

Abstract
FANCJ/BRIP1 encodes a helicase that has been implicated in the maintenance of genomic stability. Here, to better understand FANCJ function in DNA damage responses, we have examined the regulation of its cellular localization. FANCJ nuclear foci assemble spontaneously during S phase and are induced by various stresses. FANCJ foci colocalize with the replication fork following treatment with hydroxyurea, but not spontaneously. Using FANCJ mutants, we find that FANCJ helicase activity and the capacity to bind BRCA1 are both involved in FANCJ recruitment. Given similarities to the recruitment of another Fanconi anemia protein, FANCD2, we tested for colocalization of FANCJ and FANCD2. Importantly, these proteins show substantial colocalization, and FANCJ promotes the assembly of FANCD2 nuclear foci. This process is linked to the proper localization of FANCJ itself since both FANCJ and FANCD2 nuclear foci are compromised by FANCJ mutants that abrogate its helicase activity or interaction with BRCA1. Our results suggest that FANCJ is recruited in response to replication stress and that FANCJ/BRIP1 may serve to link FANCD2 to BRCA1.
Introduction
Genomic instability is associated with the development of cancer and with a poorer prognosis. Many tumor suppressor proteins function in cellular pathways that maintain a stable genome by responding to DNA damage (Risinger and Groden 2004). FANCJ/BRIP1 has been implicated in DNA damage responses through its interaction with the protein encoded by the BRCA1 breast cancer susceptibility gene (Cantor et al. 2001) and by being a Fanconi anemia gene (Levitus et al. 2005; Levran et al. 2005; Litman et al. 2005).
FANCJ was first identified by a physical interaction with BRCA1, which is mediated by phosphorylation of FANCJ at S990 (Yu et al. 2003; Xie et al. 2010). Accordingly, this interaction is disrupted in the S990A mutant of FANCJ. Like BRCA1, FANCJ has been identified as a breast cancer susceptibility protein (Seal et al. 2006). It appears that FANCJ functions downstream of BRCA1 in human cells. For example, BRCA1 is required for the assembly of FANCJ nuclear foci (Cantor et al. 2001), but it has not been demonstrated that this is mediated through a physical interaction between the proteins. Unlike human FANCJ, homologues in chicken and Caenorhabditis elegans lack the conserved Ser990-X-X-Phe motif and presumably do not interact with BRCA1 (Bridge et al. 2005; Youds et al. 2008).
Human FANCJ has a helicase domain (Cantor et al. 2001). As in other helicases, mutation of a conserved lysine at residue 52 (K52R), which is involved in ATP hydrolysis (Cantor et al. 2001), abrogates the helicase activity of FANCJ (Cantor et al. 2004; Gupta et al. 2005). Two mutations of FANCJ associated with early onset breast cancer disrupt its helicase activity (Cantor et al. 2004), suggesting the potential importance of this activity to the function of FANCJ as a tumor suppressor. FANCJ displays preferential binding to a forked duplex substrate and has 5′ to 3′ helicase activity (Gupta et al. 2005). Both the K52R and S990A mutants of FANCJ can alter cellular sensitivity to DNA damage (Peng et al. 2007; Xie et al. 2010), suggesting a role in DNA damage responses for FANCJ helicase activity and its capacity to bind BRCA1.
Fanconi anemia (FA) is associated with a predisposition to cancer, including leukemia and various solid tumors, as well as bone marrow failure and assorted congenital anomalies (Mathew 2006; Taniguchi and D’Andrea 2006). Cells from FA patients are characterized by chromosome instability, both spontaneously and in response to DNA interstrand crosslinking agents such as mitomycin C (MMC). There are 14 identified FA genes, and eight of the encoded proteins (FANC- A, B, C, E, F, G, L, and M) are required for the monoubiquitination of the FA proteins FANCD2 (Garcia-Higuera et al. 2001; Taniguchi and D’Andrea 2006; Wang 2007) and FANCI (Sims et al. 2007; Smogorzewska et al. 2007). Given that a non-ubiquitinable FANCD2 mutant confers no resistance to MMC (Garcia-Higuera et al. 2001; Taniguchi et al. 2002b; Montes de Oca et al. 2005), it appears that monoubiquitination of this protein is critical to the FA pathway.
Among the other FA proteins, FANCJ, along with FANCD1/BRCA2, FANCN/PALB2, and FANCO/RAD51C, is not required for FANCD2 monoubiquitination (Howlett et al. 2002; Levitus et al. 2004; Bridge et al., 2005; Litman et al. 2005; Reid et al. 2007; Vaz et al. 2010). For this reason, it has been suggested that FANCJ functions downstream of monoubiquitinated FANCD2 (Bridge et al. 2005; Cantor and Andreassen 2006). It should be noted, in this context, that BRCA1 has not been identified as a FA gene (Cantor and Andreassen 2006). Consistent with a role in DNA damage responses, BRCA1, FANCJ, and FANCD2 are all required for cellular resistance to MMC (Garcia-Higuera et al. 2001; Moynahan et al. 2001; Peng et al. 2007) and, to varying degrees, ionizing radiation (IR) (Scully et al. 1999; Taniguchi et al. 2002b; Peng et al. 2006).
Various DNA damage response proteins are recruited in response to DNA damage or replication stress (Andreassen et al. 2006). The mechanism and pattern of recruitment can provide important insights into the function of a particular protein in DNA damage responses. It is known that FANCJ foci are inducible in response to IR and that these foci colocalize with DNA damage response proteins such as BRCA1 and RPA (Cantor et al. 2001; Peng et al. 2006; Gupta et al. 2007). The localization of FANCJ to DNA damage foci further implicates it in DNA damage responses, but the regulation and role of this localization are not well understood.
Here, to better understand FANCJ function, we have examined its recruitment. We provide quantitative evidence that FANCJ foci assemble in response to a variety of stresses and that FANCJ is recruited to blocked replication forks in response to treatment with hydroxyurea. These results suggest that FANCJ may be recruited to lesions that impede the replication fork. In agreement with this possibility, we find that the ATR checkpoint kinase, which is an important mediator of the replication-stress response (Cimprich and Cortez 2008), influences DNA damage-induced assembly of FANCJ foci. Further, we have utilized the FANCJ-K52R and FANCJ-S990A mutants to determine how FANCJ is recruited to nuclear foci. We find that the capacity of FANCJ to bind to BRCA1, and its helicase activity, is important for its localization. Interestingly, we also find that both mutants are associated with a defect in the assembly of FANCD2 nuclear foci, both spontaneously and in response to DNA damage induced by MMC. Thus, proper localization of FANCJ is associated with efficient recruitment of FANCD2. Together, our results suggest that FANCJ may have a role in linking FANCD2 recruitment to BRCA1. This is of interest since each of these proteins is required for the maintenance of genomic stability.
Materials and methods
Cell culture
EUFA30-hTERT fibroblasts (Levitus et al. 2005), which were kindly provided by Dr. Hans Joenje (Vrije Universiteit Medical Center), PD20 cells containing the pMMP vector or corrected with wild-type FANCD2, and MCF7 cells were grown in DMEM supplemented as described previously (Garcia-Higuera et al. 2001; Litman et al. 2005; Zhang et al. 2009). Also, PD845 and PD846 fibroblasts (Mankad et al. 2006) immortalized with hTERT were grown as described for EUFA30-hTERT cells (Litman et al. 2005). Cells were kept at 37°C in a humid incubator.
