

Abstract
Early menopause or premature ovarian insufficiency (POI) is a common cause of infertility in women and affects about one per cent of young women. This disorder has significant psychological sequelae and major health implications. Its relevance has increased in recent years due to the fact that age of motherhood is being delayed in developed countries, with the risk of having either primary ovarian insufficiency or less possibilities of pregnancy. The main characteristics are absence of ovulation, amenorrhoea and high levels of serum gonadothropins (hypergonadotropic hypogonadism). Although the aetiology remains uncertain in most cases, several rare specific causes have been elucidated. Potential causes for POI are iatrogenic (ovarian surgery, radiotherapy or chemotherapy), environmental factors, viral infections, metabolic and autoinmune diseases, and genetic alterations. Because of the association with other autoimmune diseases, close follow up is recommended in patients with POI. The traditional indicators to evaluate ovarian ageing are age, serum hormonal levels, anti-Mullerian hormone, antral follicle count, and ultrasonography of ovaries. Hormone replacement therapy remains the mainstay of treatment, and the best chance of achieving a pregnancy is through oocyte donation. This article aims to present an overview of potential causes, clinical manifestations, and treatment options of POI.
Keywords: Early menopause, hypergonadotropic, hypogonadism, ovarian dysfunction, ovarian failure, ovarian physiology, premature ovarian failure
Introduction
The women fertility reduces in parallel with the ageing1. Premature ovarian insufficiency (POI), commonly referred to as premature ovarian failure (POF) is a disorder characterized by dysfunction of the ovary before 40 years of age. The main characteristics include absence of ovulation, amenorrhoea and high levels of serum gonadothropins (hypergonadotropic hypogonadism, HH). POF is not uncommon; its incidence is estimated to be as high as 1 in 100 by the age of 40, and 1 in 1000 by the age of 30 years1,2.
As early as in 1930s, physicians noted abnormally elevated urinary gonadotropin levels in “premature menopause”. In 1950, Atria3 observed 20 patients with “precocious menopause” and defined the basic clinical features of POI, including amenorrhoea before the age of 40, clinical manifestations of hypoestrogenism, an association with viral sickness, and the efficacy of hormonal treatment.
Ovarian ageing resulting in ovarian failure and menopause is a continous4. Menopause generally occurs around 51 yr of age, with an age range varying between 40 and 60 yr1,2,5. The World Health Organization (WHO) defines menopause as the permanent cessation of menstruation due to the loss of ovarian follicular activity5. The final menstrual period is retrospectively assigned after 12 months of amenorrhoea. During the Stages of Reproductive Aging Workshop (STRAW) the criteria were formulated to distinguish the various stages of reproductive ageing, based on menstrual cycle pattern and follicle stimulating hormone (FSH) levels: (i) the (early, broad and late) reproductive stage, (ii) the (early and late) menopausal transition, and (iii) the (early and late) postmenopause. Cycles in the early menopausal transition are characterized by elevated but variable early follicular phase FSH levels and low anti-Müllerian hormone (AMH) levels and antral follicle count (AFC)6. Menstrual cycles in the late menopausal transition are described by increased variability in cycle length, extreme variations in hormonal levels, and increased prevalence of anovulation6.
POI is defined as the occurrence of amenorrhoea (for 4 months or more) before the age of 40 in women, accompanied with an increase of serum FSH to menopausal level (usually over 40 IU/l, obtained at least 1 month apart) and estradiol levels less than 50 pg/ml (which signifies hypoestrogenism)7. Another indicator is the AMH, whose serum levels can help assess the state of follicular senescence, which is a possible predictor of risk for POI8. Howewer, there are presently no single screening test that can predict a woman's reproductive lifespan. Serum hormonal levels, AMH, AFC and ultrasonography of ovaries, together with age are traditional signs to evaluate ovarian ageing9. The conception of ovarian reserve is the assessment of the reproductive ability as a function of the number and quality of residual oocytes. Decreased or diminished ovarian reserve (DOR) describes patients of reproductive age having regular menses whose response to ovarian stimulation or fertility is reduced compared with women of similar age. DOR is different from menopause or POI10.
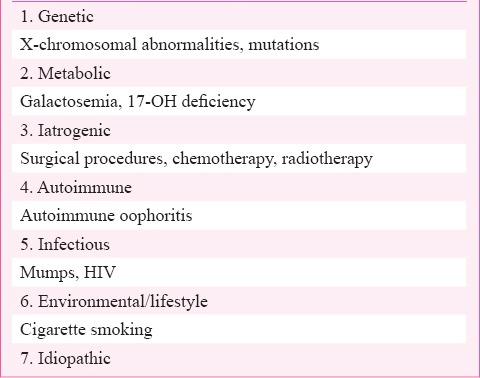
As yet, there are no well-established diagnostic criteria but differential diagnosis is needed to discount pregnancy, and other underlying conditions causing secundary amenorrhoea such as polycystic ovary syndrome (PCOS), hypothalamic disease, or uncontrolled diabetes mellitus11. Potential causes for POF are iatrogenic (ovarian surgery, radiotherapy or chemotherapy), environmental factors, viral infections, metabolic and autoinmune diseases, and genetic alterations, although its origin is idiopathic, and probably genetic in most cases12 (Table). This article provides an overview of potential aetiologies, clinical manifestations, and treatment modalities of POI.
