

Researchers provide a conceptual framework to understand current knowledge of the fundamentals of cancer metabolism.
Keywords: Cancer, metabolism, mitochondria, glycolysis, ROS, oncogenes
Abstract
Tumors reprogram pathways of nutrient acquisition and metabolism to meet the bioenergetic, biosynthetic, and redox demands of malignant cells. These reprogrammed activities are now recognized as hallmarks of cancer, and recent work has uncovered remarkable flexibility in the specific pathways activated by tumor cells to support these key functions. In this perspective, we provide a conceptual framework to understand how and why metabolic reprogramming occurs in tumor cells, and the mechanisms linking altered metabolism to tumorigenesis and metastasis. Understanding these concepts will progressively support the development of new strategies to treat human cancer.
INTRODUCTION AND OVERARCHING PRINCIPLES
Cancer metabolism is one of the oldest areas of research in cancer biology, predating the discovery of oncogenes and tumor suppressors by some 50 years. The field is based on the principle that metabolic activities are altered in cancer cells relative to normal cells, and that these alterations support the acquisition and maintenance of malignant properties. Because some altered metabolic features are observed quite generally across many types of cancer cells, reprogrammed metabolism is considered a hallmark of cancer (1, 2). Precisely how metabolism becomes reprogrammed in cancer cells, whose functions or malignant properties are enabled by these activities, and how to exploit metabolic changes for therapeutic benefit are among the key questions driving research in the field.
This review covers several fundamental principles in cancer metabolism, with the goal of introducing non-experts to the concepts motivating ongoing research. With the explosion of research in cancer metabolism over the past decade, no single review can possibly cover it all. The sections below highlight some of the essential, recent papers supporting these core principles. An overarching theme in cancer metabolism is that reprogrammed activities improve cellular fitness to provide a selective advantage during tumorigenesis. Most of the classical examples of reprogrammed activities either support cell survival under stressful conditions or allow cells to grow and proliferate at pathologically elevated levels. Three of these—altered bioenergetics, enhanced biosynthesis, and redox balance—are discussed at length below. It logically flows that if these activities provide benefit to the malignant cell, then some of them might be suitable therapeutic targets. This rendering of cancer metabolism is supported by many examples in which inhibition of an enhanced metabolic activity results in impaired growth of experimental tumors (3, 4). In some cases, the particular metabolic liabilities of cancer cells have been translated into effective therapies in human cancer. Asparaginase, an enzyme that converts the amino acid asparagine to aspartic acid and ammonia, is an essential component of treatment for acute lymphoblastic leukemia (ALL) (5). Because of their high rates of protein synthesis and poor ability to synthesize asparagine de novo, ALL cells require a constant supply of asparagine from the plasma. This supply is essentially eliminated by systemic administration of asparaginase. Ultimately, effective metabolic therapy will require defining the stage of tumor progression in which each pathway provides its benefit to the cancer cell. Some activities first become essential very early in tumorigenesis as the nascent tumor begins to experience nutrient limitations (6). In other cases, altered pathways may be dispensable in primary tumors but essential for metastasis (7, 8). Because new therapeutic targets are nominated from simple experimental models like cultured cells, it will be essential to define their context-specific roles in biologically accurate models of tumor initiation and progression.
METABOLIC REPROGRAMMING AND ONCOMETABOLITES IN CANCER
Altered metabolic activity supports anabolic growth during nutrient-replete conditions, catabolism to support cell survival during nutrient limitation, and fortification of redox homeostatic systems to counteract the metabolic effects of oncogene activation, tumor suppressor loss, and other stresses (9). Discovery and characterization of reprogrammed activities may provide opportunities to image tumor tissue noninvasively, predict tumor behavior, and prevent tumor progression by blocking essential pathways. It is important to differentiate “metabolic reprogramming” from “oncometabolites,” two terms widely used in the recent cancer metabolism literature (10). We propose that the term metabolic reprogramming be used to describe conventional metabolic pathways whose activities are enhanced or suppressed in tumor cells relative to benign tissues as a consequence of tumorigenic mutations and/or other factors. Oncometabolite is a relatively new term that refers to metabolites whose abundance increases markedly in tumors. We suggest that this term be reserved for metabolites for which (i) there is a clear mechanism connecting a specific mutation in the tumor to accumulation of the metabolite, and (ii) there is compelling evidence for involvement of the metabolite in the development of malignancy.
The classical example of a reprogrammed metabolic pathway in cancer is the Warburg effect or aerobic glycolysis (11). Glycolysis is a physiological response to hypoxia in normal tissues, but Otto Warburg in the 1920s observed that tumor slices and ascites cancer cells constitutively take up glucose and produce lactate regardless of oxygen availability, an observation that has been seen in many types of cancer cells and tumors (12). The increase in glycolytic flux allows glycolytic intermediates to supply subsidiary pathways to fulfill the metabolic demands of proliferating cells (11). Like glycolytic intermediates, tricarboxylic acid (TCA) cycle intermediates are also used as precursors for macromolecule synthesis (13). Their utilization in biosynthetic pathways requires that carbon be resupplied to the cycle so that intermediate pools are maintained; pathways that “refill” the cycle are termed anaplerotic pathways, and they generate TCA cycle intermediates that can enter the cycle at sites other than acetyl-CoA (coenzyme A) (14). Two activities that provide anaplerotic fluxes in cancer cells are glutaminolysis, which produces α-ketoglutarate from glutamine, and pyruvate carboxylation, which produces oxaloacetate from glucose/pyruvate. Oxidation of the branched-chain amino acids (BCAAs) isoleucine and valine also provides an anaplerotic flux in some tissues.
Despite the incredible genetic and histological heterogeneity of tumors, malignancy seems to involve the common induction of a finite set of pathways to support core functions like anabolism, catabolism, and redox balance (15). The general induction of these pathways may reflect their regulation by signaling pathways that are commonly perturbed in cancer cells (Fig. 1). Normal cells, upon stimulation by growth factors, activate phosphatidylinositol 3-kinase (PI3K) and its downstream pathways AKT and mammalian target of rapamycin (mTOR), thereby promoting a robust anabolic program involving increased glycolytic flux and fatty acid synthesis through activation of hypoxia-inducible factor–1 (HIF-1) and sterol regulatory element–binding protein (SREBP), respectively (16). Tumor cells very frequently contain mutations that allow the PI3K-AKT-mTOR network to achieve high levels of signaling with minimal dependence on extrinsic stimulation by growth factors (17). Many of the best-characterized oncogenes and tumor suppressors reside in the PI3K-AKT-mTOR network, and aberrant activation of this pathway is among the most frequent alterations seen in a diverse set of cancers.
Fig. 1. Signaling pathways that regulate cancer metabolism.
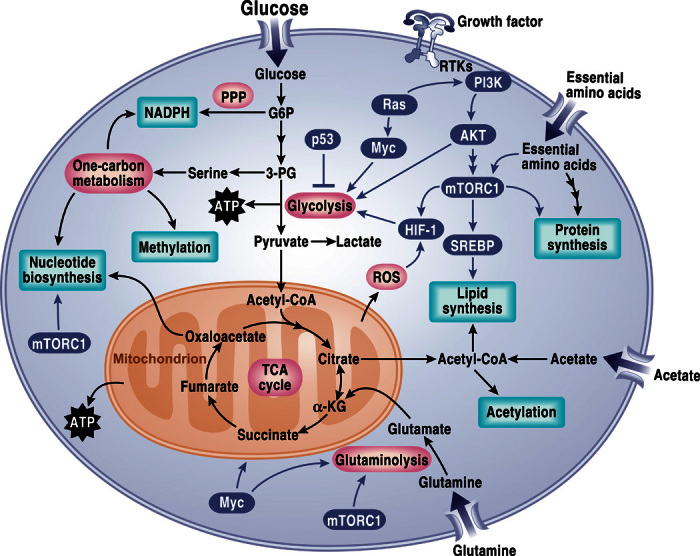
Tumor cells have aberrant activation of mTORC1 that induces an anabolic growth program resulting in nucleotide, protein, and lipid synthesis. Loss of tumor suppressors like p53 or activation of oncogenes like MYC further promotes anabolism through transcriptional regulation of metabolic genes. Metabolism controls signaling through regulating reactive oxygen species (ROS), acetylation, and methylation. PPP, pentose phosphate pathway; G6P, glucose-6-phosphate; 3-PG, 3-phosphoglycerate; ATP, adenosine 5´-triphosphate; mTORC1, mTOR complex 1; α-KG, α-ketoglutarate; RTK, receptor tyrosine kinase.
Another commonly deregulated pathway in cancer is gain of function of MYC by chromosomal translocations, gene amplification, and single-nucleotide polymorphisms. MYC increases the expression of many genes that support anabolic growth, including transporters and enzymes involved in glycolysis, fatty acid synthesis, glutaminolysis, serine metabolism, and mitochondrial metabolism (18). Oncogenes like Kras, which is frequently mutated in lung, colon, and pancreatic cancers, co-opt the physiological functions of PI3K and MYC pathways to promote tumorigenicity. Aside from oncogenes, tumor suppressors such as the p53 transcription factor can also regulate metabolism (19). The p53 protein–encoding gene TP53 (tumor protein p53) is mutated or deleted in 50% of all human cancers. The tumor-suppressive functions of p53 have been ascribed to execution of DNA repair, cell cycle arrest, senescence, and apoptosis. However, recent studies indicate that p53 tumor-suppressive actions might be independent of these canonical p53 activities but rather dependent on the regulation of metabolism and oxidative stress (20, 21). Loss of p53 increases glycolytic flux to promote anabolism and redox balance, two key processes that promote tumorigenesis (19).
A salient feature of many tumors is that they reside in a low-oxygen environment (hypoxia) ranging from 0 to 2% O2 because the tumor cell proliferation rate often exceeds the rate of new blood vessel formation (angiogenesis) (22). The metabolic adaptation to hypoxia is coordinated by HIF-1, which induces metabolic genes involved in increasing glycolytic flux (23). Some tumors display constitutive activation of HIF-1 under normoxic conditions through a variety of mechanisms, including (i) hyperactivation of mTORC1, (ii) loss of von Hippel–Lindau, (iii) accumulation of ROS, and (iv) accumulation of the TCA cycle metabolites succinate or fumarate due to cancer-specific mutations in succinate dehydrogenase (SDH) or fumarate hydratase (FH), respectively (24).
The robust coordinated induction of metabolic pathways that support tumorigenesis by combination of deregulation of PI3K-AKT-mTOR signaling pathways, loss of tumor suppressors, and activation of oncogenes alleviates the necessity of having mutations or amplifications in metabolic enzymes per se. Thus, examples of metabolic enzyme deregulation through genetic mutation are rare. One example is the elevated expression of phosphoglycerate dehydrogenase (PHGDH) due to amplification in a fraction of breast cancer and melanoma (25, 26). PHGDH catalyzes the conversion of the glycolytic intermediate 3-phosphoglycerate to 3-phosphohydroxypyruvate in the first step of the serine biosynthesis pathway. Serine metabolism supplies methyl groups to the one-carbon and folate pools contributing to nucleotide synthesis, methylation reactions, and NADPH (reduced nicotinamide adenine dinucleotide phosphate) production (27). Inhibiting serine biosynthesis by silencing PHGDH in cells with high levels of this enzyme results in growth suppression, and the enzyme displays oncogenic properties in gain of function assays (25, 26).
The other examples of mutational deregulation of metabolic enzymes are those that generate oncometabolites. The current list of true oncometabolites is short (28). The term is most commonly and appropriately applied to d-2-hydroxyglutarate (D2HG), a reduced form of the TCA cycle intermediate α-ketoglutarate. D2HG is scarce in normal tissues but rises to millimolar concentrations in tumors with mutations in isocitrate dehydrogenase 1 or 2 (IDH1 or IDH2). These mutations occur commonly in gliomas, acute myelogenous leukemias (AMLs), and other types of cancer (29–31). D2HG and its relationship to mutant IDH1 and IDH2 have been reviewed extensively elsewhere (32). The most relevant point here is that D2HG production requires a neomorphic enzyme activity imparted to IDH1/IDH2 by specific active-site mutations (33, 34). High levels of D2HG interfere with the function of dioxygenases requiring α-ketoglutarate as a cosubstrate. These include prolyl hydroxylases, cytosine hydroxylases, and histone demethylases, whose inhibition influences gene expression in part via an altered epigenetic state characterized by a failure to express differentiation programs (35–41). Thus, although D2HG arises from an alteration of the metabolic network, its role in cancer seems to depend on nonmetabolic effects. Currently, D2HG is being used as a biomarker for disease monitoring, and inhibitors specific to mutants IDH1/IDH2 are in clinical trials for AML and solid tumors.
The metabolite 2HG also exists as the enantiomer l-2HG (L2HG), which is not produced by mutant forms of IDH1/IDH2. This metabolite arises from the noncanonical activity of various dehydrogenases, including malate dehydrogenase and lactate dehydrogenase, whose promiscuous behavior reduces α-ketoglutarate to L2HG (42–44). L2HG may be oxidized back to α-ketoglutarate by a FAD-linked enzyme, L2HG dehydrogenase (L2HGDH). L2HGDH deficiency, also called L2HG aciduria, is a rare neurometabolic disease of childhood caused by the inheritance of biallelic mutations in the gene encoding L2HGDH (45). Affected children have seizures, mental retardation, white matter abnormalities of the brain, and systemically elevated levels of L2HG. Remarkably, a number of these children have developed malignant brain tumors (46), providing an early clue to the significance of D2HG in IDH1/IDH2-mutant gliomas and raising the question of whether L2HG is also an oncometabolite. L2HG and D2HG exhibit different effects on dioxygenase function (38), suggesting that the sensitivity of a particular tissue to the presence of either metabolite may depend on which dioxygenases are expressed. Recent work has demonstrated modest accumulation of L2HG in cells experiencing hypoxia or electron transport chain (ETC) dysfunction (42, 47) and in human renal cell carcinomas, which frequently display epigenetic silencing of L2HGDH (48). It is unknown whether reducing L2HG levels in these settings will promote cellular differentiation or suppress tumor progression.
The principle that oncometabolites exert their effects outside of the conventional metabolic network also applies to the other two molecules that can reasonably be called oncometabolites: fumarate and succinate (28). Both are TCA cycle intermediates found throughout the body, but some tumors accumulate massive levels of fumarate and/or succinate as a consequence of loss-of-function mutations in FH or the SDH complex, respectively (49–51). Although these mutations markedly reprogram metabolism by impairing TCA cycle flux, the extent to which fumarate and succinate participate in cancer development likely involves their nonmetabolic functions (28). Like D2HG, evidence indicates that succinate and fumarate interfere with dioxygenase activity, supporting the notion that a general property of oncometabolites is the ability to regulate epigenetics (52, 53). PHGDH overexpression and mutations in IDH1/IDH2, SDH, and FH all alter metabolite levels that control epigenetics (54). Several other metabolites, including acetyl-CoA, α-ketoglutarate, and S-adenosylmethionine also participate in epigenetic reprogramming, and time will tell whether genetically specific alterations of these metabolites in tumors promote tumorigenesis (54). Some metabolites, notably fumarate, covalently bind to sulfhydryl groups in glutathione, proteins, and peptides, altering their function and perhaps accounting for another mechanism by which oncometabolites promote or perpetuate malignant phenotypes (55–58).
