

Abstract
Neurotransmitters (NTs) are endogenous signaling molecules which play an important role in regulating various physiological processes in animals. Detection of these chemical messengers is often challenging due to their low concentration levels and fast degradation rate in vitro. In order to address these challenges, herein we employed in vivo microdialysis (MD) sampling to study neurotransmitters in the crustacean model Cancer borealis. Multi-faceted separation tools, such as capillary electrophoresis (CE) and ion mobility mass spectrometry were utilized in this work. Small molecules were separated by different mechanisms and detected by matrix-assisted laser desorption/ionization mass spectrometric imaging (MALDI-MSI). Performance of this separation-based MSI platform was also compared to liquid chromatography-electrospray ionization mass spectrometry (LC-ESI-MS). By utilizing both MALDI and ESI MS, a total of 208 small molecule neurotransmitters and metabolites were identified, of which 39 were identified as signaling molecules secreted in vivo. In addition, the inherent property of sub micro-scale sample consumption using CE enables shorter time of MD sample collection. Temporal resolution of in vivo MD was improved by approximately 10-fold compared to LC-ESI-MS, indicating the significant advantage of applying separation-assisted MALDI MS imaging platform.
Keywords: capillary electrophoresis, neurotransmitter, MALDI-MS, small molecule identification, CE-MSI, ion mobility, untargeted metabolomics
1 Introduction
Monitoring signaling molecules in circulatory systems can provide important insight into functional behavior related to environmental change, drug effects, and disease states [1–3]. In vivo microdialysis (MD) is a powerful sampling technique that allows direct collection of signaling molecules from extracellular space with minimum disturbance to their physiological conditions [4, 5]. Usually, MD probes consist of two pieces of microporous tubings sheathed in a semi-permeable membrane that is plugged at one end. With a MD probe implanted into an area of interest within an animal, molecules that fall below the molecular-weight cutoff (MWCO) threshold of the probe will passively diffuse across the membrane according to their concentration gradients. Perfusion through the membrane, typically performed at 0.1–3.0 μL/min, generates a stream of dialysate that can be analyzed for compounds of interest [6, 7]. The MD sampling technique has been applied to a wide variety of tissues and organs in the body including liver, heart, skin, blood, placenta, stomach, and ear, with analytes of interest ranging from low molecular weight substances including amino acids and metabolites, to higher molecular substances such as neuropeptides [8, 9]. The advantages of MD benefits a variety of applications in neurochemical and clinical studies [10–12].
Tremendous challenges confront scientists attempting in vivo neurochemical measurements. For example, neurotransmitters may change in concentration rapidly requiring high temporal resolution detection capability. Additionally, low endogenous levels of many compounds present in vivo, typically at nM-pM range [13], can make the detection of these molecules difficult. Despite the unique advantage of in vivo sampling by MD, the low recovery rate of molecules also poses additional challenges for analysis. To address these problems, researchers have made great advancements in both MD sampling strategies and downstream analytical platforms. Attempts to increase MD recovery have been made by various laboratories. One successful technique, affinity-enhanced MD, improves recovery by using affinity agents added to the perfusion fluid to capture analytes of interest thereby increased the amount of target analytes recovered [14–18]. High temporal resolution, defined as the shortest time duration over which a dynamic change event can be observed, is critical for the functional study of targeted compounds. To be able to accurately correlate the concentration of analytes with behavior or stimuli, the analysis time must be shorter than the duration of measured events. In MD experiments, temporal resolution is limited by the downstream analytical sensitivity of the instrument platform used to study the termporal events. Analytical techniques like capillary electrophoresis (CE) with laser-induced fluorescence detection have been successfully coupled with MD aiming to improve temporal resolution [19–23]. It often necessitates additional sample work-up in order to render the analytes fluorescent. The coupling of CE with mass spectrometry provides enhanced sensitivity without the need for sample derivatization to detect low abundance molecules [24]. Analysis of MD samples by CE can provide improved temporal resolution compared to LC, as smaller amount of sample is needed for CE separation due to its capability of handling sub-μL volume of samples. Dialysate can be either online or off-line loaded and separated by a CE system for analysis of neurotransmitters (NTs), amino acids, peptides, and biomarker detection [25–27]. Decoupling sampling from analytical measurement can be highly desirable. In the case of offline CE-MS, analytical separation can be coupled to dialysate collection to increase throughput and reduce sample losses from pipetting and non-specific adsorption to the wall of fraction collection vials. The CE eluate can then be analyzed on a mass spectrometry platform to facilitate the on-the-fly measurement of several NTs and metabolite compounds at a single MD time point.