Retroviral transduction of FA-J cells
K52R and S990A mutations were introduced into the FANCJ cDNA in pBluescriptKS (+/−) using a QuikChange II XL Site-Directed Mutagenesis Kit (Stratagene) according to the manufacturer’s instructions. Mutations were verified by sequencing. Following introduction of Xho I (5′) and Not I (3′) restrictions sites by PCR, these cDNAs were cloned into the pOZ retroviral vector containing a C-terminal Flag-HA tag (Nakatani and Ogryzko 2003). Generation of retroviruses, transduction, and selection of cells using magnetic beads conjugated to IL2 (Upstate) was as previously described (Fan et al. 2009). Cells were analyzed in the first or second week after selection.
siRNA and transfection
Expression of targeted genes was knocked down by transient transfection with siRNAs directed against FANCJ (5′-GTACAGTACCCCACCTTAT) (Litman et al. 2005), BRCA1 (5′- GGAACCTGTCTCCACAAAG) (Vandenberg et al. 2003), ATR (5′-CCTCCGTGATGTTGCTTGA), and GFP (5′-CGGCAAGCTGACCCTGAAGTTCAT) (Andreassen et al. 2004). Cells were plated at least 16 h prior to transfection to yield approximately 30% confluency at the time of transfection. SiRNA oligos were transfected by Oligofectamine according to the manufacturer’s instructions (Invitrogen). To obtain maximal RNA interference, cells were transfected with siRNA again 24 h after the initial siRNA transfection. Cells were analyzed 48 h after the second transfection.
Immunofluorescence microscopy
Cells were grown on poly-D-lysine coated glass coverslips for a minimum of 16 h prior to experimental manipulation, and unless otherwise noted, cells were fixed with 2% paraformaldehyde for 20 min, permeabilized with 0.2% Triton X-100 for 3 min, washed, and incubated with primary and secondary antibodies as previously described (Fan et al. 2009). FANCD2, FANCJ, BRCA1, and γ-H2AX were detected with E35 rabbit antiserum (Garcia-Higuera et al. 2001), 2G7 mouse monoclonal antibodies (Cantor et al. 2001) (a kind gift of Dr. Sharon Cantor, University of Massachusetts), rabbit polyclonal antibodies (Cell Signaling), and Ser139 rabbit polyclonal antibodies (Upstate), respectively. FANCJ-Flag-HA fusion proteins were detected with M2 anti-Flag antibodies (Sigma). Coverslips were mounted over Vectashield containing DAPI and were sealed with fingernail polish.
For double-labeling with anti-PCNA and anti-FANCJ antibodies, cells were fixed with 2% paraformaldehyde in PBS for 20 min at RT, washed once with PBS, fixed 10 min with methanol at −20°C, and then incubated with 0.2% Triton X-100 in PBS for 2 min. Cells were then washed with PBS prior to incubation with primary antibodies, as described above. PCNA was detected with a mouse antibody (PC10, Santa Cruz).
For determination of the S phase index, FA-J cells were incubated with 30 μM bromodeoxyuridine (BrdU) for 10 min, then fixed with 2% paraformaldehyde, permeabilized, treated with 2 N HCl for antibody access, neutralized, and incubated with anti-BrdU, antibody as previously described (Andreassen et al. 2004).
Three counts of 150 or more cells each was made for each treatment condition, unless otherwise noted, and averages and standard deviations were calculated. P values were calculated using Student’s t test. Labeled cells were observed with a Zeiss Axiovert 200M microscope, and images were collected with a Hamamatsu Camera using Openlab software (Improvision). Images were processed into figures using Photoshop (Adobe).
Immunoblotting
Lysates were electrophoresed by SDS-PAGE (6% poly-acrylamide, bis-acrylamide). Proteins were then transferred to nitrocellulose, and membranes blocked and incubated with primary antibodies, as previously described (Taniguchi et al. 2002a). Antibodies included anti-FANCD2 (E35), E47 rabbit anti-FANCJ (a kind gift of Dr. Sharon Cantor), anti-BRCA1 (Upstate), anti-ATR (Sigma), anti-Flag (Sigma), rabbit anti-histone H2A (acidic patch) (Sigma), anti-γ-tubulin (Sigma), and C4 anti-actin antibodies (a kind gift of Dr. James Lessard, Cincinnati Children’s Research Foundation). Membranes were washed, incubated with HRP-linked secondary antibodies (Amersham), and detected by chemiluminescence (Amersham) as previously described (Taniguchi et al. 2002a).
Nuclear Fractionation
Tert-immortalized EUFA-30 FA-J fibroblasts that contained the empty pOZ vector alone, or which were corrected by expression of FANCJ, were fractionated as described previously (Bogliolo et al. 2007). Briefly, an equivalent number of cells was collected for each cell type, and cells were washed twice with PBS pre-chilled to 4°C. Cells were resuspended in 10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 0.1% Triton X-100, 1 mM dithiothreitol, 10 mM NaF, 1 mM Na3VO4, and Complete Protease Inhibitor Cocktail (Roche). Cells were incubated for 5 min on ice. Cytoplasmic and soluble nuclear proteins were collected in the supernatant following centrifugation (1,300×g, 4 min). Nuclei were then lysed in 3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and protease inhibitors (Roche) for 30 min on ice. Soluble nuclear proteins were collected by centrifugation (1,700×g, 4 min) and were pooled with the previous fraction. Together, this fraction contained soluble proteins. The pellet was washed once in the buffer described above. The pellet, which contained chromatin proteins, was resuspended in SDS sample buffer and was sonicated.
G2/M Accumulation
EUFA30 fibroblasts containing the empty vector or wild-type FANCJ were plated to be subconfluent for the course of the experiment. After 24 h, cells were left untreated or were exposed to 0.35 μg/ml melphalan for an additional 48 h. Cells were collected, fixed, and incubated with RNAse A and propidium iodide as previously described (Andreassen et al. 2004). For analysis, 5×103 cells were measured using a FacsCalibur instrument (Becton-Dickinson), aggregates were gated out, and the percentage of cells in G2/M was calculated using ModFit software.
Results
We conducted an analysis of FANCJ nuclear foci by immunofluorescence microscopy to gain insight into its potential functions and manner of regulation (Fig. 1). While a previous analysis reported that IR induces FANCJ foci (Peng et al. 2006), our quantitative analyses demonstrate that FANCJ foci were also induced by other stresses in MCF7 mammary adenocarcinoma cells. An example of foci assembled in untreated cells and following treatment with MMC is shown in Fig. 1a. Quantification shows that both MMC and hydroxyurea (HU) induced FANCJ foci, as did exposure to IR (Fig. 1b). Thus, FANCJ foci are assembled in response to a variety of DNA damages and replication stress.
Fig. 1.

FANCJ nuclear foci assemble spontaneously in S phase cells, are induced by various stresses, and are recruited to blocked replication forks. a Examples of FANCJ nuclear foci (green) in untreated populations of MCF7 cells or following treatment of cells with 0.5 μM MMC for 20 h. Nuclei were stained with DAPI (blue) for visualization. b Quantification of the percentage of MCF7 cells that displayed five or more FANCJ foci in untreated populations, following exposure to 0.5 μM MMC or 2 mM HU for 7 h, or at 7 h following exposure to 5 Gy IR. c Examples of FANCJ nuclear foci (red) and PCNA foci (green) in untreated MCF7 cells or following treatment with 2 mM HU for 20 h. The degree of colocalization is demonstrated in the merged image. d The percentage of S phase or non-S phase cells, identified by the presence or absence of nuclear PCNA signal, respectively, that displayed five or more FANCJ foci. e The percentage of cells with five or more FANCJ nuclear foci that displayed colocalization with PCNA foci in untreated MCF7 cells, or at 2 or 7 h of treatment with 2 mM HU. PCNA is a marker for the replication fork. Quantitative values represent the average of three counts of 150 or more cells each±standard deviation (b, d–e). The levels of FANCJ foci, or colocalization of FANCJ and PCNA foci, were significantly increased (p<0.01) following treatment with HU or DNA damage in b and e, respectively
Importantly, and in agreement with a previous report (Peng et al. 2006), spontaneous FANCJ foci were detected in a subset of untreated MCF7 mammary adenocarcinoma cells (Fig. 1a, b). We considered the possibility that these spontaneous foci might be cell cycle-linked (Fig. 1c, d). Since certain DNA repair processes, such as HR, are coupled to S phase progression (Rothkamm et al. 2003), we examined whether spontaneous FANCJ foci assemble strictly at this stage of the cell cycle. For this purpose, we examined cells that were double-labeled with antibodies to FANCJ and to PCNA. PCNA foci assemble during S phase at the replication fork (Bravo and Macdonald-Bravo 1985). As shown in the example in Fig. 1c, and by quantification in Fig. 1d, nearly all cells that contained spontaneous FANCJ foci were in S phase and not at other stages of the cell cycle, as determined by the presence or absence of nuclear PCNA.
