

Abstract
Identification of the usefulness of lipid-based formulations (LBFs) for delivery of poorly water-soluble drugs is at date mainly experimentally based. In this work we used a diverse drug data set, and more than 2,000 solubility measurements to develop experimental and computational tools to predict the loading capacity of LBFs. Computational models were developed to enable in silico prediction of solubility, and hence drug loading capacity, in the LBFs. Drug solubility in mixed mono-, di-, triglycerides (Maisine 35-1 and Capmul MCM EP) correlated (R2 0.89) as well as the drug solubility in Carbitol and other ethoxylated excipients (PEG400, R2 0.85; Polysorbate 80, R2 0.90; Cremophor EL, R2 0.93). A melting point below 150 °C was observed to result in a reasonable solubility in the glycerides. The loading capacity in LBFs was accurately calculated from solubility data in single excipients (R2 0.91). In silico models, without the demand of experimentally determined solubility, also gave good predictions of the loading capacity in these complex formulations (R2 0.79). The framework established here gives a better understanding of drug solubility in single excipients and of LBF loading capacity. The large data set studied revealed that experimental screening efforts can be rationalized by solubility measurements in key excipients or from solid state information. For the first time it was shown that loading capacity in complex formulations can be accurately predicted using molecular information extracted from calculated descriptors and thermal properties of the crystalline drug.
Keywords: lipid-based formulations, solubility prediction, loading capacity, molecular properties, in silico prediction
Introduction
The demand for formulations that facilitate drug solubilization is growing due to the increasing number of poorly water-soluble drug molecules emerging from drug discovery programs. A sufficient dissolution and solubility must be attained in the gastrointestinal tract (GIT) for orally delivered drugs to be available for absorption. Poor GIT solubility substantially limits effective drug development and, in the worst case, leads to late termination of the candidate drug.1,2 Lipid-based formulations (LBFs) are a means to circumvent the low solubility issues associated with lipophilic drugs. In contrast to conventional dosage forms, the drug is typically predissolved in LBFs to overcome the hurdle of dissolution in the GIT.3,4 Extensive research efforts have focused on optimizing in vitro lipolysis experiments and altering formulation composition to keep the drug solubilized and available for absorption.5−7 Nevertheless, these studies have not particularly focused on the molecular mechanism governing drug loading into LBFs per se, which is an equally important aspect to achieve successful drug delivery.8
Less than 4% of all orally administered drugs on the market use LBF dosage forms.3 Part of the reason is likely because of the complex nature of lipid-based drug delivery systems, i.e., systems consisting of mixtures of oils, surfactants, and cosolvents in different proportions.9,10 Moreover, the LBF development process to date is mainly experimentally based, where the drug is screened for solubility in numerous excipients.8,11,12 This process is time and resource intensive, and demands that there are large amounts of the drug available for screening purposes. More importantly, this procedure is not based on an understanding of the molecular interactions between drug and formulation components, with the result that suboptimal formulations might be selected.
Traditionally, phase diagrams have been used to compose miscible formulations and the LBFs have subsequently been experimentally determined for drug loading capacity.13 To understand what defines drug solubility in cosolvent and lipid mixtures a few other approaches have been applied. One of the early models for prediction of solubilization in cosolvent/water mixtures was the log–linear model of Yalkowsky and co-workers.14,15 Recent studies on lipid systems have used similar approaches to estimate drug solubility in complex lipid mixtures.16−18 Based on four model compounds, it was shown that the sum of the loading capacity in the LBF is equal to the solubility of each included excipient, when compensated for the fraction of the excipient in the formulation.17 The weakness of this methodology is that it requires experimental measurements, which limits the applicability for rapid estimation of LBF loading capacity.
Solubility in binary mixtures of cosolvent and water has also been modeled mathematically in different ways. One such quantitative structure–property relationship (QSPR) model was developed, on a data set of 122 drugs, to predict solubility in polyethylene glycol (PEG400)/water mixtures. The QSPR model developed at each volume fraction (25%, 50%, 75% PEG/water) had an R2 < 0.9 and a root-mean square error (RMSE) of <0.5 log unit for the training set. These QSPR models were solely based on calculated descriptors, and the important descriptors reflected weight, volume, density, radius of gyration, number of rotatable bonds, hydrogen-bond donors, and hydrogen-bond acceptors.19 Another study on the same data set performed a stepwise linear regression analysis which included the melting point (Tm) as a descriptor. Although Tm contributed to the solubility estimation in pure PEG400 (R2 0.71, RMSE 0.55), there was a minimal decrease in model performance when the solid state descriptor was excluded (R2 0.69, RMSE 0.58).20 We previously applied a similar methodology to predict solubility in PEG400 which resulted in a model with comparable accuracy (R2 0.62, RMSE 0.44). The inclusion of Tm as a descriptor in that model did not improve the performance.21
Although methodologies exist to calculate solubility in cosolvents and lipid mixtures, there is not yet any model to accurately predict solubility—and hence loading capacity—in complex mixtures of lipid excipients without the demand for experimental work. The aim of the current study was to identify molecular characteristics of poorly soluble compounds that would define solubility in lipids, surfactants, and cosolvents commonly used in LBFs. Furthermore, we aimed to develop tools to enable rational experimental screening and computational prediction of loading capacity in the LBFs.
Experimental Section
Material
All drug compounds were purchased from Sigma-Aldrich (St. Louis, USA) except acitretin (Ontario Chemicals Inc., Canada), candesartan, and candesartan cilexetil (Angene Ltd., China), danazol (Coral Drugs IVT, India), fenofibric acid (Labratoreo Chimico Internazionale, Italy), halofantrine, (SmithKline Beecham Pharmaceuticals, India), and itraconazole (Lee Pharma Ltd., India). Felodipine was a gift from AstraZeneca (Mölndal, Sweden). The excipients soybean oil (SBO), Cremophor EL, Cremophor ELP, polysorbate 80 (PS80), Carbitol, and PEG400 were purchased from Sigma-Aldrich (St. Louis, USA). Captex 355 (Captex) and Capmul MCM EP (Capmul) (Abitec, Janesville, WI, USA) were generous gifts from Barentz (Denmark, Copenhagen), and Maisine 35-1 (Maisine) was kindly donated by Gattefossé (Lyon, France). The average molecular weights used in calculation of mol per mol solubility values were as follows: SBO, 873.3 g/mol; Maisine, 489.7 g/mol; Captex, 504.9 g/mol; Capmul, 248.4 g/mol; Cremophor EL and Cremophor ELP, 2424.4 g/mol; PS80, 1310.0 g/mol; PEG400 400.0 g/mol; and Carbitol, 134.2 g/mol.
Data Set Selection and Characteristics
In this study 35 orally delivered compounds were investigated for their solubility in excipients that are commonly used in LBFs. The data set was selected to cover compounds that are potential candidates of LBFs (reflected with a logP ≥ 2), while still being structurally diverse (Table 1), and expected to give adequate range in solubility in the excipients. Solubility of all 35 compounds was determined in four LBF excipients: Maisine (long-chain mono-, di-, triglyceride), Capmul (medium-chain mono-, di-, triglyceride), Cremophor EL (surfactant), and Carbitol (cosolvent). A smaller number of compounds (n = 25) were determined in Cremophor ELP (surfactant) to investigate differences in solvation capacity between the Cremophor EL and Cremophor ELP. The latter is a refined version of Cremophor EL containing less water (Cremophor EL ≤3% w/w, Cremophor ELP ≤0.5% w/w). All excipients were stored under argon gas to avoid oxidation and water uptake. Seven of the compounds were studied in SBO (long-chain triglyceride), Captex (medium-chain triglyceride), PS80 (surfactant), and PEG400 (cosolvent). For the remaining 28 compounds, we used the solubility data in these excipients reported in our previous publication.21 Nine selected model compounds were determined for loading capacity in four formulations representative of the lipid formulation classification system (LFCS) types II, IIIA, IIIB, and IV (Figure 1).