In this work, we employed CE separation coupled to in vivo MD of Jonah crab (Cancer borealis) to study secreted signaling molecules such as NTs and metabolites in crustacean hemolymph. Instead of analyzing CE fractions by ESI-MS, we incorporated MALDI mass spectrometric imaging (MSI) with CE separation for small molecule measurement. MALDI was selected because the technique has a higher tolerance to salts and impurities which is a benefit when interrogating dialysate samples. Therefore, CE separated dialysate from crab does not need to be further desalted and cleaned up, reducing sample loss associated with additional manipulation. Neurotransmitters have lower molecular masses than peptides or proteins that often makes identifying isobaric molecules by accurate mass matching alone challenging especially when it is possible to have multiple metabolic features of similar or same m/z within one scan even after CE separation. To address this challenge, a second dimension of separation was applied to the gas-phase analytes with ion mobility mass spectrometry (IMS). With IMS, molecular features within MD-CE fractions of crustacean hemolymph can be further separated based on their gas-phase ion mobility drift times. With the flexibility of integrating multi-faceted MS platforms, metabolic coverage of crustacean hemolymph was significantly improved compared to using a single platform. Sensitivity and feature identification performance of this MD-CE-MSI platform was evaluated by comparing to a LC-MS approach utilizing a high-mass accuracy and high-resolution orbitrap mass analyzer (e.g., Q Exactive) [28–30].
2 Materials and methods
2.1 Chemicals and materials
Acetic acid, ammonium hydroxide, acetone, acetonitrile (ACN), methanol (MeOH), ammonium bicarbonate, and urea were purchased from Fisher Scientific (Pittsburgh, PA). α-Cyano-4-hydroxycinnamic acid (CHCA) and formic acid (FA) were from Sigma Aldrich (St. Louis, MO). Fused-silica capillary with 75 μm i.d. and 365 μm o.d. was purchased from Polymicro Technologies (Phoenix, AZ). A TM-Sprayer from HTX Technologies (Carrboro, NC) was used to spray MALDI matrix (10 mg/mL CHCA in 50% ACN). Millipore C18 Ziptip column was used for sample cleaning, and all water used in this study was doubly distilled on a Millipore filtration system (Bedford, MA).
2.2 In vivo Microdialysis
CMA/20 Elite probes with 4 mm membranes of polyarylethersulfone (PAES) were purchased from CMA Microdialysis (Harvard Apparatus, Holliston, MA, USA). A KD Harvard 22 (Harvard Apparatus, Holliston, MA, USA), and a Pump 11 Elite Nanomite Syringe Pump (Harvard Apparatus, Holliston, MA, USA) were used to drive perfusate through MD probes and tubing. Probes consisted of a 6,000 Da MWCO tip were rinsed with crab saline prior to implantation.
Jonah crabs, Cancer borealis, were purchased from Ocean Resources, Inc. (Sedgwick, ME, USA) and The Fresh Lobster Company (Gloucester, MA, USA). Crabs were maintained in an artificial seawater tank at 10~13 °C and were allowed to adjust to the tanks for at least one week after shipment before performing microdialysis. Details of animal housing procedures were described elsewhere [17]. Animals were housed, treated and sacrificed following the animal care protocol in accordance with the University of Wisconsin-Madison’s animal care guidelines. The surgery of microdialysis was adapted from previous publications [31]. After the probe was surgically implanted in the crab, the animal was allowed to recover for at least 24 h before dialysate was collected for MS analysis. Physiological crab saline (440 mM NaCl; 11 mM KCl; 13 mM CaCl2; 26 mM MgCl2; 10 mM HEPES acid; pH 7.4, adjusted with NaOH) was used as perfusion solution. The flow rate was set to be 0.5 μL/min by a programmable syringe pump. Dialysate samples were collected with a refrigerated fraction collector (BASi HoneyComb, Bioanalytical Systems, Inc. Indianapolis, IN, USA). The dialysate fractions were concentrated ~10 fold in a SpeedVac (Thermo Fisher Scientific, Waltham, MA, USA) prior to downstream analysis.
2.3 CE-MSI
The interface for coupling CE to mass spectrometric imaging instrumentation was modified from previous work in our group [32, 33]. Briefly, a 60 cm long with 75 μm i.d./365 μm o.d. fused-silica capillary was pretreated and used for CE separation. A fracture was made near the outlet of the capillary and coated with cellulose acetate membrane to form an ion permeable channel. The inlet end of CE column was placed 15 cm higher than outlet to maintain siphoning flow after retraction by pressure.
