

Abstract
Tyrosine O-sulfation is a common protein post-translational modification that regulates many biological processes, including leukocyte adhesion and chemotaxis. Many peptides with therapeutic potential also contain a sulfotyrosine residue(s). We report a one-step synthesis of Fmoc-fluorosulfated tyrosine. An efficient Fmoc solid-phase peptide synthesis strategy is then introduced for incorporating the fluorosulfated tyrosine residue into peptides-of-interest. Standard simultaneous peptide-resin cleavage and removal of the acid-labile side-chain protecting groups affords the crude peptides containing fluorosulfated tyrosine. Basic ethylene glycol, serving as solvent and reactant, transforms the fluorosulfated tyrosine peptides into sulfotyrosine peptides in high yield.
Keywords: fluorosulfate, peptides, SuFex, sulfotyrosine, sulfur
SuFEx reaction enables sulfotyrosine peptide synthesis
A facile Fmoc solid-phase synthesis of fluorosulfated tyrosine peptides is reported, enabled by a one-step synthesis of Fmoc-fluorosulfated tyrosine. An efficient ethylene glycolysis method for the transformation of side-chain deprotected fluorosulfated tyrosine peptides to sulfotyrosine peptides is central to the efficiency of this methodology.
Tyrosine O-sulfation is a common enzymatic post-translational modification that occurs while the secreted and transmembrane proteome traffics through the Golgi compartment of the cell.[1] Phosphorylation and sulfation of tyrosine (Tyr) similarly modulate protein-protein interactions and affect conformational changes within a protein.[2]
Currently, one of several approaches can be used for the solid-phase peptide synthesis (SPPS) of protein fragments and polypeptides comprising sulfotyrosine (sY) residues. In the oldest approach, Fmoc Tyr–OSO3− +Na (Fmoc = (9H-fluoren-9-ylmethoxy)-carbonyl) is simply coupled into the growing peptide;[3] however, subsequent couplings can be challenging and direct incorporation of more than one sY residue often compromises resin swelling, impeding further amino acid coupling steps.[3c, 3d] Moreover, acidic deprotection conditions often lead to desulfation. In an alternative method, the neopentyl (Np; (CH3)3CCH2-) group is used to protect the sY residue as a neutral sulfate diester, i.e., as Tyr-O-SO2-ONp.[2, 4] The Fmoc-Tyr(OSO3Np)-OH building block is obtained by a 4-step synthesis in 66% overall yield and is currently commercially available. Fmoc-based SPPS is used to incorporate the Np-protected sY diester building block into the peptide-of-interest. After cleavage of the peptide from the 2-chlorotrityl resin and removal of the standard side chain protecting groups, the Np group is removed in 1–2 M ammonium acetate at 37 °C over 6–12 h.[2] Another option employs a 5-step strategy to synthesize a dichlorovinyl sulfate ester protected sY residue that is incorporated by SPPS into the desired peptide employing an Fmoc strategy.[5] Resin cleavage and side-chain protecting group cleavage are performed as usual (95:2.5:2.5 = trifluoroacetic acid (TFA):triisopropylsilane (TIPS):H2O or 82.5:5:5:5:2.5 = TFA:phenol:H2O:thioanisole:1,2-Ethanedithiol(EDT)) and then the dichlorovinyl sulfate ester protecting group(s) is removed in solution by hydrogenolysis (balloon) using 30 weight % of 10% Pd/C, H2 and 9 equiv. of ammonium formate in methanol at 25 °C for 1 h, minimizing desulfation.[5] The hydrogenolysis step precludes the incorporation of cysteine (Cys) residues into the peptides. An even more recent strategy involves the Fmoc-based solid phase synthesis of peptides containing Tyr residues with distinct phenol protecting groups.