Table.
Aetiological factors of premature ovarian failure
Anatomy and Physiology
Determination of follicle number
The stock of primordial follicles (oocytes surrounded by granulosa cells) in the ovaries, established before birth, has to supply the reproductive needs of a woman for the lifetime. The number of primordial follicles having potential to develop into a fertilizable oocyte is one component of the ovarian reserve13. At about 20 wk of foetal life, a maximum number of about 7 million germ cells is reached14,15. At birth and at menarche, about 1,000,000 and 3,00,000 are left, respectively, of which only 400-500 follicles will develop fully and ovulate over the next 35-40 years. At a mean age of 37-38 yr, only about 25,000 resting follicles are present in the ovaries. Menstrual cycles become irregular at about age 45, which is on average six years at the (pre) menopause6.
It is uncertain why so many germ cells are lost during the development of the primordial follicle pool. Once shaped, primordial follicles are recruited throughout life to enter folliculogenesis. The ovarian reserve can be influenced at multiple points during development and folliculogenesis: (i) primordial germ cell migration and proliferation; (ii) oocyte entry into meiosis I, synapsis, recombination and arrest in dictyate stage; (iii) transition from oocyte clusters to primordial follicles; (iv) primordial follicle activation; and (v) deficiencies in folliculogenesis or ovulation. The first three phases occur at the foetal and therefore, are not likely to be relevant in clinical treatment of infertility. The current and long-held dogma has been that the number of primordial follicles at birth represents the total ovarian reserve for the entire length of reproductive life; howewer, contradictory information suggest that oocyte “stem cells” continuosly contribute to the ovarian reserve13,16. The remarkably good result of in vitro fertilization (IVF) in (pre)menopausal women when employing oocytes of young donors further highlights that the oocyte rather than the endometrium determines the age-dependent fertility loss17.
Only in the case of fecundation is the second division completed and therefore, all stages are under the influence of certain signals, and the entire process may last for many decades. It is suggested that oocyte derived factors, such as growth differentiation factor 9 (GDF9) or bone morphogenetic protein 15 (BMP15) may be effective in regulating folliculogenesis18.
Endocrinological factors in ovarian ageing
From the time of puberty onward, the GnRH (gonadotropin releasing hormone) pulse generator communicates with the pituitary gland through the pulsatile secretion of GnRH, which in turn communicates with the ovaries through the secretion of FSH and luteinizing hormone (LH). At each level of this communication system, multiple neuro endocrine molecules orchestrate precise positive and negative feedback loops, toensure regular opportunities for fertilization19,20. Glutamate and norepinephrine generate major excitatory signals, while γ-amino butyric acid (GABA) and endogenous opioids generate inhibitory signals19. Recently discovered factors, such as the RF-amide peptide superfamily [kisspeptins, 26/43RFa, gonadotropin-inhibiting hormone (GnIH), RF-releasing peptides (RFRP)] also play a role19.
Reproductive ageing is considered as a dysregulation of the GnRH pulse generator by a progressive lack of neurochemical control from other brain centers15,20. One of the first signals of this change is the monotropic increase of FSH during the early follicular phase, which produces the acceleration of follicle depletion15. Then, the interruption in the development of dominant follicles or withdrawal of estrogen without corpus luteum function causes extended cycles and anovulatory haemorrhages19. FSH acts on granulosa cells through its receptor (FSHr) to upregulate Cyp19a1 (commonly named as aromatase) and hydroxysteroid (17-β) dehydrogenase 1 to stimulate estradiol production. FSH is critical for protection of follicular atresia, improvement of granulosa cell proliferation, and stimulates the upregulation of luteinizing hormone receptors (LHr) in these cells. Furthermore, an additional growth hormone, insuline-like growth factor 1 (IGF1), increases granulosa cell responsiveness to FSH13. As a result of the decreasing cohort of antral follicles, first inhibin B secretion reduces, then estradiol, and finally inhibin A secretion. This results in variations in the feedback mechanisms. The endocrine control of luteal phase does not significantly change with advancing age. In case ovulation still happens, the secrection of estradiol, progesterone, and inhibin A from the corpus luteum appear unchanged19,21.