CE eluate was deposited directly onto a customized MALDI plate (Thermo Scientific, San Jose, CA, USA) whose movement was mechanically controlled by a syringe pump at a speed of 3.7 mm/min. After the CE trace dried, CHCA matrix (10 mg/mL) was applied to the plate using a TM- sprayer to generate a homogenous trace on the plate. The parameters were carefully adjusted and set as follows: matrix flow rate: 200 mL/min; nozzle velocity: 1200 mm/min; temperature: 85 °C; gas pressure: 13 psi; line spacing: 3mm; number of layers: 2; dry time: 1 min. The matrix covered CE trace was then analyzed using a MALDI LTQ Orbitrap XL (Thermo Scientific, San Jose, CA, USA) with imaging experimental set up. In order to improve multiplexing and throughput the instrument method consisted of a full MS scan from m/z 100–900 followed by three tandem mass scans set up in parallel. By setting up raster and spiral movements of MALDI laser beam, orbitrap full scans were acquired every 100 μm with 11 μJ MALDI laser energy, followed by three parallel targeted tandem mass scans at 50 um with HCD collision energy ranging from 35 to 55. The imaging data was then processed using ImageQuest software (Thermo Scientific).
2.4 MALDI-ion mobility mass spectrometry
Data was acquired on a MALDI-IM-MS enabled instrument (Synapt G2 Q-TOF, Waters, Milford, MA, USA) with a Nd: YAG laser at a repetition rate of 200 Hz. External calibration was performed using Glu-1-Fibrinopeptide B (Glu-Fib) standard of 1.0 μM before each experiment. The MD-CE fractions of crustacean hemolymph were deposited on a MALDI-Synapt target plate every 30s under the same CE experimental condition. Mass spectra were acquired in sensitivity mode and over a mass range of m/z 100 to 900. Laser energy attenuation was 300 (arbitrary units). Ion mobility separation was performed at a drift gas pressure of 2.30 Torr using nitrogen gas, a wave velocity of 650 m/s, and a wave height of 40.0 V.
2.5 LC-MS analysis
A LC-MS experiment was also performed in order to compare the performance of separation-assisted MALDI platform. In the LC-MS experiment, an automated combination of positive and negative ionization polarities was utilized to achieve comprehensive identification of small molecule metabolites. This extended feature of fast polarity switching on the mass analyzer enables unbiased detection of metabolites in complex biological samples [28]. For LC-MS experiments, microdialysate was collected for five hours, desalted by C18 Ziptip, and concentrated into 30 μL H2O containing 0.1% FA. A Dionex UltiMate 3000 LC system was coupled with a Q Exactive Orbitrap mass spectrometer (Thermo Scientific). With 5μL injection of the desalted sample, metabolite separation was achieved with a Phenomenex biphenyl column (75.1 μm × 150 mm, 1.7 μm, 100 Å) heated to 30°C at a flow rate of 0.3 mL/min. Mobile phase A (MPA) was 0.1% FA in H2O and mobile phase B (MPB) was 0.1% FA in MeOH. The 12 min binary gradient was set as follows: 0–5 min, 0–2% MPB; 5–10 min, 2%–50% MPB; 10–12min, 90% MPB. Full MS scans were acquired from m/z 70 to 1000 for both positive and negative ESI modes. Orbitrap resolution was 70 K (m/z 400), automatic gain control (AGC) target was 1×106, and maximum injection time (IT) was 100 ms. Raw data files were processed by SIEVETM software for peak alignment and framing to generate a list of detected mass features.