Table 1. Physicochemical Properties of Selected Model Compoundsa.
| compound | Mw (Da) | logP | Tm (°C) | ΔSf (J/mol·K) | TPSA | A/B/N/Am | pKab |
|---|---|---|---|---|---|---|---|
| acitretin | 326.5 | 5.6 | 221 | 115 | 47 | A | 4.2 |
| albendazole | 265.4 | 3.1 | 203 | 95 | 92 | Am | 3.7, 9.926 |
| bezafibrate | 361.9 | 3.8 | 185 | 110 | 76 | A | 3.643 |
| candesartan | 440.5 | 4.6 | 178 | nd | 119 | Am | 2.1, 3.3, 4.544 |
| candesartan cilexetil | 610.7 | 7.4 | 167 | 116 | 143 | Am | 3.0, 2.2 |
| carbamazepine | 236.3 | 2.7 | 173c | nd | 48 | N | na |
| cinnarizine | 368.6 | 5.5 | 119 | 103 | 6 | B | 7.526 |
| clofazimine | 473.4 | 6.9 | 222 | 74 | 42 | B | 9.045 |
| clotrimazole | 344.9 | 5.2 | 142 | 77 | 18 | B | 5.2 |
| danazol | 337.5 | 4.9 | 227 | 63 | 46 | N | na |
| diflunisal | 250.2 | 3.1 | 213 | 76 | 58 | A | 3.145 |
| dipyridamole | 504.7 | 2.5 | 166 | 72 | 145 | B | 6.226 |
| disulfiram | 296.6 | 4.6 | 67 | 84 | 121 | B | 2.5 |
| ethinylestradiol | 296.4 | 4.9 | 183 | 62 | 40 | A | 10.3 |
| felodipine | 384.3 | 3.6 | 143 | 73 | 65 | N | na |
| fenbendazole | 299.4 | 3.8 | 226 | 123 | 92 | B | 5.146 |
| fenofibrate | 360.9 | 5.1 | 79 | 85 | 53 | N | na |
| fenofibric acid | 318.8 | 4.1 | 184 | 99 | 64 | A | 3.5 |
| glibenclamide | 494.1 | 4.1 | 174 | 99 | 122 | A | 5.926 |
| griseofulvin | 352.8 | 2.2 | 217 | 87 | 71 | N | na |
| halofantrine | 500.5 | 8.2 | 77 | 89 | 23 | B | 9.2 |
| haloperidol | 375.9 | 3.9 | 151 | 133 | 41 | B | 8.626 |
| indomethacin | 357.8 | 4.2 | 160 | 80 | 69 | A | 3.926 |
| itraconazole | 705.7 | 6.5 | 166 | 128 | 105 | B | 3.945 |
| mefenamic acid | 241.3 | 4.0 | 230 | 74 | 49 | A | 4.445 |
| naproxen | 230.3 | 2.8 | 155 | 75 | 47 | A | 4.226 |
| niclosamide | 327.1 | 3.6 | 231 | 87 | 95 | A | 10.3, 8.1 |
| noscapine | 413.5 | 3.0 | 175 | 80 | 76 | B | 5.9 |
| perphenazine | 404.0 | 4.2 | 94 | 86 | 60 | B | 7.828 |
| praziquantel | 312.5 | 2.7 | 139 | 72 | 41 | N | na |
| progesterone | 314.5 | 3.6 | 128 | 63 | 34 | N | na |
| saquinavir | 670.9 | 3.9 | nd | nd | 167 | B | 7.447 |
| sulfasalazine | 398.4 | 2.0 | 255 | 99 | 146 | A | 10.9, 8.0, 2.448 |
| tolfenamic acid | 261.7 | 4.1 | 213 | 78 | 49 | A | 4.126 |
| toltrazuril | 425.4 | 6.1 | 192 | 89 | 111 | A | 8.2 |
Molecular weight (Mw), logP (displays AlogP), and TPSA (displays TPSA(Tot)) were calculated with DragonX 6.0.16 (Talete, Italy). Melting point (Tm) and entropy of fusion (ΔSf) were determined with differential scanning calorimetry (see Experimental Section). Abbreviations: acid (A); base (B); neutral in the pH range 2–12 (N); ampholyte (Am).
pKa data was collected from the literature or, if not available in the literature, predicted through ADMET Predictor v7.1 (Lancaster, CA).
Tm of carbamazepine corresponds to form III (see Results).
Figure 1.

Assembled type II–IV formulations used for loading capacity determinations of nine model compounds. The percent of excipient corresponds to % w/w.
Differential Scanning Calorimetry Experiments
The compound onset of melting (Tm) and heat of fusion (ΔHf) were measured with differential scanning calorimetry (DSC) (DSC Q2000, TA Instruments, Japan), coupled to an automatic cooling system. The instrument was calibrated for temperature and enthalpy using indium. Approximately 1–3 mg of each compound was weighed into a nonhermetic aluminum pan and heated at 10 °C/min to 20 °C above the literature Tm. The nitrogen gas flow rate was set to 50 mL/min. All measurements were performed in triplicate. Tm and ΔHf were used to calculate the entropy of fusion (ΔSf) from Gibbs free energy of fusion. From the extracted solid state data the ideal solubility (log Xic) was calculated for each compound.22
Drug Solubility in Single Excipients
A small-scale shake-flask method was used to measure the drug solubility in all excipients.21 Approximately 150% of the estimated soluble amount of the compounds was weighed into a glass vial to which 750 mg of excipient was added. Vials were sealed with a lid, vortexed, and placed on a plate shaker (incubator at 37 °C) for the time of the study. After 24, 48, and 72 h (or longer if required) the vials were centrifuged (Eppendorf centrifuge 5810R) at 37 °C, 2800g for 30 min. Immediately after centrifugation 20–30 mg of the supernatant was transferred to a 5 mL volumetric glass flask and diluted with methanol except for SBO, which is insoluble in methanol and therefore was diluted in isopropanol. The drug suspensions were thereafter dispersed thoroughly by vortexing the vials and returned to the incubator at 37 °C. The samples were kept until after the analysis to guarantee that solubility could be determined at later time points. Equilibrium solubility was the value at which the solubility differed less than 10% between two consecutive time points. Compounds were quantified in a 96-well UV-plate reader (Tecan, Safire2) at compound-specific wavelengths. For samples with too low sensitivity to be detected by UV, LC–MS/MS (Waters Xevo TQ, Milford, MA) was used. The LC–MS/MS analytical conditions can be found in Table SI1. For all analytical techniques, accuracy of the analysis was controlled by a quality control (prepared from a separate stock solution), and triplicate recovery samples with a known concentration of drug compound.
Formulation Composition, Self-Emulsification, and Solubility
LBFs were assembled from the excipients in this study and those investigated in a previous study.21 We aimed to design formulations representative of the different types in the LFCS.9,10 The formulation ingredients were preheated to 37 °C to facilitate handling and to obtain homogeneous mixing. The excipients were added to a test tube in predefined fractions (%, w/w) and vortexed thoroughly. Formulations were defined as stable when there was no sign of phase separation after incubation at 37 °C for a week followed by centrifugation (37 °C, 2800g, 30 min, Eppendorf centrifuge 5810R). Stable formulations were tested for self-emulsification by dispersing 1 g of formulation in 39 mL (37 °C) of ultrapure water Millipore (Billerica, MA, USA), i.e., conditions depicting a worst-case scenario. Resulting homogeneous clear or milky dispersions were considered to be self-emulsified.23,24 The final LBFs are presented in Figure 1. Cinnarizine, disulfiram, fenbendazole, fenofibrate, fenofibric acid, halofantrine, noscapine, progesterone, and tolfenamic acid were chosen as model compounds to be investigated for loading capacity in the LBFs. These compounds represent a wide range in physicochemical properties (Table 1) and solubility in the single excipients (Tables 2 and 3). Formulations were prepared the day before the solubility experiments, in order to equilibrate for at least 12 h; determinations were carried out as described for the single excipients.
Table 2. Experimentally Determined Thermodynamic Solubility in Glycerides at 37°Ca.
