophrenia [57,58,62]. Of particular interest is the finding in one model, in which the activity of the transdominant DISC1 peptide could be artificially regulated (Figure 2), that affecting DISC1 function during early postnatal development was sufficient to induce behavioral abnormalities in the adult animal [59]. This is in line with the neurodevelopmental hypothesis of schizophrenia and consistent with the apparent role DISC1 is thought to have.
Mouse models based on associations with common polymorphisms
The Val158Met polymorphism in the Comt gene
A common human polymorphism in the 22q11 COMT gene, the Val158Met polymorphism, affects the stability and thereby the activity of the enzyme. This polymorphism has been implicated in many diseases and disease traits including schizophrenia [67,68]. As is the case for many common human polymorphisms, the original association has not always been replicated [69]. However, the Val allele, which confers higher enzymatic activity, seems to be robustly associated with poor performance on working memory and attentional-set shifting tasks, compared with the less active Met allele [68,70,71]. There are also several studies identifying differences in brain activation between genotypes [72]. It is usually assumed that increased COMT activity results in reduced levels of extracellular dopamine in the cortex. In line with this idea, the [11C]NNC D1-specific positron emission tomography (PET) ligand has revealed an increase in D1 receptor density in the PFC of Val allele carriers, which might reflect a compensation for decrease extracellular dopamine levels [73]. Patients with schizophrenia also show an upregulation in cortical D1 receptor availability that correlates with deficits in working memory [74] (but see also Ref. [75]). Because animal studies in rodents and monkeys have found that working memory is sensitive to changes in prefrontal D1 receptor activation, alterations in prefrontal dopamine signaling might be responsible for cognitive deficits of schizophrenia [76]. Understanding the biological relevance of modulated COMT activity might, therefore, provide key inroads into the cognitive symptoms.
To model the increase in enzymatic activity observed in human Val allele carriers, transgenic mice have been generated in which COMT was overexpressed in the brain. As predicted from human studies, increased COMT activity affected executive function including working memory and attentional-set shifting negatively [77]. Amphetamine rescued some of the cognitive deficit, implying that an increase in dopamine release can compensate for COMT overactivity. These mice can now be used to understand the underlying biology of the observed behavioral deficits. For example, they can address the question of whether increased enzymatic COMT activity really reduces extracellular dopamine levels in the cortex, as assumed, and, if so, does it lead to a compensatory upregulation of D1 receptors. Modulation of COMT activity has been postulated as a drug therapy for the cognitive deficits of schizophrenia. To this end, transgenic COMT mice might be useful for testing the efficacy of COMT inhibitors, once suitably brain penetrant versions are available.
Neuregulin–ErbB signaling
Several linkage studies in independent populations have identified chromosome 8p as a locus involved in schizophrenia (see meta-analysis in Refs [78,79]). Subsequent fine mapping, haplotype-association analysis and disequilibrium testing identified neuregulin 1 (NRG1) as a strong candidate gene [80,81]. More than 80 SNPs within the gene have now been associated with schizophrenia, either alone or within a haplotype involving multiple SNPs, some of which have been associated with specific endophenotypes such as hypofrontality and decreased premorbid IQ [82]. There are also reports of genetic association for one of NRG1s functional receptors, v-erb-a erythroblastic leukemia viral oncogene homolog 4 (ERBB4) [83]. Expression of both ligand and receptor has been reported to be altered in postmortem cortical tissue from patients with schizophrenia [81]. As with all proposed susceptibility targets for schizophrenia, there are also negative association and expression studies for both genes, but the convergence of the positive findings builds a strong enough case to investigate the function of NRG1–ERBB4 signaling in mice. Several mouse mutations have been published including knockdowns of NRG1 and ERBB receptors, in addition to targeted mutations of specific domains such as the transmembrane, cystein-rich, epidermal growth factor and immunoglobulin-like domains. The behavioral phenotypes of several Nrg1 and ErbB4 mutants have recently been compared [84]. Heterozygous Nrg1 knockout mice, in addition to mice with deletions within the Nrg1 gene, are hyperactive, as are ErbB4 mutant mice; by contrast, ErbB2 and ErbB3 heterozygous mice seem to be behaviorally normal [85,86]. So far, prefrontal-dependent cognition has not been extensively studied in these mouse models, with the exceptions of working memory, which was found to be intact in heterozygous transmembrane NRG1 mutant mice [87] but impaired in heterozygous type III NRG1 mutant mice [88]. More cognitive studies using several prefrontal-dependent tasks are needed to study the function of NRG1 in cognition.