Genetic causes
Genetic causes are considered as major factor in determining age at menopause in general population, and are described in 7 per cent of POF occurrence12,22. The chromosomal and genetic aberrations mostly involve X-chromosome, yet findings of autosomal involvement are reported22. Even though a large number of related genes have been found, with some understanding of their pathogenesis, the precise genetic mechanisms are often uncertain23.
Sex chromosome abnormalities
Aneuploidies: In various reports, chromosomal abnormalities, namely aneuploidies and rearrangements have been established as the most common causes of POI, corroborating the importance of cytogenetic testing and genetic advising24. Loss of one X-chromosome as X monosomy [Turner syndrome (TS)], in which most women present with gonadal dysgenesis with primary amenorrhoea and loss of ovarian reserve before puberty. Employing fluorescent in situ hybridization (FISH) and several specific marker to the short arm of X-chromosome including DXS1058, DXS6810, DXS1302 and ZXDB, deletion of Xp11.2-p22.1 was submitted as a critical zone linked to TS and POI24. In adittion, X-mosaicisms (45, XO, 45, XO/46, XX, 46, XX/47, XXX) are currently the most common chromosomal defect with incidences ranging from 5 to 40 per cent. Women in this group can present menstruation during several years before developing a complete POI12,25. The wide variation in the occurrence of chromosomal disorders possibly represents patient referral bias.
One candidate gene for gonadal dysgenesis in these patients is USP9X (ubiquitin-specific protease 9), which escapes the X-inactivation process and is located on chromosome Xp11.4, a critical region for ovarian development11. Other candidate genes include ZFX (zinc finger protein, X-linked) and BMP15 (bone morphogenetic protein 15). As for other aneuploidies, POI may be related with trisomy X (47, XXX) and tetrasomy (48, XXXX).
Mutations: The FMR1 (Fragile X mental retardation 1) premutation of CGG replicates with incidence of 1:800 in males and 1:100-200 in women, is accepted as the most important gene associated with POI26,27; is located on Xq27.323 and produces an autosomal dominant genetic disorder called fragile X syndrome, distinguished by mental retardation and other symptom such as hyperactivity or attention and emotional problems12,27. Premutation alleles befall in 7 per cent of sporadic POI and 21 per cent in familial POI, considerably higher than in general population28. A critical region from Xq13.3 to Xq27 has been characterized for ovarian development and function29,30. Powell et al31, employing molecular techniques, found a second gene (POF-2) of paternal origin situated more proximal to the Xq locus at Xq13.3-q21.1. Region Xq 21.3-Xq27 is considered premature ovarian failure 1 (POF-1). Other potential POF genes located on X- long arm are Diaphanous homolog 2 (DIAH2) related with cytoskeleton and implicated in oogenesis and Dachshund homolog 2 (DACH2)32. The surveillance suggested that transcendent genes for normal ovarian function are situated on both arms of the X-chromosome. Autosomal genes whose mutations cause some of the syndromes associated with the development of POF are: FSHR, GNAS, FOXL2, GALT, AIRE, STAR, CYP17A1, CYP19A1, eIF2B, NOG, ATM, POLG, PMM1, BMPR1B and GJA412.
Metabolic disorders
Classical galactosaemia is triggered by mutation in the galactose-1-phosphate uridyl- transferase (GALT) gene located on chromosome 9p1333 and is a complex, life-threatening disease occurring during the first week of life and comprises various clinical abnormalities which, in the absence of a galactose-restricted diet, result to liver failure in the second half of the first week of life and death by acute liver and kidney failure within a few days34. In some cases presenting with a milder phenotype, the diagnosis will be made later in childhood when mental retardation, cataract and hepatomegalia will be detected. In other cases, galactosaemia might be completely asymptomatic, when a sufficient residual GALT activity is still continued.
Clinically, patients present with hypergonadotrophic hypogonadism in a contex of either primary amenorrhoea with pubertal retardation or secondary amenorrhoea which may start at any age and progress to POI. Howewer, the ovarian dysfunction may present transiently as a gonadotrophin-resistant syndrome described by an alternation of periods with hypergonadotrophic failure and ovulatory cycles35. The mutations that completely eliminate GALT activity, such as the homozygous Q188 mutation, are responsible for a poor prognosis, whereas mutations which allow a remaining GALT activity are less like to induce long-term complications36.
Mechanisms and timing of follicle development disruption are still not clear. It has been hypothesized that the accumulation of galactose and its toxic products of metabolism (galactose-I-phosphate and galactitol) after birth (since toxic metabolites in foetus should be cleared rapidly by maternal enzymes) leads to direct ovarian impairment36. Glycosylation defects have been hypothesized to account for some of the neurological long-term complications of galactosaemia36.