| compound | Maisine 35-1 (mg/g) | soybean oil (mg/g) | Capmul MCM EP (mg/g) | Captex 355 (mg/g) |
|---|---|---|---|---|
| acitretin | 0.62 ± 0.06 | 0.12 ± 0.01 | 1.54 ± 0.10 | 0.24 ± 0.01 |
| albendazole | 3.86 ± 0.21 | 0.17 ± 0.01 | 3.17 ± 0.10 | 0.39 ± 0.05 |
| bezafibrate | 1.55 ± 0.05 | ≤0.20 | 6.36 ± 0.26 | 0.22 ± 0.01 |
| candesartan | ≤0.41 | ≤0.03 | 3.19 ± 0.14 | 0.01 ± 0.00 |
| candesartan c | 1.99 ± 0.19 | 0.31 ± 0.01 | 9.07 ± 0.38 | 0.71 ± 0.02 |
| carbamazepine | 30.0 ± 1.66 | 1.42 ± 0.16 | 55.5 ± 3.23 | 2.56 ± 0.12 |
| cinnarizine | 29.0 ± 0.78 | 30.6 ± 0.88 | 33.5 ± 1.12 | 42.0 ± 1.71 |
| clofazimine | 11.7 ± 0.24 | 9.51 ± 0.21 | 16.3 ± 0.44 | 13.3 ± 0.38 |
| clotrimazole | 75.4 ± 8.95 | 15.1 ± 0.76 | 136.7 ± 5.94 | 18.8 ± 0.59 |
| danazol | 14.7 ± 0.79 | 3.89 ± 0.15 | 28.5 ± 1.11 | 7.41 ± 0.91 |
| diflunisal | 31.6 ± 0.99 | 7.86 ± 0.59 | 53.4 ± 1.29 | 16.5 ± 1.73 |
| dipyridamole | 3.02 ± 0.16 | 0.07 ± 0.01 | 12.3 ± 0.36 | 0.14 ± 0.02 |
| disulfiram | 49.0 ± 3.05 | 21.4 ± 1.07 | 47.2 ± 2.05 | 54.7 ± 2.39 |
| ethinylestradiol | 22.1 ± 1.83 | 13.4 ± 0.47 | 56.3 ± 1.76 | 44.0 ± 3.13 |
| felodipine | 37.7 ± 1.37 | 9.59 ± 0.67 | 80.2 ± 2.22 | 26.4 ± 1.75 |
| fenbendazole | 0.69 ± 0.11 | 0.09 ± 0.01 | 1.28 ± 0.09 | 0.19 ± 0.00 |
| fenofibrate | 78.6 ± 4.38 | 79.9 ± 3.92 | 102.3 ± 3.05 | 168.8 ± 15.7 |
| fenofibric acid | 7.38 ± 0.22 | 0.80 ± 0.05 | 14.5 ± 0.50 | 1.97 ± 0.05 |
| glibenclamide | 0.52 ± 0.14 | ≤0.05 | 5.49 ± 0.26 | 0.06 ± 0.01 |
| griseofulvin | 2.43 ± 0.05 | 0.50 ± 0.06 | 5.56 ± 0.21 | 1.00 ± 0.04 |
| halofantrine | 71.2 ± 4.58 | 52.8 ± 6.30 | 52.6 ± 3.01 | 99.6 ± 2.82 |
| haloperidol | 3.57 ± 0.16 | 1.22 ± 0.13 | 13.4 ± 0.51 | 2.42 ± 0.08 |
| indomethacin | 13.0 ± 0.88 | 2.02 ± 0.05 | 28.6 ± 1.30 | 4.80 ± 0.34 |
| itraconazole | 2.26 ± 0.16 | 0.09 ± 0.01 | 5.60 ± 0.67 | 0.18 ± 0.00 |
| mefenamic acid | 5.38 ± 0.40 | 1.87 ± 0.12 | 9.63 ± 0.40 | 2.72 ± 0.04 |
| naproxen | 19.5 ± 1.42 | 5.49 ± 0.71 | 40.3 ± 1.19 | 9.61 ± 0.14 |
| niclosamide | 3.97 ± 0.19 | 1.38 ± 0.11 | 10.1 ± 0.40 | 2.82 ± 0.11 |
| noscapine | 5.36 ± 0.61 | 1.91 ± 0.15 | 7.23 ± 0.20 | 3.37 ± 0.11 |
| perphenazine | 92.5 ± 5.80 | 16.7 ± 1.72 | 192.4 ± 5.63 | 27.6 ± 2.50 |
| praziquantel | 85.9 ± 5.32 | nd | 184.9 ± 12.4 | 13.0 ± 0.48 |
| progesterone | 66.2 ± 4.87 | 30.4 ± 1.28 | 99.1 ± 2.94 | 36.9 ± 1.15 |
| saquinavir | ≥123 | 2.34 ± 0.09 | ≥288 | 7.09 ± 0.14 |
| sulfasalazine | 0.16 ± 0.02 | 0.01 ± 0.00 | 3.16 ± 0.06 | 0.02 ± 0.00 |
| tolfenamic acid | 14.1 ± 1.18 | 3.81 ± 0.23 | 26.0 ± 1.45 | 7.24 ± 0.54 |
| toltrazuril | 7.90 ± 1.03 | 0.64 ± 0.04 | 11.8 ± 0.59 | 2.06 ± 0.15 |
| min | 0.16 | 0.01 | 1.28 | 0.01 |
| max | ≥123 | 79.9 | ≥288 | 168.8 |
Abbreviations: Candesartan cilexetil (candesartan c).
Table 3. Experimentally Determined Thermodynamic Solubility in Surfactants and Cosolvents at 37 °Ca.
| compound | Cremophor EL (mg/g) | PS80 (mg/g) | Carbitol (mg/g) | PEG400 (mg/g) |
|---|---|---|---|---|
| acitretin | 2.85 ± 0.14 | 3.09 ± 0.03 | 3.52 ± 0.20 | 1.07 ± 0.01 |
| albendazole | 2.75 ± 0.14 | 2.09 ± 0.12 | 5.99 ± 0.19 | ≥5.75 |
| bezafibrate | 37.9 ± 5.23 | 31.8 ± 1.20 | 43.0 ± 1.83 | 36.0 ± 1.71 |
| candesartan | 11.3 ± 0.67 | 5.04 ± 0.94 | 20.5 ± 1.26 | 12.9 ± 1.33 |
| candesartan c | 26.4 ± 1.46 | 26.3 ± 0.54 | 77.0 ± 2.04 | 25.9 ± 0.86 |
| carbamazepine | 34.5 ± 1.06 | 34.6 ± 1.45 | 82.9 ± 1.80 | 73.9 ± 4.71 |
| cinnarizine | 22.7 ± 1.83 | 29.3 ± 0.92 | 54.3 ± 2.76 | 19.5 ± 0.89 |
| clofazimine | 15.9 ± 0.83 | 15.6 ± 1.04 | 22.8 ± 0.54 | 13.2 ± 0.61 |
| clotrimazole | 44.1 ± 3.01 | 51.1 ± 3.18 | 135.0 ± 3.57 | 74.5 ± 5.92 |
| danazol | 29.8 ± 0.50 | 30.1 ± 4.37 | 83.2 ± 1.97 | 35.8 ± 2.04 |
| diflunisal | 157.9 ± 6.54 | 119.7 ± 12.4 | 286.3 ± 13.4 | 169.2 ± 27.3 |
| dipyridamole | 9.57 ± 0.29 | 8.31 ± 0.37 | 43.1 ± 2.40 | 16.9 ± 0.80 |
| disulfiram | 88.8 ± 3.66 | 55.7 ± 3.32 | 162.6 ± 8.46 | 69.6 ± 3.26 |
| ethinylestradiol | 163.9 ± 10.1 | ≥99.4 | 247.1 ± 8.93 | ≥163 |
| felodipine | 125.1 ± 6.23 | 45.2 ± 4.36 | 217.2 ± 16.0 | 43.9 ± 6.25 |
| fenbendazole | 2.07 ± 0.09 | 1.90 ± 0.05 | 4.13 ± 0.28 | 2.83 ± 0.07 |
| fenofibrate | 101.3 ± 5.76 | 102.3 ± 3.15 | 201.7 ± 11.1 | 65.1 ± 4.94 |
| fenofibric acid | 58.4 ± 2.59 | 57.1 ± 2.49 | 92.8 ± 4.81 | 54.8 ± 2.69 |
| glibenclamide | 11.2 ± 1.01 | 10.5 ± 0.22 | 21.1 ± 0.46 | 8.25 ± 0.32 |
| griseofulvin | 9.70 ± 0.36 | 8.13 ± 0.27 | 18.9 ± 0.99 | 13.5 ± 0.60 |
| halofantrine | 27.9 ± 0.79 | 25.0 ± 1.60 | 79.3 ± 7.90 | 5.73 ± 0.38 |
| haloperidol | 11.3 ± 0.82 | 7.06 ± 0.64 | 27.2 ± 1.82 | 12.3 ± 0.92 |
| indomethacin | 71.6 ± 4.57 | 116.1 ± 8.33 | 187.4 ± 7.08 | 134.6 ± 11.3 |
| itraconazole | 1.92 ± 0.29 | 1.42 ± 0.19 | 6.30 ± 0.55 | 2.47 ± 0.08 |
| mefenamic acid | 30.7 ± 0.89 | 26.2 ± 0.65 | 43.6 ± 1.52 | 25.1 ± 0.86 |
| naproxen | 119.9 ± 2.97 | 106.6 ± 4.31 | 183.0 ± 4.92 | 132 ± 2.87 |
| niclosamide | 44.1 ± 1.75 | 30.2 ± 1.77 | 49.6 ± 2.62 | 61.7 ± 1.72 |
| noscapine | 12.3 ± 0.25 | 14.3 ± 0.91 | 25.6 ± 1.00 | 19.4 ± 0.94 |
| perphenazine | 71.7 ± 3.04 | 76.6 ± 4.35 | 232.6 ± 13.4 | 98.4 ± 4.32 |
| praziquantel | 45.4 ± 3.51 | 19.2 ± 1.20 | 116.9 ± 14.8 | 28.0 ± 1.08 |
| progesterone | 41.1 ± 1.27 | 28.3 ± 2.19 | 67.02 ± 1.36 | 17.1 ± 0.71 |
| saquinavir | ≥45.9 | 48.9 ± 2.15 | ≥317 | ≥300 |
| sulfasalazine | 13.1 ± 1.02 | 11.4 ± 0.63 | 16.4 ± 0.72 | 11.8 ± 0.58 |
| tolfenamic acid | 70.6 ± 3.87 | 57.3 ± 7.05 | 85.5 ± 4.29 | 46.8 ± 7.00 |
| toltrazuril | 19.7 ± 3.11 | 11.1 ± 1.29 | 50.2 ± 2.47 | 18.3 ± 2.27 |
| min | 1.92 | 1.42 | 3.52 | 1.07 |
| max | 163.9 | 119.7 | ≥317 | ≥300 |
Abbreviations: Candesartan cilexetil (candesartan c).