BIOENERGETICS
Otto Warburg’s hypothesis that cancer cells take up glucose and generate a substantial amount of lactate in the presence of ambient oxygen due to impaired mitochondrial function led to the widely held misconception that cancer cells rely on glycolysis as their major source of ATP (59, 60). Today, it is clear that cancer cells exhibit aerobic glycolysis due to activation of oncogenes, loss of tumor suppressors, and up-regulation of the PI3K pathway, and that one advantage of high glycolytic rates is the availability of precursors for anabolic pathways (2, 61). Warburg’s observation that tumors display a high rate of glucose consumption has been validated in many human cancers with fluorodeoxyglucose positron emission tomography, which uses a radioactive glucose analog to image glucose uptake in tumors and adjacent normal tissue. Nevertheless, many studies have demonstrated that the great majority of tumor cells have the capacity to produce energy through glucose oxidation (that is, the process by which glucose-derived carbons enter the TCA cycle and are oxidized to CO2, producing ATP through oxidative phosphorylation). Furthermore, limiting glycolytic ATP production by inhibiting the activity of pyruvate kinase fails to prevent tumorigenesis, suggesting that the major role of glycolysis is not to supply ATP (62). Moreover, mitochondrial metabolism is necessary for cancer cell proliferation and tumorigenesis (63–65). Thus, despite their high glycolytic rates, most cancer cells generate the majority of ATP through mitochondrial function, with the likely exception of tumors bearing mutations in enzymes involved in mitochondrial respiration (for example, SDH and FH) (66). Nevertheless, cells harboring mutations in FH or SDH still rely on mitochondrial metabolism by metabolically rewiring themselves to provide the necessary TCA cycle intermediates and ROS for cell proliferation (55, 67–70).
In addition to pyruvate derived from glycolysis, fatty acids and amino acids can supply substrates to the TCA cycle to sustain mitochondrial ATP production in cancer cells (Fig. 2). The breakdown of fatty acids (β-oxidation) in the mitochondria generates acetyl-CoA and the reducing equivalents NADH and FADH2, which are used by the ETC to produce mitochondrial ATP. The amino acid glutamine can generate glutamate and subsequently α-ketoglutarate to fuel the TCA cycle through a series of biochemical reactions termed glutaminolysis (71). Furthermore, the BCAAs isoleucine, valine, and leucine, which are elevated in plasma of patients with pancreatic cancers, can be converted into acetyl-CoA and other organic molecules that also enter the TCA cycle (72). The metabolic flexibility afforded by multiple inputs into the TCA cycle allows cancer cells to adequately respond to the fuels available in the changing microenvironment during the evolution of the tumor (9). A combination of the local tumor microenvironment and oncogenic lesions is likely to dictate the fuel used by mitochondria to sustain tumor growth.
Fig. 2. Metabolic pathways under nutrient-replete and nutrient-deprived conditions.
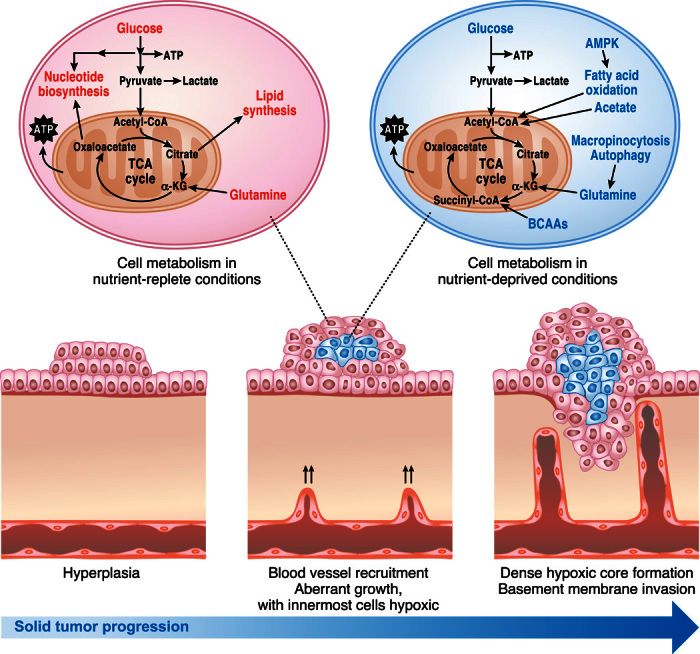
Accessibility to nutrients within solid tumors is regulated by proximity to the vasculature. Cells located adjacent to the vasculature use nutrients and oxygen to fuel anabolic pathways that support proliferation. However, cells distant from the vasculature have diminished accessibility to nutrients and oxygen and may engage in alternative forms of metabolism including oxidation of fatty acids and BCAAs as well as macromolecular degradation through autophagy and macropinocytosis to support cell viability.
Solid tumors contain significant heterogeneity of perfusion, such that many tumor cells reside in nutrient- and oxygen-poor environments. Cancer cells have therefore adapted multiple mechanisms to sustain mitochondrial function for survival. For example, the mitochondrial ETC can function optimally at oxygen levels as low as 0.5% (73). Moreover, hypoxic tumor cells (<2% O2) can continue to cycle and use glutamine as a fuel for oxidative ATP production (74–76). Kras-driven pancreatic cancer cells in nutrient-depleted conditions use proteins scavenged from the extracellular space to produce glutamine and other amino acids to fuel the TCA cycle for cell survival and growth (Fig. 2) (77). Furthermore, if pyruvate oxidation to acetyl-CoA is compromised by hypoxia or ETC impairment, glutamine can provide acetyl-CoA as a biosynthetic precursor to sustain tumor growth (69, 78, 79).
When tumor cells become nutrient-deprived, they adapt to the microenvironment by decreasing their demand for ATP. The resultant increase in ATP availability maintains an adequate ATP/ADP (adenosine 5´-diphosphate) ratio to drive unfavorable biological reactions. The anabolic kinase mTOR, discussed in greater detail below, drives the energetically demanding growth of tumor cells. This kinase is inhibited when amino acids and oxygen levels are diminished (80). Furthermore, decreased mTOR activity increases autophagic flux. In oncogenic Kras- or Braf-driven non–small-cell lung cancer (NSCLC) cells, autophagy provides an intracellular glutamine supply to sustain mitochondrial function (81, 82). To survive the hypoxic tumor microenvironment, cancer cells also diminish their demand for ATP by decreasing highly demanding ATP-dependent processes, such as running the Na/K-dependent adenosine triphosphatase. If diminishing ATP demand is not sufficient to maintain ATP/ADP ratio, the rise in ADP activates adenylate kinase, a phosphotransferase enzyme that buffers the fall in ATP levels by converting two ADP molecules into adenosine 5´-monophosphate (AMP) and ATP (83). The rise in AMP during nutrient deprivation triggers the activation of AMP kinase (AMPK), which activates catabolic pathways like fatty acid oxidation to stimulate ATP production (84). In conditions of metabolic stress, certain Ras-driven cancer cells scavenge lipids to support ATP production (85). Ovarian cancer cells use fatty acids from neighboring adipocytes to fuel mitochondrial ATP production (86). Thus, there are multiple mechanisms by which cancer cells maintain their ATP/ADP ratio to sustain viability in nutrient- and oxygen-poor environments.
BIOSYNTHESIS OF MACROMOLECULES
Biosynthetic or anabolic pathways are an essential aspect of cancer metabolism because they enable cells to produce the macromolecules required for replicative cell division and tumor growth. As a general theme, these pathways involve the acquisition of simple nutrients (sugars, essential amino acids, etc.) from the extracellular space, followed by their conversion into biosynthetic intermediates through core metabolic pathways like glycolysis, the PPP, the TCA cycle, and nonessential amino acid synthesis, and finally the assembly of larger and more complex molecules through ATP-dependent processes (Fig. 3). The three macromolecular classes most commonly studied in cancer metabolism are proteins, lipids, and nucleic acids, which comprise approximately 60, 15, and 5% of the dry mass of mammalian cells, respectively. Evidence indicates that biosynthesis of all three classes is under the control of the same signaling pathways that govern cell growth and are activated in cancer via tumorigenic mutations, particularly PI3K-mTOR signaling.
Fig. 3. Anabolic pathways that promote growth.

Glucose metabolism generates glycolytic intermediates that can supply subsidiary pathways including the hexosamine pathway, PPP, and one-carbon metabolism, all of which support cell growth. Mitochondrial TCA cycle intermediates such as oxaloacetate (OAA) and citrate are used to generate cytosolic aspartate and acetyl-CoA for nucleotide and lipid synthesis, respectively. Mitochondria also generate H2O2 and acetyl-CoA for redox signaling and acetylation, respectively. NADPH is used to drive anabolic reactions and to maintain antioxidant capacity. Cytosolic sources of NADPH include the oxidative PPP, IDH1, and enzymes from one-carbon metabolism including MTHFD1. Mitochondrial sources of NADPH include MTHFD2, MTHF2L, and IDH2. HK2, hexokinase 2; G6PDH, glucose-6-phosphate dehydrogenase; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; LDH, lactate dehydrogenase; ACLY, ATP citrate lyase; GLS, glutaminase; SHMT, serine hydroxymethyltransferase; MTHFD2, methylenetetrahydrofolate dehydrogenase 2; MTHFD2L, MTHFD2-like; ACSS2, acyl-CoA synthetase short-chain family member 2; THF, tetrahydrofolate.
Protein biosynthesis is highly regulated and requires access to a full complement of essential and nonessential amino acids. Cancer cells and other cells under the influence of growth factor signaling express surface transporters that allow them to acquire amino acids from the extracellular space (87). This not only provides cells with the raw materials needed for protein synthesis but also allows them to maintain activity of the mTOR signaling system, specifically mTORC1. mTORC1 is stimulated by the presence of amino acids and activates protein synthesis via its effects on translation and ribosome biogenesis (80). Most nonessential amino acids are produced through transamination reactions, which transfer the amino group from glutamate to a ketoacid. Proliferating cancer cells take up glutamine and convert it to glutamate through a variety of deamidation and transamidation reactions, most notably the mitochondrial amidohydrolase glutaminase (71). Together, these enzymes generate a large intracellular glutamate pool available for nonessential amino acid synthesis. Both glutamine uptake and glutaminase activity are stimulated by mTORC1, providing glutamate for transamination reactions and/or maintenance of the TCA cycle, which also contributes to amino acid synthesis. Furthermore, when the intracellular glutamine supply exceeds the cell’s demands, glutamine can be exported in exchange for essential amino acids to stimulate mTORC1 and protein synthesis (88). Thus, growth conditions in which glutamine and essential amino acids are abundant enable mTORC1-mediated activation of protein synthesis.
When nutrients are scarce, cells have access to a number of catabolic pathways to degrade macromolecules and resupply key pools of intracellular metabolic intermediates. Protein degradation pathways have been characterized extensively as mechanisms to supply amino acids in cancer cells. Intracellular proteins and other macromolecules can be recycled through autophagy, a highly regulated process through which proteins and organelles are delivered to the lysosome and degraded (89). Autophagy is an essential survival pathway during nutrient or growth factor deprivation, and genetic studies demonstrate that it contributes to some forms of cancer in mice (90, 91). However, because autophagy supplies amino acids through protein degradation, it does not serve as a source of net protein synthesis. It is also potently suppressed by mTORC1. Macropinocytosis allows cells to internalize proteins and other components of the extracellular milieu and deliver them for degradation via the endocytic pathway. Under conditions of nutrient depletion, macropinocytosis supplies both nitrogen and carbon to central metabolic pathways (92). Evidence indicates that extracellular protein degradation, like autophagy, is suppressed by mTORC1 (93). Suppressing pathways of protein degradation may help maximize rates of net protein synthesis when extracellular amino acids are available and mTORC1 is active.
Tumor cells rapidly produce fatty acids for membrane biosynthesis, lipidation reactions, and cellular signaling. Fatty acid synthesis requires sources of acetyl-CoA and reducing power in the form of cytosolic NADPH; effective fatty acid synthesis therefore requires integration with other pathways of carbon and redox metabolism. In most cultured cells, glucose is the most prominent acetyl-CoA source for fatty acid synthesis (94, 95). Glutamine and acetate have been demonstrated to provide alternative carbon sources when access to glucose-derived acetyl-CoA is impaired by hypoxia or mitochondrial dysfunction (69, 78, 79, 96). Leucine degradation can also supply acetyl-CoA in some cell lines (97). The relative importance of these nutrients for fatty acid synthesis in vivo is unknown, although early studies suggested that most fatty acyl carbon in experimental tumors is derived from glucose (98, 99). Isotopic tracing experiments designed to assess the cytosolic NADPH pool have recently suggested that most NADPH used for fatty acid synthesis arises from the PPP (100, 101).
Transcription of genes involved in fatty acid synthesis is regulated by the SREBP-1 transcription factor (102). SREBP-1 regulates not only the enzymes needed to convert acetyl-CoA into fatty acids but also the enzymes of the PPP and pathways required to convert acetate and glutamine into acetyl-CoA (103). This transcription factor therefore regulates genes encoding proteins that catalyze or facilitate fatty acid synthesis. In lipid-replete conditions, SREBP-1’s transcriptional activity is suppressed by its sequestration in the endoplasmic reticulum. Under conditions of sterol depletion, proteolytic cleavage releases the transcriptionally active domain, which travels to the nucleus and binds to sterol response elements in the promoters of lipogenic genes (104).
In cancer cells with constitutively high rates of fatty acid synthesis, additional mechanisms help keep SREBP-1 in a transcriptionally active state. mTORC1 signaling, via its effector S6 kinase (S6K), activates a transcriptional program that includes both SREBP-1 and the related protein SREBP-2, which regulates transcription of genes in sterol biosynthesis (103). Both SREBP-1 and SREBP-2 are required for mTORC1-mediated cell proliferation. The mechanism of SREBP activation by mTORC1 is incompletely understood but involves nuclear entry of the phosphatidic acid phosphatase Lipin-1, which enhances nuclear SREBP abundance and activity on the promoters of lipogenic genes (105).