[6] These protecting groups are selectively removed while the peptide is still attached to the resin and the phenol is subjected to tyrosine O-sulfation employing sulfuryl imidazolium salt treatment (8 equiv./phenol functional group). Acidic cleavage of the peptide from the resin and removal of the standard side chain protecting groups are followed by removal of the 2,2,2-trichloroethyl protecting group via catalytic hydrogenation using Pd(OH)2 on carbon to afford the sY peptide-of-interest.[6a]
Herein we report a short and efficient route to sY-containing peptides, wherein Fmoc-protected fluorosulfated tyrosine (Y(OSO2F)) is incorporated into the peptide-of-interest via a Fmoc solid phase synthesis strategy, either manually or by use of a peptide synthesizer. Like other sulfur(VI) fluorides, aromatic fluorosulfates are redox stable and hence do not serve as halogenation agents.[7] They are also very stable toward hydrolysis under neutral and acidic conditions, and moreover survive in basic milieu (e.g., phosphate buffer at pH 10).[7] However, the ArOSO2-F linkage becomes reactive in the presence of an appropriate nucleophile only if the reaction conditions meet the stringent needs for the departure of the “F-” from its covalent link to the S(VI)-center.[7–8] In the case at hand, the contrast between high stability alongside the activatable sulfur(VI) fluoride exchange (SuFEx) reactivity pushes this latest click reaction to the very top; only the CuAAC process is still standing in this rarified territory of click chemistry.[7–9] Click reactions are defined as processes that proceed under operationally simple conditions and generate products in high yields with minimal requirements for purification.[7, 9]
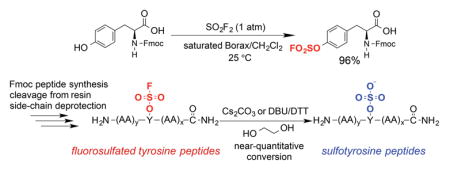
The ease of obtaining the Fmoc-protected Y(OSO2F) SPPS building block and the high stability of aromatic fluorosulfates enables the efficient synthesis of peptides containing the Ar-O-SO2F side chain using an Fmoc chemistry strategy. The Fmoc-Y(OSO2F)-OH amino acid (1) used in SPPS is prepared in one step in 96% yield by reacting commercially available Fmoc-protected Tyr and sulfuryl fluoride (gas) in a biphasic solvent system (CH2Cl2/saturated aqueous Borax buffer; Scheme 1). The synthesis was performed on a 5 g scale and is expected to be amenable to scaling up. After removal of CH2Cl2 at reduced pressure and addition of 1 M HCl, the precipitated product is removed by filtration, washed with water, dried, and used without further purification in SPPS. The Fmoc primary amine protecting group is removed during each SPPS cycle (Scheme 2) using 2-methyl-piperidine (2-MP)[5b, 10] to avoid a small but observable reaction between piperidine and the fluorosulfate functionality that lowered the yield and purity of the desired Y(OSO2F)-containing peptides.[11] The fluorosulfate functional group is stable under the standard acidic Rink amide resin-peptide cleavage conditions (95:2.5:2.5 = TFA:TIPS:H2O) used to liberate the side chain deprotected peptide from the resin. The resin-free Y(OSO2F) substructure(s) in the peptide-of-interest is then converted into the sY functionality by employing ethylene glycol as reactant and solvent along with a base (Cs2CO3, or 1,8-diazabicyclo[5.4.0]undec-7-ene(DBU)) (Scheme 3).
Scheme 1.

Synthesis of Fmoc-fluorosulfated tyrosine 1.
Scheme 2.

Overview of Y(OSO2F) peptide synthesis
Scheme 3.

Overview of arylfluorosulfate hydrolysis to afford sY peptides.