Chemotherapy irradiation and environmental toxins
Chemotherapy and radiotherapy remain the cornerstone of cancer treatment. The efficacy of chemotherapeutic agents depends upon their ability to destroy rapidly dividing cells. These produce DNA defects as well as oxidative damage in somatic and germ cells. Persistent unrepaired DNA double-strand breaks activate apoptotic death in oocytes37. The clinical impact of chemotherapeutic drugs on the ovary is variable ranging form no effect to complete ovarian atrophy. The degree of damage is dependent upon the type of the chemotherapeutic agent used, dose given, age of the patient and her baseline ovarian reserve. The prepubertal ovary is less susceptible to damage by chemotherapeutic agents while older women hava a lower ovarian reserve and therefore, are more susceptible to POF38.
Alkylating agent cyclophosphamide is a non-cell cycle-specific drug that is cytotoxic even to resting cells, and results in up to 40 per cent risk of ovarian failure at childbearing age38. Alkylating agents are reported to be of high risk of gonadotoxicity, while vinca alkaloids, anthracyclic antibiotics, and antimetabolites are of relatively low risk. Histological examination of ovaries in females after treatment with chemotherapeutic drugs showed blood vessel damage, cortical fibrosis and reduced follicle numbers22.
Human oocyte is susceptible to damage after radiation, with an estimated median lethal dose (LD50) of >2Gy. Damage to the ovary by radiotherapy is due to the age of the patient and dose of the ovarian exposure. The effective sterilizing dose (ESD) is the dose of fractionated radiotherapy at wich POI occurs immediately after treatment in 97.5 per cent of patients. ESD reduces with rising age, being 20.3 Gy at birth, 18.4 Gy at 10 yr, 16.5 at 20 yr, and 14.3 Gy at 30 yr, with only 6 Gy being required to cause POI in women over 40 yr old39.
All women who desire to preserve fertility should be advised and informed about currently available fertility preservation options by fertility specialists. Recommendations should be individualized and should not violate the ethical principles. Gonadotropin-relasing hormone agonist (GnRHa) is often used in combination with chemotherapy causing suprression of the gonadotropin levels to prepubertal levels and reduces utero-ovarian perfusion40, these actions are believed to protect the follicles from apoptosis.
Chen et al41 concluded that the use of GnRHa should be proposed in women of reproductive age receiving chemotherapy and should be given before or during treatment. GnRHa should begin at least 10 days before the beginning of chemotherapy because of the initial flare-up effect and should continue till two weeks after the end of chemotherapy42. Currently, ovarian tissue cyopreservation, embryo cryopreservation, oocyte cryopreservation and in vitro maturation are considered and these can be reommended in selected patients and should be offered only by centres with the necessary laboratory and surgical capability43. After POI prevention contraception should be considered in sexually active women44.
Environmental toxins resulting in oocyte damage might cause POI. Smoking is the most widely studied toxin that alters ovarian function by accelerating follicular atrophy and atresia through increased apoptosis in primordial germ cells45. Polycyclic aromatic hydrocarbons (PaHs), toxic chemicals in tobacco, induce aromatic hydorcarbon receptor (Ahr)-driven expression of Bax in oocytes, followed by apoptosis22. Endocrine disruptors, heavy metals, solvents, insecticides, plastics and industrial chemicals, were associated with adverse reproductive outcomes and ovarian failure in animals. Howewer, the underlying mechanisms were not yet fully clarified and contradictory results were found in human23.
Immunology of POI
Autoinmune disease is characterized by autoreactive T-cells and the presence of organ and non-organ-specific autoantibodies46,47. There are three different situations of autoinmune ovarian insufficiencies: associated with adrenal autoimmunity, non-adrenal autoimmunity and isolated idiopathic POI48.
Initial data estimated that 10 per cent of patients with Addison's disease would develop POI 5-14 years before adrenal disorder. Addison's disease infrequently develops in isolation, with the majority of patients having other associated endocrine disorders. The strongest association of POI is with autoinmune Addison's disease in the context of two types of autoimmune polyendocrine syndromes (APS)46,47. Type 1 [autoimmune polyendocrinopathy candidiasis ectodermal dystrophy (APECED)] characterized as hypoparathyroidism, adrenal failure, and chronic mucocutaneous candidiasis occurs in young children. POI in the form of primary amenorrhoea develops in 60 per cent of type I patients and type II (a polygenic syndrome with autoinmune Addison's disease with adrenal insufficiency and other autoimmune illness without hypoparathyroidism)46,48 occurs in a much broader age range (third to fourth decade) and the incidence of POI is variable.
POI may also be associated with localized or systemic non-adrenal disorders49,50. Between 10 and 30 per cent of women with POI have a simultaneous auto immune disease, the most commonly reported being hypothyroidism49,50, and the most clinically important hypoadrenalism; as well concurrent with hypoparathyroidism51, hypophysitis, type 1 diabetes mellitus, and non-endocrine autoimmune haemolytic anaemia, pernicious anaemia, vitiligo, alopecia areata, celiac disease, inflammatory bowel diseases, primary biliary cirrhosis, glomerulonephritis, multiple sclerosis, Sjogren's syndrome52 and myasthenia gravis48 have been reported.