Mouse models based on specific hypotheses: the dopamine hypothesis, increased density of striatal D2 receptors
Based on seminal experiments by Arvid Carlsson and others, Jacques Van Rossum proposed forty years ago that overstimulation of dopamine receptors could be part of the etiology of schizophrenia (for a historical review, see Ref. [89]). The strongest support for the hypothesis comes from the work of Solomon Snyder and Philip Seeman, who found that the efficacies of antipsychotic medications correlate directly with their occupancy of dopamine receptors [90,91]. More recently, brain imaging studies have found both an increase in the release of dopamine and an increase in the density and occupancy of the D2 receptor in the striatum of patients with schizophrenia [92–95]. Several studies suggest that in at least a subpopulation of patients the observed increase in D2 receptor binding might be determined genetically [96–99].
To study the causal relationship between increased D2 receptor density in the striatum and schizophrenia endophenotypes, D2 receptors have been selectively overexpressed in the striatum of the mouse [21] (Figure 3). These mice were created using the bi-transgenic tetracycline-sensitive expression system [100], which provides temporal control of transgene expression. Mice with D2 receptor overexpression selectively in the striatum show impairments in prefrontal-dependant cognitive processes, which are also affected in schizophrenia. Furthermore, these animals display a decrease in dopamine turnover and an increase in D1 receptor activation in the PFC that could be responsible for the cognitive deficits [18,21]. In addition to the defect in prefrontal functioning, these mice show a deficit in incentive motivation [101] that relates to the negative symptoms. Most interestingly, the cognitive, but not the motivational, deficits persisted long after expression of the transgene has been switched off. In fact, expression during prenatal development is sufficient to cause cognitive deficits in adulthood.
Figure 3.

Overexpression (OE) of dopamine D2 receptors (D2R) selectively in the striatum leads to cognitive and negative-like symptoms. (a) Selective overexpression of dopamine D2 receptors in the striatum using the tTA system (see legend for Figure 2). Although the tTA transcription factor is expressed in the whole forebrain, the tetO-D2R construct response is restricted to the striatum with very limited expression outside of the striatum (gene on) [21]. The transgene can be switched off by feeding the mice doxycycline (Dox; gene off). (b) Cognitive symptoms: excess D2 receptors in the striatum leads to impairments in two prefrontal-dependent cognitive endophenotypes that are also impaired in patients with schizophrenia: working memory and conditioned associative learning [18,21]. Neither deficit is reversed by switching off the transgene in the adult animal. Negative symptoms: an excess of D2 receptors in the striatum also induces impairment in incentive motivation. When required to lever press for food rewards in a progressive ratio schedule, striatal D2R-overexpressing mice stop performing much sooner than control littermates [101]. In contrast to the cognitive deficits, this deficit is reversed by switching off the transgene in the adult animal.
These findings suggest three important new ideas. First, hyperfunctioning of the mesostriatal dopamine pathway could be primarily involved in the etiology of the cognitive symptoms by affecting the functioning of the PFC. Second, the reason that treatment by means of pharmacological blockade of D2 receptors has minimal, if any, beneficial effects on the cognitive symptoms of schizophrenia is because antipsychotic medication is given too late, long after irreversible compensatory changes have taken place during development. Third, the cognitive and negative symptoms might share some etiological components, which is consistent with the observation that the severity of these two types of symptoms strongly correlates in patients.
Concluding remarks
In this review we illustrated how mouse models are being used to study the biology that might underlie some of the cognitive endophenotypes of schizophrenia. We have focused on genetic mouse models that attempt to recapitulate three specific types of molecular alterations observed in schizophrenia: (i) rare structural mutations, (ii) common polymorphisms and (iii) striatal dopaminergic hyperfunction. Our discussion was not intended to be complete and we apologize for omitting other models that are interesting in this context.
What have we learned from these mouse models? Most importantly we have learned that there might be a causal link between genetic or molecular alterations observed in schizophrenia and cognitive endophenotypes of schizophrenia. Whereas genetic studies in humans are mainly correlative, the presented genetic mouse models demonstrate that they can be used to study the consequences of specific genetic or molecular alterations. The fact that several Disc1 mouse models show enlarged ventricles and deficits in working memory suggests that Disc1 could also be causally involved in the generation of these endophenotypes in patients that carry the t(1;11) translocation. Moreover, the presented models give insight into possible mechanisms by which molecular alterations affect cognition. One model showed that upregulation of D2 receptors in the striatum leads to deficits in prefrontal-dependent behavior, possibly by affecting dopamine signaling in the PFC. This interplay between the striatal and prefrontal dopamine systems might not only exist in the mouse, but could also be involved in the generation of the cognitive deficits in patients with schizophrenia.
Although genetic mouse models of schizophrenia and psychiatric disorders in general have been generated for over a decade now, the current generation of studies is proving to be more informative. This is due to several factors: the identification of new genetic risk alleles in humans, the technical advances in mouse genetics (better spatial and temporal resolution) and the development of the field of cognitive endophenotyping in mice. The last point is very important because it provides confidence in the concept of modeling cognitive endophenotypes of schizophrenia in mice.