3 Results and discussion
3.1 In vivo MD sampling coupled offline to CE-MSI technique (MD-CE-MSI) for untargeted metabolomics study
Previously, our group has employed the MD sampling technique on crabs to study neuropeptide content in the circulating hemolymph [17, 31, 34]. However, the study of NTs in the model crustacean system, which is an essential component of neuromodulators in the nervous system, has been lacking. One of the advantages of MD sampling is that it yields simpler samples due to the size-defined MWCO dialysis membrane which excludes large molecules such as proteins and large neuropeptides. In the MD-CE-MSI experiments, dialysate was collected for two hours. The resulting dialysate was concentrated ten folds before CE-MSI analysis. Loading amount of the sample was 0.2~0.4 μL, indicating shorter time intervals can be achieved to meet the requirement of monitoring physiological changes. By coupling CE with MALDI-MS, an increased flexibility for the independent optimization of CE and MS experiments was realized. In addition, CE fractions are available for reanalysis and further biochemical characterization on the same spots or places if required. In MD-CE-MSI experiments, a syringe pump was used to drive crab saline through MD probes at a flow rate of 0.5 μL/min. In order to fully preserve the resolution from the CE separation, we have introduced trace level MSI incorporated with CE [32, 33]. CE eluent was continuously deposited onto a horizontally moving MALDI target plate. To create a uniform and homogenous matrix layer on the acquired CE trace, CHCA (10 mg/mL) was applied to the CE trace using a TM sprayer. With higher tolerance to contaminants and impurities using MALDI, desalting steps prior to MALDI Orbitrap analysis were omitted thereby increasing the recovery rate of target analytes.
3.2 Multiplex imaging set up of MD-CE-MSI
MD-CE-MSI data was acquired in the small molecule range m/z 100–900 Da on a MALDI Orbitrap analyzer and processed using ImageQuest software. In a MD-CE-MSI data, each analyte appears as a colored image extracted away from background ions. Fig. 1D shows the total ion count (TIC) of the imaging data for the CE separation from 2 min to 20 min. The signals in the TIC trace represent both analytes of interest and matrix background peaks. Unlike matrix ions, target analytes featured specific migration times, indicated by the colored region in the CE trace displayed in Fig. 1 (A, B and C). Fig. 1A displays the extracted region of interest (ROI) for 5-hydroxyindoleacetic acid (5-HIAA) migrated at 11 min, the main metabolite of serotonin through enzymatic conversion by monoamine oxidase-A [35]. Levels of 5-HIAA have been associated with aggressive behavior and autistic spectrum disorders [36]. Fig. 1B displays the CE migration time (14 min) for aminobenzoic acid, one of the most abundant molecules found in crustaean hemolymph. It is a substrate of enzyme anthranilate hydroxylase in benzoate degradation via hydroxylation pathway, also known as a component of tryptophan metabolism [37]. In Fig. 1C shows ROI of dopamine observed at 12.5 min in CE-MD trace, which plays a number of important roles in brain, body as well as elsewhere in biology [38]. The MALDI mass spectra of each representative metabolite and their corresponding structures are displayed in Fig. 1E, 1F, 1G, respectively.
Figure 1. MALDI Orbitrap imaging of small molecule metabolites separated by CE.

(A, B, C) 2D images of 5-HIAA, aminobenzoic acid and dopamine, respectively. (D) Image of total ion current (TIC) of the acquired MD-CE trace. Color bars on the right showing relative intensities of image signals. (E, F, G) Mass spectra and structures of 5-HIAA, aminobenzoic acid dopamine, respectively.
The HMDB database searching was performed with 5 ppm error tolerance. From the suggested compounds, each one of the identities was manually examined so as to keep only endogenous metabolites in the results. Metabolites from plants or drugs, exogenous metabolites and food additives were filtered out. Furthermore, experiments of tandem mass fragmentation were carried out to determine the structure of the high abundant metabolites displayed in the CE-MSI trace. Fig. 2 displays how MS/MS confirmation data for dopamine was acquired during a MD-CE-MSI acquisition. In order to collect MS and MS/MS mass spectra for a given image, raster and spiral movements for the MALDI laser beam were used. This acquisition method is displayed graphically in Fig. 2C Fig. 2A shows the MS/MS ROI for dopamine at CE migration time of 12.5 min. Fig. 2B displays the ROI data in a three-dimensional format which includes image width information and signal intensity. Fig. 2D and 2E show that the experimental precursor and fragment ions observed for dopamine matched well with those resulting from the dopamine standard. With the ability of collecting MS/MS data on MD-CE prepared samples, analytes were identified with higher confidence and revealed the potential of studying neurotransmitters in a more targeted way. However, ambiguity still existed when assigning the identities of some neurotransmitters, as many small molecules share the exact same m/z even with mass accuracy less than 3 ppm. For one m/z at a particular CE migration time, it was possible that multiple metabolic features were present. In order to address this problem, we employed a second dimension for separation, ion mobility, combined with MALDI MS (MALDI-IMS).
Figure 2. Illustration of multiplexed MD-CE-MSI on MALDI Orbitrap instrument.