Antiovarian antibodies
A plausible hypothesis for autoimmune oophoritis is a selective involvement of developing follicles, sparing primordial follicle in early phase, with increased ovarian size with luteinized cyst53. This autoinmune disease is described by serum ovarian autoantibodies detected mainly by indirect immunofluorescence53 and enzyme-linked immunosorbent assay (ELISA). Antigens involved in autoimmune oophoritis, such as zona pellucida/oocyte54, granulosa cells, theca cells52, corpus luteum and steroidogenic enzymes: 17 α-hydroxylase (17α-OH), cytocrome P450 side-chain cleavage enzyme (p450 scc) and 21-hydroxylase47,53 have been reported as the markers of ovarian autoinmunity.
Infections
Mumps oophoritis has been considered to be a cause of POI. True incidence of post-oophoritis ovarian failure is uncertain. In the majoritiy of affected women, return ovarian function occurs following recovery55,56. HIV infection or the corresponding antiretroviral treatment may impair ovarian functions and fertility, and end in POI57. There are also anecdotal reports of viral and microbial infection, such as varicella, cytomegalovirus, malaria being followed by POI56,58. The true frequency of ovarian failure due to viral illness is uncertain.
Treatment and prognosis
After corroborating the diagnosis of POI, karyotyping and analysis of FMR1 premutation should be done to exclude major genetic causes. It is widely accepted that the mainstay of treatment of POI is hormone replacement therapy (HRT), at least until the average age of natural menopause. These women have an increased risk of premature death, mainly from cardiovascular disease, and POI has a potentially devasting influence on bone, with a decreasing bone mineral density, osteoporosis, and increased risk of fracture. Other reasons of morbidity comprise dementia, cognitive decrease and Parkinsonism59.
Induction of puberty is mandatory in young women with pre-pubertal POI, to enable the pubertal growth spurt as well as for development of secondary sexual characteristics, but uterine morphology is often insufficient with failure to accomplish normal volume and configuration, and often a low success rate on oocyte donation programmes60. An extensive variety of treatment modalities are reported in post-pubertal young women, but, there is no clear evidence of best practice61. Physiological Sexual Steroids Replacement (pSSR) appears to recover uterine morphology and parameters of uterine fuction; howewer, both, age at POI and underlying aetiology seem to play a role in the success of the medication.
Pregnancies have been shown to occur in women with POI62. Ovulation was reported in 20 per cent of patients with POI who were observed succesively over a 4-6-month period, but forecasting the probability of spontaneous remission in a specific individual is currently not feasible. Assisted reproductive technique with oocyte donation and in vitro fertilization should be recommended. Besides medical treatment, professional and family support is essential as POI can also adversely affect emotional health, provoke stress, loss of self-esteem, social isolation, increased shyness and anxiety.
References
- 1.Emre SY, Balik KI, Bauthan O. Ovarian aging and premature ovarian failure. J Turk Ger Gynecol Assoc. 2014;15:190–6. doi: 10.5152/jtgga.2014.0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fenton AJ. Premature ovarian insufficiency: Pathogenesis and management. J Midlife Health. 2015;6:147–53. doi: 10.4103/0976-7800.172292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atria A. Early menopause and hormone therapy. Rev Med Chil. 1950;78:373–7. [PubMed] [Google Scholar]
- 4.Pouresmaelli F, Fazeli Z. Premature ovarian failure: A critical condition in the reproductive potential with various genetic causes. Int J Fertil Steril. 2014;8:1–12. [PMC free article] [PubMed] [Google Scholar]
- 5.World Health Organization (WHO). Research on the menopause in the 1990s. Report of a WHO Scientific Group. World Health Organ Tech Rep Ser. 1996;866:1–107. [PubMed] [Google Scholar]
- 6.Harlow SD, Gass M, Hall JE, Lobo R, Maki P, Rebar RW, et al. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. J Clin Endocrinol Metab. 2012;97:1159–68. doi: 10.1210/jc.2011-3362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin M, Yu YQ, Huang HF. An update on primary ovarian insufficiency. Sci China Life Sci. 2012;55:677–86. doi: 10.1007/s11427-012-4355-2. [DOI] [PubMed] [Google Scholar]