What can we learn from mouse models in the future? We believe that two areas of research will in particular benefit from the use of mouse models.
The study of gene–gene interactions: according to the common disease–common allele model of schizophrenia, alterations in the function of any individual gene alone do not result in schizophrenia. Even in the common disease–rare allele model, no fully penetrant mutation has yet been identified. By crossing individual genetically modified mice with each other it will be possible to determine how different genes functionally interact.
The study of gene–environment interactions: even in combination, genetic variations are not sufficient to cause schizophrenia, as evidenced by 50% discordance in homozygotic twins. This fact, along with decades of epidemiological studies, highlights the importance of environmental contributions to the development of the disease. Gene–environment interactions have been studied in patients with psychosis [102]; however, they are difficult to perform in humans because the genetic background and the environmental conditions cannot be rigorously controlled [103]. This, however, can be done in mice, and indeed increased risk for schizophrenia caused by prenatal infection has been modeled in mice. Cytokine induction during pregnancy produces offspring with behavioral and morphological abnormalities in adulthood that are similar to schizophrenia endophenotypes [104]. In the future, the consequences of prenatal infections or other environmental insults can be studied in genetically modified mice. These studies will be of great importance in understanding how genes and the environment interact. Moreover, they might identify risk factors that populations with a specific genetic predisposition might want to avoid.
Acknowledgments
This work was supported by NARSAD (www.narsad.org), the National Institute of Mental Health Silvio O. Conte Center for Schizophrenia Research (www.nimh.nih.gov) and the Lieber Center for Schizophrenia Research. We also are grateful to Harold and Shari Levy whose generous contributions have supported our schizophrenia research.
References
- 1.Burmeister M, et al. Psychiatric genetics: progress amid controversy. Nat Rev Genet. 2008;9:527–540. doi: 10.1038/nrg2381. [DOI] [PubMed] [Google Scholar]
- 2.Allen NC, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 3.Walsh T, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 4.Stefansson H, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Consortium, T.I.S. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu B, et al. Strong association of de novo copy number mutations with sporadic schizophrenia. Nat Genet. 2008;40:880–885. doi: 10.1038/ng.162. [DOI] [PubMed] [Google Scholar]
- 7.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–645. doi: 10.1176/appi.ajp.160.4.636. [DOI] [PubMed] [Google Scholar]
- 8.Brown MS, Goldstein JL. Expression of the familial hypercholesterolemia gene in heterozygotes: mechanism for a dominant disorder in man. Science. 1974;185:61–63. doi: 10.1126/science.185.4145.61. [DOI] [PubMed] [Google Scholar]
- 9.Walters JT, Owen MJ. Endophenotypes in psychiatric genetics. Mol Psychiatry. 2007;12:886–890. doi: 10.1038/sj.mp.4002068. [DOI] [PubMed] [Google Scholar]
- 10.Kraepelin E. Dementia Praecox and Paraphrenia. Livingstone: 1919. [Google Scholar]
- 11.Malhotra AK, et al. A functional polymorphism in the COMT gene and performance on a test of prefrontal cognition. Am J Psychiatry. 2002;159:652–654. doi: 10.1176/appi.ajp.159.4.652. [DOI] [PubMed] [Google Scholar]
- 12.Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994;6:348–357. doi: 10.1176/jnp.6.4.348. [DOI] [PubMed] [Google Scholar]
- 13.Liddle PF, Morris DL. Schizophrenic syndromes and frontal lobe performance. Br J Psychiatry. 1991;158:340–345. doi: 10.1192/bjp.158.3.340. [DOI] [PubMed] [Google Scholar]
- 14.Nuechterlein KH, et al. Identification of separable cognitive factors in schizophrenia. Schizophr Res. 2004;72:29–39. doi: 10.1016/j.schres.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 15.Fuster JM. Animal Neuropsysiology. In: Fuster JM, editor. The prefrontal cortex: Anatomy, physiology and neuropsychology of the frontal lobe. 3rd. Lippincott-Raven; 1997. pp. 66–102. [Google Scholar]
- 16.Birrell JM, Brown VJ. Medial frontal cortex mediates perceptual attentional set shifting in the rat. J Neurosci. 2000;20:4320–4324. doi: 10.1523/JNEUROSCI.20-11-04320.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markowitsch HJ, Pritzel M. Comparative analysis of prefrontal learning functions in rats, cats, and monkeys. Psychol Bull. 1977;84:817–837. [PubMed] [Google Scholar]