(A) 2D imaging of dopamine identified in MD-CE-MSI. (B) 3D imaging of dopamine identified in MD-CE-MSI. The x axis in the 3D image represents CE separation time from 2 to 20 min. Y axis is the width of the CE image that is 0.6 mm. Z axis showed relative abundance of the extracted ion from TIC. (C) Illustration of laser movement in the multiplexed method. Orbitrap full scan was acquired every 100 μm shown in pink color, followed by three tandem mass fragmentation at 50 μm. (D) MS/MS fragmentation of dopamine standard acquired under the same instrumental condition acquired on MALDI Orbitrap. (E) MS/MS spectrum of observed dopamine in MD-CE-MSI.
3.3 MALDI-IMS for separation of isobaric molecules
Ion mobility (IM) brings an additional dimension to MALDI-MS experiments by adding the capability of separating different compound families from one another. The ion mobility cell, positioned between quadrupole and time-of-flight (TOF) analyzer in Synapt G2 instrument, allows the separation of isobaric compounds with similar m/z based on their collisional cross section, gas phase conformation and charge, which cannot be differentiated by single mass criterion [39–41]. Different ion conformations have different ion cross sections and result in different drift times. This particular property makes MS comparable to LC separation and allows for separation and identification of chemical families such as matrix, lipids, and small molecules by their drift time inside the ion mobility cell. Here MD-CE fractions of crustacean hemolymph were further separated by ion mobility mass spectrometry. As observed in the averaged mass spectrum of one fraction migrated at 11 min measured with MALDI-IMS in Fig. 3A, many compounds had the same m/z, but it is possible to discriminate between them according to their drift time. Fig. 3B showed the driftscope image corresponding to the 3D visualization of detected signals. The mass scale was the y-axis, the drift time was x-axis, and the color scale represents signal intensity constituting the third dimension of the driftscope image. Target analytes were filtered from background at a minimum intensity threshold of 1,000 counts visualized in this 3D mapping Fig. 3C. As shown in Fig. 3, isobaric ions were successfully separated by their different conformations in the mobility cell.
Figure 3. Ion mobility separation of MD-CE fractions.

(A) Total ion chromatogram of MD-CE fraction with migration time at 11 min which is further separated by ion mobility mass spectrometry. (B) Image from driftscope showed drift times of small molecules from m/z 100 to 900. (C) As shown in 3D driftscope image, isobaric molecules in one MD-CE fraction were successfully separated by their drift times.
The zoomed image in driftscope Fig. 4 illustrated how the added drift time differentiated isobaric small molecules from diverse classes. Taking the metabolite of m/z 191.0468 as an example, four different small molecules showed on driftsope in the mass range of 1 Da, indicated by red dots in the white circle in the enlarged driftscope image visualization Fig. 4A and 4B. Three of them shared the same m/z of 191.0468, and were successfully separated by their mobility in gas phase. Although only one peak was observed in the mass spectrum, it actually contained three different compounds. Through HMDB MS search with maximum 10 ppm mass error for MALDI-TOF analyzer, these ions were 2,3-diaminosalizylic acid found in blood, sodiated adduct of pyrroloylglycine which is the biomarker of hyperlipidemia, and 1-(2-Furanyl)-1-pentanone which appears to be an additive found in animal food. The other metabolite co-migrated in this MD-CE fraction was a protonated ion m/z 191.1280, which was matched as beta-damascenone belonging to lipid class. Therefore, food additive should be excluded from dicovery results, while the other three remained. This example illustrated how ion mobility differentiates a large number of isobaric molecules by their IM drift time and helps to refine the dicovery data. Approximately 20% more peaks have been detected with the addition of ion mobility separation and they were marked by solid dots in supplementary data Table. S1.
Figure 4. Ion mobility separation of small molecules in different classes with identical mass.

(A) Zoom in figure of four different compounds eluted in one chromatographic peak. (B) Heat map of isobaric compounds filtered from background. (C) Chromatographic peak shared by four different molecules.