Both fatty acids and lipids can also be acquired from the extracellular space to supply membrane biosynthesis. PI3K signaling activates fatty acid uptake and suppresses fatty acid oxidation, thereby maximizing lipogenesis in proliferating cells under the control of growth factors (106). Lipid uptake may acquire further importance during conditions of metabolic stress, when the ability to meet oncogene-driven demands for biosynthesis is compromised. The ability to scavenge lysophospholipids (lipid intermediates containing a glycerophosphate backbone with one acyl chain) is required for maximal cancer cell growth during hypoxia, which suppresses de novo fatty acid synthesis from glucose (85). Furthermore, in cancer cells with constitutive mTORC1 signaling, hypoxia induces a state of dependence on access to extracellular desaturated fatty acids to maintain endoplasmic reticulum integrity in support of protein biosynthesis (107). Notably, SREBP-1 was first identified as the transcription factor responsible for expression of the low-density lipoprotein receptor (LDLR) (108), implying that enhanced de novo lipogenesis occurs concomitantly with enhanced import of lipids from the extracellular space. These parallel pathways appear to be essential in glioma, where oncogenic activation of epidermal growth factor receptor (EGFR) signaling stimulates SREBP-1 and expression of LDLR (109). These cancer cells are highly sensitive to inhibitors of fatty acid and cholesterol biosynthesis. Inhibition of the EGFR-PI3K signaling axis but not of mTORC1 suppresses nuclear translocation of SREBP-1 in glioma cells with oncogenic EGFR, suggesting an alternate, mTORC1-independent mode of SREBP-1 activation in glioma cells (109). This transcriptional program includes LDLR expression and induces reliance on cholesterol uptake to maintain the intracellular pool (110). Impairing intracellular cholesterol availability by activating liver X receptor induced cell death both in culture and in vivo, suggesting a pharmacological approach to silence lipogenic programs in glioma (110).
Purine and pyrimidine nucleotides are required for synthesis of RNA and DNA. De novo biosynthesis of nucleotides is complex, requiring input from several pathways in a coordinated fashion. The phosphoribosylamine backbone of these molecules is produced from ribose-5-phosphate, an intermediate of the PPP, and an amide donation reaction using glutamine as a substrate (111). The purine and pyrimidine bases are constructed from various nonessential amino acids and methyl groups donated from the one-carbon/folate pool. The TCA cycle contributes oxaloacetate, which is transaminated to aspartate, an intermediate required to synthesize both purine and pyrimidine bases. Conversion of ribonucleotides to deoxynucleotides by ribonucleotide reductase requires a source of NADPH. Well-characterized mechanisms of feedback inhibition exist to prevent excessive accumulation of nucleotides, and mutations interrupting these mechanisms can produce pathological accumulation of nucleotide intermediates (for example, precipitation of uric acid crystals in gout).
Clearly, nucleotide biosynthesis is a targetable vulnerability in cancer cells because nucleoside analogs and antifolates have been a mainstay of chemotherapeutic regimens for decades (112). However, relatively little is known about how oncogenic signaling interfaces with nucleotide biosynthesis. It is likely that the effects of numerous signaling pathways on glucose and amino acid metabolism influence the availability of precursors for purines and pyrimidines. In the case of mTORC1, evidence points to a distinct mechanism by which activation of the signaling pathway enables nucleotide biosynthesis. The mTORC1 effector ribosomal S6K1 phosphorylates the trifunctional enzyme CAD (carbamoyl-phosphate synthetase 2, aspartate transcarbamoylase, dihydroorotase), which catalyzes the first three steps of pyrimidine synthesis (113). Phosphorylation on CAD S1859 is required for mTORC1-dependent stimulation of pyrimidine biosynthesis (113). Additional work is needed to determine how other aspects of de novo nucleotide synthesis, purine and pyrimidine salvage pathways, and accessory activities like folate metabolism are regulated in cancer cells in support of cell proliferation.
REDOX BALANCE
ROS are intracellular chemical species that contain oxygen and include the superoxide anion (O2−), hydrogen peroxide (H2O2), and the hydroxyl radical (OH·) (114). The mitochondria and cytosolic NADPH oxidases (NOXs) produce O2− from the one-electron reduction of oxygen (115). O2− is converted into H2O2 by the enzymatic activity of superoxide dismutase 1 or 2, which are localized to the cytosol or mitochondrial matrix, respectively. H2O2 is subsequently detoxified to water by the enzymatic activity of mitochondrial and cytosolic peroxiredoxins (PRXs), which, as a consequence, undergo H2O2-mediated oxidation of their active-site cysteines (116). Thioredoxin (TXN), thioredoxin reductase (TrxR), and the reducing equivalent NADPH reduce oxidized PRXs to complete the catalytic cycle (117). Glutathione peroxidases (GPXs) can also convert H2O2 to water in the mitochondrial matrix and cytosol through H2O2-mediated oxidation of reduced glutathione (GSH) (118). Glutathione reductase (GR) and NADPH reduce oxidized glutathione (GSSG) back to GSH. Additionally, catalase, an abundant antioxidant in peroxisomes, can detoxify H2O2 to water without any cofactors. However, in the presence of ferrous or cuprous ions, H2O2 can become OH· and quickly cause the oxidation of lipids, proteins, and DNA, resulting in cellular damage. NADPH is required to maintain multiple antioxidant defense systems. The cytosol has multiple sources of NADPH generation, including the oxidative PPP, malic enzyme 1, IDH1, and one-carbon metabolism. NADPH generation in the mitochondria, in part, is controlled by one-carbon metabolism and IDH2.
Historically, ROS have been thought of as lethal metabolic by-products of cellular respiration and protein folding. However, studies over the past two decades have unveiled a previously underappreciated role of ROS in cellular signaling. Low levels of ROS, particularly H2O2, can reversibly oxidize the cysteine residues of proteins to positively regulate cell proliferation and cellular adaptation to metabolic stress (119). As H2O2 levels increase, however, cell death signaling pathways are initiated, and H2O2 is converted to OH·, which can directly damage DNA, proteins, and lipids. Cancer cells have an increased rate of spatially localized mitochondria- and NOX-dependent ROS production compared to normal cells. This allows for the proximal activation of signaling pathways [PI3K and mitogen-activated protein kinase/extracellular signal–regulated kinase (MAPK/ERK)] and transcription factors [HIF and nuclear factor κB (NF-κB)] necessary for tumorigenesis. The cancer cell–specific increased rate of spatially localized ROS production is due to a combination of oncogenic lesions and the tumor microenvironment. For example, the activation of oncogenes, PI3K signaling pathway induction, and hypoxia (low-oxygen levels) stimulate the increased rate of ROS production from the mitochondria and NOXs in cancer cells (120–122). Thus, mitochondria-targeted antioxidants and NOX inhibitors can prevent cancer cell proliferation, hypoxic activation of HIF, tumorigenesis, and metastasis (64, 123–125).
The increased localized ROS in cancer cells, which activates signaling pathways and transcription factors to promote tumorigenesis, needs to be buffered from reaching levels of ROS that incur cellular damage by the increased expression of antioxidant proteins (126). Thus, cancer cells have higher levels of ROS scavenging enzymes than normal cells, preventing ROS-mediated activation of death-inducing pathways like c-Jun N-terminal kinase (JNK) and p38 MAPK and oxidation of lipids, proteins, and DNA, resulting in irreversible damage and cell death. One mechanism by which cancer cells increase their antioxidant capacity is by activating the transcription factor nuclear factor (erythroid-derived 2)–related factor-2 (NRF2) (127). Specifically, NRF2 is activated following disruption of the interaction of NRF2 with its binding partner Kelch-like ECH-associated protein 1 (KEAP1). Critical cysteine residues within KEAP1 can undergo oxidation, succination, and glutathionylation, thereby inhibiting the KEAP1-NRF2 interaction, leading to the proteasomal degradation of NRF2. Additionally, NRF2 activation can occur independently of KEAP1 (128). Once activated, NRF2 induces the transcription of many antioxidant proteins including GPXs and TXNs as well as enzymes involved in GSH synthesis and cysteine import through the cysteine/glutamate antiporter. Furthermore, to maintain the antioxidant capacity of GPXs and TXNs, NADPH is required. NRF2 plays an important role in activating enzymes that increase cytosolic NADPH levels. NRF2 also regulates the serine biosynthesis pathway, generating NADPH in the mitochondria, which is critical for redox balance under hypoxic conditions (129, 130). Therefore, inactivating NRF2 or disabling antioxidant proteins in cancer cells would allow for the accumulation of excessive amounts of ROS to levels that initiate toxicity and reduce tumorigenesis (128, 131, 132).
During tumorigenesis and metastasis, redox homeostasis is required (Fig. 4). An emerging model of redox balance is that as a tumor initiates, the metabolic activity of cancer cells is increased, resulting in an increase in ROS production and subsequent activation of signaling pathways that support cancer cell proliferation, survival, and metabolic adaptation (126). Accordingly, to prevent toxic levels of ROS, tumor cells increase their antioxidant capacity to allow for cancer progression (133). The harsh tumor microenvironment increases ROS levels due to hypoxia, and the low glucose levels limit flux through the cytosolic oxidative PPP, thus decreasing cytosolic NADPH levels. Cells in these nutrient-deprived conditions activate AMPK to increase NADPH levels by stimulating PPP-dependent NADPH and diminishing anabolic pathways, such as lipid synthesis, that require high levels of NADPH (134, 135). ROS-dependent signaling and increased mitochondrial respiration are also necessary for tumor metastasis (124, 136). However, when tumor cells detach from a matrix, they encounter high levels of ROS that incur cellular damage and require activation of adaptive ROS-mitigating pathways to survive and grow (137, 138). The ability to up-regulate antioxidant proteins and increase flux through NADPH-producing metabolic pathways enables distant metastasis to occur (8). These findings suggest that perhaps disabling antioxidant capacity in cancer cells to raise ROS levels might be beneficial in preventing metastasis.
Fig. 4. Cancer cells maintain redox balance.
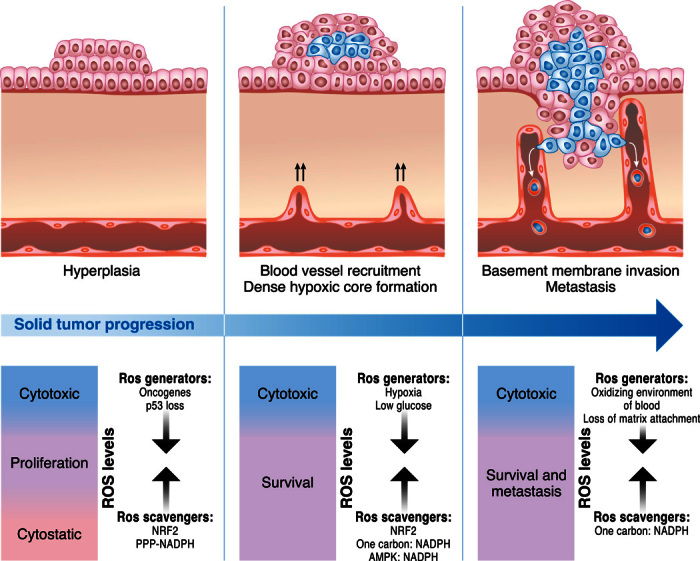
Cancer cells have increased rates of ROS production due to activation of oncogenes and loss of tumor suppressors that promote signaling pathways supporting proliferation and survival. However, cancer cells prevent the buildup of ROS to levels that incur damage by increasing antioxidant capacity through induction of NRF2-dependent genes and, in glucose replete conditions, the use of PPP to generate NADPH. As cells encounter hypoxia and low glucose due to limited vasculature accessibility, the levels of ROS further increase, requiring AMPK and one-carbon metabolism to enhance NADPH production to raise antioxidant capacity. Loss of matrix attachment and escape of cancer cells into the blood for dissemination to distant sites incur further increases in ROS levels, which require additional enhancements of antioxidant defenses to avoid cell death. It is important to note that too little ROS or too high steady-state ROS levels within cancer cells result in failure for solid tumor progression and metastasis.
TARGETING METABOLISM FOR CANCER THERAPY
There are a few things to consider when determining what makes a good metabolic target for cancer therapy. First, inhibition of some metabolic enzymes is likely to be systemically toxic because of their physiological functions in normal tissues (139). The feasibility of targeting these pathways therapeutically depends on whether systemic blockade of the pathway can be tolerated. Normal proliferating cells, such as immune cells and stem cells, also reprogram their metabolism in a manner similar to cancer cells (140, 141). Metabolic inhibitors should likely not interfere with the adaptive immune system. Nevertheless, there are excellent examples of pathways whose reprogramming does provide an adequate therapeutic window in cancer. Enhanced nucleotide and DNA synthesis in tumor cells is targeted by antifolates (methotrexate, pemetrexed, and others) (112). Although these drugs do produce toxicity in normal proliferative tissues like the intestinal epithelium and bone marrow, they are essential components of highly successful chemotherapeutic regimens. Thus, it is critical to elucidate in normal cells any toxic effects of metabolic enzyme inhibition. To circumvent this challenge, one approach is to target a metabolic enzyme in a deregulated pathway specific to cancer cells. To date, many of the genetic and pharmacologic interventions on metabolic enzymes have been conducted using human cancer cells subcutaneously injected into athymic mice. Therefore, it will be important for future experiments to not only use patient-derived xenograft (PDX) models but also make use of genetically engineered mouse cancer models and syngeneic mouse models that have intact immune systems, especially given the promising results from immunotherapy. An emerging theme is that cancer cells display metabolic plasticity and can alter their metabolic profile during the course of tumorigenesis and metastasis. Thus, it is conceivable that cancer cells could develop resistance to inhibition of a particular metabolic pathway by expressing alternate protein isoforms or up-regulating compensatory pathways. Therefore, a rational cancer therapeutic strategy should involve targeting multiple metabolic pathways simultaneously or targeting a particular metabolic pathway in combination with therapies against oncogenic or signaling pathways. Here, we highlight a few promising metabolic targets in glycolytic, one-carbon, mitochondrial, and redox metabolism.
Glycolysis was an early attractive target for cancer therapy given the clinical observation that many tumors exhibit a significant increase in glucose uptake compared with adjacent normal tissue (112). LDH-A, a metabolic enzyme that converts pyruvate (the final product of glycolysis) to lactate, was identified as the first metabolic target of the oncogene MYC (142). Genetic or pharmacologic inhibition of LDH-A has been shown to diminish MYC-driven tumors in xenograft models (143, 144). Furthermore, recent studies indicate that inhibition of LDH-A leads to the regression of established tumors in genetically engineered mouse models of NSCLC without systemic toxicity (145). Genetic ablation of LDH-A also delays the progression of myeloid leukemia (146). Thus, the increased expression of LDH-A, specifically in MYC-mutant cancer cells, may prove to be an attractive target. Another potential therapeutic target is the glycolytic protein HK2. Many tumor cells overexpress HK2, and preclinical mouse models of genetically engineered NSCLC and breast cancer demonstrate that HK2 inhibition delays tumor progression (3). Furthermore, systemic HK2 deletion in mice does not cause adverse physiological consequences. However, the effect of LDH-A and HK2 on the adaptive immune system is currently unknown. Lactate has been shown to inhibit cytotoxic T cells; thus, LDH-A inhibition may cooperate with immune checkpoint inhibitors to unleash host inflammatory T cells that will specifically attack tumor cells (147). Lactate can also reprogram macrophages to promote tumorigenesis (148). Thus, it may be efficacious to target LDH-A or HK2 in highly glycolytic tumors that overexpress these proteins.