- 18.Bach ME, et al. Transient and selective overexpression of D2 receptors in the striatum causes persistent deficits in conditional associative learning. Proc Natl Acad Sci U S A. 2008;105:16027–16032. doi: 10.1073/pnas.0807746105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bissonette GB, et al. Double dissociation of the effects of medial and orbital prefrontal cortical lesions on attentional and affective shifts in mice. J Neurosci. 2008;28:11124–11130. doi: 10.1523/JNEUROSCI.2820-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brigman JL, Rothblat LA. Stimulus specific deficit on visual reversal learning after lesions of medial prefrontal cortex in the mouse. Behav Brain Res. 2008;187:405–410. doi: 10.1016/j.bbr.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Kellendonk C, et al. Transient and selective overexpression of dopamine D2 receptors in the striatum causes persistent abnormalities in prefrontal cortex functioning. Neuron. 2006;49:603–615. doi: 10.1016/j.neuron.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 22.Touzani K, et al. Consolidation of learning strategies during spatial working memory task requires protein synthesis in the prefrontal cortex. Proc Natl Acad Sci U S A. 2007;104:5632–5637. doi: 10.1073/pnas.0611554104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodship J, et al. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobrynski LJ, Sullivan KE. Velocardiofacial syndrome, DiGeorge syndrome: the chromosome 22q11. 2 deletion syndromes. Lancet. 2007;370:1443–1452. doi: 10.1016/S0140-6736(07)61601-8. [DOI] [PubMed] [Google Scholar]
- 25.Ryan AK, et al. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botto LD, et al. A population-based study of the 22q11.2 deletion: phenotype, incidence, and contribution to major birth defects in the population. Pediatrics. 2003;112:101–107. doi: 10.1542/peds.112.1.101. [DOI] [PubMed] [Google Scholar]
- 27.McDonald-McGinn DM, et al. The 22q11.2 deletion: screening, diagnostic workup, and outcome of results; report on 181 patients. Genet Test. 1997;1:99–108. doi: 10.1089/gte.1997.1.99. [DOI] [PubMed] [Google Scholar]
- 28.Niklasson L, et al. Neuropsychiatric disorders in the 22q11 deletion syndrome. Genet Med. 2001;3:79–84. doi: 10.1097/00125817-200101000-00017. [DOI] [PubMed] [Google Scholar]
- 29.Bassett AS, et al. Clinical features of 78 adults with 22q11 deletion syndrome. Am J Med Genet A. 2005;138:307–313. doi: 10.1002/ajmg.a.30984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy KC, et al. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- 31.Pulver AE, et al. Psychotic illness in patients diagnosed with velo-cardio-facial syndrome and their relatives. J Nerv Ment Dis. 1994;182:476–478. doi: 10.1097/00005053-199408000-00010. [DOI] [PubMed] [Google Scholar]
- 32.Karayiorgou M, Gogos JA. The molecular genetics of the 22q11-associated schizophrenia. Brain Res Mol Brain Res. 2004;132:95–104. doi: 10.1016/j.molbrainres.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 33.Paylor R, Lindsay E. Mouse models of 22q11 deletion syndrome. Biol Psychiatry. 2006;59:1172–1179. doi: 10.1016/j.biopsych.2006.01.018. [DOI] [PubMed] [Google Scholar]
- 34.Lindsay EA, et al. Congenital heart disease in mice deficient for the DiGeorge syndrome region. Nature. 1999;401:379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 35.Lindsay EA, et al. Tbx1 haploinsufficieny in the DiGeorge syndrome region causes aortic arch defects in mice. Nature. 2001;410:97–101. doi: 10.1038/35065105. [DOI] [PubMed] [Google Scholar]
- 36.Merscher S, et al. TBX1 is responsible for cardiovascular defects in velo-cardio-facial/DiGeorge syndrome. Cell. 2001;104:619–629. doi: 10.1016/s0092-8674(01)00247-1. [DOI] [PubMed] [Google Scholar]
- 37.Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- 38.Paylor R, et al. Tbx1 haploinsufficiency is linked to behavioral disorders in mice and humans: implications for 22q11 deletion syndrome. Proc Natl Acad Sci U S A. 2006;103:7729–7734. doi: 10.1073/pnas.0600206103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Long JM, et al. Behavior of mice with mutations in the conserved region deleted in velocardiofacial/DiGeorge syndrome. Neurogenetics. 2006;7:247–257. doi: 10.1007/s10048-006-0054-0. [DOI] [PubMed] [Google Scholar]