We then determined whether spontaneous FANCJ foci colocalize with PCNA foci, which would indicate the association of FANCJ with the replication fork and a potential function at this site. As shown in the example in Fig. 1c, and by quantification in Fig. 1e, FANCJ foci showed only a low level of colocalization, if any, with the replication fork in untreated cells (1.8±1.3%). Similarly, MMC and IR, which unlike HU do not arrest DNA replication, yielded low levels of colocalization of FANCJ foci with PCNA foci (2.8±0.4% and 2.0±0.9% at 7 h after treatment with 0.5 μM MMC or 5 Gy IR, respectively). Interestingly, however, FANCJ foci strongly colocalized with PCNA foci within 2 h of treatment with HU and also at 7 h of treatment (80.2±2.2% of FANCJ-positive nuclei displayed colocalization at 7 h of treatment with HU). Thus, the recruitment of FANCJ following exposure to DNA damage may involve replication stress.
To further investigate whether FANCJ is recruited to DNA damage foci in response to replication stress, we depleted ATR from cells. ATR is a central regulator of the replication-stress response (Cimprich and Cortez 2008). ATR levels were depleted using a siRNA that we have described previously, which targets this protein (Andreassen et al. 2004). Depletion of ATR had no clear effect on FANCJ protein levels (Supplementary Material, Fig. S1a). However, depletion of ATR resulted in a decrease in the levels of FANCJ foci following exposure to MMC (Supplementary Material, Fig. S1b), as compared with cells transfected with a control siRNA. There was no effect on FANCJ foci in untreated cells, consistent with this protein being recruited, in part, by the ATR-mediated replication-stress response.
To better understand the molecular determinants of the assembly of FANCJ into nuclear foci, we reconstituted EUFA30 FA-J cells, which are deficient for FANCJ (Levitus et al. 2005), with wild-type FANCJ or with two different FANCJ mutants. The K52R mutant is defective for FANCJ helicase activity (Cantor et al. 2004; Gupta et al. 2006) and the non-phosphorylable S990A mutant of FANCJ does not bind to BRCA1 (Yu et al. 2003; Xie et al. 2010). Each form of FANCJ was expressed as a C-terminal fusion with a Flag-HA epitope tag. Wild-type FANCJ and each of the mutants were expressed at comparable levels (Fig. 2a) and were detected with antibodies that recognized the Flag epitope tag or FANCJ itself. Functional correction of the EUFA30 FA-J cell line by expression of wild-type FANCJ-Flag-HA was demonstrated by a clear reduction in G2/M accumulation following treatment with melphalan (Supplementary Material, Fig. S2).
Fig. 2.

The assembly of FANCJ nuclear foci involves its capacity to bind to BRCA1 and its helicase activity. a The levels of FANCJ, detected with antibodies to either FANCJ or the Flag epitope tag, in EUFA30 FA-J cells in which wild-type FANCJ, or the S990A or K52R mutants of FANCJ, were expressed. FANCJ mutants were expressed as C-terminal Flag-HA fusion proteins. FANCJ-Flag-HA was detected with antibodies to either FANCJ or the Flag epitope tag. Cells that contained the empty pOZ vector are shown for comparison. b Representative images of epitope-tagged FANCJ, detected with an antibody against the Flag peptide, for EUFA30 FA-J cells that contained the pOZ vector alone or which were corrected by expression of FANCJ-WT. Cells were exposed to 0.5 μM MMC for 20 h. An enlargement of the nucleus, which is positive for FANCJ foci, is in the inset. c Quantification of the assembly of FANCJ nuclear foci, detected with an anti-Flag antibody, in EUFA30 cells containing empty vector, wild-type FANCJ, or its mutants, both in untreated populations or following exposure to 0.5 μM MMC for 20 h. d Example of BrdU incorporation in untreated EUFA30 cells corrected with wild-type FANCJ is shown (left). Determination of the percentage of S phase cells by measuring BrdU incorporation in untreated populations of EUFA30 cells containing empty vector or each form of FANCJ described above (right). For each measurement, three counts of 150 or more cells each were made using a microscope and the average±standard deviation is shown. The levels of FANCJ foci were statistically different (p<0.01) in cells corrected with wild-type FANCJ, as compared with cells expressing no FANCJ or other forms of the protein, both in untreated populations and following exposure to MMC (c)
Detection of foci by immunofluorescence microscopy was conducted using anti-Flag antibodies. In the example shown, EUFA30 fibroblasts that contained the empty retroviral vector were devoid of foci detectable with anti-Flag antibodies (Fig. 2b). In contrast, wild-type FANCJ-Flag-HA assembled into distinct nuclear foci in a subset of cells following treatment with 0.5 μM MMC for 20 h. A full set of images, including representative examples for FANCJ-K52R and FANCJ-S990A, where only a small fraction of cells assembled FANCJ foci, is shown in Supplementary Material, Fig. S3. Quantification of the assembly of FANCJ foci by each mutant, or by wild-type FANCJ, as detected with anti-Flag antibodies, is shown in Fig. 2c. The assembly of FANCJ-S990A or FANCJ-K52R into nuclear foci was compromised, as compared with wild-type FANCJ, both in untreated populations and following exposure to MMC. We did not test these mutants for the assembly of FANCJ foci following treatment with IR or HU.
While EUFA30 FA-J cells expressing FANCJ-S990A had a slightly elevated rate of passage through S phase, as measured by BrdU incorporation (Andreassen et al. 2004), cells lacking FANCJ or expressing FANCJ-WT or FANCJ-K52R had comparable rates of S phase progression (Fig. 2d). Thus, effects on the assembly of FANCJ foci were not due to cell cycle perturbations. We conclude that the assembly of FANCJ nuclear foci involves both its binding to BRCA1 and its helicase activity.
Colocalization can yield important insight into the function of specific proteins. Given that FANCJ (Fig. 1) and another FA protein, FANCD2 (Garcia-Higuera et al. 2001; Taniguchi et al. 2002a), form spontaneous S phase foci and are induced by a variety of stresses, we assayed for colocalization of these proteins. Examples of colocalization in untreated MCF7 cells and following exposure to MMC are shown in Fig. 3a. While some dispersed nuclear signal from FANCJ and FANCD2 was detected, consistent with previous reports (Cantor et al. 2001; Garcia-Higuera et al. 2001), foci were more intense. Quantification in untreated MCF7 cells, or following exposure to MMC, HU, or IR, indicated detectable colocalization of FANCJ with FANCD2 foci in each case and is shown in Fig. 3b. The colocalization of FANCJ foci and FANCD2 foci suggests a possible functional interaction between these proteins.
Fig. 3.