Another potential glucose-dependent target is PHGDH, an enzyme in the de novo serine synthesis pathway. High levels of PHGDH have been found in a subset of human melanoma and breast cancers, and these cancer cells require PHGDH for their growth in vitro (25, 26). Serine starvation in mice diminishes tumorigenicity of p53-null cancers (149). De novo synthesis or exogenous uptake of serine can enter the mitochondria where SHMT2 converts it into glycine to generate folate intermediates (101, 150). In many cancer types, SHMT2 expression is elevated and correlates with a poor prognosis. Furthermore, the transcription factors MYC and HIF induce SHMT2 under hypoxia to promote survival (130, 151). Currently, it is not known whether targeting PHGDH, SHMT2, or other enzymes in the one-carbon metabolism pathway would be effective in delaying or regressing tumor progression in genetically engineered, PDX, or syngeneic mouse models of cancer without incurring systemic toxicity. However, given the importance of one-carbon metabolism in supporting the anabolic needs of cancer cells and its up-regulation in cancer cells, it is likely that this pathway is needed for in vivo tumor progression (152).
Mitochondrial metabolism has also emerged as a key target for cancer therapy, in part, due to the revelation that the antidiabetic drug metformin is an anticancer agent (153). Numerous epidemiological studies first suggested that diabetic patients taking metformin, to control their blood glucose levels, were less likely to develop cancer and had an improved survival rate if cancer was already present (154). Laboratory-based studies have also provided evidence that metformin may serve as an anticancer agent (155–157). Biochemists recognized that metformin reversibly inhibits mitochondrial complex I (158–160). Recent studies indicate that metformin acts as an anticancer agent by inhibiting mitochondrial ETC complex I (161). Specifically, metformin inhibits mitochondrial ATP production, inducing cancer cell death when glycolytic ATP levels diminish as a result of limited glucose availability. Metformin also inhibits the biosynthetic capacity of the mitochondria to generate macromolecules (lipids, amino acids, and nucleotides) within cancer cells (162). The remarkable safety profile of metformin is due to its uptake by organic cation transporters (OCTs), which are only present in a few tissues, such as the liver and kidney (163). Certain tumor cells also express OCTs to allow the uptake of metformin (164). However, in the absence of OCTs, tumors would not accumulate metformin to inhibit mitochondrial complex I. Ongoing clinical trials using metformin as an anticancer agent should assess the expression levels of OCTs to identify the tumors with highest expression, which are likely to be susceptible to metformin. Moreover, it is not clear whether the current antidiabetic dosing of metformin used in clinical trials allows for metformin accumulation to levels necessary to inhibit mitochondrial complex I in tumors. Thus, it is possible that metformin at doses higher than those currently used for diabetes might be more efficacious without causing toxicity. Like metformin, the biguanide phenformin exhibits anticancer properties by inhibiting mitochondrial complex I (165). In contrast to metformin, phenformin is readily transported into tumor cells and has been withdrawn from human use in most parts of the world due to its clinical increase in the incidence of lactic acidosis. However, it is worth considering phenformin as a possible cancer therapy because lactic acidosis can be monitored. Biguanide sensitivity can be improved in mice starved for serine or in tumors that have lost p53 or LKB1 (155, 166, 167). Thus, biguanides, and possibly other mitochondrial complex I inhibitors, may be effective anticancer agents.
Another potential therapeutic strategy to inhibit mitochondrial metabolism in certain tumors would be to use autophagy or glutaminase inhibitors. Autophagy provides amino acids, such as glutamine, that fuel the TCA cycle in NSCLC and pancreatic cancers, and short-term autophagy inhibition has been shown to decrease tumor progression without incurring systemic toxicity in mouse models of NSCLC (168, 169). Some tumors are addicted to using glutamine to support TCA cycle metabolism even in the absence of autophagy; thus, glutaminase inhibitors can reduce tumor burden in these models (4, 75, 170). An alternative approach is to target acetate metabolism. Although a major function of the mitochondria is to provide acetyl-CoA to the cell, cancer cells can also use acetate to support cell growth and survival during metabolic stress (hypoxia or nutrient deprivation) (96, 171). The cytosolic enzyme acetyl-CoA synthase 2 (ACCS2), which converts acetate to acetyl-CoA, is dispensable for normal development; thus, ACCS2 is a promising target of acetate metabolism. ACCS2 knockout mice do not display overt pathologies, but genetic loss of ACCS2 reduces tumor burden in models of hepatocellular carcinoma (171). Human glioblastomas can oxidize acetate and may be sensitive to inhibitors of this process (172). Thus, targeting metabolism with inhibitors of autophagy, acetate metabolism, and other pathways that supply key metabolic intermediates may be efficacious in some contexts.
Because mitochondrial inhibitors are unlikely to be effective cancer therapies as single agents, combination therapy is likely the best approach. For example, the use of metformin with the current clinical PI3K inhibitors, which reduce glucose uptake and glycolysis (173), is one approach that would impair both sources of ATP within cells. Targeted therapies against oncogenes such as KRAS, BRAF, and NOTCH1 kill a large majority of cancer cells but ultimately yield resistant cells that exhibit an increased sensitivity to inhibitors that impair mitochondrial metabolism (174–176). Cancer-initiating cells also have increased sensitivity to mitochondrial inhibitors, adding further evidence that inhibiting mitochondrial metabolism may suppress tumor recurrence (177, 178).
Furthermore, to counterbalance the increased production of ROS encountered during tumorigenesis and metastasis, cancer cells increase their antioxidant capacity (126). Thus, an additional therapeutic approach is to target redox metabolism, that is, selectively disable the antioxidant capacity of cancer cells causing ROS levels to rise and induce cancer cell death (133). The reducing equivalent NADPH is required to maintain multiple antioxidant defense systems. The cytosol has multiple sources of NADPH generation, including the oxidative PPP, malic enzyme 1, IDH1, and one-carbon metabolism. By contrast, NADPH generation in the mitochondria is controlled in part by one-carbon metabolism and IDH2. Many of these NADPH-generating systems are critical for normal cell survival and function. Nevertheless, there are two NADPH-generating systems that may serve as potential therapeutic targets. It is estimated that 400 million people worldwide are deficient in G6PDH, an enzyme in the oxidative PPP that converts NADP+ to NADPH. However, certain tumors rely on this pathway as a major source of cytosolic NADPH; therefore, it may be therapeutic to disable this pathway and induce oxidative stress and diminish tumor growth. Moreover, RNA profiling of metabolic enzymes identified the mitochondrial one-carbon metabolism protein MTHFD2, which can generate NADPH, as being highly expressed in 19 different cancer types but not in normal adult proliferating cells (152). Loss of MTHFD2 in cancer cells increases ROS levels and sensitizes the cells to oxidant-induced cell death in vitro. An interesting approach to depleting NADPH levels and increasing ROS is to administer high doses of vitamin C (ascorbate). Vitamin C is imported into cells through sodium-dependent vitamin C transporters, whereas the oxidized form of vitamin C, dehydroascorbate (DHA), is imported into cells through glucose transporters such as GLUT1 (179). When the cell takes up DHA, it is reduced back to vitamin C by glutathione (GSH), which consequently becomes GSSG. Subsequently, GSSG is converted back to GSH by NADPH-dependent GR. Because the blood is an oxidizing environment, vitamin C becomes oxidized to DHA before being taken up by the cell. Thus, high doses of vitamin C diminish the tumorigenesis of colorectal tumors that harbor oncogenic KRAS mutations and express high levels of GLUT1 by depleting the NADPH and GSH pools and consequently increasing ROS levels to induce cancer cell death (179, 180). Vitamin C administered at high doses intravenously is safe in humans and, in conjunction with conventional paclitaxel-carboplatin therapy, demonstrated a benefit in a small number of patients (181). Additional strategies to diminish GSH include the administration of buthionine sulfoximine, an irreversible inhibitor of γ-glutamylcysteine synthetase, which can be safely administered to humans and is efficacious in preclinical tumor models (182). Moreover, glutathione is a tripeptide consisting of cysteine, glutamate, and glycine. Thus, decreasing glutamate levels using glutaminase inhibitors or diminishing cysteine levels by preventing extracellular cysteine (two linked cysteine molecules) uptake can also raise ROS levels in cancer cells to induce cell death.
An important consideration is that normal stem cells are sensitive to ROS levels; thus, it is important to stratify patients on the basis of their expression levels of a particular antioxidant protein or antioxidant pathway. It is critical to determine which antioxidant pathways are likely up-regulated as a result of the high rate of ROS production within cancer cells. Many cancer types use the NRF2 pathway to maintain redox balance; therefore, targeting this pathway may provide a viable therapeutic opportunity (128). Additionally, superoxide dismutase 1 (SOD1) is overexpressed in NSCLC, and its inhibition kills human NSCLC cells and decreases the tumor burden in mouse models of NSCLC (183). Because NRF2 and SOD1 knockout mice develop normally, short-term inhibition of these pathways might be an effective way to kill cancer cells.
TECHNOLOGIES ENABLING DISCOVERY IN CANCER METABOLISM
Many recent advances in our understanding of cancer metabolism have been propelled by advanced technologies to detect metabolites and metabolic activities (184). A key concept is that quantifying metabolites (that is, metabolomics) is a more distinct form of metabolic analysis than measuring the activities of metabolic pathways [that is, metabolic flux analysis (185)]. Although these two approaches can provide complementary types of information, they are not interchangeable. One cannot infer metabolic activity from changes in metabolite levels, and altered metabolic fluxes may or may not cause changes in metabolite levels (186). Both of these approaches have provided important recent insights into cancer metabolism, and using the two techniques together provides the most complete assessment of metabolic phenotypes.
Metabolomics experiments seek to characterize and quantify the metabolites in a biological sample, usually by nuclear magnetic resonance (NMR) or, more commonly, mass spectrometry. Depending on the methods of extraction, separation, and detection, metabolomics experiments may focus on particular classes of metabolites or provide a comprehensive analysis of as many metabolites as possible. Targeted approaches typically detect a few dozen to a few hundred molecules, whereas untargeted analyses may detect more than 1000. Detecting alterations of metabolite levels in cancer can be extremely valuable. The massive accumulation of D2HG in IDH1-mutant gliomas was initially discovered through a metabolomics approach (33). Because altered metabolite levels can be detected noninvasively using 1H magnetic resonance spectroscopy (MRS), perturbed metabolite levels discovered through metabolomics can sometimes be translated into clinical diagnostic techniques. Elevated levels of lactate, choline, glycine, and other metabolites are detected by MRS in glioma. More recently, MRS techniques have been developed to monitor specific metabolic states programmed by tumor-specific mutations in metabolic enzymes. Applications include elevated 2HG in IDH1/IDH2-mutated gliomas (187) and elevated succinate in SDH-deficient paragangliomas (188).
Metabolic flux studies use isotope tracers like 13C, 15N, and 2H to track flow through metabolic pathways. Typically, a nutrient of interest is labeled by an isotope (for example, 13C-glucose) and supplied to cancer cells in the culture medium. Metabolites extracted from the culture are analyzed for isotope enrichment using mass spectrometry or NMR. The extent and distribution of labeling within informative metabolites encode information about which pathways are active in the cells. Incorporating additional data (for example, definitive rates of nutrient consumption, waste secretion, and biomass production) allows quantitative fluxes to be determined across a metabolic network.
Isotope tracing studies provide information about metabolic alterations in cancer cells that cannot be detected by metabolite levels alone. For example, hypoxia and mutations in the ETC induce a restructuring of the TCA cycle in which many of the intermediates are produced in the reverse order from the conventional form of the cycle. The key reaction in this pathway involves the reductive carboxylation of α-ketoglutarate to isocitrate in a NADPH-dependent carboxylation reaction catalyzed by IDH1 and/or IDH2. Although metabolomics experiments can detect altered levels of TCA cycle metabolites in cells using the reductive carboxylation pathway or in cells with deficiencies in pyruvate import into mitochondria, the marked restructuring of the cycle is apparent only through isotope tracing experiments, particularly experiments using 13C-glutamine as the tracer (69, 78, 79, 189–191). An example of the use of isotope tracers to identify metabolic liabilities involves the surprising discovery that a significant fraction of cellular NADPH, particularly in the mitochondria, is produced through folate metabolism (100, 101). These studies involved a sophisticated combination of 13C and 2H tracers, coupled with quantitative measurements of metabolic flux.
Several recent studies have begun to use stable isotopes to investigate metabolism in intact tumors. Because these isotopes do not undergo radioactive decay, they are safe for administration to animals and human subjects. Systemic administration of 13C-labeled nutrients through either boluses or continuous infusions has been shown to generate substantial labeling of glycolytic and TCA cycle intermediates in tumors. In mice bearing orthotopic transplants of high-grade human gliomas, continuous infusion of 13C-glucose was demonstrated to produce steady-state labeling of metabolites from the TCA cycle within the tumor, enabling the assessment of several metabolic pathways (192). Here, tumors with diverse oncogenotypes oxidized glucose-derived pyruvate in the mitochondria and synthesized glutamine from glucose carbon. In contrast to most cultured glioma cell lines, these tumors did not demonstrate significant levels of 13C-glutamine oxidation in vivo, and primary cell lines derived from the tumors did not require glutamine for survival or proliferation. In another study, metabolism of 13C-glucose and 13C-glutamine in autochthonous models of MYC- or MET-driven tumorigenesis revealed that metabolic phenotypes depend not only on the tumor’s genetic driver but also on the tissue or origin. MYC but not MET stimulated glutamine catabolism in liver tumors, whereas MYC-driven lung tumors expressed glutamine synthetase and accumulated glutamine (193). Thus, in vivo isotope tracing can detect metabolic activities of intact tumors and characterize some of the factors that specify the metabolic phenotype.
Administration of 13C-labeled nutrients has also proven to be valuable in human cancer (172, 194–197). Fan et al. (196) used 13C-glucose to demonstrate that human non–small cell lung tumors metabolize glucose through glycolysis and the TCA cycle concurrently, with metabolites from both pathways demonstrating higher levels of labeling in tumors relative to adjacent lung tissue. In a subsequent study, these investigators demonstrated that the anaplerotic enzyme pyruvate carboxylase (PC) was highly expressed in lung tumors and contributed to 13C labeling in TCA cycle intermediates (195). Enhanced glucose oxidation involving both PC and pyruvate dehydrogenase (PDH) was demonstrated in a separate cohort of non–small cell lung tumors, in which formal analysis of metabolic fluxes was used to complement measurements of 13C labeling (197). An important conclusion from these studies, and from a similar study in mice bearing KRAS-driven tumors (198), is that non–small cell lung tumors demonstrate higher levels of both glycolysis and glucose oxidation relative to adjacent, benign lung. This finding sharply contrasts with the frequently invoked “switch” from oxidative metabolism to glycolysis in malignant tissue, commonly used to explain the Warburg effect (Fig. 5A). Rather, the data support a model in which the amplitude of both pathways is increased simultaneously, perhaps through increased substrate delivery and enzyme expression in tumor cells (Fig. 5B). It is also significant that human tumors exhibit substantial heterogeneity of metabolic phenotypes, both between tumors and even within distinct regions of the same tumor (197). The extent of glucose-dependent labeling of TCA cycle intermediates is predicted by noninvasive assessment of tumor perfusion by magnetic resonance imaging, providing an approach to identify areas of regional metabolic heterogeneity in human cancer (197).