- 40.Stark KL, et al. Altered brain microRNA biogenesis contributes to phenotypic deficits in a 22q11-deletion mouse model. Nat Genet. 2008;40:751–760. doi: 10.1038/ng.138. [DOI] [PubMed] [Google Scholar]
- 41.Paterlini M, et al. Transcriptional and behavioral interaction between 22q11.2 orthologs modulates schizophrenia-related phenotypes in mice. Nat Neurosci. 2005;8:1586–1594. doi: 10.1038/nn1562. [DOI] [PubMed] [Google Scholar]
- 42.St Clair D, et al. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- 43.Blackwood DH, et al. Schizophrenia and affective disorders–cosegregation with a translocation at chromosome 1q42 that directly disrupts brain-expressed genes: clinical and P300 findings in a family. Am J Hum Genet. 2001;69:428–433. doi: 10.1086/321969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Porteous DJ, Millar JK. Disrupted in schizophrenia 1: building brains and memories. Trends Mol Med. 2006;12:255–261. doi: 10.1016/j.molmed.2006.04.009. [DOI] [PubMed] [Google Scholar]
- 45.Cannon TD, et al. Association of DISC1/TRAX haplotypes with schizophrenia, reduced prefrontal gray matter, and impaired short- and long-term memory. Arch Gen Psychiatry. 2005;62:1205–1213. doi: 10.1001/archpsyc.62.11.1205. [DOI] [PubMed] [Google Scholar]
- 46.Hodgkinson CA, et al. Disrupted in schizophrenia 1 (DISC1): association with schizophrenia, schizoaffective disorder, and bipolar disorder. Am J Hum Genet. 2004;75:862–872. doi: 10.1086/425586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kamiya A, et al. Recruitment of PCM1 to the centrosome by the cooperative action of DISC1 and BBS4: a candidate for psychiatric illnesses. Arch Gen Psychiatry. 2008;65:996–1006. doi: 10.1001/archpsyc.65.9.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Millar JK, et al. Yeast two-hybrid screens implicate DISC1 in brain development and function. Biochem Biophys Res Commun. 2003;311:1019–1025. doi: 10.1016/j.bbrc.2003.10.101. [DOI] [PubMed] [Google Scholar]
- 49.Millar JK, et al. DISC1 and PDE4B are interacting genetic factors in schizophrenia that regulate cAMP signaling. Science. 2005;310:1187–1191. doi: 10.1126/science.1112915. [DOI] [PubMed] [Google Scholar]
- 50.Miyoshi K, et al. Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol Psychiatry. 2003;8:685–694. doi: 10.1038/sj.mp.4001352. [DOI] [PubMed] [Google Scholar]
- 51.Morris JA, et al. DISC1 (Disrupted-In-Schizophrenia 1) is a centrosome-associated protein that interacts with MAP1A, MIPT3, ATF4/5 and NUDEL: regulation and loss of interaction with mutation. Hum Mol Genet. 2003;12:1591–1608. doi: 10.1093/hmg/ddg162. [DOI] [PubMed] [Google Scholar]
- 52.Ozeki Y, et al. Disrupted-in-Schizophrenia-1 (DISC-1): mutant truncation prevents binding to NudE-like (NUDEL) and inhibits neurite outgrowth. Proc Natl Acad Sci U S A. 2003;100:289–294. doi: 10.1073/pnas.0136913100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sawamura N, et al. Nuclear DISC1 regulates CRE-mediated gene transcription and sleep homeostasis in the fruit fly. Mol Psychiatry. 2008;13:1138–1148. doi: 10.1038/mp.2008.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kamiya A, et al. A schizophrenia-associated mutation of DISC1 perturbs cerebral cortex development. Nat Cell Biol. 2005;7:1167–1178. doi: 10.1038/ncb1328. [DOI] [PubMed] [Google Scholar]
- 55.Duan X, et al. Disrupted-In-Schizophrenia 1 regulates integration of newly generated neurons in the adult brain. Cell. 2007;130:1146–1158. doi: 10.1016/j.cell.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Austin CP, et al. Expression of disrupted-in-schizophrenia-1, a schizophrenia-associated gene, is prominent in the mouse hippocampus throughout brain development. Neuroscience. 2004;124:3–10. doi: 10.1016/j.neuroscience.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 57.Hikida T, et al. Dominant-negative DISC1 transgenic mice display schizophrenia-associated phenotypes detected by measures translatable to humans. Proc Natl Acad Sci U S A. 2007;104:14501–14506. doi: 10.1073/pnas.0704774104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pletnikov MV, et al. Inducible expression of mutant human DISC1 in mice is associated with brain and behavioral abnormalities reminiscent of schizophrenia. Mol Psychiatry. 2008;13:173–186, 115. doi: 10.1038/sj.mp.4002079. [DOI] [PubMed] [Google Scholar]