Statistics and Model Development
The solubility determinations were performed in triplicate, and the solubility values are presented as means ± standard deviation. The coefficient of determination (R2) was used to ensure the goodness-of-fit for standard curves and simple correlations. Multivariate data analysis (Simca 13.0.2.0, Umetrics, Sweden) was applied to investigate the influence of molecular structure and physiochemical properties on solubility and whether the loading capacity of the LBFs could be predicted from molecular properties. First, Corina 3.49 (Molecular Networks, Erlangen, Germany) was used to convert SMILES strings into three-dimensional structures, which then were used as input for calculation of molecular descriptors with DragonX 6.0.16 (Talete, Italy). The descriptors were blinded to avoid selection bias, followed by removal of skewed descriptors, mean centering, and scaling to unity of variance. This led to a matrix consisting of 1660 variables. To reduce the descriptor matrix and remove strongly linearly correlated descriptors before the variable selection, a script was used (R, 3.2.0, Vienna, Austria) to exclude those correlating ≥|0.9|. The compound data set was sorted into training (Tr) and test (Te) sets. Strong outliers identified in the principal component analysis (PCA) and the distance-to-the-model-of-X (DModX) plot of the data set were excluded from the training set; instead, these were placed in the test set to avoid distortions in the model. Similarly, compounds for which an exact solubility value not could be determined were placed in the test set. An additional criterion for the test set was that it was well distributed over the chemical space of the training set in the PCA plot. The solubility in the logarithm form of mol compound/mol excipient was used as response. Partial least-squares projection to latent structures (PLS) was then used to identify trends, predict quantitative response values, and understand differences between the included excipients. The PLS models were developed with a standardized protocol from our group.25,26 A variable selection procedure removed nonsignificant descriptors, decreased model complexity, and increased model interpretability and robustness. The first exclusion step removed all variables except the 100 most important for the response. Thereafter additional variables were removed based on the variable importance to projection (VIP) and the loading plot, and monitored by the leave-one-out (using 7 groups), and cross-validated R2 (Q2). If the exclusion of the variable had no effect or increased the Q2, it was permanently eliminated from the model. The variable selection procedure was repeated until no further descriptors could be removed without lowering Q2. All PLS models were validated by root-mean square error of the estimate (RMSEE) calculations of the training and test sets and permutation tests (100 iterations).
Calculation and Prediction of Loading Capacity in Lipid-Based Formulations
The loading capacities of the LBFs were calculated for the nine model compounds by taking use of eq 1:
| 1 |
where SLBF is the total drug loading in the formulation and equal to the sum of the solubility in the pure excipient (Se), multiplied by the weight percentage of that excipient in the formulation (We).14,17 In other words, the drug loading capacity of the formulation is the sum of the drug solubility in all included excipients normalized by the contributing weight fraction of the excipients in the formulation. The above equation was modified from the original log–linear equation;14,15 here the sum of the weighted mean of the solubility is used instead of the weighed geometric mean. Our ultimate aim was to provide computational tools for prediction of solubility in LBFs, with no requirement for prior experimental screening of solubility in lipid excipients. For that purpose the predicted solubility values from the PLS models were similarly summed by making use of eq 1 and thereafter compared to the experimental values.
Results
Solid State Characterization
DSC was used to provide the following solid state data: Tm (°C), ΔHf (J/g), ΔSf (J/mol·K), and calculation of ideal solubility at 37 °C (log Xic). The ideal solubility has previously been identified to be closely related to solubility of crystalline organic nonelectrolytes in aqueous systems,22 and was therefore included as a descriptor in this work. The four solid state related properties determined from the DSC thermograms were used, together with calculated molecular descriptors, as variables in the in silico model development. The DSC thermograms generated easily interpretable and sharp peaks for all compounds except for candesartan, carbamazepine, and saquinavir. For candesartan the Tm was confirmed to be 178 °C in a capillary melting point apparatus (Electrothermal, England). Saquinavir gradually liquefied by this approach starting at ∼100 °C. No sharp melting point was observed, and at high temperature, the compound decomposed. Hence, for saquinavir the Tm could not be determined. The DSC thermograms of carbamazepine showed a melting peak at 173 °C (form III) upon which an exothermic crystallization to form I (181 °C) occurred that subsequently melted at 191 °C.27 Hence, the solubility data reported herein is for carbamazepine form III. Candesartan, carbamazepine, and saquinavir were not included in the PLS model development, since all four solid state descriptors were required as input for modeling.
Drug Solubility in Single Excipients
Thirty-five poorly water-soluble drugs were selected and measured for solubility in five commonly used LBF excipients. The compounds were selected to be as diverse in chemical properties as possible while remaining suitable LBF candidates. The data set had the following physicochemical properties: lipophilicity (reflected by the calculated octanol/water partition coefficient, AlogPoct) 2.0 to 8.2, molecular weight 230.3 to 705.7 g/mol, and Tm 67–255 °C. In addition, the compounds were selected to be representative examples of acids, bases, and nonionizable compounds (Table 1).
Equilibrium solubility in the excipients varied 10,000-fold, from as low as 0.01 mg/g up to >300 mg/g. The general ranking order of drug solubility in the excipients (mg/g) was long-chain triglyceride < medium-chain triglyceride < surfactant < cosolvent (Tables 2 and 3). The mixed mono-, di-, triglycerides (Maisine and Capmul) had higher solvation capacity than the corresponding triglycerides (SBO and Captex, respectively) on an mg/g scale (Table 2). In general the solubility was slightly higher in Cremophor EL than Cremophor ELP, but the rank order of compound solubility was similar (Spearman rank coefficient 0.97) (Table SI2). For 10 of the compounds the solubility differed more than 10 mg/g in these two surfactants. In the cosolvents, the drug solubility was higher in Carbitol than PEG400 for all compounds investigated except for niclosamide (Table 3).
In our previous study, there was a high correlation between SBO and Captex when the solubility was converted to a mol per mol scale.21 The extended data set in this study (n = 35) verified this trend (R2 0.99) (Figure 2A). Intriguingly, the mixed mono-, di-, triglycerides (Maisine and Capmul) also displayed equal solvation capacities on a mol per mol scale (R2 0.89) (Figure 2A). Further, the solubility in ethoxylated solvents was highly correlated as exemplified with the strong correlations between Carbitol and PEG400 (R2 0.85), PS80 (R2 0.90), and Cremophor EL (R2 0.93) (Figure 2B). Hence this confirms the importance of the ethoxylation for the solvation capacity of these excipients. In addition, on a mol/mol scale, the surfactants (PS80 and Cremophor EL) had a 2- to10-fold greater solvation capacity than what was obtained in the ethoxylated cosolvents (PEG400 and Carbitol).
Figure 2.