Fig. 5. Relationship between glycolysis and oxidative phosphorylation in cancer cells.
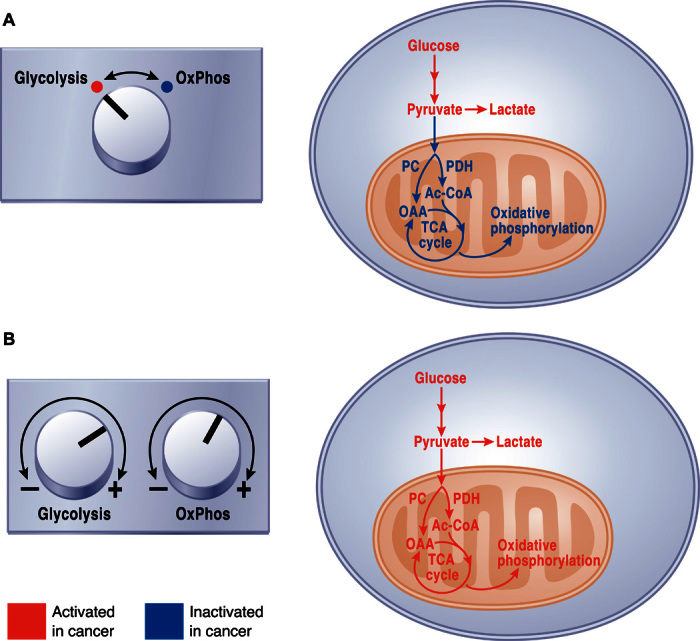
(A) A common view of cancer cell metabolism invokes a switch from glucose oxidation in normal tissues toward glycolysis and suppressed oxidative phosphorylation (OxPhos) in cancer. (B) Analysis of metabolic activity in intact tumors from humans and mice argues against a switch. Rather, tumors appear to enhance both glycolysis and glucose oxidation simultaneously relative to surrounding tissue.
Metabolomics and metabolic flux analysis can be integrated with functional genomics to identify and understand metabolic vulnerabilities in cancer cells. This approach has produced several good examples of screens that identified potential therapeutic targets while stimulating entirely new lines of investigation in cancer cell biology. For example, the serine biosynthetic enzyme PHGDH was first identified as a metabolic vulnerability in breast cancer cells through a large-scale in vivo short hairpin RNA screen targeting thousands of metabolic enzymes (25). PHGDH is frequently amplified at the genomic level in breast tumors and melanomas and exhibits oncogene-like features in cell culture (25, 26). Subsequent work on serine biosynthesis, much of it involving metabolomics and metabolic flux analysis, has uncovered novel functions and liabilities of this pathway in cancer cell growth and stress resistance (129, 150, 151). Combining functional screens with metabolic analysis can also identify context-specific vulnerabilities that may be therapeutically actionable. A CRISPR (clustered regularly interspaced short palindromic repeats)–based loss-of-function screen identified GOT1, the cytosolic aspartate aminotransferase, as conditionally essential for survival during treatment with the ETC inhibitor phenformin (199). Isotope labeling then demonstrated that ETC blockade caused the direction of this enzyme to reverse from aspartate consumption in untreated cells to aspartate synthesis during ETC blockade (200). In addition to the discovery of synthetic lethality between ETC and GOT1 inhibition, these studies led to the novel biological concept that a major function of the ETC in proliferating cells is to support the synthesis of aspartate for nucleotide and protein synthesis (199, 200).
CONCLUSIONS AND CURRENT CHALLENGES
Substantial progress has been made in the past decade toward understanding the mechanisms, biological consequences, and liabilities associated with metabolic reprogramming in cancer. Several common themes have emerged from this research (Box 1). First, metabolic reprogramming is essential for the biology of malignant cells, particularly their ability to survive and grow by using conventional metabolic pathways to produce energy, synthesize biosynthetic precursors, and maintain redox balance. Second, metabolic reprogramming is the result of mutations in oncogenes and tumor suppressors, leading to activation of PI3K and mTORC1 signaling pathways and transcriptional networks involving HIFs, MYC, and SREBP-1. Third, alterations in metabolite levels can affect cellular signaling, epigenetics, and gene expression through posttranslational modifications such as acetylation, methylation, and thiol oxidation. Fourth, taken together, studies on cultured cells have demonstrated a remarkable diversity of anabolic and catabolic pathways in cancer, with induction of autophagy and utilization of extracellular lipids and proteins complementing the classical pathways like glycolysis and glutaminolysis. We have exited the period when cancer metabolism could be considered synonymous with the Warburg effect.
Box 1.
Key Principles and Lessons Learned
Reprogrammed metabolic pathways are essential for cancer cell survival and growth.
Frequently reprogrammed activities include those that allow tumor cells to take up abundant nutrients and use them to produce ATP, generate biosynthetic precursors and macromolecules, and tolerate stresses associated with malignancy (for example, redox stress and hypoxia).
An emerging class of reprogrammed pathways includes those allowing cancer cells to tolerate nutrient depletion by catabolizing macromolecules from inside or outside the cell (for example, autophagy, macropinocytosis, and lipid scavenging).
Reprogramming may be regulated intrinsically by tumorigenic mutations in cancer cells or extrinsically by influences of the microenvironment.
Oncometabolites (for example, 2HG) accumulate as a consequence of genetic changes within a tumor and contribute to the molecular process of malignant transformation.
Many metabolites exert their biological effects outside of the classical metabolic network, affecting signal transduction, epigenetics, and other functions.
New approaches to assess metabolism in living tumors in humans and mice may improve our ability to understand how metabolic reprogramming is regulated and which altered pathways hold opportunities to improve care of cancer patients.
Several challenges will likely shape research over the next decade. First, the studies cited above were performed primarily in cancer cell lines rather than intact tumors. These straightforward experimental models have been highly informative about the molecular mechanisms of metabolic reprogramming, particularly those linking aberrant signaling to altered metabolic fluxes. But it is challenging (perhaps impossible) to model an accurate tumor microenvironment in culture. Direct analysis of metabolic fluxes in intact tumors should begin to play a more prominent role in the field and may prove essential in determining precisely how to deploy metabolic inhibitors in clinical trials. Along these lines, it is remarkable that some tumor cell metabolic vulnerabilities observed in vivo are absent from cultured cell models (198) and that metabolic phenotypes are inconsistent even across single solid tumors in patients (197). Developing rational therapeutic strategies will be aided by learning how to derive metabolic information efficiently and comprehensively from both preclinical and clinical models of intact tumor growth. A further challenge for these in vivo studies will be to develop analytical or computational approaches to deconvolute the distinct metabolic phenotypes of discrete cell types (cancer cells, cancer-associated fibroblasts, lymphocytes, and endothelial cells) within solid tumors. This may allow us to understand the metabolic cooperativity among populations of cells within a tumor and whether metabolic reprogramming of stromal cells provides therapeutic opportunities. Second, by far the best recent candidate for a targetable, tumor-specific metabolic activity is the neomorphic function of mutant IDH1/IDH2. This has stimulated intense interest in finding other metabolic alterations for which the therapeutic window may be wide enough for real clinical opportunities. Third, although we have learned a great deal about the metabolic pathways that support cancer cell proliferation, we know much less about the metabolism that supports survival of nonproliferating tumor cells, which constitute the bulk of the malignant cells in most solid tumors. Along these lines, the metabolism of tumor-initiating cells/cancer stem cells is just now beginning to be investigated, and it will be of major interest to devise strategies to target metabolism in these cells. Finally, we still know relatively little about metabolic interactions between tumor and host. This area has the potential for enormous impact on public health. It is clear that obesity and diabetes, both of which are reaching epidemic proportions in the developed world, increase cancer risk, but we lack insight into how to break these links.
Acknowledgments
We are grateful to J. Schaffer for illustrating the figures. Funding: This work was supported by NIH grants RO1 CA12306708 (to N.S.C.) and RO1 CA157996 to (R.J.D.). Author contributions: N.S.C. wrote the abstract, bioenergetics, redox, and targeting metabolism for cancer therapy sections. R.J.D. wrote the introduction, biosynthesis, technology, Box 1, and conclusion sections. N.S.C. and R.J.D. both wrote the metabolic reprogramming and oncometabolites sections. Competing interests: R.J.D. is on the Advisory Boards of Agios Pharmaceuticals and Peloton Therapeutics. N.S.C. declares no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
REFERENCES AND NOTES
- 1.Hanahan D., Weinberg R. A., Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Pavlova N. N., Thompson C. B., The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patra K. C., Wang Q., Bhaskar P. T., Miller L., Wang Z., Wheaton W., Chandel N., Laakso M., Muller W. J., Allen E. L., Jha A. K., Smolen G. A., Clasquin M. F., Robey R. B., Hay N., Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell 24, 213–228 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shroff E. H., Eberlin L. S., Dang V. M., Gouw A. M., Gabay M., Adam S. J., Bellovin D. I., Tran P. T., Philbrick W. M., Garcia-Ocana A., Casey S. C., Li Y., Dang C. V., Zare R. N., Felshera D. W., MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. U.S.A. 112, 6539–6544 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clavell L. A., Gelber R. D., Cohen H. J., Hitchcock-Bryan S., Cassady J. R., Tarbell N. J., Blattner S. R., Tantravahi R., Leavitt P., Sallan S. E., Four-agent induction and intensive asparaginase therapy for treatment of childhood acute lymphoblastic leukemia. N. Engl. J. Med. 315, 657–663 (1986). [DOI] [PubMed] [Google Scholar]
- 6.Yun J., Rago C., Cheong I., Pagliarini R., Angenendt P., Rajagopalan H., Schmidt K., Willson J. K., Markowitz S., Zhou S., Diaz L. A. Jr, Velculescu V. E., Lengauer C., Kinzler K. W., Vogelstein B., Papadopoulos N., Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 325, 1555–1559 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loo J. M., Scherl A., Nguyen A., Man F. Y., Weinberg E., Zeng Z., Saltz L., Paty P. B., Tavazoie S. F., Extracellular metabolic energetics can promote cancer progression. Cell 160, 393–406 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piskounova E., Agathocleous M., Murphy M. M., Hu Z., Huddlestun S. E., Zhao Z., Leitch A. M., Johnson T. M., DeBerardinis R. J., Morrison S. J., Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boroughs L. K., DeBerardinis R. J., Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 17, 351–359 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ward P. S., Thompson C. B., Metabolic reprogramming: A cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lunt S. Y., Vander Heiden M. G., Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Koppenol W. H., Bounds P. L., Dang C. V., Otto Warburg’s contributions to current concepts of cancer metabolism. Nat. Rev. Cancer 11, 325–337 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Ahn C. S., Metallo C. M., Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 3, 1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Owen O. E., Kalhan S. C., Hanson R. W., The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 277, 30409–30412 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Cantor J. R., Sabatini D. M., Cancer cell metabolism: One hallmark, many faces. Cancer Discov. 2, 881–898 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dibble C. C., Manning B. D., Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 15, 555–564 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yuan T. L., Cantley L. C., PI3K pathway alterations in cancer: Variations on a theme. Oncogene 27, 5497–5510 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stine Z. E., Walton Z. E., Altman B. J., Hsieh A. L., Dang C. V., MYC, metabolism, and cancer. Cancer Discov. 5, 1024–1039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kruiswijk F., Labuschagne C. F., Vousden K. H., p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 16, 393–405 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Jiang L., Kon N., Li T., Wang S. J., Su T., Hibshoosh H., Baer R., Gu W., Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li T., Kon N., Jiang L., Tan M., Ludwig T., Zhao Y., Baer R., Gu W., Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 149, 1269–1283 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jain R. K., Munn L. L., Fukumura D., Dissecting tumour pathophysiology using intravital microscopy. Nat. Rev. Cancer 2, 266–276 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Semenza G.L., Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaelin W. G. Jr, Ratcliffe P. J., Oxygen sensing by metazoans: The central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Possemato R., Marks K. M., Shaul Y. D., Pacold M. E., Kim D., Birsoy K., Sethumadhavan S., Woo H.-K., Jang H. G., Jha A. K., Chen W. W., Barrett F. G., Stransky N., Tsun Z.-Y., Cowley G. S., Barretina J., Kalaany N. Y., Hsu P. P., Ottina K., Chan A. M., Yuan B., Garraway L. A., Root D. E., Mino-Kenudson M., Brachtel E. F., Driggers E. M., Sabatini D. M., Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Locasale J. W., Grassian A. R., Melman T., Lyssiotis C. A., Mattaini K. R., Bass A. J., Heffron G., Metallo C. M., Muranen T., Sharfi H., Sasaki A. T., Anastasiou D., Mullarky E., Vokes N. I., Sasaki M., Beroukhim R., Stephanopoulos G., Ligon A. H., Meyerson M., Richardson A. L., Chin L., Wagner G., Asara J. M., Brugge J. S., Cantley L. C., Vander Heiden M. G., Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Locasale J. W., Serine, glycine and one-carbon units: Cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang M., Soga T., Pollard P. J., Oncometabolites: Linking altered metabolism with cancer. J. Clin. Invest. 123, 3652–3658 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan H., Parsons D. W., Jin G., McLendon R., Rasheed B. A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G. J., Friedman H., Friedman A., Reardon D., Herndon J., Kinzler K. W., Velculescu V. E., Vogelstein B., Bigner D. D., IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mardis E. R., Ding L., Dooling D. J., Larson D. E., McLellan M. D., Chen K., Koboldt D. C., Fulton R. S., Delehaunty K. D., McGrath S. D., Fulton L. A., Locke D. P., Magrini V. J., Abbott R. M., Vickery T. L., Reed J. S., Robinson J. S., Wylie T., Smith S. M., Carmichael L., Eldred J. M., Harris C. C., Walker J., Peck J. B., Du F., Dukes A. F., Sanderson G. E., Brummett A. M., Clark E., McMichael J. F., Meyer R. J., Schindler J. K., Pohl C. S., Wallis J. W., Shi X., Lin L., Schmidt H., Tang Y., Haipek C., Wiechert M. E., Ivy J. V., Kalicki J., Elliott G., Ries R. E., Payton J. E., Westervelt P., Tomasson M. H., Watson M. A., Baty J., Heath S., Shannon W. D., Nagarajan R., Link D. C., Walter M. J., Graubert T. A., DiPersio J. F., Wilson R. K., Ley T. J., Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang M. R., Kim M. S., Oh J. E., Kim Y. R., Song S. Y., Seo S. I., Lee J. Y., Yoo N. J., Lee S. H., Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int. J. Cancer 125, 353–355 (2009). [DOI] [PubMed] [Google Scholar]