- 8.Visser JA, Schipper I, Laven JS, Themmen AP. Anti-Mullerian hormone: an ovarian reserve marker in primary ovarian insufficiency. Nat Rev Endocrinol. 2012;8:331–41. doi: 10.1038/nrendo.2011.224. [DOI] [PubMed] [Google Scholar]
- 9.Younis JS. Ovarian aging and implications for fertility female health. Minerva Endocrinol. 2012;37:41–57. [PubMed] [Google Scholar]
- 10.Practice Committee of American Society for Reproductive Medicine. Testing and interpreting measures of ovarian reserve: a committee opinion. Fertil Steril. 2015;103:e9–19. doi: 10.1016/j.fertnstert.2014.12.093. [DOI] [PubMed] [Google Scholar]
- 11.Kovanci E, Schutt AK. Premature ovarian failure: clinical presentation and treatment. Obstet Gynecol Clin North Am. 2015;42:153–61. doi: 10.1016/j.ogc.2014.10.004. [DOI] [PubMed] [Google Scholar]
- 12.Fortuño C, Labarta E. Genetics of primary ovarian insufficiency: a review. Assist Reprod Genet. 2014;31:1573–85. doi: 10.1007/s10815-014-0342-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wood MA, Rajkovic A. Genomic markers of ovarian reserve. Semin Reprod Med. 2013;31:399–415. doi: 10.1055/s-0033-1356476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grive KJ, Freiman RN. The developmental origins of the mammalian ovarian reserve. Development. 2015;142:2554–63. doi: 10.1242/dev.125211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Finch CE. The menopause and aging, a comparative perspective. J Steroid Biochem Mol Biol. 2014;142:132–41. doi: 10.1016/j.jsbmb.2013.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johnson J, Canning J, Kaneko T, Pru JK, Tilly JL. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature. 2004;428:145–50. doi: 10.1038/nature02316. [DOI] [PubMed] [Google Scholar]
- 17.Crawford NM, Steiner AZ. Age-related infertility. Obstet Gynecol Clin North Am. 2015;42:15–25. doi: 10.1016/j.ogc.2014.09.005. [DOI] [PubMed] [Google Scholar]
- 18.Andreu-Vieyra C, Lin YN, Matzuk MM. Mining the oocyte transcriptome. Trends Endocrinol Metab. 2006;17:136–43. doi: 10.1016/j.tem.2006.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Hale GE, Robertson DM, Burger HG. The perimenopausal woman: endocrinology and management. J Steroid Biochem Mol Biol. 2014;142:121–31. doi: 10.1016/j.jsbmb.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 20.Soni M, Hogervorst E. Premature ovarian insufficiency and neurological function. Minerva Endocrinol. 2014;39:189–99. [PubMed] [Google Scholar]
- 21.deKoning CH, McDonell J, Themmen AP, de Jong FH, Homburg R, Lambalk CB. The endocrine and follicular growth dynamics throughout the menstrual cycle in women with consistently or variably elevated early follicular phase FSH compared with controls. Hum Reprod. 2008;23:1416–23. doi: 10.1093/humrep/den092. [DOI] [PubMed] [Google Scholar]
- 22.Chapman C, Cree L, Shelling AN. The genetics of premature ovarian failure: current perspectives. Int J Womens Health. 2015;23:799–810. doi: 10.2147/IJWH.S64024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goswami D, Conway GS. Premature ovarian failure. Hum Reprod Update. 2005;11:391–410. doi: 10.1093/humupd/dmi012. [DOI] [PubMed] [Google Scholar]
- 24.Jiao X, Qin C, Li J, Qin Y, Gao X, Zhang B, et al. Cytogenetic analysis of 531 Chinese women with premature ovarian failure. Hum Reprod. 2012;21:2201–7. doi: 10.1093/humrep/des104. [DOI] [PubMed] [Google Scholar]
- 25.Welt CK. Primary ovarian insufficiency: a more accurate term for premature ovarian failure. Clin Endocrinol. 2008;68:499–509. doi: 10.1111/j.1365-2265.2007.03073.x. [DOI] [PubMed] [Google Scholar]
- 26.Gleicher N, Weghofer A, Barad DH. A pilot study of pre- mature ovarian senescence: I. Correlation of triple CGG repeats on the FMR1 gene to ovarian reserve parameters FSH and anti-Müllerian hormone. Fertil Steril. 2009;91:1700–6. doi: 10.1016/j.fertnstert.2008.01.098. [DOI] [PubMed] [Google Scholar]