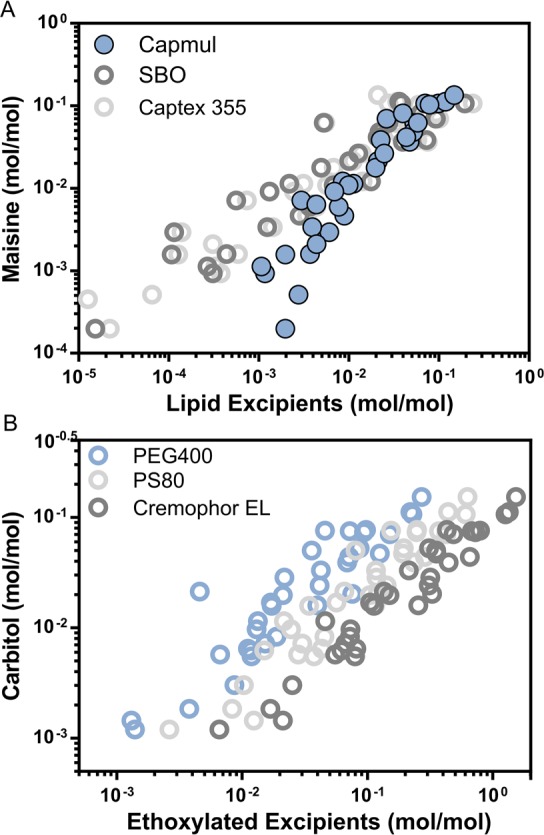
Relation between solubility in single excipients. (A) Mixed mono-, di-, triglycerides. (B) Ethoxylated surfactants and cosolvents.
We also investigated the relationship between drug solubility in single excipients and melting temperature. The compounds with Tm below 150 °C in general displayed solubility values greater than 10 mg/g in SBO (Figure 3), and a similar trend was observed for all studied glycerides. In Captex and Maisine, a Tm below 150 °C corresponded to solubility values above 20 mg/g and in Capmul above 40 mg/g (Figure SI1). Moreover, it was observed that compounds that did not follow this general trend had a high melting point in combination with low entropy of fusion. Hence, the large and structurally diverse data set yet again confirmed the importance of solid state limitations of high-melting compounds not only in water-based solvents,22,28 but also for solubility in lipids. However, this data set showed no such trends for solubility in surfactants and cosolvents (Figure SI1).
Figure 3.
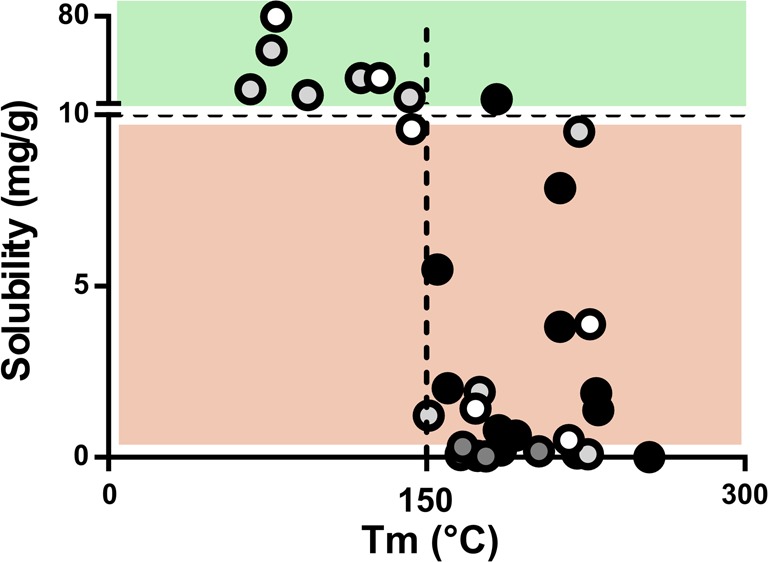
Solubility of drug compounds in soybean oil compared to melting point. Compounds with a Tm below 150 °C in general displayed solubility greater than 10 mg/g in this triglyceride (upper green area). Black circle (acid), dark gray circle (ampholyte), light gray circle (base), and white circle (neutral).
Another trend was that acidic compounds in general displayed lower solubility in the lipids than the bases and neutral compounds did, but this was not observed in the surfactants and cosolvents. One reason may be that acidic compounds are less prone to dissolve in the lipids because of the low fraction free fatty acid present in the lipids. However, for this particular data set, a further explanation may be that the acids had relatively high melting points.
Computational Prediction of Drug Solubility in Lipid Excipients
We developed eight PLS models (SBO, Maisine, Captex, Capmul, Cremophor EL, PS80, Carbitol, and PEG400) to analyze which molecular properties were most important for describing thermodynamic solubility in commonly used lipid excipients and to allow for fast prediction of loading capacity in LBFs. All eight PLS models used 2–3 principal components and 4–6 variables, and produced statistically strong models (R2 0.81–0.94 and Q2 0.78–0.91). Importantly, they predicted the test sets well (Table 4). In this work, the logarithm form of the ideal solubility was included in the model development, and this term was found necessary to allow highly accurate models to be developed for all excipients (lipids, surfactants, and cosolvents). This descriptor was the most important descriptor in all excipient models, except the PEG400 model in which it was the third most important descriptor. Previous studies have also found that descriptors related to crystal lattice energy are of less importance for solubility predictions in PEG400 systems.20,21
Table 4. Statistics of and Descriptors Used in the Final Developed PLS Models of Drug Solubility (log (mol drug/mol excipient)) in Single Excipientsa.
| SBO | Maisine | Captex | Capmul | Cremophor EL | PS80 | Carbitol | PEG400 | |
|---|---|---|---|---|---|---|---|---|
| R2 | 0.93 | 0.93 | 0.94 | 0.94 | 0.85 | 0.86 | 0.87 | 0.81 |
| Q2 | 0.91 | 0.91 | 0.91 | 0.90 | 0.81 | 0.81 | 0.83 | 0.78 |
| RMSEETr | 0.28 (n = 25) | 0.19 (n = 25) | 0.25 (n = 25) | 0.15 (n = 25) | 0.23 (n = 25) | 0.22 (n = 25) | 0.21 (n = 25) | 0.27 (n = 25) |
| RMSEETe | 0.23 (n = 4) | 0.46 (n = 7) | 0.37 (n = 6) | 0.35 (n = 7) | 0.33 (n = 7) | 0.38 (n = 7) | 0.27 (n = 7) | 0.40 (n = 6) |
| log Xicb | + | + | + | + | + | + | + | + |
| TPSA (tot) | – | – | – | |||||
| nN | – | – | – | |||||
| JGI6 | + | + | + | |||||
| SAacc | – | |||||||
| B10[C-O] | – | – | ||||||
| B04[N-O] | – | |||||||
| B07[N-O] | + | |||||||
| HATS7e | + | |||||||
| HATS6i | + | |||||||
| GATS1p | – | |||||||
| GATS8s | + | + | + | |||||
| MATS7e | + | |||||||
| Mor18ib | – | – | – | – | ||||
| Mor26i | – | |||||||
| R6e+ | + | + | + | + | ||||
| CMC-80 | + | |||||||
| TDB06s | + | |||||||
| G2s | + |
A plus sign (+) indicates a positive influence on the solubility, and a minus sign (−) indicates a negative effect on the solubility.
Descriptor with a negative range.
Total polar surface area (TPSA (tot)), number of nitrogens (nN), and surface area of hydrogen bond acceptors (SAacc) negatively influenced solubility in the lipid excipients, thus identifying the detrimental effect of polar groups on the solubility in these systems. In addition to these easily interpreted molecular properties, topological charge distribution (JGI6) was of importance in three of the lipid PLS models. JGI6 positively impacted solubility, i.e., the distribution of topological charges is an advantage for lipid solubility. Cremophor EL is a surfactant with similarities in its molecular structure to the lipids, and also in this model nN was found to be a descriptor that negatively impacted the solubility. Other significant descriptors in the surfactant and cosolvent models were related to ionization potential, size, and electronegativity. Mor18i and Mor26i belong to the 3D-MORSE descriptors and contain information about both size and ionization state; for solubility in surfactant and cosolvent excipients, larger structures without ionization potential appear to be unfavorable. Additionally, electronegative atoms positively influenced solubility (R6e+). For detailed description of the calculated descriptors, see Table SI3. Table 4 depicts the descriptor trends. The descriptors of the lipid models (SBO, Maisine, Captex) are similar, and likewise the cosolvent models (Carbitol, PEG400) are based on comparable descriptors. Interestingly, Cremophor EL and PS80 share several descriptors with the cosolvents, yet again emphasizing the importance of the ethoxylated chain of the surfactants on the final solubility. In addition, the in silico models revealed that Capmul, the most surface active lipid of those investigated, has descriptors in common with both the lipids and the surfactants.