- 32.Losman J.-A., Kaelin W. G. Jr, What a difference a hydroxyl makes: Mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev. 27, 836–852 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dang L., White D. W., Gross S., Bennett B. D., Bittinger M. A., Driggers E. M., Fantin V. R., Jang H. G., Jin S., Keenan M. C., Marks K. M., Prins R. M., Ward P. S., Yen K. E., Liau L. M., Rabinowitz J. D., Cantley L. C., Thompson C. B., Vander Heiden M. G., Su S. M., Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 465, 966 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ward P. S., Patel J., Wise D. R., Abdel-Wahab O., Bennett B. D., Coller H. A., Cross J. R., Fantin V. R., Hedvat C. V., Perl A. E., Rabinowitz J. D., Carroll M., Su S. M., Sharp K. A., Levine R. L., Thompson C. B., The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting α-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Figueroa M. E., Abdel-Wahab O., Lu C., Ward P. S., Patel J., Shih A., Li Y., Bhagwat N., Vasanthakumar A., Fernandez H. F., Tallman M. S., Sun Z., Wolniak K., Peeters J. K., Liu W., Choe S. E., Fantin V. R., Paietta E., Löwenberg B., Licht J. D., Godley L. A., Delwel R., Valk P. J., Thompson C. B., Levine R. L., Melnick A., Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell 18, 553–567 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lu C., Ward P. S., Kapoor G. S., Rohle D., Turcan S., Abdel-Wahab O., Edwards C. R., Khanin R., Figueroa M. E., Melnick A., Wellen K. E., O’Rourke D. M., Berger S. L., Chan T. A., Levine R. L., Mellinghoff I. K., Thompson C. B., IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Losman J.-A., Looper R. E., Koivunen P., Lee S., Schneider R. K., McMahon C., Cowley G. S., Root D. E., Ebert B. L., Kaelin W. G. Jr, (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koivunen P., Lee S., Duncan C. G., Lopez G., Lu G., Ramkissoon S., Losman J. A., Joensuu P., Bergmann U., Gross S., Travins J., Weiss S., Looper R., Ligon K. L., Verhaak R. G. W., Yan H., Kaelin W. G. Jr, Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Turcan S., Rohle D., Goenka A., Walsh L. A., Fang F., Yilmaz E., Campos C., Fabius A. W. M., Lu C., Ward P. S., Thompson C. B., Kaufman A., Guryanova O., Levine R., Heguy A., Viale A., Morris L. G. T., Huse J. T., Mellinghoff I. K., Chan T. A., IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483, 479–483 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chowdhury R., Yeoh K. K., Tian Y.-M., Hillringhaus L., Bagg E. A., Rose N. R., Leung I. K. H., Li X. S., Woon E. C. Y., Yang M., McDonough M. A., King O. N., Clifton I. J., Klose R. J., Claridge T. D. W., Ratcliffe P. J., Schofield C. J., Kawamura A., The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu W., Yang H., Liu Y., Yang Y., Wang P., Kim S.-H., Ito S., Yang C., Wang P., Xiao M.-T., Liu L.-X., Jiang W.-., Liu J., Zhang J.-., Wang B., Frye S., Zhang Y., Xu Y.-., Lei Q.-., Guan K.-L., Zhao S.-., Xiong Y., Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Intlekofer A. M., Dematteo R. G., Venneti S., Finley L. W. S., Lu C., Judkins A. R., Rustenburg A. S., Grinaway P. B., Chodera J. D., Cross J. R., Thompson C. B., Hypoxia induces production of L-2-hydroxyglutarate. Cell Metab. 22, 304–311 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rzem R., Vincent M.-F., Van Schaftingen E., Veiga-da-Cunha M., L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J. Inherit. Metab. Dis. 30, 681–689 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Oldham W. M., Clish C. B., Yang Y., Loscalzo J., Hypoxia-mediated increases in L-2-hydroxyglutarate coordinate the metabolic response to reductive stress. Cell Metab. 22, 291–303 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Topçu M., Jobard F., Halliez S., Coskun T., Yalçinkayal C., Gerceker F. O., Wanders R. J. A., Prud’homme J.-F., Lathrop M., Özguc M., Fischer J., L-2-Hydroxyglutaric aciduria: Identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum. Mol. Genet. 13, 2803–2811 (2004). [DOI] [PubMed] [Google Scholar]
- 46.Aghili M., Zahedi F., Rafiee E., Hydroxyglutaric aciduria and malignant brain tumor: A case report and literature review. J. Neurooncol 91, 233–236 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Mullen A. R., Hu Z., Shi X., Jiang L., Boroughs L. K., Kovacs Z., Boriack R., Rakheja D., Sullivan L. B., Linehan W. M., Chandel N. S., DeBerardinis R. J., Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 7, 1679–1690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shim E.-H., Livi C. B., Rakheja D., Tan J., Benson D., Parekh V., Kho E.-Y., Ghosh A. P., Kirkman R., Velu S., Dutta S., Chenna B., Rea S. L., Mishur R. J., Li Q., Johnson-Pais T. L., Guo L., Bae S., Wei S., Block K., Sudarshan S., l-2-Hydroxyglutarate: An epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov. 4, 1290–1298 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tomlinson I. P. M., Alam N. A., Rowan A. J., Barclay E., Jaeger E. E. M., Kelsell D., Leigh I., Gorman P., Lamlum H., Rahman S., Roylance R. R., Olpin S., Bevan S., Barker K., Hearle N., Houlston R. S., Kiuru M., Lehtonen R., Karhu A., Vilkki S., Laiho P., Eklund C., Vierimaa O., Aittomäki K., Hietala M., Sistonen P., Paetau A., Salovaara R., Herva R., Launonen V., Aaltonen L. A. Multiple Leiomyoma Consortium , Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat. Genet. 30, 406–410 (2002). [DOI] [PubMed] [Google Scholar]
- 50.Gottlieb E., Tomlinson I. P. M., Mitochondrial tumour suppressors: A genetic and biochemical update. Nat. Rev. Cancer 5, 857–866 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Baysal B. E., Ferrell R. E., Willett-Brozick J. E., Lawrence E. C., Myssiorek D., Bosch A., van der Mey A., Taschner P. E. M., Rubinstein W. S., Myers E. N., Richard C. W. III, Cornelisse C. J., Devilee P., Devlin B., Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science 287, 848–851 (2000). [DOI] [PubMed] [Google Scholar]
- 52.Laukka T., Mariani C. J., Ihantola T., Cao J. Z., Hokkanen J., Kaelin W. G. Jr, Godley L. A., Koivunen P., Fumarate and succinate regulate expression of hypoxia-inducible genes via TET enzymes. J. Biol. Chem. 291, 4256–4265 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiao M., Yang H., Xu W., Ma S., Lin H., Zhu H., Liu L., Liu Y., Yang C., Xu Y., Zhao S., Ye D., Xiong Y., Guan K.-L., Inhibition of α-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 26, 1326–1338 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaelin W. G. Jr, McKnight S. L., Influence of metabolism on epigenetics and disease. Cell 153, 56–69 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sullivan L. B., Martinez-Garcia E., Nguyen H., Mullen A. R., Dufour E., Sudarshan S., Licht J. D., Deberardinis R. J., Chandel N. S., The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol. Cell 51, 236–248 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Adam J., Hatipoglu E., O’Flaherty L., Ternette N., Sahgal N., Lockstone H., Baban D., Nye E., Stamp G. W., Wolhuter K., Stevens M., Fischer R., Carmeliet P., Maxwell P. H., Pugh C. W., Frizzell N., Soga T., Kessler B. M., El-Bahrawy M., Ratcliffe P. J., Pollard P. J., Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: Roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 20, 524–537 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bardella C., El-Bahrawy M., Frizzell N., Adam J., Ternette N., Hatipoglu E., Howarth K., O’Flaherty L., Roberts I., Turner G., Taylor J., Giaslakiotis K., Macaulay V. M., Harris A. L., Chandra A., Lehtonen H. J., Launonen V., Aaltonen L. A., Pugh C. W., Mihai R., Trudgian D., Kessler B., Baynes J. W., Ratcliffe P. J., Tomlinson I. P., Pollard P. J., Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J. Pathol. 225, 4–11 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Ooi A., Wong J.-C., Petillo D., Roossien D., Perrier-Trudova V., Whitten D., Min B. W. H., Tan M.-H., Zhang Z., Yang X. J., Zhou M., Gardie B., Molinié V., Richard S., Tan P. H., Teh B. T., Furge K. A., An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20, 511–523 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Warburg O., On respiratory impairment in cancer cells. Science 124, 269–270 (1956). [PubMed] [Google Scholar]
- 60.Warburg O., On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 61.Dang C. V., Links between metabolism and cancer. Genes Dev. 26, 877–890 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Israelsen W. J., Dayton T. L., Davidson S. M., Fiske B. P., Hosios A. M., Bellinger G., Li J., Yu Y., Sasaki M., Horner J. W., Burga L. N., Xie J., Jurczak M. J., DePinho R. A., Clish C. B., Jacks T., Kibbey R. G., Wulf G. M., Di Vizio D., Mills G. B., Cantley L. C., Vander Heiden M. G., PKM2 isoform-specific deletion reveals a differential requirement for pyruvate kinase in tumor cells. Cell 155, 397–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Joshi S., Tolkunov D., Aviv H., Hakimi A. A., Yao M., Hsieh J. J., Ganesan S., Chan C. S., White E., The genomic landscape of renal oncocytoma identifies a metabolic barrier to tumorigenesis. Cell Rep. 13, 1895–1908 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Weinberg F., Hamanaka R., Wheaton W. W., Weinberg S., Joseph J., Lopez M., Kalyanaraman B., Mutlu G. M., Budinger G. R. S., Chandel N. S., Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. U.S.A. 107, 8788–8793 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Martinez-Reyes I., Diebold L. P., Kong H., Schieber M., Huang H., Hensley C. T., Mehta M. M., Wang T., Santos J. H., Woychik R., Dufour E., Spelbrink J. N., Weinberg S. E., Zhao Y., DeBerardinis R. J., Chandel N. S., TCA cycle and mitochondrial membrane potential are necessary for diverse biological functions. Mol. Cell 61, 199–209 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zu X. L., Guppy M., Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 313, 459–465 (2004). [DOI] [PubMed] [Google Scholar]
- 67.Lussey-Lepoutre C., Hollinshead K. E. R., Ludwig C., Menara M., Morin A., Castro-Vega L.-J., Parker S. J., Janin M., Martinelli C., Ottolenghi C., Metallo C., Gimenez-Roqueplo A.-P., Favier J., Tennant D. A., Loss of succinate dehydrogenase activity results in dependency on pyruvate carboxylation for cellular anabolism. Nat. Commun. 6, 8784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cardaci S., Zheng L., MacKay G., van den Broek N. J. F., MacKenzie E. D., Nixon C., Stevenson D., Tumanov S., Bulusu V., Kamphorst J. J., Vazquez A., Fleming S., Schiavi F., Kalna G., Blyth K., Strathdee D., Gottlieb E., Pyruvate carboxylation enables growth of SDH-deficient cells by supporting aspartate biosynthesis. Nat. Cell Biol. 17, 1317–1326 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mullen A. R., Wheaton W. W., Jin E. S., Chen P.-H., Sullivan L. B., Cheng T., Yang Y., Linehan W. M., Chandel N. S., DeBerardinis R. J., Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481, 385–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Guzy R., Sharma B., Bell E., Chandel N., Schumacker P., Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol. Cell. Biol. 28, 718–731 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hensley C. T., Wasti A. T., DeBerardinis R. J., Glutamine and cancer: Cell biology, physiology, and clinical opportunities. J. Clin. Invest. 123, 3678–3684 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mayers J. R., Wu C., Clish C. B., Kraft P., Torrence M. E., Fiske B. P., Yuan C., Bao Y., Townsend M. K., Tworoger S. S., Davidson S. M., Papagiannakopoulos T., Yang A., Dayton T. L., Ogino S., Stampfer M. J., Giovannucci E. L., Qian Z. R., Rubinson D. A., Ma J., Sesso H. D., Gaziano J. M., Cochrane B. B., Liu S., Wactawski-Wende J., Manson J. E., Pollak M. N., Kimmelman A. C., Souza A., Pierce K., Wang T. J., Gerszten R. E., Fuchs C. S., Vander Heiden M. G., Wolpin B. M., Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat. Med. 20, 1193–1198 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chandel N., Budinger G. R. S., Choe S. H., Schumacker P. T., Cellular respiration during hypoxia. Role of cytochrome oxidase as the oxygen sensor in hepatocytes. J. Biol. Chem. 272, 18808–18816 (1997). [DOI] [PubMed] [Google Scholar]
- 74.Fan J., Kamphorst J. J., Mathew R., Chung M. K., White E., Shlomi T., Rabinowitz J. D., Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 9, 712 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., Tsukamoto T., Rojas C. J., Slusher B. S., Zhang H., Zimmerman L. J., Liebler D. C., Slebos R. J. C., Lorkiewicz P. K., Higashi R. M., Fan T. W. M., Dang C. V., Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15, 110–121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Le A., Stine Z. E., Nguyen C., Afzal J., Sun P., Hamaker M., Siegel N. M., Gouw A. M., Kang B.-h., Yu S.-H., Cochran R. L., Sailor K. A., Song H., Dang C. V., Tumorigenicity of hypoxic respiring cancer cells revealed by a hypoxia–cell cycle dual reporter. Proc. Natl. Acad. Sci. U.S.A. 111, 12486–12491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kamphorst J. J., Nofal M., Commisso C., Hackett S. R., Lu W., Grabocka E., Vander Heiden M. G., Miller G., Drebin J. A., Bar-Sagi D., Thompson C. B., Rabinowitz J. D., Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 75, 544–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Metallo C. M., Gameiro P. A., Bell E. L., Mattaini K. R., Yang J., Hiller K., Jewell C. M., Johnson Z. R., Irvine D. J., Guarente L., Kelleher J. K., Vander Heiden M. G., Iliopoulos O., Stephanopoulos G., Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wise D. R., Ward P. S., Shay J. E. S., Cross J. R., Gruber J. J., Sachdeva U. M., Platt J. M., DeMatteo R. G., Simon M. C., Thompson C. B., Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U.S.A. 108, 19611–19616 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Laplante M., Sabatini D. M., mTOR signaling in growth control and disease. Cell 149, 274–293 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Guo J. Y., Chen H.