Five sY-containing peptides, 2–6 (Table 1), were prepared using this optimized Fmoc SPPS strategy followed by arylfluorosulfate ethylene glycolysis. The peptides were purified by high performance liquid chromatography (HPLC) as explained in more detail below. The peptide DADEsYL-NH2 (2) comprises a sequence in the epidermal growth factor receptor (EGFR), which when tyrosine O-sulfated is expected to be a good inhibitor of protein tyrosine phosphatase 1B.[12] The monosulfated peptide YEsYLDYDF-NH2 (3) and the trisulfated peptide sYEsYLDsYDF-NH2 (4) correspond to residues 5–12 of mature P-selectin glycoprotein ligand 1 (PGSL-1) that binds to P-selectin and plays an important role in the rolling adhesion of leukocytes on vascular endothelium.[3c, 13] Disulfated peptide TTPDsYGHsYDDKDTLDLNTPVDK-NH2 (5) is a substructure of C5aR, a classical G-protein-coupled receptor that is implicated in many inflammatory diseases.[14] The tetrasulfated peptide DADSENSSFsYsYsYDsYLDEVAF-NH2 (6) corresponds to residues 14–33 of chemokine receptor D6, which scavenges extracellular pro-inflammatory CC chemokines and suppresses inflammation and tumorigenesis.[15] The respectable isolated yields of peptides 4–6, which contain multiple sY residues, reflects the efficiencies of incorporating Y(OSO2F) and arylfluorosulfate ethylene glycolysis in different peptide sequences (Table 1).
Table 1.
Amino acid sequences and isolated yields of sY and Y(OSO2F) peptides.
| No. | Protein Subsequence | Amino Acid Sequence | Isolated Yield |
|---|---|---|---|
| 2 | EGFR(988–993) | DADEsYL-NH2 | 67% |
| 3 | PGSL-1(5–12) | YEsYLDYDF-NH2 | 54% |
| 4 | PGSL-1(5–12) | sYEsYLDsYDF-NH2 | 58% |
| 5 | C5aR(7–28) | TTPDsYGHsYDDKDTLDLNTPVDK-NH2 | 54% |
| 6 | D6(14–33) | DADSENSSFsYsYsYDsYLDEVAF-NH2 | 36% |
| 7 | EGFR(988–993) | DADEY(OSO2F)L-NH2 | 64% |
| 8 | CXCR4(19–30) | GDY(OSO2F)DSMKEPCFR-NH2 | 40% |
| 9 | CXCR4(19–30) | GDsYDSMKEPCFR-NH2 | 35% |
For all SPPS couplings, including the coupling of amino acid 1, we used 5 equiv. of the appropriate side chain protected amino acid preactivated with HCTU/HOBt/DIPEA (1:1:1) for 30 min. The activated amino acid was added to the resin-bound primary amine with stirring or shaking for a coupling period of 30–60 min. Every Fmoc protecting group was removed employing 3 applications of 20% 2-MP in dimethylformamide or N-methyl-2-pyrrolidone (alternative solvent) for 10 min. We used and prefer the 95:2.5:2.5 = TFA:TIPS:H2O deprotection solution (25 °C, 180 min) to cleave the peptide-of-interest off the Rink resin and to liberate the standard side chain protecting groups; however, the other cleavage/deprotection cocktail mentioned above was used without a noticeable change in the purity of the crude peptide generated. The Y(OSO2F)-containing peptides can be easily purified by reverse phase (RP)-HPLC, exemplified by DADEY(OSO2F)L-NH2 (7) in a 64% yield. Arylfluorosulfate ethylene glycolysis of 7 using Cs2CO3 revealed complete conversion to 2, without any noticeable additional peak in the analytical HPLC chromatogram (Figure 1A, S1 and S2). However, there is no need to purify the crude Y(OSO2F) peptides before ethylene glycolysis. The crude Y(OSO2F)-containing peptides can be directly subjected to arylfluorosulfate ethylene glycolysis using Cs2CO3 as the base.[16] The sY peptides were then purified by semi-preparative RP-HPLC using a C18 column and a 20 mM ammonium acetate/CH3CN mobile phase gradient (minimizes desulfation by maintaining a near neutral pH). Using this approach, sY peptides 2–6 were obtained in 36–67% yield (Table 1) after RP-HPLC purification.
Figure 1.