- 27.Wittenberger MD, Hagerman RJ, Sherman SL, McConkie-Rosell A, Welt CK, Rebar RW, et al. The FMR1 premutation and reproduction. Fertil Steril. 2007;87:456–65. doi: 10.1016/j.fertnstert.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 28.Murray A, Ennis S, Morton N. No evidence for parent of origin influencing premature ovarian failure in fragile X premutation carriers. Am J Hum Genet. 2000;67:253–4. doi: 10.1086/302963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baronchelli S, Villa N, Redaelli S, Lissoni S, Saccheri F, Panzeri E, et al. Investigating the role of X chromosome breakpoints in premature ovarian failure. Mol Cytogenet. 2012;5:32. doi: 10.1186/1755-8166-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Toniolo D, Rizzolio F. X chromosome and ovarian failure. Semin Reprod Med. 2007;25:264–71. doi: 10.1055/s-2007-980220. [DOI] [PubMed] [Google Scholar]
- 31.Powell CM, Taggart RT, Drumheller TC, Wangsa D, Qian C, Nelson LM, et al. Molecular and cytogenetic studies of an Xi; autosome translocation in a patient with premature ovarian failure and review of the literature. Am J Med Genet. 1994;52:19–26. doi: 10.1002/ajmg.1320520105. [DOI] [PubMed] [Google Scholar]
- 32.Bilgin EM, Covanci E. Gentics of premature ovarian failure. Curr Opin Obstet Gynecol. 2015;27:167–74. doi: 10.1097/GCO.0000000000000177. [DOI] [PubMed] [Google Scholar]
- 33.Viggiano E, Marabotti A, Burlina AP, Cazzorla C, D’Apice MR, Giordano L, et al. Clinical and molecular spectra in galactosemic patients from neonatal screening in northeastern Italy: structural and functional characterization of new variations in the galactose-1-phosphate uridyltransferase (GALT) gene. Gene. 2015;559:112–8. doi: 10.1016/j.gene.2015.01.013. [DOI] [PubMed] [Google Scholar]
- 34.Timson DJ. The molecular basis of galactosemia - Past, present and future. Gene. 2015 doi: 10.1016/j.gene.2015.06.077. pii: S03781119(15) 00801-X. [DOI] [PubMed] [Google Scholar]
- 35.van Erven B, Gubbels CS, van Golde RJ, Dunselman GA, Derhaag JG, de Wert G, et al. Fertility preservation in female classic galactosemia patients. Orphanet J Rare Dis. 2013;8:107. doi: 10.1186/1750-1172-8-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ryan EL, Lynch ME, Taddeo E, Gleason TJ, Epstein MP, Fridovich-Keil JL. Cryptic residual GALT activity is a potential modifier of scholastic outcome in school age children with classic galactosemia. J Inherit Metab Dis. 2013;36:1049–61. doi: 10.1007/s10545-012-9575-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, Jasin M. Distinct DNA-damage-dependent and independent response drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci Usa. 2005;102:737–42. doi: 10.1073/pnas.0406212102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sonigo C, Sermondade N, Benard J, Benoit A, Shore J, Sifer C, et al. The past, present and future of fertility preservation in cancer patients. Future Oncol. 2015 Sep 11; doi: 10.2217/fon.15.152. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Wallace WH, Thomson AB, Saran F, Kelsey TW. Predicting age of ovarian faiure after radiation to a field that includes the ovaries. Int J Radiat Oncol Biol Phys. 2005;62:738–44. doi: 10.1016/j.ijrobp.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 40.Meirow D, Dor J, Kauffman B, Shrim A, Rabinovici J, Schiff E, et al. Cortical fibrosis and blood-vessels damage in human ovaries exposed to chemotherapy. Potential mechanisms of ovarian injury. Hum Reprod. 2007;22:1622–33. doi: 10.1093/humrep/dem027. [DOI] [PubMed] [Google Scholar]
- 41.Chen H, Li J, Cui T, Hu L. Adjuvant gonadotropin-releasing hormone analogues for the prevention of chemotherapy induced premature ovarian failure in premenopausal women. Cochrane Database Syst Rev. 2011;11:CD008018. doi: 10.1002/14651858.CD008018.pub2. [DOI] [PubMed] [Google Scholar]
- 42.Castelo-Branco C, Nomdedeu B, Camus A, Mercadal S, Martínez de Osaba MJ, Balasch J. Use of gonadotropin-releasing hormone agonists in patients with Hodgkin's disease for preservation of ovarian function and reduction of gonadotoxicity related to chemotherapy. Fertil Steril. 2007;87:702–5. doi: 10.1016/j.fertnstert.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 43.Mahajan N. Fertility preservation in female cancer patients: An overview. J Hum Reprod Sci. 2015;8:3–13. doi: 10.4103/0974-1208.153119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Castelo-Branco C, Rabanal A, Nomdedeu B, Durán M, Arigita M, Balasch J. Unintended pregnancy after gonadal failure chemoprevention with gonadotropin-releasing hormone agonist in women with hematologic malignancies. Fertil Steril. 2009;92:1260–3. doi: 10.1016/j.fertnstert.2009.02.009. [DOI] [PubMed] [Google Scholar]