Calculation and Prediction of Solubility in Lipid-Based Formulations
All four formulation types met the requirements of self-emulsification and absence of phase separation (Figure 1). The loading capacity of the formulations ranged from 1.7 mg/g to 93.0 mg/g in type II, 1.3 mg/g to 167.1 mg/g in type IIIA, 2.8 mg/g to 166.0 mg/g in type IIIB, and 1.9 mg/g to 92.9 mg/g in type IV. This extensive data set confirms earlier studies,16−18 i.e., the total loading capacity of a complex formulation can be calculated by proportionately summing up the solubility of the compound in the single excipients (eq 1). This trend was regardless of formulation type or whether the compound displayed low, medium, or high solubility in the LBFs (R2 0.91) (Figure 4A). Predicted solubility values from the developed PLS models and eq 1 were also in agreement with experimental loading capacity of the formulations (R2 of 0.79 for all formulations). The predictions were slightly better for the lipid-rich LBFs (II and IIIA) (R2 0.82) (Figure 4B), than the formulations with higher quantities of surfactants and cosolvents (IIIB and IV) (R2 0.79) (Figure 4C). The better accuracy of the predictions in the lipid-rich formulations was not unexpected, given the model performance of the lipid models compared to the surfactant and cosolvent models (Table 4). If the weighted geometric mean of the solubility is used instead (log–linear equation), comparable accuracy in the predictions is achieved. The use of either the weighed mean or the geometric mean of the solubility for this purpose has been discussed elsewhere.17
Figure 4.

Experimentally determined solubility plotted against calculated and predicted solubility through eq 1 in four types of LBFs. (A) Experimental solubility in LBF type II–IV compared to calculated solubility. (B) Experimental solubility in lipid rich LBFs (type II and IIIA) compared to predicted solubility. (C) Experimental solubility in surfactant/cosolvent rich LBFs (type IIIB and IV) compared to predicted solubility.
Discussion
Loading capacity in LBFs is a critical factor during formulation development, but it is still poorly understood which molecular features determine solubility in commonly used LBF excipients. A log–linear relationship was originally identified for solubility in cosolvent/water systems.14 In this work, we used a linear model to calculate solubility in complex lipid, surfactant, and cosolvent mixtures. Our aim was to establish tools that rapidly and accurately inform formulators of the potential use of LBFs as delivery systems for poorly water-soluble drugs and, ultimately, develop computational tools for solubility prediction in single excipients and complex formulations. Such models are highly warranted as they would provide flexible and rapid predictions of loading capacity in any new LBF for which excipient in silico models exist. We also hoped to gain a mechanistic understanding of the molecular features important for drug solvation in lipid excipients and formulations.
Our results strengthen the concept of obtaining solubility data in key excipients. As we have proposed previously, such data can be used to estimate solubility in similar excipients.21 In other words, solubility determination in key excipients reflecting one triglyceride, one mixed mono-, di-, triglyceride, and one ethoxylated excipient can be used to estimate solubility in similar excipients. Using ten structurally diverse drugs, Thi et al. observed that the ranking order of excipients (i.e., the amount of drug dissolved on a mg/g scale) in general was long-chain oil < medium-chain oil < surfactant.29 Our database with solubility values for 35 compounds in 9 excipients confirms the trend reported by Thi et al. We also observed that mixed mono-, di-, triglyceride glycerides (Maisine and Capmul) overall had higher solvation capacities than the corresponding triglycerides (SBO and Captex, respectively) on a mg/g scale (Table 2). Notably, compounds that fall outside this general rank trend in solubility are commonly used model compounds in the study of LBFs. For instance, cinnarizine, halofantrine, and fenofibrate are more soluble in Captex than Capmul and less soluble in the surfactants than in the medium-chain lipids. We speculate that these particular drug molecules are compounds that thrive in lipids and their aliphatic chain more than other, similarly lipophilic ones. Although these three drugs have different molecular profiles and reflect both basic and neutral drugs (see, e.g., Table 1), they are all low melting compounds with fairly high logP and low or modest polar surface area. Moreover, the three compounds possess some degree of elongation and flexibility that might help in covering polar groups, and thus favor lipophilic milieus. Other compounds with similar properties, e.g., disulfiram and perphenazine, also follow a similar trend and display high lipid solubility. The analysis herein stresses that precautions should be taken when choosing single model compounds, and as a suggestion the use of cinnarizine, halofantrine, and fenofibrate as general LBF model compounds should be re-evaluated. When studies all use the same model compounds, the understanding of these systems, and thus the LBF development, could be misguided. On the other hand, this analysis also revealed three optimal physicochemical properties for drug solubility in lipids: the drugs should be neutral or basic, have a low to intermediate Tm (preferably <150 °C), and have few polar groups.
Mixtures of lipid, surfactants, and cosolvent are likely to form stable microemulsions in the formulation itself. These will have some degree of short-range order, e.g., reversed micelles and lamellar phases, whereas any long-range order is stochastic.30−33 In other words, the microemulsions that form are distinguishable from the homogeneous solution because they are isotropic over short distances. These types of excipients are prone to water uptake which may impact the microstructures in pure excipients and mixtures and thus distort solubility determinations. We chose to work with off-the-shelf excipients (stored under argon gas) to mimic the laboratory situation. In a previous work, we had determined the amount of water in PEG400 to be minimal (0.32% w/w).21 Nevertheless, water sorption may be solvent dependent and could increase or decrease drug solubility, depending on the properties of the particular drug.30,34 Through determinations of solubility in Cremophor EL (≤3% w/w water) and Cremophor ELP (≤0.5% w/w water) of a larger drug data set, we have confirmed that the compound solubility follows the same rank order in the two, but the impact of a small amount of water is indeed compound dependent. Overall the solubility was higher in the surfactant containing more water. Water has been observed to increase the solvation capacity of other organic solvents.35,36 For example, the solubility of N-methylbenzamide increases with more than 50% in water-saturated tricaprylin compared to dry tricaprylin.34
The predictive power of the developed models is in line with similar models for prediction of drug water solubility.37 Our models represent the first computer-aided tools that can predict drug solubility in various types of excipients, ranging from triglycerides to cosolvents, and allow calculation of loading capacity in complex formulations. To further challenge the developed computational formulation models, a set of five compounds (clofazime, dipyridamole, griseofulvin, mefenamic acid, naproxen) was measured for their loading capacity in an additional type IV formulation (50% w/w PEG400; 50% w/w PS80). The calculated solubility (eq 1) was in excellent agreement with the experimental (R2 1.0), and with only a slight decrease in predictability using the computer approach (R2 0.9). Solid state properties were of major importance for solubility in all excipient models; in fact, we were not able to develop well performing cosolvent models without inclusion of such descriptors. Even though the inclusion of solid state descriptors allowed development of predictive cosolvent models, Tm alone did not give a threshold value for solubility as was the case for the lipid excipients. Based on our data set it is suggested that it would be better to make solid state modifications to enhance the solubility of high-melting compounds rather than using lipid rich LBFs (I-IIIA). Moreover, polar groups negatively influenced solubility in lipids, as seen from the negative contribution of polar surface area, number of nitrogens, and surface area of hydrogen bond acceptors. The modeling work also revealed that, when electronegative atoms are present, the provided possibility to distribute the topological charge is positive for lipid solubility. Taken together, suitable candidates for a lipid-rich LBF (I-IIIA) would be lipophilic, low-melting, have few polar groups, and have a capacity to distribute charges. Examples meeting these criteria are fenofibrate and perphenazine. In contrast, more “hydrophilic” compounds (lower end of the logP scale investigated here) could benefit from formulation in less lipophilic LBFs (IIIB-IV), in which the polar descriptors were unimportant. For solubility in these systems, the important descriptors are related to ionization state, electronegativity, size, and shape. Hence, suitable candidates in such formulations would be polar but still lipophilic, and low-to-high melting, e.g., diflunisal and niclosamide.
In this work we showed that the weighted average of the solubility in single excipients could be used to predict loading capacity in complex lipid, surfactant, and cosolvent systems. Drug solubility is clearly linked to drug localization, and we speculate that this equation might be valid due to stable micro- or nanostructures formed in these mixtures. In the colloidal structures, or “excipient clusters”, the drug reaches the same local maximum solubility as in the pure excipients, and hence the loading capacity of the LBF is the sum of the solubility in the included excipients. MD simulations of similar excipients, glyceride surfactant, and mixed di- and triglyceride systems show that the excipients reside in clusters.38 When there are no or only trace amounts of water, reversed micellar structures form.39 Experimental characterization of such systems also confirms these reversed micellar structures.40−42 We are now embarking on studies in which microscopic observation of excipient mixtures will be combined with MD simulations, to provide a deeper mechanistic understanding of the molecular interactions in these complex systems.