FANCJ and FANCD2 foci partially colocalize in untreated MCF7 cells and following exposure to various types of DNA damage. a Examples in which FANCJ (green) and FANCD2 nuclear foci (red) colocalized in untreated populations of MCF7 cells or following treatment of cells with 0.5 μM MMC for 20 h. b Quantification of the percentage of MCF7 cells with FANCJ foci that displayed three or more colocalized FANCD2 foci in untreated populations, following exposure to 0.5 μM MMC or 2 mM HU for 7 h, or at 7 h following exposure to 5 Gy IR. Results represent the average of three counts of 150 or more cells each±standard deviation
Since FANCJ has been proposed to function downstream of FANCD2 (Bridge et al. 2005; Cantor and Andreassen 2006), and given the observed colocalization of FANCJ and FANCD2 foci (Fig. 3), we sought to determine whether the assembly of FANCJ foci might be dependent upon FANCD2. Thus, we compared PD20 FA-D2 cells that lacked FANCD2 and their counterparts corrected by expression of FANCD2 but found no difference in FANCJ foci following treatment with MMC (Supplementary Material, Fig. S4).
Next, to determine whether the assembly of FANCD2 foci is instead dependent upon FANCJ, we examined the assembly of FANCD2 foci in two different FANCJ-deficient cell lines (EUFA30 and IFAR943/1) from FA-J patients (Levitus et al. 2005; Levran et al. 2005). Both EUFA30 and IFAR943/1 cells displayed undetectable endogenous FANCJ, or low levels relative to PD845 (FA-A) and PD846 (normal control) cells (Fig. 4a). FANCJ-Flag-HA expression in EUFA30 and IFAR943/1 cells was detected with antibodies directed against either FANCJ or HA (Fig. 4a). FANCD2 was monoubiquitinated in both of these cell lines, and this was induced by treatment with MMC. FANCJ was not required for FANCD2 monoubiquitination since the levels were similar with or without correction with wild-type FANCJ. In contrast, the PD845 FA-A cell line displayed a low level of FANCD2 monoubiquitination in untreated populations, but no increase was observed following treatment with MMC.
Fig. 4.

The assembly of FANCD2 foci is compromised in FA-J cells. a Levels of endogenous FANCJ were detected with the mouse monoclonal 2G7 antibody in tert-immortalized fibroblasts from a normal control individual (PD846), a FA-A patient (PD845), and two different FA-J patients (EUFA30 and IFAR943/1). The FA-J cells contained vector alone or were corrected by the expression of FANCJ. Expression of FANCJ-Flag-FANCJ in transduced FA-J cell lines was detected with HA.11 mouse anti-HA antibody. Both monoubiquitinated (-L) and unubiquitinated FANCD2 (-S) were detected with E35 antibody. An equivalent amount of protein was loaded from each sample and actin is shown as a loading control. b Examples of FANCD2 foci detected by immunofluorescence microscopy in EUFA30 (FA-J) cells expressing wild-type FANCJ, or containing only the empty pOZ-C vector, or in PD845 (FA-A) cells. An enlargement of the FANCD2-positive cell from the EUFA30+Vector example (cell at upper left) is in the inset for better visualization. Cells were treated with 0.5 μM MMC for 20 h and nuclei were detected by counterstaining with DAPI. c Quantification of the percentage of EUFA30 or IFAR943/1 FA-J cells with FANCD2 foci, either with or without genetic correction. Foci were also quantified in untreated PD845 and PD846 cells, or those exposed to 0.5 μM MMC for 20 h. d Quantification of the percentage of EUFA-30 cells containing empty vector alone, wild-type FANCJ, FANCJ-S990A, or FANCJ-K52R, which displayed FANCD2 nuclear foci, both in untreated populations or following treatment with 0.5 μM MMC for 20 h. Values represent the average of three blind counts of 150 or more cells each±standard deviation from unidentified slides (c–d). For each particular FAJ cell line levels of FANCD2 foci were statistically different (p<0.04) in cells corrected with wild-type FANCJ, as compared with the absence of FANCJ or expression of other forms of the protein (c–d)
Representative images suggest that the assembly of FANCD2 foci was more efficient in FA-J cells (EUFA30) corrected by the expression of FANCJ than in uncorrected FA-J cells containing the pOZ empty vector (Fig. 4b). For example, fewer FA-J cells displayed FANCD2 foci than did corrected FA-J cells. Also, within positive cells, FANCD2 foci were generally brighter following reintroduction of FANCJ. A fibroblast line (PD845) from a FA patient with biallelic mutation of FANCA, which unlike FANCJ is required for FANCD2 monoubiquitination (Garcia-Higuera et al. 2001; Levitus et al. 2004), was also examined for comparison. In contrast to FA-J cells, the PD845 cell line was completely deficient for the assembly of FANCD2 foci (Fig. 4b).
Quantification demonstrates that both EUFA30 and IFAR943/1 FA-J cells displayed some FANCD2 foci. Reconstitution with FANCJ enhanced the assembly of FANCD2 foci, however, either without treatment or in response to treatment with 0.5 μM MMC for 20 h (Fig. 4c). The levels of FANCD2 foci in corrected FA-J cell lines were similar to those observed in PD846 cells from a normal control individual. In contrast, PD845 cells were completely deficient for the assembly of FANCD2 foci, either with or without MMC treatment (Fig. 4c). Thus, FANCJ promotes the efficient assembly of FANCD2 foci but does so in a manner that is distinguishable from deficient monoubiquitination of FANCD2.
Next, to determine molecular requirements for the role of FANCJ in promoting the assembly of FANCD2 foci, we utilized EUFA30 FA-J fibroblasts reconstituted with wild-type FANCJ or with the two different FANCJ mutants. Quantification demonstrates that unlike wild-type FANCJ, neither the S990A mutant nor the K52R mutant of FANCJ, corrected the defective assembly of FANCD2 foci present in EUFA30 FA-J fibroblasts (Fig. 4d). The percentage of FA-J cells with FANCD2 foci was the same for cells containing the empty vector alone and for those expressing FANCJ-K52R or FANCJ-S990A, either without treatment or following exposure to 0.5 μM MMC for 20 h. The assembly of FANCD2 foci following treatment with IR or HU was not measured in FA-J cells reconstituted with FANCJ-S990A or FANCJ-K52R. Again, as shown by measuring BrdU incorporation (Fig. 2d), these differences were not due to an altered rate of cell cycle progression. Since the efficient assembly of FANCJ foci also required both FANCJ helicase activity and FANCJ binding to BRCA1 (Fig. 2c), normal assembly of FANCD2 foci may be linked to the recruitment of FANCJ.
FANCD2 must be monoubiquitinated to assemble into nuclear foci in chromatin (Montes de Oca et al. 2005). Thus, we hypothesized that FANCJ may regulate FANCD2 foci by promoting the association of monoubiquitinated FANCD2 with chromatin. To test this possibility, we separated nuclei from FA-J cells, which contained the empty vector or which were corrected with FANCJ, into soluble and chromatin fractions. FANCJ was detected in the corrected cells and was associated with both the soluble and chromatin-containing fractions from untreated cells (Fig. 5a). Most of the unubiquitinated FANCD2 (-S) was present in soluble fractions (containing both cytoplasmic and nuclear proteins) (lanes 1 and 3), either with or without correction of the FANCJ-deficiency. In contrast, monoubiquitinated FANCD2 (-L) was predominantly detected in chromatin-containing fractions (lanes 2 and 4). Interestingly, monoubiquitinated FANCD2 associated with chromatin less efficiently in uncorrected FA-J cells (FA-J+Vector) than in corrected FA-J cells. This can be seen by the increased percentage of total monoubiquitinated FANCD2, identified in the boxes, which is present in the soluble fractions from uncorrected FA-J cells (for example, compare lane 1 to lane 2, and lane 3 to lane 4).
Fig. 5.