Monitoring the ethylene glycol-mediated hydrolysis of purified Y(OSO2F) peptides 7 and 8, affording sY peptides 2 and 9, respectively, by RP-HPLC using 20 mM ammonium acetate/CH3CN mobile phases. Purified 7 (gray line in A) and 8 (gray line in B) dissolved in ethylene glycol were analyzed by analytical RP-HPLC with tR = 22.8 and 24.7 min, respectively. After adding Cs2CO3 for 7 and DBU/DTT for 8 and stirring for 120 min, the samples were analyzed using the same gradient. sY Peptides 2 and 9 eluted at tR = 17.5 and 21.3 min, respectively. The absorption in B between 5 and 17 min is from DBU and DTT.
In the optimization of the hydrolysis of Y(OSO2F)-containing peptide 7 to sY-containing peptide 2, we observed significant desulfation of sY in the presence of base in aqueous solutions. In addition, upon treating peptide 7 with Cs2CO3 dissolved in methanol, we observed the apparent methylation of peptide 7, presumably owing to the formation of a Tyr-O-SO2-OCH3 intermediate, which appears to transfer a methyl group to a neighboring carboxylate side chain (Figure S3). Neighboring acidic residues are common at sites of protein tyrosine sulfation, thus it is important to solve this problem.[1b, 1c, 17] While utilization of methanol/NH3(2M)/Cs2CO3 attenuated methylation, it was still observed. Utilizing Cs2CO3 dissolved in ethanol resulted in peptide ethylation, consistent with formation of a Tyr-O-SO2-OCH2CH3 intermediate (Figure S4). With Cs2CO3 dissolved in isopropanol or tertiary butyl alcohol, no reaction occurred. Notably, while ethylene glycol/Cs2CO3 and 1,4 butanediol/Cs2CO3 combinations afforded quantitative lysis with no side reactions, 1,3 propanediol/Cs2CO3 afforded < 50 % yield of sY peptide 2 and numerous side products (Figure S5). We hypothesized that efficient cyclic ether formation is key to the mechanism of arylfluorosulfate ethylene glycolysis (Scheme 4). To support this hypothesis, we explored the ethylene glycolysis of the small molecule Ph-O-SO2-F (5 mmoles), employing Cs2CO3 or DBU as the base. This relatively large-scale ethylene glycolysis reaction generated gaseous ethylene oxide, whose identity was confirmed by 1H and 13C NMR employing a distillation-like capture in cold CDCl3 (Figure S6–S8).
Scheme 4.

Proposed mechanism of arylfluorosulfate ethylene glycolysis
The crude peptide GDY(OSO2F)DSMKEPCFR-NH2 (8) (Table 1), containing Cys and methionine residues, was successfully synthesized using the SPPS strategy and side chain deprotection/resin cleavage approach outlined above. Peptide 8 was then HPLC purified in an isolated yield of 40 % (based on resin loading) in order to optimize the hydrolysis strategy for affording sY peptides containing Cys, in this case peptide GDsYDSMKEPCFR-NH2 (9; Table 1). This was done because the ethylene glycolysis/Cs2CO3 method generated a S CH2-CH2-OH functional group on the Cys side chain of 9, as discerned by high resolution mass spectrometry (Figure S9).[18] The optimized protocol for the ethylene glycolysis of peptide 8 employed 5% DBU as the base in ethylene glycol containing 0.5% dithiothreitol (DTT). This ethylene glycolysis solution converted 8 into 9, without any discernable byproduct based on HPLC reaction monitoring (Figure 1B, S10 and S11). Adding DTT was the key to minimizing the ethylene oxide-derived thiol alkylation mentioned above. This approach afforded sY peptide 9 in 35% isolated yield (Table 1) based on resin loading. Peptide 9 comprises residues 19–30 of CXCR4, which is crucial for embryonic development and has been implicated in cancer metastasis and HIV infection.[19]
In summary, we have described a one-step synthesis of the Fmoc-Y(OSO2F) amino acid used without purification for the synthesis of Y(OSO2F)-containing peptides. We demonstrate that the Fmoc synthesis of Y(OSO2F)-containing peptides is both practical and efficient. Standard side chain deprotection and resin cleavage solutions perform well. Two different fluorosulfate ethylene glycolysis protocols are introduced for the efficient production of sY peptides depending on whether the peptide lacks or contains a Cys residue. The facile synthesis described herein takes advantage of the unique reactivity of sulfur(VI) fluorides. Our approach can easily be implemented by commercial and academic peptide synthesis facilities, since the Fmoc-Y(OSO2F) amino acid is commercially available.