- 59.Li W, et al. Specific developmental disruption of disrupted-in-schizophrenia-1 function results in schizophrenia-related phenotypes in mice. Proc Natl Acad Sci U S A. 2007;104:18280–18285. doi: 10.1073/pnas.0706900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shen S, et al. Schizophrenia-related neural and behavioral phenotypes in transgenic mice expressing truncated Disc1. J Neurosci. 2008;28:10893–10904. doi: 10.1523/JNEUROSCI.3299-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schurov IL, et al. Expression of disrupted in schizophrenia 1 (DISC1) protein in the adult and developing mouse brain indicates its role in neurodevelopment. Mol Psychiatry. 2004;9:1100–1110. doi: 10.1038/sj.mp.4001574. [DOI] [PubMed] [Google Scholar]
- 62.Clapcote SJ, et al. Behavioral phenotypes of Disc1 missense mutations in mice. Neuron. 2007;54:387–402. doi: 10.1016/j.neuron.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 63.Koike H, et al. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci U S A. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kvajo M, et al. A mutation in mouse Disc1 that models a schizophrenia risk allele leads to specific alterations in neuronal architecture and cognition. Proc Natl Acad Sci U S A. 2008;105:7076–7081. doi: 10.1073/pnas.0802615105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ma L, et al. Cloning and characterization of Disc1, the mouse ortholog of DISC1 (Disrupted-in-Schizophrenia 1) Genomics. 2002;80:662–672. doi: 10.1006/geno.2002.7012. [DOI] [PubMed] [Google Scholar]
- 66.Ishizuka K, et al. Evidence that many of the DISC1 isoforms in C57BL/6J mice are also expressed in 129S6/SvEv mice. Mol Psychiatry. 2007;12:897–899. doi: 10.1038/sj.mp.4002024. [DOI] [PubMed] [Google Scholar]
- 67.Chen J, et al. Functional analysis of genetic variation in catechol-O-methyltransferase (COMT): effects on mRNA, protein, and enzyme activity in postmortem human brain. Am J Hum Genet. 2004;75:807–821. doi: 10.1086/425589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tunbridge EM, et al. Catechol-o-methyltransferase, cognition, and psychosis: Val158Met and beyond. Biol Psychiatry. 2008;60:141–151. doi: 10.1016/j.biopsych.2005.10.024. [DOI] [PubMed] [Google Scholar]
- 69.Munafo MR, et al. Lack of association of the COMT (Val158/ 108 Met) gene and schizophrenia: a meta-analysis of case-control studies. Mol Psychiatry. 2005;10:765–770. doi: 10.1038/sj.mp.4001664. [DOI] [PubMed] [Google Scholar]
- 70.Egan MF, et al. Effect of COMT Val108/158 Met genotype on frontal lobe function and risk for schizophrenia. Proc Natl Acad Sci U S A. 2001;98:6917–6922. doi: 10.1073/pnas.111134598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bruder GE, et al. Catechol-O-methyltransferase (COMT) genotypes and working memory: associations with differing cognitive operations. Biol Psychiatry. 2005;58:901–907. doi: 10.1016/j.biopsych.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 72.Heinz A, Smolka MN. The effects of catechol O-methyltransferase genotype on brain activation elicited by affective stimuli and cognitive tasks. Rev Neurosci. 2006;17:359–367. doi: 10.1515/revneuro.2006.17.3.359. [DOI] [PubMed] [Google Scholar]
- 73.Slifstein M, et al. COMT genotype predicts cortical-limbic D1 receptor availability measured with [(11)C]NNC112 and PET. Mol Psychiatry. 2008;13:821–827. doi: 10.1038/mp.2008.19. [DOI] [PubMed] [Google Scholar]
- 74.Abi-Dargham A, et al. Prefrontal dopamine D1 receptors and working memory in schizophrenia. J Neurosci. 2002;22:3708–3719. doi: 10.1523/JNEUROSCI.22-09-03708.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Okubo Y, et al. Decreased prefrontal dopamine D1 receptors in schizophrenia revealed by PET. Nature. 1997;385:634–636. doi: 10.1038/385634a0. [DOI] [PubMed] [Google Scholar]
- 76.Goldman-Rakic PS, et al. Targeting the dopamine D1 receptor in schizophrenia: insights for cognitive dysfunction. Psychopharmacology (Berl) 2004;174:3–16. doi: 10.1007/s00213-004-1793-y. [DOI] [PubMed] [Google Scholar]
- 77.Papaleo F, et al. Genetic dissection of the role of catechol-O-methyltransferase in cognition and stress reactivity in mice. J Neurosci. 2008;28:8709–8723. doi: 10.1523/JNEUROSCI.2077-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Badner JA, Gershon ES. Meta-analysis of whole-genome linkage scans of bipolar disorder and schizophrenia. Mol Psychiatry. 2002;7:405–411. doi: 10.1038/sj.mp.4001012. [DOI] [PubMed] [Google Scholar]