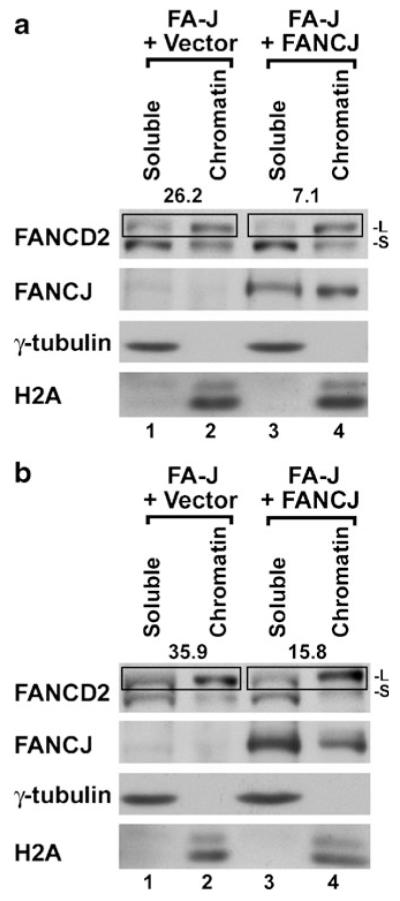
FANCJ promotes the association of monoubiquitinated FANCD2 with chromatin. a Untreated FA-J cells (EUFA30) expressing FANCJ-Flag-HA, or which contained the empty vector alone, were separated into soluble and chromatin-containing fractions. Representative results for untreated cells (a), or those exposed to 0.5 μM MMC for 20 h (b), are shown. Two FANCD2 bands were detected by immunoblotting, including the slower migrating monoubiquitinated (-L) form and the faster migrating unubiquitinated (-S) form. Signal for monoubiquitinated FANCD2 in detergent-soluble and chromatin fraction pairs are enclosed within boxes. The percentage of FANCD2-L present in the soluble fraction for each pair is shown above the box as a measure of the efficiency of its association with chromatin. A lower number represents a tighter association with chromatin. γ-tubulin and H2A are markers for the soluble and chromatin fractions, respectively. These markers demonstrate efficient fractionation of both uncorrected and corrected FA-J cells, either with or without DNA damage. An equivalent number of cells were fractionated for each sample
As another measure of the influence of FANCJ on the association of monoubiquitinated FANCD2 with chromatin, we also examined cells following the induction of DNA damage by MMC (Fig. 5b). Higher levels of monoubiquitinated FANCD2 were observed, either with or without correction for FANCJ (there was more FANCD2-L in lanes 2 and 4 versus FANCD2-S in lanes 1 and 3 when comparing MMC-treated and untreated cells). Under such conditions, FANCJ again promoted the association of monoubiquitinated FANCD2 with chromatin (compare lane 1 with lane 2 and lane 3 to lane 4). A role for FANCJ in promoting the association of monoubiquitinated FANCD2 with chromatin was consistently observed in three experiments. Following treatment with MMC, 29.8±6.1% of monoubiquitinated FANCD2 associated with chromatin in uncorrected cells versus 13.0±3.1% in cells corrected by expression of FANCJ. This difference is statistically significant (p<0.02).
As an additional measure of whether FANCJ regulates the assembly of FANCD2 foci, we also depleted FANCJ using a siRNA in MCF7 cells. Substantial depletion of FANCJ is demonstrated in Fig. 6a. Suppression of FANCJ did not substantially alter FANCD2 monoubiquitination, however. Quantification demonstrates that suppression of FANCJ resulted in partial inhibition of the assembly of FANCD2 foci, relative to controls transfected with a siRNA against GFP, both in untreated populations or following exposure to 0.5 μM MMC for 20 h (Fig. 6b). Depletion of FANCJ also inhibited FANCD2 foci in MCF7 cells exposed to IR or HU (Supplementary Material, Fig. S5). FANCJ may therefore be involved in regulating the assembly of FANCJ foci in response to a variety of DNA damage or replication stresses.
Fig. 6.

FANCJ is involved in the recruitment of FANCD2 to DNA double-strand breaks. a Immunoblots to detect FANCD2, FANCJ, or BRCA1 in extracts from untreated MCF7 cells transfected with a control siRNA directed against GFP, or with siRNAs against either FANCJ or BRCA1. Both unubiquitinated (-S) and monoubiquitinated (-L) FANCD2 were detected with E35 antibody. Actin is shown as a loading control. b Quantification of the assembly of FANCD2 nuclear foci in MCF7 cells transfected with siGFP (control), or with siRNAs targeting FANCJ or BRCA1. Cells were left untreated or were incubated with 0.5 μM MMC for 20 h. c An example of the colocalization of FANCD2 foci (red) with DNA double-strand breaks, detected with an antibody against γ-H2AX (green), in MCF7 cells at 7 h following exposure to 5 Gy IR. Colocalization is evident in the merged image. Nuclei were identified by staining with DAPI. d Quantification of the percentage of MCF7 cells transfected with the indicated siRNAs that displayed γ-H2AX foci at 7 h after exposure to 5 Gy IR. e Quantification of the percentage of MCF7 cells, transfected with indicated siRNAs which displayed colocalization of five or more FANCD2 and γ-H2AX foci after exposure to IR. Average values were calculated from three counts of 150 or more cells each and are displayed±standard deviation (b, e). The levels of FANCD2 foci and its colocalization with γ-H2AX foci were statistically different (p<0.01) in cells depleted of FANCJ or BRCA1, as compared with cells transfected with the control siRNA (b, e)
Consistent with our finding that a FANCJ mutant that is defective for binding to BRCA1 does not support efficient assembly of FANCD2 foci (Fig. 4d), we find that depletion of BRCA1 suppresses the assembly of FANCD2 foci to a similar degree as depletion of FANCJ (Fig. 6b). Importantly, as observed previously (Cantor et al. 2001; Gupta et al. 2007), BRCA1 also had a role in the assembly of FANCJ foci (data not shown). These results, along with defects in FANCD2 foci associated with the FANCJ-S990A mutant (Fig. 4d), support the possibility that BRCA1 and FANCJ cooperate in regulating the assembly of FANCD2 foci.
As an assay of the requirement for FANCJ in targeting FANCD2 to sites of DNA damage, we examined MCF7 cells, with or without depletion of FANCJ, for the colocalization of FANCD2 foci with γ-H2AX foci at 7 h following exposure to 5 Gy IR. Foci formed by γ-H2AX are indicative of DNA double-strand breaks in DNA, such as those induced by IR (Rothkamm and Lobrich 2003). FANCD2 and γ-H2AX foci displayed colocalization after treatment with IR in untransfected MCF7 cells (Fig. 6c). The majority of untreated cells did not display FANCD2 foci or colocalization of FANCD2 foci with γ-H2AX foci (data not shown).
Depletion of FANCJ or BRCA1 resulted in a decrease in the colocalization of FANCD2 foci with γ-H2AX foci following exposure to IR, but had no effect on the assembly of γ-H2AX foci (Fig. 6d, e). Taken together, our results suggest that FANCJ, as well as FANCJ helicase activity and the capacity of FANCJ to bind to BRCA1, have a role in regulating the recruitment of FANCD2 to sites of DNA damage.
Discussion
While it is well established that FANCJ has structure-specific helicase activity in vitro (Gupta et al. 2005), to better understand its functions in DNA damage responses, we have focused here on the regulation of the cellular localization of FANCJ. We have found that FANCJ foci are induced by a variety of stresses, including MMC and IR, and that FANCJ is recruited to replication forks blocked by HU. Thus, FANCJ recruitment in response to DNA damage may involve replication stress. It is already known that lesions such as ICLs and DSBs activate ATR, which is indicative of a replication-stress response (Andreassen et al. 2004; Lin et al. 2004).