Conclusion
In conclusion, the tools developed here rationalize the identification of LBFs as a useful formulation strategy for poorly water-soluble drug molecules. First, melting point measurements can identify the use of formulations that include large amounts of oils, i.e., LFCS I-IIIA. Tm < 150 °C was a good indicator of reasonable solubility in glycerides and can serve as a baseline for the selection of LBFs as potential enabling formulations. However, drugs with higher melting points might be well solubilized in LBFs containing high quantities of cosolvents and surfactants; we found no relationship between Tm and solubility in such solvents. Second, solubility in key excipients can be estimated based on solubility in similar excipients. For example, measurements in PEG400 can be used to estimate solubility in other ethoxylated excipients such as Cremophor EL, PS80, and Carbitol. Third, the loading capacity of LBFs can be calculated or predicted from solubility in single excipients. Our study is the first to develop computational models for prediction of loading capacity in LBFs based on calculated molecular descriptors and experimentally determined solid state properties. These models accurately predicted the loading capacity in four complex lipid formulations, allowing the identification of suitable LBFs from a rapid DSC measurement (using a few mgs only) and easily calculated molecular descriptors. The computational models presented here provide advantages in speed and simplicity and increase our understanding of the molecular features important for loading capacity in any new LBF.
Acknowledgments
We thank the European Research Council (Grant 638965) and the Swedish Research Council (Grants 621-2011-2445 and 621-2014-3309) for financial support. We are grateful to Elena Diesch, Angelica Friberg, and Paulina Jakubiak for skillful experimental assistance. We are thankful to Simulations Plus (Lancaster, CA) for providing us with a reference site license for the software ADMET Predictor.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.molpharmaceut.5b00704.
Analytical details, thermodynamic solubility in Cremophor ELP, explanation of DragonX 6.0.16 descriptors, and solubility melting point relationships (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Benet L. Z.; Wu C.-Y.; Custodio J. M. Predicting drug absorption and the effects of food on oral bioavailability. Bull. Technol. Gattefossé. 2006, 99, 9–16. [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–890. 10.1038/nrd2445. [DOI] [PubMed] [Google Scholar]
- Hauss D. J. Oral lipid-based formulations. Adv. Drug Delivery Rev. 2007, 59, 667–676. 10.1016/j.addr.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Porter C. J. H.; Trevaskis N. L.; Charman W. N. Lipids and lipid-based formulations: optimizing the oral delivery of lipophilic drugs. Nat. Rev. Drug Discovery 2007, 6, 231–248. 10.1038/nrd2197. [DOI] [PubMed] [Google Scholar]
- Williams H. D.; Sassene P.; Kleberg K.; Calderone M.; Igonin A.; Jule E.; Vertommen J.; Blundell R.; Benameur H.; Müllertz A.; Pouton C. W.; Porter C. J. Toward the Establishment of Standardized In Vitro Tests for Lipid-Based Formulations, Part 3: Understanding Supersaturation Versus Precipitation Potential During the In Vitro Digestion of Type I, II, IIIA, IIIB and IV Lipid-Based Formulations. Pharm. Res. 2013, 30, 3059–3076. 10.1007/s11095-013-1038-z. [DOI] [PubMed] [Google Scholar]
- Williams H. D.; Anby M. U.; Sassene P.; Kleberg K.; Bakala-N’Goma J.; Calderone M.; Jannin V.; Igonin A.; Partheil A.; Marchaud D.; Jule E.; Vertommen J.; Maio M.; Blundell R.; Benameur H.; Carrière F.; Müllertz A.; Pouton C. W.; Porter C. J. H. Toward the establishment of standardized in vitro tests for lipid-based formulations, Part 2: The effect of bile salt concentration and drug loading on the performance of Type I, II, IIIA, IIIB and IV formulations during in vitro digestion. Mol. Pharmaceutics 2012, 9 (11), 3286–3300. 10.1021/mp300331z. [DOI] [PubMed] [Google Scholar]
- Williams H. D.; Sassene P.; Kleberg K.; Bakala-N’Goma J.-C.; Calderone M.; Jannin V.; Igonin A.; Partheil A.; Marchaud D.; Jule E.; Vertommen J.; Maio M.; Blundell R.; Benameur H.; Carrière F.; Müllertz A.; Porter C. J. H.; Pouton; Colin W. Toward the establishment of standardized in vitro tests for lipid-based formulations, Part 1: Method parameterization and comparison of in vitro digestion profiles across a range of representative formulations. J. Pharm. Sci. 2012, 101 (9), 3360–3380. 10.1002/jps.23205. [DOI] [PubMed] [Google Scholar]
- Chen X. Q.; Gudmundsson O.; Hageman M. Application of Lipid-Based Formulations in Drug Discovery. J. Med. Chem. 2012, 55 (18), 7945–7956. 10.1021/jm3006433. [DOI] [PubMed] [Google Scholar]
- Pouton C. W. Lipid formulations for oral administration of drugs: non-emulsifying, self-emulsifying and ‘self-microemulsifying’ drug delivery systems. Eur. J. Pharm. Sci. 2000, 11 (Suppl. 2), 93–98. 10.1016/S0928-0987(00)00167-6. [DOI] [PubMed] [Google Scholar]
- Pouton C. W. Formulation of poorly water-soluble drugs for oral administration: Physicochemical and physiological issues and the lipid formulation classification system. Eur. J. Pharm. Sci. 2006, 29, 278–287. 10.1016/j.ejps.2006.04.016. [DOI] [PubMed] [Google Scholar]
- Wyttenbach N.; Alsenz J.; Grassmann O. Miniaturized assay for solubility and residual solid screening (SORESOS) in early drug development. Pharm. Res. 2007, 24, 888–898. 10.1007/s11095-006-9205-0. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Jain A.; Papoutsakis D.; Dannenfelser R.; Panicucci R.; Garad S. Selection of oral bioavailability enhancing formulations during drug discovery. Drug Dev. Ind. Pharm. 2012, 38 (2), 235–247. 10.3109/03639045.2011.602406. [DOI] [PubMed] [Google Scholar]
- Pouton C. W. Formulation of self-emulsifying drug delivery systems. Adv. Drug Delivery Rev. 1997, 25, 47–58. 10.1016/S0169-409X(96)00490-5. [DOI] [Google Scholar]
- Yalkowsky S. H.; Flynn G. L.; Amidon G. L. Solubility of nonelectrolytes in polar solvents. J. Pharm. Sci. 1972, 61, 983–984. 10.1002/jps.2600610643. [DOI] [PubMed] [Google Scholar]
- Yalkowsky S. H.; Valvani S. C.; Amidon G. L. Solubility of nonelectrolytes in polar solvents IV: Nonpolar drugs in mixed solvents. J. Pharm. Sci. 1976, 65, 1488–1494. 10.1002/jps.2600651018. [DOI] [PubMed] [Google Scholar]
- Prajapati H. N.; Dalrymple D. M.; Serajuddin A. T. M. A comparative evaluation of mono-, di-and triglyceride of medium chain fatty acids by lipid/surfactant/water phase diagram, solubility determination and dispersion testing for application in pharmaceutical dosage form development. Pharm. Res. 2012, 29, 285–305. 10.1007/s11095-011-0541-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sacchetti M.; Nejati E. Prediction of Drug Solubility in Lipid Mixtures from the Individual Ingredients. AAPS PharmSciTech 2012, 13 (4), 1103–11091–7. 10.1208/s12249-012-9830-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S. V.; Patel S. Prediction of the solubility in lipidic solvent mixture: Investigation of the modeling approach and thermodynamic analysis of solubility. Eur. J. Pharm. Sci. 2015, 77, 161–169. 10.1016/j.ejps.2015.06.014. [DOI] [PubMed] [Google Scholar]
- Rytting E.; Lentz K.; Chen X.-Q.; Qian F.; Venkatesh S. A. Quantitative Structure-Property Relationship for Predicting Drug Solubility in PEG 400/Water Cosolvent Systems. Pharm. Res. 2004, 21, 237–244. 10.1023/B:PHAM.0000016237.06815.7a. [DOI] [PubMed] [Google Scholar]