Supplementary Material
Acknowledgments
We thank the Skaggs Institute for Chemical Biology and NIH GM051105 (JWK) for financial support.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.a) Huttner WB. Annu Rev Physiol. 1988;50:363–376. doi: 10.1146/annurev.ph.50.030188.002051. [DOI] [PubMed] [Google Scholar]; b) Kehoe JW, Bertozzi CR. Chem Biol. 2000;7:R57–61. doi: 10.1016/s1074-5521(00)00093-4. [DOI] [PubMed] [Google Scholar]; c) Seibert C, Sakmar TP. Biopolymers. 2008;90:459–477. doi: 10.1002/bip.20821. [DOI] [PubMed] [Google Scholar]
- 2.Simpson LS, Zhu JZ, Widlanski TS, Stone MJ. Chem Biol. 2009;16:153–161. doi: 10.1016/j.chembiol.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Yagami T, Shiwa S, Futaki S, Kitagawa K. Chem Pharm Bull. 1993;41:376–380. doi: 10.1248/cpb.41.376. [DOI] [PubMed] [Google Scholar]; b) Kitagawa K, Futaki S, Yagami T, Sumi S, Inoue K. Int J Pept Protein Res. 1994;43:190–200. doi: 10.1111/j.1399-3011.1994.tb00522.x. [DOI] [PubMed] [Google Scholar]; c) Koeller KM, Smith ME, Wong CH. Bioorg Med Chem. 2000;8:1017–1025. doi: 10.1016/s0968-0896(00)00041-9. [DOI] [PubMed] [Google Scholar]; d) Young T, Kiessling LL. Angew Chemie. 2002;41:3449–3451. doi: 10.1002/1521-3773(20020916)41:18<3449::AID-ANIE3449>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 4.Simpson LS, Widlanski TS. J Am Chem Soc. 2006;128:1605–1610. doi: 10.1021/ja056086j. [DOI] [PubMed] [Google Scholar]
- 5.a) Ali AM, Hill B, Taylor SD. J Org Chem. 2009;74:3583–3586. doi: 10.1021/jo900122c. [DOI] [PubMed] [Google Scholar]; b) Ali AM, Taylor SD. Angew Chemie. 2009;48:2024–2026. doi: 10.1002/anie.200805642. [DOI] [PubMed] [Google Scholar]; c) Ali AM, Taylor SD. J Pept Sci. 2010;16:190–199. doi: 10.1002/psc.1220. [DOI] [PubMed] [Google Scholar]
- 6.a) Liu X, Malins LR, Roche M, Sterjovski J, Duncan R, Garcia ML, Barnes NC, Anderson DA, Stone MJ, Gorry PR, et al. ACS Chem Biol. 2014;9:2074–2081. doi: 10.1021/cb500337r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Taleski D, Butler SJ, Stone MJ, Payne RJ. Chem Asian J. 2011;6:1316–1320. doi: 10.1002/asia.201100232. [DOI] [PubMed] [Google Scholar]
- 7.Dong J, Krasnova L, Finn MG, Sharpless KB. Angew Chemie. 2014;53:9430–9448. doi: 10.1002/anie.201309399. [DOI] [PubMed] [Google Scholar]
- 8.Dong J, Sharpless KB, Kwisnek L, Oakdale JS, Fokin VV. Angew Chemie. 2014;53:9466–9470. doi: 10.1002/anie.201403758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kolb HC, Finn MG, Sharpless KB. Angew Chemie. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 10.Hachmann J, Lebl M. J Comb Chem. 2006;8:149. doi: 10.1021/cc050123l. [DOI] [PubMed] [Google Scholar]
- 11.When we used piperidine in the SPPS of peptide 7 we could purify the piperidine addition byproduct for peptide 7. See the Supporting Information for details.