Additionally, we have identified molecular determinants of FANCJ localization, including a role for FANCJ helicase activity and for the capacity of FANCJ to bind BRCA1. Each of these mutants alters cellular sensitivity to DNA damage (Xie et al. 2010), suggesting the potential importance of the localization of this protein. It is interesting, in this context, that FANCJ and BRCA1, to which FANCJ binds, are both associated with inherited breast cancer (Seal et al. 2006). Further, several mutants identified in breast cancer patients affect FANCJ helicase activity in vitro (Cantor et al. 2004; Gupta et al. 2005). Thus, the localization of FANCJ may be important for its function as a tumor suppressor.
We find a novel function for FANCJ in promoting the recruitment of another Fanconi anemia protein, FANCD2, to sites of DNA damage. Thus, rather than FANCJ being dependent on monoubiquitinated FANCD2, these proteins may cooperate in DNA damage responses. Interestingly, we also find that FANCJ helicase activity and its ability to bind to BRCA1 are both involved in the recruitment of FANCD2 to nuclear foci. This is important because this suggests that FANCJ may link the behavior of monoubiquitinated FANCD2 to BRCA1, which has a role in DNA damage signaling and in coordinating DNA damage responses (Foray et al. 2003; Kitagawa et al. 2004; Lin et al. 2004).
Regulation of FANCJ recruitment in response to DNA damage
While it was already known that FANCJ nuclear foci are induced by IR, we provide quantitative evidence that they are induced by multiple stresses, including MMC and HU. This result is consistent with previous studies suggesting diverse functions for FANCJ, including a role in mediating resistance to both IR and MMC (Litman et al. 2005; Peng et al. 2006), and in unwinding G quadruplex DNA that can impede DNA replication and cause genomic instability (Wu et al. 2008).
Several lines of evidence suggest that FANCJ foci are assembled at sites of damage. For example, it has been demonstrated previously that FANCJ foci colocalize with γ-H2AX foci at DSBs in response to IR (Peng et al. 2006). Also, FANCJ colocalizes with BRCA1, a key protein recruited to sites of DNA damage, in response to DNA damage (Cantor et al. 2001).
Here, we have elucidated key molecular determinants of the recruitment of FANCJ to DNA damage foci. While it was known that BRCA1 is required for the assembly of FANCJ foci (Cantor et al. 2001; Gupta et al. 2007), it was unknown whether this is through a direct or indirect mechanism. By observing a defect in FANCJ foci assembly by the S990A mutant, which is deficient for binding to BRCA1, our results suggest that BRCA1 may directly regulate the recruitment of FANCJ through a physical interaction. Because phosphorylation of FANCJ at S990 appears to be mediated by a cyclin-dependent kinase (Yu et al. 2003), a dependence upon the interaction with BRCA1 may dictate that FANCJ foci assemble spontaneously during S phase, as we observe. Further, FANCJ is required for DNA repair by HR (Litman et al. 2005). Since HR is restricted to S phase and G2 (Rothkamm et al. 2003), regulation by cell cycle-linked phosphorylation may serve to restrict FANCJ recruitment and function in HR to these phases. Additionally, the assembly of FANCJ foci is largely abrograted in the K52R helicase-dead mutant. Perhaps binding to BRCA1 might determine the site of FANCJ localization, while FANCJ helicase activity might promote enhanced recruitment of FANCJ by unwinding DNA proximal to the lesion.
We find that FANCJ does not colocalize with the replication fork spontaneously in untreated cells and therefore may not play a role in normal DNA replication. But, FANCJ is strongly recruited to sites of blocked replication. We suggest that this may permit recruitment of FANCJ to any lesion that impedes or slows replication of DNA, presumably for a function in repairing the lesion. Support for such a role for FANCJ comes from a recent report that deficiency for FANCJ in human cells results in genomic deletions of potentially difficult to replicate G/C tracts (London et al. 2008).
Consistent with a role for FANCJ in responding to replication stress, ATR influences the assembly of FANCJ foci in response to DNA damage induced by MMC. Since depletion of ATR only partially inhibits DNA damage-induced assembly of FANCJ foci, it appears that other unknown factors may also be involved. It should be noted that no potential sites of ATR-dependent phosphorylation on FANCJ have been reported. Interestingly, it has recently been reported that FANCJ may also have a role in signaling ATR-dependent checkpoints (Gong et al. 2010). Further work will be required to determine how FANCJ is both regulated by ATR and can also mediate a critical function of ATR.
FANCJ is involved in the recruitment of FANCD2 to sites of DNA damage
Our results suggest a novel function for FANCJ in recruiting FANCD2 to damaged chromatin in human cells. This is based upon our observation that FANCJ is involved in the normal assembly of FANCD2 foci, both in untreated cells and following exposure to exogenous DNA damage. Deficiency for FANCJ in two different FA-J cell lines, or following siRNA-mediated depletion of FANCJ in MCF7 cells, compromises the assembly of FANCD2 foci (Figs. 4c and 6b; Supplementary Material, Fig. S5). Also, by measuring colocalization of FANCD2 foci with γ-H2AX foci, a marker of DSBs (Rogakou et al. 1999; Rothkamm and Lobrich 2003), in MCF7 cells depleted of FANCJ, we demonstrate that FANCJ is involved in the recruitment of FANCD2 to DSBs following exposure to IR (Fig. 6e). Further, FANCD2 foci assemble in chromatin (Montes de Oca et al. 2005), and we have discovered that FANCJ promotes the association of monoubiquitinated FANCD2 with chromatin, both in untreated cells and following exposure to MMC (Fig. 5). Interestingly, another DNA damage response factor, XPF-ERCC1, also promotes the association of monoubiquitinated FANCD2 with chromatin (Bhagwat et al. 2009).
The FA nuclear core complex and FANCI are required for FANCD2 monoubiquitination (Garcia-Higuera et al. 2001; Taniguchi et al. 2002a; Sims et al. 2007; Smogorzewska et al. 2007). Thus, a deficiency in any of the eight identified FA nuclear core complex proteins or FANCI completely abrogates the assembly of FANCD2 foci (Garcia-Higuera et al. 2001; Taniguchi et al. 2002a; Sims et al. 2007). In contrast, we find that FA-J cells, which lack FANCJ, display a compromised, but partial assembly, of FANCD2 foci. This phenotype appears to distinguish the FA-J complementation group from other FA complementation groups. Unlike FANCJ-deficient cells, FANCD2 foci are not compromised in FA-D1 cells, which are deficient for BRCA2 (Wang et al. 2004).
Since FANCJ is not required for FANCD2 monoubiquitination, it has been suggested that FANCJ functions downstream of FANCD2 monoubiquitination. Our results demonstrate that the assembly of FANCJ foci is not dependent upon FANCD2 (Suppl. Fig. S4). Instead, FANCJ may act as a co-factor that promotes recruitment of monoubiquitinated FANCD2 to chromatin. In this manner, FANCJ may not be absolutely required for the assembly of FANCD2 foci, and this may result in the partial assembly of FANCD2 foci that appears to be characteristic of FA-J cells.
FANCJ helicase activity is required for the efficient assembly of monoubiquitinated FANCD2 into DNA damage foci. In this light, it should be noted that we (data not shown), and others (Litman et al. 2005), have not found a physical complex between FANCJ and FANCD2. We have examined soluble extracts and chromatin-derived extracts, both from untreated cells and following treatment with MMC. FANCJ helicase activity could either unwind DNA to unmask a binding site for monoubiquitinated FANCD2 or could generate a FANCD2 binding site by promoting processing of the damaged DNA. Thus, an interaction between FANCJ and FANCD2 may not be required for the recruitment of monoubiquitinated FANCD2 to damaged chromatin.