- 79.Lewis CM, et al. Genome scan meta-analysis of schizophrenia and bipolar disorder, part II: schizophrenia. Am J Hum Genet. 2003;73:34–48. doi: 10.1086/376549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Stefansson H, et al. Neuregulin 1 and susceptibility to schizophrenia. Am J Hum Genet. 2002;71:877–892. doi: 10.1086/342734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Harrison PJ, Law AJ. Neuregulin 1 and schizophrenia: genetics, gene expression, and neurobiology. Biol Psychiatry. 2006;60:132–140. doi: 10.1016/j.biopsych.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 82.Hall J, et al. A neuregulin 1 variant associated with abnormal cortical function and psychotic symptoms. Nat Neurosci. 2006;9:1477–1478. doi: 10.1038/nn1795. [DOI] [PubMed] [Google Scholar]
- 83.Nicodemus KK, et al. Further evidence for association between ErbB4 and schizophrenia and influence on cognitive intermediate phenotypes in healthy controls. Mol Psychiatry. 2006;11:1062–1065. doi: 10.1038/sj.mp.4001878. [DOI] [PubMed] [Google Scholar]
- 84.Duffy L, et al. Behavioral profile of a heterozygous mutant mouse model for EGF-like domain neuregulin 1. Behav Neurosci. 2008;122:748–759. doi: 10.1037/0735-7044.122.4.748. [DOI] [PubMed] [Google Scholar]
- 85.Gerlai R, et al. Heregulin, but not ErbB2 or ErbB3, heterozygous mutant mice exhibit hyperactivity in multiple behavioral tasks. Behav Brain Res. 2000;109:219–227. doi: 10.1016/s0166-4328(99)00175-8. [DOI] [PubMed] [Google Scholar]
- 86.Golub MS, et al. Behavioral characteristics of a nervous system-specific erbB4 knock-out mouse. Behav Brain Res. 2004;153:159–170. doi: 10.1016/j.bbr.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 87.O'Tuathaigh CM, et al. Phenotypic characterization of spatial cognition and social behavior in mice with ‘knockout’ of the schizophrenia risk gene neuregulin 1. Neuroscience. 2007;147:18–27. doi: 10.1016/j.neuroscience.2007.03.051. [DOI] [PubMed] [Google Scholar]
- 88.Chen YJ, et al. Type III neuregulin-1 is required for normal sensorimotor gating, memory-related behaviors, and corticostriatal circuit components. J Neurosci. 2008;28:6872–6883. doi: 10.1523/JNEUROSCI.1815-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baumeister AA, Francis JL. Historical development of the dopamine hypothesis of schizophrenia. J Hist Neurosci. 2002;11:265–277. doi: 10.1076/jhin.11.3.265.10391. [DOI] [PubMed] [Google Scholar]
- 90.Seeman P, et al. Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci U S A. 1975;72:4376–4380. doi: 10.1073/pnas.72.11.4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Creese I, et al. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science. 1976;192:481–483. doi: 10.1126/science.3854. [DOI] [PubMed] [Google Scholar]
- 92.Laruelle M. Imaging dopamine transmission in schizophrenia. A review and meta-analysis. Q J Nucl Med. 1998;42:211–221. [PubMed] [Google Scholar]
- 93.Abi-Dargham A, et al. Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A. 2000;97:8104–8109. doi: 10.1073/pnas.97.14.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Seeman P, Kapur S. Schizophrenia: more dopamine, more D2 receptors. Proc Natl Acad Sci U S A. 2000;97:7673–7675. doi: 10.1073/pnas.97.14.7673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Frankle WG. Neuroreceptor imaging studies in schizophrenia. Harv Rev Psychiatry. 2007;15:212–232. doi: 10.1080/10673220701679812. [DOI] [PubMed] [Google Scholar]
- 96.Lawford BR, et al. The C/C genotype of the C957T polymorphism of the dopamine D2 receptor is associated with schizophrenia. Schizophr Res. 2005;73:31–37. doi: 10.1016/j.schres.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 97.Hanninen K, et al. Association between the C957T polymorphism of the dopamine D2 receptor gene and schizophrenia. Neurosci Lett. 2006;407:195–198. doi: 10.1016/j.neulet.2006.08.041. [DOI] [PubMed] [Google Scholar]
- 98.Hoenicka J, et al. C957T DRD2 polymorphism is associated with schizophrenia in Spanish patients. Acta Psychiatr Scand. 2006;114:435–438. doi: 10.1111/j.1600-0447.2006.00874.x. [DOI] [PubMed] [Google Scholar]
- 99.Monakhov M, et al. Association study of three polymorphisms in the dopamine D2 receptor gene and schizophrenia in the Russian population. Schizophr Res. 2008;100:302–307. doi: 10.1016/j.schres.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 100.Mayford M, et al. Control of memory formation through regulated expression of a CaMKII transgene. Science. 1996;274:1678–1683. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 101.Drew MR, et al. Transient overexpression of striatal D2 receptors impairs operant motivation and interval timing. J Neurosci. 2007;27:7731–7739. doi: 10.1523/JNEUROSCI.1736-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Caspi A, et al. Moderation of the effect of adolescent-onset cannabis use on adult psychosis by a functional polymorphism in the catechol-O-methyltransferase gene: longitudinal evidence of a gene X environment interaction. Biol Psychiatry. 2005;57:1117–1127. doi: 10.1016/j.biopsych.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 103.Caspi A, Moffitt TE. Gene–environment interactions in psychiatry: joining forces with neuroscience. Nat Rev Neurosci. 2006;7:583–590. doi: 10.1038/nrn1925. [DOI] [PubMed] [Google Scholar]