-Y., Mathew R., Fan J., Strohecker A. M., Karsli-Uzunbas G., Kamphorst J. J., Chen G., Lemons J. M. S., Karantza V., Coller H. A., DiPaola R. S., Gelinas C., Rabinowitz J. D., White E., Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 25, 460–470 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Strohecker A. M., White E., Autophagy promotes BrafV600E-driven lung tumorigenesis by preserving mitochondrial metabolism. Autophagy 10, 384–385 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lanning N. J., Looyenga B. D., Kauffman A. L., Niemi N. M., Sudderth J., DeBerardinis R. J., MacKeigan J. P., A mitochondrial RNAi screen defines cellular bioenergetic determinants and identifies an adenylate kinase as a key regulator of ATP levels. Cell Rep. 7, 907–917 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Hardie D. G., Schaffer B. E., Brunet A., AMPK: An energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 26, 190–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kamphorst J. J., Cross J. R., Fan J., de Stanchina E., Mathew R., White E. P., Thompson C. B., Rabinowitz J. D., Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U.S.A. 110, 8882–8887 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nieman K. M., Kenny H. A., Penicka C. V., Ladanyi A., Buell-Gutbrod R., Zillhardt M. R., Romero I. L., Carey M. S., Mills G. B., Hotamisligil G. S., Yamada S. D., Peter M. E., Gwin K., Lengyel E., Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 17, 1498–1503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.McCracken A. N., Edinger A. L., Nutrient transporters: The Achilles’ heel of anabolism. Trends Endocrinol. Metab. 24, 200–208 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nicklin P., Bergman P., Zhang B., Triantafellow E., Wang H., Nyfeler B., Yang H., Hild M., Kung C., Wilson C., Myer V. E., MacKeigan J. P., Porter J. A., Wang Y. K., Cantley L. C., Finan P. M., Murphy L. O., Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Galluzzi L., Pietrocola F., Levine B., Kroemer G., Metabolic control of autophagy. Cell 159, 1263–1276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.White E., The role for autophagy in cancer. J. Clin. Invest. 125, 42–46 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galluzzi L., Pietrocola F., Bravo-San Pedro J. M., Amaravadi R. K., Baehrecke E. H., Cecconi F., Codogno P., Debnath J., Gewirtz D. A., Karantza V., Kimmelman A., Kumar S., Levine B., Maiuri M. C., Martin S. J., Penninger J., Piacentini M., Rubinsztein D. C., Simon H.-U., Simonsen A., Thorburn A. M., Velasco G., Ryan K. M., Kroeme G.r, Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Commisso C., Davidson S. M., Soydaner-Azeloglu R. G., Parker S. J., Kamphorst J. J., Hackett S., Grabocka E., Nofal M., Drebin J. A., Thompson C. B., Rabinowitz J. D., Metallo C. M., Vander Heiden M. G., Bar-Sagi D., Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Palm W., Park Y., Wright K., Pavlova N. N., Tuveson D. A., Thompson C. B., The utilization of extracellular proteins as nutrients is suppressed by mTORC1. Cell 162, 259–270 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yoo H., Stephanopoulos G., Kelleher J. K., Quantifying carbon sources for de novo lipogenesis in wild-type and IRS-1 knockout brown adipocytes. J. Lipid Res. 45, 1324–1332 (2004). [DOI] [PubMed] [Google Scholar]
- 95.DeBerardinis R. J., Mancuso A., Daikhin E., Nissim I., Yudkoff M., Wehrli S., Thompson C. B., Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc. Natl. Acad. Sci. U.S.A. 104, 19345–19350 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Schug Z. T., Peck B., Jones D. T., Zhang Q., Grosskurth S., Alam I. S., Goodwin L. M., Smethurst E., Mason S., Blyth K., McGarry L., James D., Shanks E., Kalna G., Saunders R. E., Jiang M., Howell M., Lassailly F., Thin M. Z., Spencer-Dene B., Stamp G., van den Broek N. J. F., Mackay G., Bulusu V., Kamphorst J. J., Tardito S., Strachan D., Harris A. L., Aboagye E. O., Critchlow S. E., Wakelam M. J. O., Schulze A., Gottlieb E., Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Green C. R., Wallace M., Divakaruni A. S., Phillips S. A., Murphy A. N., Ciaraldi T. P., Metallo C. M., Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 12, 15–21 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kannan R., Lyon I., Baker N., Dietary control of lipogenesis in vivo in host tissues and tumors of mice bearing Ehrlich ascites carcinoma. Cancer Res. 40, 4606–4611 (1980). [PubMed] [Google Scholar]
- 99.Ookhtens M., Kannan R., Lyon I., Baker N., Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am. J. Physiol. 247, R146–R153 (1984). [DOI] [PubMed] [Google Scholar]
- 100.Fan J., Ye J., Kamphorst J. J., Shlomi T., Thompson C. B., Rabinowitz J. D., Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Lewis C. A., Parker S. J., Fiske B. P., McCloskey D., Gui D. Y., Green C. R., Vokes N. I., Feist A. M., Vander Heiden M. G., Metallo C. M., Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol. Cell 55, 253–263 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Horton J. D., Goldstein J. L., Brown M. S., SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Invest. 109, 1125–1131 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Düvel K., Yecies J. L., Menon S., Raman P., Lipovsky A. I., Souza A. L., Triantafellow E., Ma Q., Gorski R., Cleaver S., Vander Heiden M. G., MacKeigan J. P., Finan P. M., Clish C. B., Murphy L. O., Manning B. D., Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39, 171–183 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goldstein J. L., DeBose-Boyd R. A., Brown M. S., Protein sensors for membrane sterols. Cell 124, 35–46 (2006). [DOI] [PubMed] [Google Scholar]
- 105.Peterson T. R., Sengupta S. S., Harris T. E., Carmack A. E., Kang S. A., Balderas E., Guertin D. A., Madden K. L., Carpenter A. E., Finck B. N., Sabatini D. M., mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 146, 408–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Deberardinis R. J., Lum J. J., Thompson C. B., Phosphatidylinositol 3-kinase-dependent modulation of carnitine palmitoyltransferase 1A expression regulates lipid metabolism during hematopoietic cell growth. J. Biol. Chem. 281, 37372–37380 (2006). [DOI] [PubMed] [Google Scholar]
- 107.Young R. M., Ackerman D., Quinn Z. L., Mancuso A., Gruber M., Liu L., Giannoukos D. N., Bobrovnikova-Marjon E., Diehl J. A., Keith B., Simon M. C., Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev. 27, 1115–1131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Yokoyama C., Wang X., Briggs M. R., Admon A., Wu J., Hua X., Goldstein J. L., Brown M. S., SREBP-1, a basic-helix-loop-helix-leucine zipper protein that controls transcription of the low density lipoprotein receptor gene. Cell 75, 187–197 (1993). [PubMed] [Google Scholar]
- 109.Guo D., Prins R. M., Dang J., Kuga D., Iwanami A., Soto H., Lin K. Y., Huang T. T., Akhavan D., Hock M. B., Zhu S., Kofman A. A., Bensinger S. J., Yong W. H., Vinters H. V., Horvath S., Watson A. D., Kuhn J. G., Robins H. I., Mehta M. P., Wen P. Y., DeAngelis L. M., Prados M. D., Mellinghoff I. K., Cloughesy T. F., Mischel P. S., EGFR signaling through an Akt-SREBP-1–dependent, rapamycin-resistant pathway sensitizes glioblastomas to antilipogenic therapy. Sci. Signal. 2, ra82 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Guo D., Reinitz F., Youssef M., Hong C., Nathanson D., Akhavan D., Kuga D., Amzajerdi A. N., Soto H., Zhu S., Babic I., Tanaka K., Dang J., Iwanami A., Gini B., DeJesus J., Lisiero D. D., Huang T. T., Prins R. M., Wen P. Y., Robins H. I., Prados M. D., DeAngelis L. M., Mellinghoff I. K., Mehta M. P., James C. D., Chakravarti A., Cloughesy T. F., Tontonoz P., Mischel P. S., An LXR agonist promotes glioblastoma cell death through inhibition of an EGFR/AKT/SREBP-1/LDLR–dependent pathway. Cancer Discov. 1, 442–456 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Stincone A., Prigione A., Cramer T., Wamelink M. M. C., Campbell K., Cheung E., Olin-Sandoval V., Grüning N.-M., Krüger A., Tauqeer Alam M., Keller M. A., Breitenbach M., Brindle K. M., Rabinowitz J. D., Ralser M., The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 90, 927–963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Vander Heiden M. G., Targeting cancer metabolism: A therapeutic window opens. Nat. Rev. Drug Discov. 10, 671–684 (2011). [DOI] [PubMed] [Google Scholar]
- 113.Ben-Sahra I., Howell J. J., Asara J. M., Manning B. D., Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339, 1323–1328 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Murphy M. P., How mitochondria produce reactive oxygen species. Biochem. J. 417, 1–13 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Brand M. D., The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Rhee S. G., Woo H. A., Kil I. S., Bae S. H., Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 287, 4403–4410 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Cox A. G., Winterbourn C. C., Hampton M. B., Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 425, 313–325 (2010). [DOI] [PubMed] [Google Scholar]
- 118.Murphy M. P., Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid. Redox Signal. 16, 476–495 (2012). [DOI] [PubMed] [Google Scholar]
- 119.Finkel T., From sulfenylation to sulfhydration: What a thiolate needs to tolerate. Sci. Signal. 5, pe10 (2012). [DOI] [PubMed] [Google Scholar]
- 120.Cheung E. C., Lee P., Ceteci F., Nixon C., Blyth K., Sansom O. J., Vousden K. H., Opposing effects of TIGAR- and RAC1-derived ROS on Wnt-driven proliferation in the mouse intestine. Genes Dev. 30, 52–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Irani K., Xia Y., Zweier J. L., Sollott S. J., Der C. J., Fearon E. R., Sundaresan M., Finkel T., Goldschmidt-Clermont P. J., Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275, 1649–1652 (1997). [DOI] [PubMed] [Google Scholar]
- 122.Chandel N. S., Maltepe E., Goldwasser E., Mathieu C. E., Simon M. C., Schumacker P. T., Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. U.S.A. 95, 11715–11720 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Orr A. L., Vargas L., Turk C. N., Baaten J. E., Matzen J. T., Dardov V. J., Attle S. J., Li J., Quackenbush D. C., Goncalves R. L. S., Perevoshchikova I. V., Petrassi H. M., Meeusen S. L., Ainscow E. K., Brand M. D., Suppressors of superoxide production from mitochondrial complex III. Nat. Chem. Biol. 11, 834–836 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Porporato P. E., Payen V. L., Pérez-Escuredo J., De Saedeleer C. J., Danhier P., Copetti T., Dhup S., Tardy M., Vazeille T., Bouzin C., Feron O., Michiels C., Gallez B., Sonveaux P., A mitochondrial switch promotes tumor metastasis. Cell Rep. 8, 754–766 (2014). [DOI] [PubMed] [Google Scholar]
- 125.Munson J. M., Fried L., Rowson S. A., Bonner M. Y., Karumbaiah L., Diaz B., Courtneidge S. A., Knaus U. G., Brat D. J., Arbiser J. L., Bellamkonda R. V., Anti-invasive adjuvant therapy with imipramine blue enhances chemotherapeutic efficacy against glioma. Sci. Transl. Med. 4, 127ra36 (2012). [DOI] [PubMed] [Google Scholar]
- 126.Chandel N. S., Tuveson D. A., The promise and perils of antioxidants for cancer patients. N. Engl. J. Med. 371, 177–178 (2014). [DOI] [PubMed] [Google Scholar]
- 127.Jaramillo M. C., Zhang D. D., The emerging role of the Nrf2–Keap1 signaling pathway in cancer. Genes Dev. 27, 2179–2191 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.DeNicola G. M., Karreth F. A., Humpton T. J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K. H., Yeo C. J., Calhoun E. S., Scrimieri F., Winter J. M., Hruban R. H., Iacobuzio-Donahue C., Kern S. E., Blair I. A., Tuveson D. A., Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.DeNicola G. M., Chen P.-H., Mullarky E., Sudderth J. A., Hu Z., Wu D., Tang H., Xie Y., Asara J. M., Huffman K. E., Wistuba I. I., Minna J. D., DeBerardinis R. J., Cantley L. C., NRF2 regulates serine biosynthesis in non–small cell lung cancer. Nat. Genet. 47, 1475–1481 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ye J., Fan J., Venneti S., Wan Y.-W., Pawel B. R., Zhang J., Finley L. W. S., Lu C., Lindsten T., Cross J. R., Qing G., Liu Z., Simon M. C., Rabinowitz J. D., Thompson C. B., Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4, 1406–1417 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Harris I. S., Treloar A. E., Inoue S., Sasaki M., Gorrini C., Lee K. C., Yung K. Y., Brenner D., Knobbe-Thomsen C. B., Cox M. A., Elia A., Berger T., Cescon D. W., Adeoye A., Brüstle A., Molyneux S. D., Mason J. M., Li W. Y., Yamamoto K., Wakeham A., Berman H. K., Khokha R., Done S. J., Kavanagh T. J., Lam C.-W., Mak T. W., Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 27, 211–222 (2015). [DOI] [PubMed] [Google Scholar]
- 132.Garama D. J., Harris T. J., White C. L., Rossello F. J., Abdul-Hay M., Gough D. J., Levy D. E., A synthetic lethal interaction between glutathione synthesis and mitochondrial reactive oxygen species provides a tumor-specific vulnerability dependent on STAT3. Mol. Cell. Biol. 35, 3646–3656 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Gorrini C., Harris I. S., Mak T. W., Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013). [DOI] [PubMed] [Google Scholar]
- 134.Saito Y., Chapple R. H., Lin A., Kitano A., Nakada D., AMPK protects leukemia-initiating cells in myeloid leukemias from metabolic stress in the bone marrow. Cell Stem Cell 17, 585–596 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jeon S.-M., Chandel N. S., Hay N., AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 485, 661–665 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.LeBleu V. S., O’Connell J. T., Gonzalez Herrera K. N., Wikman H., Pantel K., Haigis M. C., de Carvalho F. M., Damascena A., Domingos Chinen L. T., Rocha R. M., Asara J. M., Kalluri R., PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 16, 992–1003 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Schafer Z. T., Grassian A. R., Song L., Jiang Z., Gerhart-Hines Z., Irie H. Y., Gao S., Puigserver P., Brugge J. S., Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Jiang L., Shestov A. A., Swain P., Yang C., Parker S. J., Wang Q. A., Terada L. S., Adams N. D., McCabe M. T., Pietrak B., Schmidt S., Metallo C. M., Dranka B. P., Schwartz B., DeBerardinis R. J., Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Erez A., DeBerardinis R. J., Metabolic dysregulation in monogenic disorders and cancer—Finding method in madness. Nat. Rev. Cancer 15, 440–448 (2015). [DOI] [PubMed] [Google Scholar]
- 140.Pearce E. L., Poffenberger M. C., Chang C.-H., Jones R. G., Fueling immunity: Insights into metabolism and lymphocyte function. Science 342, 1242454 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ito K., Suda T., Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 15, 243–256 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shim H., Dolde C., Lewis B. C., Wu C.-S., Dang G., Jungmann R. A., Dalla-Favera R., Dang C. V., c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. U.S.A. 94, 6658–6663 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Fantin V. R., St-Pierre J., Leder P., Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell 9, 425–434 (2006). [DOI] [PubMed] [Google Scholar]