- 12.a) Desmarais S, Jia Z, Ramachandran C. Arch Biochem Biophys. 1998;354:225–231. doi: 10.1006/abbi.1998.0691. [DOI] [PubMed] [Google Scholar]; b) Glover NR, Tracey AS. Biochem Cell Biol. 1999;77:469–486. [PubMed] [Google Scholar]
- 13.a) Pouyani T, Seed B. Cell. 1995;83:333–343. doi: 10.1016/0092-8674(95)90174-4. [DOI] [PubMed] [Google Scholar]; b) Sako D, Comess KM, Barone KM, Camphausen RT, Cumming DA, Shaw GD. Cell. 1995;83:323–331. doi: 10.1016/0092-8674(95)90173-6. [DOI] [PubMed] [Google Scholar]; c) Rodgers SD, Camphausen RT, Hammer DA. Biophysical J. 2001;81:2001–2009. doi: 10.1016/S0006-3495(01)75850-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monk PN, Scola AM, Madala P, Fairlie DP. Br J Pharmacol. 2007;152:429–448. doi: 10.1038/sj.bjp.0707332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Blackburn PE, Simpson CV, Nibbs RJ, O’Hara M, Booth R, Poulos J, Isaacs NW, Graham GJ. Biochemical J. 2004;379:263–272. doi: 10.1042/BJ20031266. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weber M, Blair E, Simpson CV, O’Hara M, Blackburn PE, Rot A, Graham GJ, Nibbs RJ. Mol Biol Cell. 2004;15:2492–2508. doi: 10.1091/mbc.E03-09-0634. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) McCulloch CV, Morrow V, Milasta S, Comerford I, Milligan G, Graham GJ, Isaacs NW, Nibbs RJ. J Biol Chem. 2008;283:7972–7982. doi: 10.1074/jbc.M710128200. [DOI] [PubMed] [Google Scholar]
- 16.All peptides were soluble in ethylene glycol in the concentration we used (10 mg/ml). Addition of Cs2CO3 made the solubilization process faster. The amount of Cs2CO3 used for hydrolysis varied with the peptide sequence. See the Supporting Information for details.
- 17.a) Lin WH, Larsen K, Hortin GL, Roth JA. J Biol Chem. 1992;267:2876–2879. [PubMed] [Google Scholar]; b) Teramoto T, Fujikawa Y, Kawaguchi Y, Kurogi K, Soejima M, Adachi R, Nakanishi Y, Mishiro-Sato E, Liu MC, Sakakibara Y, et al. Nat Commun. 2013;4:1572. doi: 10.1038/ncomms2593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The molecular weight of the peptide byproduct generated using ethylene glycol/Cs2CO3 glycolysis is 44 Daltons heavier than the molecular weight of peptide 9. Similarly, we used a tripeptide containing Cys residue to capture the ethylene oxide generated during arylfluorosulfate ethylene glycolysis and obtained the ethylene oxide byproduct in the context of this peptide as well. See the Supporting Information for more details.
- 19.a) Veldkamp CT, Seibert C, Peterson FC, Sakmar TP, Volkman BF. J Mol Biol. 2006;359:1400–1409. doi: 10.1016/j.jmb.2006.04.052. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Seibert C, Veldkamp CT, Peterson FC, Chait BT, Volkman BF, Sakmar TP. Biochemistry. 2008;47:11251–11262. doi: 10.1021/bi800965m. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Veldkamp CT, Seibert C, Peterson FC, De la Cruz NB, Haugner JC, 3rd, Basnet H, Sakmar TP, Volkman BF. Sci Signal. 2008;1:ra4. doi: 10.1126/scisignal.1160755. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.