The assembly of FANCJ foci is largely abrogated by the S990A and K52R mutants of FANCJ, which are deficient for binding to BRCA1 and for FANCJ helicase activity, respectively (Fig. 2). Importantly, neither of these mutants support the normal assembly of FANCD2 foci either. Thus, the correct localization of FANCJ may be important for its function in regulating FANCD2 recruitment. We propose that FANCJ has a general role in recruiting FANCD2 to sites of DNA damage, but this function is not required for resistance to MMC. The S990A mutant of FANCJ displays normal sensitivity to MMC, unlike deficiency for the entire FANCJ protein or other FA proteins (Litman et al. 2005; Peng et al. 2007).
FANCJ may functionally link BRCA1 to FANCD2
We find here that BRCA1 and FANCJ may function in a pathway involved in regulating the assembly of FANCD2 nuclear foci. This is based upon the results of two different experiments. First, the assembly of FANCD2 foci is compromised in a FA-J cell line expressing the S990A mutant of FANCJ (Fig. 4d). Second, siRNA-mediated suppression of FANCJ or BRCA1 yields an equivalent decrease in the assembly of FANCD2 foci in MCF7 cells (Fig. 6b). The latter result suggests that FANCJ and BRCA1 have equivalent roles in regulating the assembly of FANCD2 foci. Earlier reports, based upon siRNA-mediated suppression of BRCA1 in HeLa cells and the analysis of BRCA1-deficient cancer cell lines (Vandenberg et al. 2003; Burkitt and Ljungman 2007), support the conclusion that BRCA1 is involved in the assembly of FANCD2 foci. Since BRCA1 mediates the assembly of FANCJ foci, perhaps BRCA1 functions upstream of FANCJ in regulating the assembly of FANCD2 foci in human cells. Together, our results suggest the possibility that FANCJ provides a link between BRCA1 and FANCD2. These proteins might cooperate within this network to promote maintenance of genomic stability since each of these proteins is required individually.
Supplementary Material
Acknowledgements
We thank Dr. Hans Joenje (Vrije Universiteit Medical Center), Dr. Sharon Cantor (University of Massachusetts Medical School), Dr. James Lessard (Cincinnati Children’s Research Foundation), and Dr. Yoshihiro Nakatani (Dana-Farber Cancer Institute) for EUFA30 cells, anti-FANCJ antibodies, anti-actin antibodies, and the pOZ retroviral vector, respectively. This work was supported by NIH R01 HL085587 (to P.R.A.) and by an award from an Institutional Research Grant from the American Cancer Society to the University of Cincinnati.
Footnotes
Electronic supplementary material The online version of this article (doi:10.1007/s00412-010-0285-6) contains supplementary material, which is available to authorized users.
Contributor Information
Fan Zhang, Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Research Foundation, 3333 Burnet Ave. ML S7.203, Cincinnati, OH 45229, USA.
Qiang Fan, Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Research Foundation, 3333 Burnet Ave. ML S7.203, Cincinnati, OH 45229, USA.
Keqin Ren, Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Research Foundation, 3333 Burnet Ave. ML S7.203, Cincinnati, OH 45229, USA.
Arleen D. Auerbach, Laboratory of Human Genetics and Hematology, The Rockefeller University, New York, NY 10021, USA
Paul R. Andreassen, Division of Experimental Hematology and Cancer Biology, Cincinnati Children’s Research Foundation, 3333 Burnet Ave. ML S7.203, Cincinnati, OH 45229, USA; Department of Pediatrics, University of Cincinnati College of Medicine, Cincinnati, OH 45229, USA
References
- Andreassen PR, D’Andrea AD, Taniguchi T. ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev. 2004;18:1958–1963. doi: 10.1101/gad.1196104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreassen PR, Ho GP, D’Andrea AD. DNA damage responses and their many interactions with the replication fork. Carcinogenesis. 2006;27:883–892. doi: 10.1093/carcin/bgi319. [DOI] [PubMed] [Google Scholar]
- Bhagwat N, Olsen AL, Wang AT, Hanada K, Stuckert P, Kanaar R, D’Andrea A, Niedernhofer LJ, McHugh PJ. XPF-ERCC1 participates in the Fanconi anemia pathway of cross-link repair. Mol Cell Biol. 2009;29:6427–6437. doi: 10.1128/MCB.00086-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogliolo M, Lyakhovich A, Callen E, Castella M, Cappelli E, Ramirez MJ, Creus A, Marcos R, Kalb R, Neveling K, Schindler D, Surralles J. Histone H2AX and Fanconi anemia FANCD2 function in the same pathway to maintain chromosome stability. EMBO J. 2007;26:1340–1351. doi: 10.1038/sj.emboj.7601574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo R, Macdonald-Bravo H. Changes in the nuclear distribution of cyclin (PCNA) but not its synthesis depend on DNA replication. EMBO J. 1985;4:655–661. doi: 10.1002/j.1460-2075.1985.tb03679.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K. The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet. 2005;37:953–957. doi: 10.1038/ng1627. [DOI] [PubMed] [Google Scholar]
- Burkitt K, Ljungman M. Compromised Fanconi anemia response due to BRCA1 deficiency in cisplatin-sensitive head and neck cancer cell lines. Cancer Lett. 2007;253:131–137. doi: 10.1016/j.canlet.2007.01.017. [DOI] [PubMed] [Google Scholar]
- Cantor SB, Andreassen PR. Assessing the link between BACH1 and BRCA1 in the FA pathway. Cell Cycle. 2006;5:164–167. doi: 10.4161/cc.5.2.2338. [DOI] [PubMed] [Google Scholar]
- Cantor SB, Bell DW, Ganesan S, Kass EM, Drapkin R, Grossman S, Wahrer DC, Sgroi DC, Lane WS, Haber DA, Livingston DM. BACH1, a novel helicase-like protein, interacts directly with BRCA1 and contributes to its DNA repair function. Cell. 2001;105:149–160. doi: 10.1016/s0092-8674(01)00304-x. [DOI] [PubMed] [Google Scholar]
- Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM. The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA. 2004;101:2357–2362. doi: 10.1073/pnas.0308717101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616–627. doi: 10.1038/nrm2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q, Zhang F, Barrett B, Ren K, Andreassen PR. A role for monoubiquitinated FANCD2 at telomeres in ALT cells. Nucleic Acids Res. 2009;37:1740–1754. doi: 10.1093/nar/gkn995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foray N, Marot D, Gabriel A, Randrianarison V, Carr AM, Perricaudet M, Ashworth A, Jeggo P. A subset of ATM- and ATR-dependent phosphorylation events requires the BRCA1 protein. EMBO J. 2003;22:2860–2871. doi: 10.1093/emboj/cdg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gong Z, Kim JE, Leung CC, Glover JN, Chen J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010;37:438–446. doi: 10.1016/j.molcel.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Sharma S, Sommers JA, Jin Z, Cantor SB, Brosh RM., Jr Analysis of the DNA substrate specificity of the human BACH1 helicase associated with breast cancer. J Biol Chem. 2005;280:25450–25460. doi: 10.1074/jbc.M501995200. [DOI] [PubMed] [Google Scholar]
- Gupta R, Sharma S, Doherty KM, Sommers JA, Cantor SB, Brosh RM., Jr Inhibition of BACH1 (FANCJ) helicase by backbone discontinuity is overcome by increased motor ATPase or length of loading strand. Nucleic Acids Res. 2006;34:6673–6683. doi: 10.1093/nar/gkl964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta R, Sharma S, Sommers JA, Kenny MK, Cantor SB, Brosh RM., Jr FANCJ (BACH1) helicase forms DNA damage inducible foci with replication protein A and interacts physically and functionally with the single-stranded DNA-binding protein. Blood. 2007;110:2390–2398. doi: 10.1182/blood-2006-11-057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Kitagawa R, Bakkenist CJ, McKinnon PJ, Kastan MB. Phosphorylation of SMC1 is a critical downstream event in the ATM-NBS1-BRCA1 pathway. Genes Dev. 2004;18:1423–1438. doi: 10.1101/gad.1200304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitus M, Rooimans MA, Steltenpool J, Cool NF, Oostra AB, Mathew CG, Hoatlin ME, Waisfisz Q, Arwert F, de Winter JP, Joenje H. Heterogeneity in Fanconi anemia: evidence for 2 new genetic subtypes. Blood. 2004;103:2498–2503. doi: 10.1182/blood-2003-08-2915. [DOI] [PubMed] [Google Scholar]