3.4 LC-MS approach for comprehensive investigation and comparison to MALDI platform
Finally, the performance of multi-staged MALDI platform was further evaluated by comparing it to a LC-MS approach. We utilized the automated combination of positive and negative ionization mode mass spectra derived from fast polarity switching that has been widely used in lipidomics and drug metabolomics area [42, 43]. This extension enables detection of metabolites in complex biological samples and provided complementary identification of untargeted metabolomics [44, 45]. Owing to fast polarity switching approach, two total ion chromatograms as well as averaged mass spectrum clearly revealed its benefit of complementary recognition gained from the integration of two ionization modes as shown in Fig. 5 (A, B and C). Resultant data was aligned by SIEVE™ for peak detection and processed through HMDB, yielding 137 metabolites identified in total. Among them, 83 were detected exclusively by the LC-ESI approach. For MALDI-MS platform integrated with CE imaging and ion mobility, 71 were uniquely found as summarized in Fig. 5D. A list of the identified signaling molecules and metabolites was provided in supporting information Table S1. In CE analysis, molecules migrated through capillary based on their mass and charge. Metabolites that are predominantly polar and highly charged were better detected in CE. In LC analysis, the amount of sample loaded onto the LC column was substantially higher than in CE, thus lower abundance molecules were better identified in LC tecnique. Therefore, these two techniques were complementary and the combined use of the CE/IM-MALDI-MSI and LC-MS platforms resulted in significatly increased signaling molecule and metabolite coverage.
Figure 5. Untargeted LC-MS analysis of hemolymph metabolites.
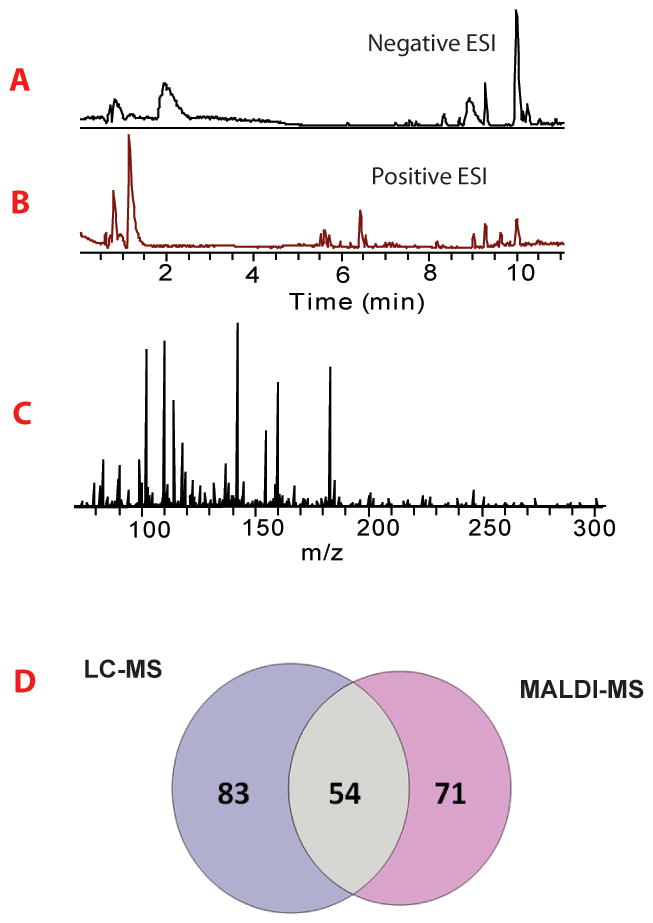
(A) Base peak ion chromatogram acquired in negative ion mode. (B) Base peak chromatogram acquired in positive ion mode. (C) Averaged mass spectrum of detected mass features. (D) Venn diagram showed comparison of metabolomics identification of MALDI-MSI and LC-MS platform. 83 metabolites were uniquely identified by LC-MS, while 71 metabolites were exclusively found by the MALDI-MSI platform.
4 Concluding remarks
In summary, we applied micro-separation approaches, CE and ion mobility, coupled with multiplexed MALDI imaging platform for NT and metabolite identification. LC-MS was also used for evaluation and comprehensive investigation, yielding 208 small molecule metabolites identified in total. In addition, these molecules were classified into 11 biochemical families detailed in Table S1. Among them, the ones belonging to necleosides, amino acids, peptides and analogues, amines and their metabolites are considered highly significant for further studies due to their potential roles in signaling. More importantly, the improved temporal resolution with CE-MSI platform, with 5 min dialysate loaded into the CE column, comparing to 1h dialysate loaded into the LC-MS system, enables more accurate monitoring of NT release and dynamic changes, providing a more powerful tool for functional studies.
Supplementary Material
Acknowledgments
We thank Dr. Robert Sturm for critical reading an early draft of this manuscript. This work is supported by the National Institutes of Health grants (1R01DK071801 and S10RR029531). SJ gratefully thanks a former Li Research Group member Dr. Zichuan Zhang for scientific discussions and making helpful suggestions. LL acknowledges an H. I. Romnes Faculty Research Fellowship and a Vilas Distinguished Achievement Professorship with funding provided by the Wisconsin Alumni Research Foundation and University of Wisconsin-Madison School of Pharmacy.