- Ghafourian T.; Bozorgi A. H. A. Estimation of drug solubility in water, PEG 400 and their binary mixtures using the molecular structures of solutes. Eur. J. Pharm. Sci. 2010, 40, 430–440. 10.1016/j.ejps.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Persson L. C.; Porter C. J.; Charman W. N.; Bergström C. A. Computational Prediction of Drug Solubility in Lipid Based Formulation Excipients. Pharm. Res. 2013, 30 (12), 3225–3237. 10.1007/s11095-013-1083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yalkowsky S. H.; Valvani S. C. Solubility and partitioning I: Solubility of nonelectrolytes in water. J. Pharm. Sci. 1980, 69, 912–922. 10.1002/jps.2600690814. [DOI] [PubMed] [Google Scholar]
- Craig D. Q. M.; Barker S. A.; Banning D.; Booth S. W. An investigation into the mechanisms of self-emulsification using particle size analysis and low frequency dielectric spectroscopy. Int. J. Pharm. 1995, 114, 103–110. 10.1016/0378-5173(94)00222-Q. [DOI] [Google Scholar]
- Kommuru T. R.; Gurley B.; Khan M. A.; Reddy I. K. Self-emulsifying drug delivery systems (SEDDS) of coenzyme Q10: formulation development and bioavailability assessment. Int. J. Pharm. 2001, 212, 233–246. 10.1016/S0378-5173(00)00614-1. [DOI] [PubMed] [Google Scholar]
- Bergström C. A. S.; Charman S. A.; Nicolazzo J. A. Computational Prediction of CNS Drug Exposure Based on a Novel In Vivo Dataset. Pharm. Res. 2012, 29 (11), 3131–3142. 10.1007/s11095-012-0806-5. [DOI] [PubMed] [Google Scholar]
- Fagerberg J. H.; Al-Tikriti Y.; Ragnarsson G.; Bergström C. A. S. Ethanol Effects on Apparent Solubility of Poorly Soluble Drugs in Simulated Intestinal Fluid. Mol. Pharmaceutics 2012, 9, 1942–1952. 10.1021/mp2006467. [DOI] [PubMed] [Google Scholar]
- Rustichelli C.; Gamberini G.; Ferioli V.; Gamberini M. C.; Ficarra R.; Tommasini S. Solid-state study of polymorphic drugs: carbamazepine. J. Pharm. Biomed. Anal. 2000, 23, 41–54. 10.1016/S0731-7085(00)00262-4. [DOI] [PubMed] [Google Scholar]
- Wassvik C. M.; Holmén A. G.; Bergström C. A. S.; Zamora I.; Artursson P. Contribution of solid-state properties to the aqueous solubility of drugs. Eur. J. Pharm. Sci. 2006, 29, 294–305. 10.1016/j.ejps.2006.05.013. [DOI] [PubMed] [Google Scholar]
- Thi T. D.; Van Speybroeck M.; Barillaro V.; Martens J.; Annaert P.; Augustijns P.; Van Humbeeck J.; Vermant J.; Van den Mooter G. Formulate-ability of ten compounds with different physicochemical profiles in SMEDDS. Eur. J. Pharm. Sci. 2009, 38, 479–488. 10.1016/j.ejps.2009.09.012. [DOI] [PubMed] [Google Scholar]
- Rane S.; Cao Y.; Anderson B. Quantitative Solubility Relationships and the Effect of Water Uptake in Triglyceride/Monoglyceride Microemulsions. Pharm. Res. 2008, 25, 1158–1174. 10.1007/s11095-007-9500-4. [DOI] [PubMed] [Google Scholar]
- Kon-No K.; Jin-No T.; Kitahara A. Solubility, critical aggregating or micellar concentration, and aggregate formation of nonionic surfactants in nonaqueous solutions. J. Colloid Interface Sci. 1974, 49, 383–389. 10.1016/0021-9797(74)90382-8. [DOI] [Google Scholar]
- Eicke H. F.Surfactants in nonpolar solvents; Aggregation and micellization. In Micelles; Topics in Current Chemistry, Vol. 87; Springer: Berlin Heidelberg, 1980; Chapter 2, pp 85–145. [DOI] [PubMed] [Google Scholar]
- Rane S. S.; Anderson B. D. What determines drug solubility in lipid vehicles: Is it predictable?. Adv. Drug Delivery Rev. 2008, 60, 638–656. 10.1016/j.addr.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Marra M.; Anderson B. D. Predictive relationships for the effects of triglyceride ester concentration and water uptake on solubility and partitioning of small molecules into lipid vehicles. J. Pharm. Sci. 2004, 93, 2768–2779. 10.1002/jps.20126. [DOI] [PubMed] [Google Scholar]
- Abraham M. H.; Zissimos A. M.; Acree W. E. Jr Partition of solutes from the gas phase and from water to wet and dry di-n-butyl ether: a linear free energy relationship analysis. Phys. Chem. Chem. Phys. 2001, 3, 3732–3736. 10.1039/b104682a. [DOI] [Google Scholar]
- Abraham M. H.; Zissimos A. M.; Acree W. E. Jr Partition of solutes into wet and dry ethers; an LFER analysis. New J. Chem. 2003, 27, 1041–1044. 10.1039/b303016d. [DOI] [Google Scholar]
- Norinder U.; Bergström C. A. Prediction of ADMET properties. ChemMedChem 2006, 1, 920–937. 10.1002/cmdc.200600155. [DOI] [PubMed] [Google Scholar]
- Benson S. P.; Pleiss J. r. Molecular Dynamics Simulations of Self-Emulsifying Drug-Delivery Systems (SEDDS): Influence of Excipients on Droplet Nanostructure and Drug Localization. Langmuir 2014, 30, 8471–8480. 10.1021/la501143z. [DOI] [PubMed] [Google Scholar]
- Warren D.; King D.; Benameur H.; Pouton C.; Chalmers D. Glyceride Lipid Formulations: Molecular Dynamics Modeling of Phase Behavior During Dispersion and Molecular Interactions Between Drugs and Excipients. Pharm. Res. 2013, 30, 3238–3253. 10.1007/s11095-013-1206-1. [DOI] [PubMed] [Google Scholar]
- Shrestha L. K.; Sato T.; Aramaki K. Intrinsic parameters for structural variation of reverse micelles in nonionic surfactant (glycerol [small alpha]-monolaurate)/oil systems: a SAXS study. Phys. Chem. Chem. Phys. 2009, 11, 4251–4259. 10.1039/b818072e. [DOI] [PubMed] [Google Scholar]
- Gulik-Krzywicki T.; Larsson K. An electron microscopy study of the L2-phase (microemulsion) in a ternary system: Triglyceride/monoglyceride/water. Chem. Phys. Lipids 1984, 35, 127–132. 10.1016/0009-3084(84)90018-5. [DOI] [Google Scholar]
- Parris N.; Joubran R. F.; Lu D. P. Triglyceride Microemulsions: Effect of Nonionic Surfactants and the Nature of the Oil. J. Agric. Food Chem. 1994, 42, 1295–1299. 10.1021/jf00042a008. [DOI] [Google Scholar]
- Bones J.; Thomas K.; Nesterenko P. N.; Paull B. On-line preconcentration of pharmaceutical residues from large volume water samples using short reversed-phase monolithic cartridges coupled to LC-UV-ESI-MS. Talanta 2006, 70, 1117–1128. 10.1016/j.talanta.2006.02.026. [DOI] [PubMed] [Google Scholar]
- Tosco P.; Rolando B.; Fruttero R.; Henchoz Y.; Martel S.; Carrupt P.; Gasco A. Physicochemical Profiling of Sartans: A Detailed Study of Ionization Constants and Distribution Coefficients. Helv. Chim. Acta 2008, 91, 468–482. 10.1002/hlca.200890051. [DOI] [Google Scholar]
- Bergström C. A. S.; Andersson S. B. E.; Fagerberg J. H.; Ragnarsson G.; Lindahl A. Is the full potential of the biopharmaceutics classification system reached?. Eur. J. Pharm. Sci. 2014, 57, 224–231. 10.1016/j.ejps.2013.09.010. [DOI] [PubMed] [Google Scholar]
- Danaher M.; De Ruyck H.; Crooks S. R. H.; Dowling G.; O’Keeffe M. Review of methodology for the determination of benzimidazole residues in biological matrices. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2007, 845, 1–37. 10.1016/j.jchromb.2006.07.046. [DOI] [PubMed] [Google Scholar]
- Fridén M.; Winiwarter S.; Jerndal G.; Bengtsson O.; Wan H.; Bredberg U.; Hammarlund-Udenaes M.; Antonsson M. Structure–Brain Exposure Relationships in Rat and Human Using a Novel Data Set of Unbound Drug Concentrations in Brain Interstitial and Cerebrospinal Fluids. J. Med. Chem. 2009, 52, 6233–6243. 10.1021/jm901036q. [DOI] [PubMed] [Google Scholar]
- Box K. J.; Völgyi G.; Ruiz R.; Comer J. E.; Takács-Novák K.; Bosch E.; Ràfols C.; Rosés M. Physicochemical Properties of a New Multicomponent Cosolvent System for the pKa Determination of Poorly Soluble Pharmaceutical Compounds. Helv. Chim. Acta 2007, 90, 1538–1553. 10.1002/hlca.200790161. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.