- Levitus M, Waisfisz Q, Godthelp BC, Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, de Winter JP, Joenje H. The DNA helicase BRIP1 is defective in Fanconi anemia complementation group J. Nat Genet. 2005;37:934–935. doi: 10.1038/ng1625. [DOI] [PubMed] [Google Scholar]
- Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD. The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet. 2005;37:931–933. doi: 10.1038/ng1624. [DOI] [PubMed] [Google Scholar]
- Lin SY, Li K, Stewart GS, Elledge SJ. Human Claspin works with BRCA1 to both positively and negatively regulate cell proliferation. Proc Natl Acad Sci USA. 2004;101:6484–6489. doi: 10.1073/pnas.0401847101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB. BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell. 2005;8:255–265. doi: 10.1016/j.ccr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- London TB, Barber LJ, Mosedale G, Kelly GP, Balasubramanian S, Hickson ID, Boulton SJ, Hiom K. FANCJ is a structure-specific DNA helicase associated with the maintenance of genomic G/C tracts. J Biol Chem. 2008;283:36132–36139. doi: 10.1074/jbc.M808152200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mankad A, Taniguchi T, Cox B, Akkari Y, Rathbun RK, Lucas L, Bagby G, Olson S, D’Andrea AD, Grompe M. Natural gene therapy in monozygotic twins with Fanconi anemia. Blood. 2006;107:3084–3090. doi: 10.1182/blood-2005-07-2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25:5875–5884. doi: 10.1038/sj.onc.1209878. [DOI] [PubMed] [Google Scholar]
- Montes de Oca R, Andreassen PR, Margossian SP, Gregory RC, Taniguchi T, Wang X, Houghtaling S, Grompe M, D’Andrea AD. Regulated interaction of the Fanconi anemia protein, FANCD2, with chromatin. Blood. 2005;105:1003–1009. doi: 10.1182/blood-2003-11-3997. [DOI] [PubMed] [Google Scholar]
- Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001;61:4842–4850. [PubMed] [Google Scholar]
- Nakatani Y, Ogryzko V. Immunoaffinity purification of mammalian protein complexes. Methods Enzymol. 2003;370:430–444. doi: 10.1016/S0076-6879(03)70037-8. [DOI] [PubMed] [Google Scholar]
- Peng M, Litman R, Jin Z, Fong G, Cantor SB. BACH1 is a DNA repair protein supporting BRCA1 damage response. Oncogene. 2006;25:2245–2253. doi: 10.1038/sj.onc.1209257. [DOI] [PubMed] [Google Scholar]
- Peng M, Litman R, Xie J, Sharma S, Brosh RM, Jr, Cantor SB. The FANCJ/MutLalpha interaction is required for correction of the cross-link response in FA-J cells. EMBO J. 2007;26:3238–3249. doi: 10.1038/sj.emboj.7601754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Arrifin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- Risinger MA, Groden J. Crosslinks and crosstalk: human cancer syndromes and DNA repair defects. Cancer Cell. 2004;6:539–545. doi: 10.1016/j.ccr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K, Lobrich M. Evidence for a lack of DNA double-strand break repair in human cells exposed to very low x-ray doses. Proc Natl Acad Sci USA. 2003;100:5057–5062. doi: 10.1073/pnas.0830918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothkamm K, Kruger I, Thompson LH, Lobrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol. 2003;23:5706–5715. doi: 10.1128/MCB.23.16.5706-5715.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scully R, Ganesan S, Vlasakova K, Chen J, Socolovsky M, Livingston DM. Genetic analysis of BRCA1 function in a defined tumor cell line. Mol Cell. 1999;4:1093–1099. doi: 10.1016/s1097-2765(00)80238-5. [DOI] [PubMed] [Google Scholar]
- Seal S, Thompson D, Renwick A, Elliott A, Kelly P, Barfoot R, Chagtai T, Jayatilake H, Ahmed M, Spanova K, North B, McGuffy L, Evans DG, Eccles D, Breast Cancer Collaboration (UK) Easton DF, Stratton MR, Rahman N. Truncating mutations in the Fanconi anemia J gene BRIP1 are low-penetrance breast cancer susceptibility alleles. Nat Genet. 2006;38:1239–1241. doi: 10.1038/ng1902. [DOI] [PubMed] [Google Scholar]
- Sims AE, Spiteri E, Sims RJ, 3rd, Arita AG, Lach FP, Landers T, Wurm M, Freund M, Neveling K, Hanenberg H, Auerbach AD, Huang TT. FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol. 2007;14:564–567. doi: 10.1038/nsmb1252. [DOI] [PubMed] [Google Scholar]
- Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, 3rd, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, Elledge SJ. Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell. 2007;129:289–301. doi: 10.1016/j.cell.2007.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi T, D’Andrea AD. Molecular pathogenesis of Fanconi anemia: recent progress. Blood. 2006;107:4223–4233. doi: 10.1182/blood-2005-10-4240. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD. S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood. 2002a;100:2414–2420. doi: 10.1182/blood-2002-01-0278. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, D’Andrea AD. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell. 2002b;109:459–472. doi: 10.1016/s0092-8674(02)00747-x. [DOI] [PubMed] [Google Scholar]
- Vandenberg CJ, Gergely F, Ong CY, Pace P, Mallery DL, Hiom K, Patel KJ. BRCA1-independent ubiquitination of FANCD2. Mol Cell. 2003;12:247–254. doi: 10.1016/s1097-2765(03)00281-8. [DOI] [PubMed] [Google Scholar]
- Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, Erven V, Neveling K, Endt D, Kesterton I, Autore F, Fraternali F, Freund M, Hartmann L, Grimwade D, Roberts RG, Schaal H, Mohammed S, Rahman N, Schindler D, Mathew CG. Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet. 2010;42:406–409. doi: 10.1038/ng.570. [DOI] [PubMed] [Google Scholar]
- Wang W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat Rev Genet. 2007;8:735–748. doi: 10.1038/nrg2159. [DOI] [PubMed] [Google Scholar]
- Wang X, Andreassen PR, D’Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24:5850–5862. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Shin-ya K, Brosh RM., Jr FANCJ helicase defective in Fanconia anemia and breast cancer unwinds G-quadruplex DNA to defend genomic stability. Mol Cell Biol. 2008;28:4116–4128. doi: 10.1128/MCB.02210-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Litman R, Wang S, Peng M, Guillemette S, Rooney T, Cantor SB. Targeting the FANCJ-BRCA1 interaction promotes a switch from recombination to poleta-dependent bypass. Oncogene. 2010;29:2499–2508. doi: 10.1038/onc.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youds JL, Barber LJ, Ward JD, Collis SJ, O’Neil NJ, Boulton SJ, Rose AM. DOG-1 is the Caenorhabditis elegans BRIP1/FANCJ homologue and functions in interstrand cross-link repair. Mol Cell Biol. 2008;28:1470–1479. doi: 10.1128/MCB.01641-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302:639–642. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- Zhang F, Fan Q, Ren K, Andreassen PR. PALB2 functionally connects the breast cancer susceptibility proteins BRCA1 and BRCA2. Mol Cancer Res. 2009;7:1110–1118. doi: 10.1158/1541-7786.MCR-09-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.