- 104.Smith SE, et al. Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci. 2007;27:10695–10702. doi: 10.1523/JNEUROSCI.2178-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karayiorgou M, et al. Schizophrenia susceptibility associated with interstitial deletions of chromosome 22q11. Proc Natl Acad Sci U S A. 1995;92:7612–7616. doi: 10.1073/pnas.92.17.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Mednick SA, et al. Adult schizophrenia following prenatal exposure to an influenza epidemic. Arch Gen Psychiatry. 1988;45:189–192. doi: 10.1001/archpsyc.1988.01800260109013. [DOI] [PubMed] [Google Scholar]
- 107.Murray RM, et al. Cannabis, the mind and society: the hash realities. Nat Rev Neurosci. 2007;8:885–895. doi: 10.1038/nrn2253. [DOI] [PubMed] [Google Scholar]
- 108.O'Connell P, et al. Developmental insanity or dementia praecox: was the wrong concept adopted? Schizophr Res. 1997;23:97–106. doi: 10.1016/S0920-9964(96)00110-7. [DOI] [PubMed] [Google Scholar]
- 109.Thompson PM, et al. Mapping adolescent brain change reveals dynamic wave of accelerated gray matter loss in very early-onset schizophrenia. Proc Natl Acad Sci U S A. 2001;98:11650–11655. doi: 10.1073/pnas.201243998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Davis KL, et al. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148:1474–1486. doi: 10.1176/ajp.148.11.1474. [DOI] [PubMed] [Google Scholar]
- 111.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 112.Patil ST, et al. Activation of mGlu2/3 receptors as a new approach to treat schizophrenia: a randomized Phase 2 clinical trial. Nat Med. 2007;13:1102–1107. doi: 10.1038/nm1632. [DOI] [PubMed] [Google Scholar]
- 113.Lewis DA, et al. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- 114.Shenton ME, et al. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Selemon LD, et al. Elevated neuronal density in prefrontal area 46 in brains from schizophrenic patients: application of a three-dimensional, stereologic counting method. J Comp Neurol. 1998;392:402–412. [PubMed] [Google Scholar]
- 116.Black JE, et al. Pathology of layer V pyramidal neurons in the prefrontal cortex of patients with schizophrenia. Am J Psychiatry. 2004;161:742–744. doi: 10.1176/appi.ajp.161.4.742. [DOI] [PubMed] [Google Scholar]
- 117.Glantz LA, Lewis DA. Reduction of synaptophysin immunoreactivity in the prefrontal cortex of subjects with schizophrenia. Regional and diagnostic specificity. Arch Gen Psychiatry. 1997;54:943–952. doi: 10.1001/archpsyc.1997.01830220065010. [DOI] [PubMed] [Google Scholar]
- 118.Glantz LA, Lewis DA. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry. 2000;57:65–73. doi: 10.1001/archpsyc.57.1.65. [DOI] [PubMed] [Google Scholar]
- 119.Weinberger DR, Berman KF. Prefrontal function in schizophrenia: confounds and controversies. Philos Trans R Soc Lond B Biol Sci. 1996;351:1495–1503. doi: 10.1098/rstb.1996.0135. [DOI] [PubMed] [Google Scholar]
- 120.Gogos JA, et al. The gene encoding proline dehydrogenase modulates sensorimotor gating in mice. Nat Genet. 1999;21:434–439. doi: 10.1038/7777. [DOI] [PubMed] [Google Scholar]
- 121.Mukai J, et al. Evidence that the gene encoding ZDHHC8 contributes to the risk of schizophrenia. Nat Genet. 2004;36:725–731. doi: 10.1038/ng1375. [DOI] [PubMed] [Google Scholar]
- 122.Brown AS. Prenatal infection as a risk factor for schizophrenia. Schizophr Bull. 2006;32:200–202. doi: 10.1093/schbul/sbj052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Brown AS, Susser ES. Prenatal nutritional deficiency and risk of adult schizophrenia. Schizophr Bull. 2008;34:1054–1063. doi: 10.1093/schbul/sbn096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rapoport JL, et al. Progressive cortical change during adolescence in childhood-onset schizophrenia. A longitudinal magnetic resonance imaging study. Arch Gen Psychiatry. 1999;56:649–654. doi: 10.1001/archpsyc.56.7.649. [DOI] [PubMed] [Google Scholar]