References
- 1.Kennedy RT. Curr Opin Chem Biol. 2013;17:860–867. doi: 10.1016/j.cbpa.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sebolt-Leopold JS, English JM. Nature. 2006;441:457–462. doi: 10.1038/nature04874. [DOI] [PubMed] [Google Scholar]
- 3.Maceyka M, Spiegel S. Nature. 2014;510:58–67. doi: 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sundström I, Arts J, Westerlund D, Andrén PE. The Analyst. 2010;135:405–413. doi: 10.1039/b917940b. [DOI] [PubMed] [Google Scholar]
- 5.Lee GJ, Park JH, Park HK. Neurol Res. 2008;30:661–668. doi: 10.1179/174313208X289570. [DOI] [PubMed] [Google Scholar]
- 6.Watson CJ, Venton BJ, Kennedy RT. Anal Chem. 2006;78:1391–1399. doi: 10.1021/ac0693722. [DOI] [PubMed] [Google Scholar]
- 7.OuYang C, Liang Z, Li L. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2015;1854:798–811. doi: 10.1016/j.bbapap.2014.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nandi P, Lunte SM. Anal Chim Acta. 2009;651:1–14. doi: 10.1016/j.aca.2009.07.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cebada J, Alvarado-Álvarez R, Becerra E, Neri-Bazán L, Rocha L, García U. J Neurosci Methods. 2006;153:1–7. doi: 10.1016/j.jneumeth.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 10.Timofeev I, Carpenter KLH, Nortje J, Al-Rawi PG, O’Connell MT, Czosnyka M, Smielewski P, Pickard JD, Menon DK, Kirkpatrick PJ, Gupta AK, Hutchinson PJ. Brain. 2011;134:484–494. doi: 10.1093/brain/awq353. [DOI] [PubMed] [Google Scholar]
- 11.Bossers SM, de Boer RDH, Boer C, Peerdeman SM. Acta Neurochir (Wien) 2012;155:345–353. doi: 10.1007/s00701-012-1582-z. [DOI] [PubMed] [Google Scholar]
- 12.Darvesh AS, Carroll RT, Geldenhuys WJ, Gudelsky GA, Klein J, Meshul CK, Van der Schyf CJ. Expert Opinion on Drug Discovery. 2011;6:109–127. doi: 10.1517/17460441.2011.547189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Q, Zubieta JK, Kennedy RT. Anal Chem. 2009;81:2242–2250. doi: 10.1021/ac802391b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Duo J, Stenken JA. Anal Bioanal Chem. 2010;399:773–782. doi: 10.1007/s00216-010-4170-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duo J, Stenken JA. Anal Bioanal Chem. 2010;399:783–793. doi: 10.1007/s00216-010-4333-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pettersson A, Amirkhani A, Arvidsson B, Markides K, Bergquist J. Anal Chem. 2004;76:1678–1682. doi: 10.1021/ac035305l. [DOI] [PubMed] [Google Scholar]
- 17.Schmerberg CM, Li L. Anal Chem. 2013;85:915–922. doi: 10.1021/ac302403e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H, Ou J, Wei Y, Wang H, Liu Z, Chen L, Zou H. Anal Chim Acta. 2015;883:90–98. doi: 10.1016/j.aca.2015.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Lada MW, Vickroy TW, Kennedy RT. Anal Chem. 1997;69:4560–4565. doi: 10.1021/ac970518u. [DOI] [PubMed] [Google Scholar]
- 20.Hogan BL, Lunte SM, Stobaugh JF, Lunte CE. Anal Chem. 1994;66:596–602. doi: 10.1021/ac00077a004. [DOI] [PubMed] [Google Scholar]
- 21.Zhou Y, Mabrouk OS, Kennedy RT. J Am Soc Mass Spectrom. 2013;24:1700–1709. doi: 10.1007/s13361-013-0605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang M, Hershey ND, Mabrouk OS, Kennedy RT. Anal Bioanal Chem. 2011;400:2013–2023. doi: 10.1007/s00216-011-4956-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hao L, Zhong X, Greer T, Ye H, Li L. Analyst. 2015;140:467–475. doi: 10.1039/c4an01582g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isbell TA, Strickland EC, Hitchcock J, McIntire G, Colyer CL. J Chromatogr B. 2015;980:65–71. doi: 10.1016/j.jchromb.2014.11.035. [DOI] [PubMed] [Google Scholar]