- 45.Dechanet C, Brunet C, Anahory T, Hamamah S, Hedon B, Dechaud H. Effects of cigarette smoking on female reproduction: from oocyte to embryo (Part I) Gynecol Obstet Fertil. 2011;39:559–66. doi: 10.1016/j.gyobfe.2011.07.033. [DOI] [PubMed] [Google Scholar]
- 46.La Marca A, Brozzeti A, Sighninolfi G, Marzotti S, Volpe E, Falorni A. Primary ovarian insufficiency: autoimmune causes. Curr Opin Obstet Gynecol. 2010;22:277–82. doi: 10.1097/GCO.0b013e32833b6c70. [DOI] [PubMed] [Google Scholar]
- 47.Silva CA, Yamakami LYS, Aikawa NE, Araujo DB, Carvalho JF, Bonfà E. Autoinmuneprmary ovarian insufficiency. Autoinm Rev. 2014;13:427–30. doi: 10.1016/j.autrev.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 48.Carp HJ, Selmi C, Shoenfeld Y. The autoimmune bases of infertility and pregnancy loss. J Autoimmun. 2012;38:J266–74. doi: 10.1016/j.jaut.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 49.Welt CK. Autoimmune oophoritis in the adolescent. Ann N Y Acad Sci. 2008;1135:118–22. doi: 10.1196/annals.1429.006. [DOI] [PubMed] [Google Scholar]
- 50.Wémeau JL, Proust-Lemoine E, Ryndak A, Vanhove L. Thyroid autoimmunity and polyglandular endocrine syndromes. Hormones (Athens) 2013;12:39–45. doi: 10.1007/BF03401285. [DOI] [PubMed] [Google Scholar]
- 51.Dragojević-Dikić S, Marisavljević D, Mitrović A, Dikić S, Jovanović T, Janković-Raznatović S. An immunological insight into premature ovarian failure (POF) Autoimmun Rev. 2010;9:771–4. doi: 10.1016/j.autrev.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 52.Euthymiopoulou K, Aletras AJ, Ravazoula P, Niarakis A, Daoussis D, Antonopoulos I, et al. Antiovarian antibodies in primary Sjogren's syndrome. Rheumatol Int. 2007;27:1149–55. doi: 10.1007/s00296-007-0364-z. [DOI] [PubMed] [Google Scholar]
- 53.Bakalov VK1, Anasti JN, Calis KA, Vanderhoof VH, Premkumar A, Chen S, et al. Autoimmune oophoritis as a mechanism of follicular dysfunction in women with 46, XX spontaneous premature ovarian failure. Fertil Steril. 2005;84:958–65. doi: 10.1016/j.fertnstert.2005.04.060. [DOI] [PubMed] [Google Scholar]
- 54.Haller-Kikkatalo K, Salumets A, Uibo R. Review on autoimmune reactions infemale infertility: antibodies to follicle stimulating hormone. Clin Dev Immunol. 2012;2012:1–15. doi: 10.1155/2012/762541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang H, Chen H, Qin Y, Shi Z, Zhao X, Xu J, et al. Risks associated with premature ovarian failure in Han Chinese women. Reprod Biomed Online. 2015;30:401–7. doi: 10.1016/j.rbmo.2014.12.013. [DOI] [PubMed] [Google Scholar]
- 56.Ebrahimi M, Akbari AF. Pathogenesis and causes of premature ovarina failure. An update. Int J Fertl Steril. 2011;5:54–65. [PMC free article] [PubMed] [Google Scholar]
- 57.Ohl J, Partisani M, Demangeat C, Binder-Foucard F, Nisand I, Lang JM, et al. Alterations of ovarian reserve tests in Human immunodeficiency virus (HIV)-infected women. Gynecol Obstet Fertil. 2010;38:313–7. doi: 10.1016/j.gyobfe.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 58.Panay N, Kalu E. Management of premature ovarian failure. Best Prac Res Clin Obstet Gynecol. 2009;23:129–40. doi: 10.1016/j.bpobgyn.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 59.Sassarini J, Lumsden MA, Critchley HOD. Sex hormone replacement in ovarian failure-new treatment concepts. Best Pract Res Clin Endocrinol Metab. 2015;29:105–14. doi: 10.1016/j.beem.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 60.Paterson WF, Hollman AS, Donaldson MD. Poor uterine development in Turner syndrome with oral oestrogen therapy. Clin Endocrinol. 2002;56:359–65. doi: 10.1046/j.1365-2265.2002.01477.x. [DOI] [PubMed] [Google Scholar]
- 61.Castelo-Branco C. Management of Turner syndrome in adult life and beyond. Maturitas. 2014;79:471–5. doi: 10.1016/j.maturitas.2014.08.011. [DOI] [PubMed] [Google Scholar]
- 62.Bidet M, Bachelot A, Bissauge E, Golmard JL, Gricourt S, Dulon J, et al. Resumption of ovarian function and pregnancies in 358 patients with premature ovarian failure. J Clin Endocrinol Metab. 2011;96:3864–72. doi: 10.1210/jc.2011-1038. [DOI] [PubMed] [Google Scholar]