- 25.Guihen E, O’Connor WT. Electrophoresis. 2010;31:55–64. doi: 10.1002/elps.200900467. [DOI] [PubMed] [Google Scholar]
- 26.Schiavone NM, Sarver SA, Sun L, Wojcik R, Dovichi NJ. J Chromatogr B. 2015;991:53–58. doi: 10.1016/j.jchromb.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang C, Liu H, Yang Q, Zhang L, Zhang W, Zhang Y. Anal Chem. 2003;75:215–218. doi: 10.1021/ac026187p. [DOI] [PubMed] [Google Scholar]
- 28.Kluger B, Bueschl C, Neumann N, Stückler R, Doppler M, Chassy AW, Waterhouse AL, Rechthaler J, Kampleitner N, Thallinger GG, Adam G, Krska R, Schuhmacher R. Anal Chem. 2014;86:11533–11537. doi: 10.1021/ac503290j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamada M, Kita Y, Kohira T, Yoshida K, Hamano F, Tokuoka SM, Shimizu T. J Chromatogr B. 2015;995–996:74–84. doi: 10.1016/j.jchromb.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 30.Oh HA, Kim D, Lee SH, Jung BH. J Pharm Biomed Anal. 2015;107:32–39. doi: 10.1016/j.jpba.2014.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Liang Z, Schmerberg CM, Li L. The Analyst. 2015;140:3803–3813. doi: 10.1039/c4an02016b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Ye H, Zhang Z, Xiang F, Girdaukas G, Li L. Anal Chem. 2011;83:3462–3469. doi: 10.1021/ac200708f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Z, Ye H, Wang J, Hui L, Li L. Anal Chem. 2012;84:7684–7691. doi: 10.1021/ac300628s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Behrens HL, Chen R, Li L. Anal Chem. 2008;80:6949–6958. doi: 10.1021/ac800798h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carpenter LL, Anderson GM, Siniscalchi JM, Chappell PB, Price LH. Neuropsychopharmacology. 2003;28:339–347. doi: 10.1038/sj.npp.1300025. [DOI] [PubMed] [Google Scholar]
- 36.Burgess NK, Sweeten TL, McMahon WM, Fujinami RS. J Autism Dev Disord. 2006;36:697–704. doi: 10.1007/s10803-006-0100-7. [DOI] [PubMed] [Google Scholar]
- 37.Arvadia P, Narwaley M, Whittal RM, Siraki AG. Arch Biochem Biophys. 2011;515:120–126. doi: 10.1016/j.abb.2011.07.015. [DOI] [PubMed] [Google Scholar]
- 38.Rice ME, Patel JC, Cragg SJ. Neuroscience. 2011;198:112–137. doi: 10.1016/j.neuroscience.2011.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stauber J, MacAleese L, Franck J, Claude E, Snel M, Kaletas BK, Wiel IMVD, Wisztorski M, Fournier I, Heeren RMA. J Am Soc Mass Spectrom. 2010;21:338–347. doi: 10.1016/j.jasms.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 40.Xu L, Kliman M, Forsythe JG, Korade Z, Hmelo AB, Porter NA, McLean JA. J Am Soc Mass Spectrom. 2015;26:924–933. doi: 10.1007/s13361-015-1131-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cole LM, Mahmoud K, Haywood-Small S, Tozer GM, Smith DP, Clench MR. Rapid Commun Mass Spectrom. 2013;27:2355–2362. doi: 10.1002/rcm.6693. [DOI] [PubMed] [Google Scholar]
- 42.Gallart-Ayala H, Courant F, Severe S, Antignac JP, Morio F, Abadie J, Le Bizec B. Anal Chim Acta. 2013;796:75–83. doi: 10.1016/j.aca.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 43.Shin M, Ji D, Kang S, Yang W, Choi H, Lee S. Arch Pharmacal Res. 2013;37:760–772. doi: 10.1007/s12272-013-0225-0. [DOI] [PubMed] [Google Scholar]
- 44.Guo B, Wang M, Liu Y, Zhou J, Dai H, Huang Z, Shen L, Zhang Q, Chen B. J Agric Food Chem. 2015;63:6954–6967. doi: 10.1021/acs.jafc.5b02222. [DOI] [PubMed] [Google Scholar]
- 45.Braña-Magdalena A, Leão-Martins JM, Glauner T, Gago-Martínez A. J AOAC Int. 2014;97:285–292. doi: 10.5740/jaoacint.sgebrana. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.