- 144.Le A., Cooper C. R., Gouw A. M., Dinavahi R., Maitra A., Deck L. M., Royer R. E., Vander Jagt D. L., Semenza G. L., Dang C. V., Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 2037–2042 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Xie H., Hanai J.-., Ren J.-G., Kats L., Burgess K., Bhargava P., Signoretti S., Billiard J., Duffy K. J., Grant A., Wang X., Lorkiewicz P. K., Schatzman S., Bousamra M. II, Lane A. N., Higashi R. M., Fan T. W. M., Pandolfi P. P., Sukhatme V. P., Seth P., Targeting lactate dehydrogenase-a inhibits tumorigenesis and tumor progression in mouse models of lung cancer and impacts tumor-initiating cells. Cell Metab. 19, 795–809 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wang Y.-H., Israelsen W. J., Lee D., Yu V. W. C., Jeanson N. T., Clish C. B., Cantley L. C., Vander Heiden M. G., Scadden D. T., Cell-state-specific metabolic dependency in hematopoiesis and leukemogenesis. Cell 158, 1309–1323 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Haas R., Smith J., Rocher-Ros V., Nadkarni S., Montero-Melendez T., D’Acquisto F., Bland E. J., Bombardieri M., Pitzalis C., Perretti M., Marelli-Berg F. M., Mauro C., Lactate regulates metabolic and pro-inflammatory circuits in control of T cell migration and effector functions. PLOS Biol. 13, e1002202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Colegio O. R., Chu N.-Q., Szabo A. L., Chu T., Rhebergen A. M., Jairam V., Cyrus N., Brokowski C. E., Eisenbarth S. C., Phillips G. M., Cline G. W., Phillips A. J., Medzhitov R., Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Maddocks O. D. K., Berkers C. R., Mason S. M., Zheng L., Blyth K., Gottlieb E., Vousden K. H., Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Labuschagne C. F., van den Broek N. J. F., Mackay G. M., Vousden K. H., Maddocks O. D. K., Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7, 1248–1258 (2014). [DOI] [PubMed] [Google Scholar]
- 151.Kim D., Fiske B. P., Birsoy K., Freinkman E., Kami K., Possemato R. L., Chudnovsky Y., Pacold M. E., Chen W. W., Cantor J. R., Shelton L. M., Gui D. Y., Kwon M., Ramkissoon S. H., Ligon K. L., Kang S. W., Snuderl M., Vander Heiden M. G., Sabatini D. M., SHMT2 drives glioma cell survival in ischaemia but imposes a dependence on glycine clearance. Nature 520, 363–367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Nilsson R., Jain M., Madhusudhan N., Sheppard N. G., Strittmatter L., Kampf C., Huang J., Asplund A., Mootha V. K., Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat. Commun. 5, 3128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Weinberg S. E., Chandel N. S., Targeting mitochondria metabolism for cancer therapy. Nat. Chem. Biol. 11, 9–15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Evans J. M. M., Donnelly L. A., Emslie-Smith A. M., Alessi D. R., Morris A. D., Metformin and reduced risk of cancer in diabetic patients. BMJ 330, 1304–1305 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buzzai M., Jones R. G., Amaravadi R. K., Lum J. J., DeBerardinis R. J., Zhao F., Viollet B., Thompson C. B., Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 67, 6745–6752 (2007). [DOI] [PubMed] [Google Scholar]
- 156.Memmott R. M., Mercado J. R., Maier C. R., Kawabata S., Fox S. D., Dennis P. A., Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev. Res. 3, 1066–1076 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Tomimoto A., Endo H., Sugiyama M., Fujisawa T., Hosono K., Takahashi H., Nakajima N., Nagashima Y., Wada K., Nakagama H., Nakajima A., Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 99, 2136–2141 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bridges H. R., Jones A. J. Y., Pollak M. N., Hirst J., Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem. J. 462, 475–487 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.El-Mir M.-Y., Nogueira V., Fontaine E., Avéret N., Rigoulet M., Leverve X., Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J. Biol. Chem. 275, 223–228 (2000). [DOI] [PubMed] [Google Scholar]
- 160.Owen M. R., Doran E., Halestrap A. P., Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem. J. 348 (Pt. 3), 607–614 (2000). [PMC free article] [PubMed] [Google Scholar]
- 161.Wheaton W. W., Weinberg S. E., Hamanaka R. B., Soberanes S., Sullivan L. B., Anso E., Glasauer A., Dufour E., Mutlu G. M., Budigner G. R. S., Chandel N. S., Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife 3, e02242 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Griss T., Vincent E. E., Egnatchik R., Chen J., Ma E. H., Faubert B., Viollet B., DeBerardinis R. J., Jones R. G., Metformin antagonizes cancer cell proliferation by suppressing mitochondrial-dependent biosynthesis. PLOS Biol. 13, e1002309 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Emami Riedmaier A., Fisel P., Nies A. T., Schaeffeler E., Schwab M., Metformin and cancer: From the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol. Sci. 34, 126–135 (2013). [DOI] [PubMed] [Google Scholar]
- 164.Pollak M., Overcoming drug development bottlenecks with repurposing: Repurposing biguanides to target energy metabolism for cancer treatment. Nat. Med. 20, 591–593 (2014). [DOI] [PubMed] [Google Scholar]
- 165.Birsoy K., Possemato R., Lorbeer F. K., Bayraktar E. C., Thiru P., Yucel B., Wang T., Chen W. W., Clish C. B., Sabatini D. M., Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 508, 108–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Shackelford D. B., Abt E., Gerken L., Vasquez D. S., Seki A., Leblanc M., Wei L., Fishbein M. C., Czernin J., Mischel P. S., Shaw R. J., LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell 23, 143–158 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Gravel S.-P., Hulea L., Toban N., Birman E., Blouin M.-J., Zakikhani M., Zhao Y., Topisirovic I., St-Pierre J., Pollak M., Serine deprivation enhances antineoplastic activity of biguanides. Cancer Res. 74, 7521–7533 (2014). [DOI] [PubMed] [Google Scholar]
- 168.Karsli-Uzunbas G., Guo J. Y., Price S., Teng X., Laddha S. V., Khor S., Kalaany N. Y., Jacks T., Chan C. S., Rabinowitz J. D., White E., Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov. 4, 914–927 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Son J., Lyssiotis C. A., Ying H., Wang X., Hua S., Ligorio M., Perera R. M., Ferrone C. R., Mullarky E., Shyh-Chang N., Kang Y., Fleming J. B., Bardeesy N., Asara J. M., Haigis M. C., DePinho R. A., Cantley L. C., Kimmelman A. C., Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Xiang Y., Stine Z. E., Xia J., Lu Y., O’Connor R. S., Altman B. J., Hsieh A. L., Gouw A. M., Thomas A. G., Gao P., Sun L., Song L., Yan B., Slusher B. S., Zhuo J., Ooi L. L., Lee C. G. L., Mancuso A., McCallion A. S., Le A., Milone M. C., Rayport S., Felsher D. W., Dang C. V., Targeted inhibition of tumor-specific glutaminase diminishes cell-autonomous tumorigenesis. J. Clin. Invest. 125, 2293–2306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Comerford S. A., Huang Z., Du X., Wang Y., Cai L., Witkiewicz A. K., Walters H., Tantawy M. N., Fu A., Manning H. C., Horton J. D., Hammer R. E., McKnight S. L., Tu B. P., Acetate dependence of tumors. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Mashimo T., Pichumani K., Vemireddy V., Hatanpaa K. J., Singh D. K., Sirasanagandla S., Nannepaga S., Piccirillo S. G., Kovacs Z., Foong C., Huang Z., Barnett S., Mickey B. E., DeBerardinis R. J., Tu B. P., Maher E. A., Bachoo R. M., Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Engelman J. A., Chen L., Tan X., Crosby K., Guimaraes A. R., Upadhyay R., Maira M., McNamara K., Perera S. A., Song Y., Chirieac L. R., Kaur R., Lightbown A., Simendinger J., Li T., Padera R. F., García-Echeverría C., Weissleder R., Mahmood U., Cantley L. C., Wong K.-K., Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat. Med. 14, 1351–1356 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Herranz D., Ambesi-Impiombato A., Sudderth J., Sánchez-Martín M., Belver L., Tosello V., Xu L., Wendorff A. A., Castillo M., Haydu J. E., Márquez J., Matés J. M., Kung A. L., Rayport S., Cordon-Cardo C., DeBerardinis R. J., Ferrando A. A., Metabolic reprogramming induces resistance to anti-NOTCH1 therapies in T cell acute lymphoblastic leukemia. Nat. Med. 21, 1182–1189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Viale A., Pettazzoni P., Lyssiotis C. A., Ying H., Sánchez N., Marchesini M., Carugo A., Green T., Seth S., Giuliani V., Kost-Alimova M., Muller F., Colla S., Nezi L., Genovese G., Deem A. K., Kapoor A., Yao W., Brunetto E., Kang Y., Yuan M., Asara J. M., Wang Y. A., Heffernan T. P., Kimmelman A. C., Wang H., Fleming J. B., Cantley L. C., DePinho R. A., Draetta G. F., Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature 514, 628–632 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Yuan P., Ito K., Perez-Lorenzo R., Del Guzzo C., Lee J. H., Shen C.-H., Bosenberg M. W., McMahon M., Cantley L. C., Zheng B., Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proc. Natl. Acad. Sci. U.S.A. 110, 18226–18231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Roesch A., Vultur A., Bogeski I., Wang H., Zimmermann K. M., Speicher D., Körbel C., Laschke M. W., Gimotty P. A., Philipp S. E., Krause E., Pätzold S., Villanueva J., Krepler C., Fukunaga-Kalabis M., Hoth M., Bastian B. C., Vogt T., Herlyn M., Overcoming intrinsic multidrug resistance in melanoma by blocking the mitochondrial respiratory chain of slow-cycling JARID1Bhigh cells. Cancer Cell 23, 811–825 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Janzer A., German N. J., Gonzalez-Herrera K. N., Asara J. M., Haigis M. C., Struhl K., Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, 10574–10579 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Yun J., Mullarky E., Lu C., Bosch K. N., Kavalier A., Rivera K., Roper J., Chio I. I. C., Giannopoulou E. G., Rago C., Muley A., Asara J. M., Paik J., Elemento O., Chen Z., Pappin D. J., Dow L. E., Papadopoulos N., Gross S. S., Cantley L. C., Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Chen Q., Espey M. G., Sun A. Y., Pooput C., Kirk K. L., Krishna M. C., Khosh D. B., Drisko J., Levine M., Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc. Natl. Acad. Sci. U.S.A. 105, 11105–11109 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Ma Y., Chapman J., Levine M., Polireddy K., Drisko J., Chen Q., High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Sci. Transl. Med. 6, 222ra18 (2014). [DOI] [PubMed] [Google Scholar]
- 182.Tagde A., Singh H., Kang M. H., Reynolds C. P., The glutathione synthesis inhibitor buthionine sulfoximine synergistically enhanced melphalan activity against preclinical models of multiple myeloma. Blood Cancer J. 4, e229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Glasauer A., Sena L. A., Diebold L. P., Mazar A. P., Chandel N. S., Targeting SOD1 reduces experimental non-small-cell lung cancer. J. Clin. Invest. 124, 117–128 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Oivares O., Däbritz J. H. M., King A., Gottlieb E., Halsey C., Research into cancer metabolomics: Towards a clinical metamorphosis. Semin. Cell Dev. Biol. 43, 52–64 (2015). [DOI] [PubMed] [Google Scholar]
- 185.Buescher J. M., Antoniewicz M. R., Boros L. G., Burgess S. C., Brunengraber H., Clish C. B., DeBerardinis R. J., Feron O., Frezza C., Ghesquiere B., Gottlieb E., Hiller K., Jones R. G., Kamphorst J. J., Kibbey R. G., Kimmelman A. C., Locasale J. W., Lunt S. Y., Maddocks O. D. K., Malloy C., Metallo C. M., Meuillet E. J., Munger J., A roadmap for interpreting 13C metabolite labeling patterns from cells. Curr. Opin. Biotechnol. 34, 189–201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.DeBerardinis R. J., Thompson C. B., Cellular metabolism and disease: What do metabolic outliers teach us? Cell 148, 1132–1144 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Andronesi O. C., Rapalino O., Gerstner E., Chi A., Batchelor T. T., Cahill D. P., Sorensen A. G., Rosen B. R., Detection of oncogenic IDH1 mutations using magnetic resonance spectroscopy of 2-hydroxyglutarate. J. Clin. Invest. 123, 3659–3663 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Lussey-Lepoutre C., Bellucci A., Morin A., Buffet A., Amar L., Janin M., Ottolenghi C., Zinzindohoué F., Autret G., Burnichon N., Robidel E., Banting B., Fontaine S., Cuenod C.-A., Benit P., Rustin P., Halimi P., Fournier L., Gimenez-Roqueplo A.-P., Favier J., Tavitian B., In vivo detection of succinate by magnetic resonance spectroscopy as a hallmark of SDHx mutations in paraganglioma. Clin. Cancer Res. 22, 1120–1129 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Vacanti N. M., Divakaruni A. S., Green C. R., Parker S. J., Henry R. R., Ciaraldi T. P., Murphy A. N., Metallo C. M., Regulation of substrate utilization by the mitochondrial pyruvate carrier. Mol. Cell 56, 425–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Yang C., Ko B., Hensley C. T., Jiang L., Wasti A. T., Kim J., Sudderth J., Calvaruso M. A., Lumata L., Mitsche M., Rutter J., Merritt M. E., DeBerardinis R. J., Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56, 414–424 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Schell J. C., Olson K. A., Jiang L., Hawkins A. J., Van Vranken J. G., Xie J., Egnatchik R. A., Earl E. G., DeBerardinis R. J., Rutter J., A role for the mitochondrial pyruvate carrier as a repressor of the Warburg effect and colon cancer cell growth. Mol. Cell 56, 400–413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Marin-Valencia I., Yang C., Mashimo T., Cho S., Baek H., Yang X.-L., Rajagopalan K. N., Maddie M., Vemireddy V., Zhao Z., Cai L., Good L., Tu B. P., Hatanpaa K. J., Mickey B. E., Matés J. M., Pascual J. M., Maher E. A., Malloy C. R., DeBerardinis R. J., Bachoo Robert M., Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 15, 827–837 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Yuneva M. O., Fan T. W. M., Allen T. D., Higashi R. M., Ferraris D. V., Tsukamoto T., Matés J. M., Alonso F. J., Wang C., Seo Y., Chen X., Bishop J. M., The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab. 15, 157–170 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Maher E. A., Marin-Valencia I., Bachoo R. M., Mashimo T., Raisanen J., Hatanpaa K. J., Jindal A., Jeffrey F. M., Choi C., Madden C., Mathews D., Pascual J. M., Mickey B. E., Malloy C. R., DeBerardinis R. J., Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 25, 1234–1244 2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Sellers K., Fox M. P., Bousamra M. II, Slone S. P., Higashi R. M., Miller D. M., Wang Y., Yan J., Yuneva M. O., Deshpande R., Lane A. N., Fan T. W.-M., Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J. Clin. Invest. 125, 687–698 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Fan T. W. M., Lane A. N., Higashi R. M., Farag M. A., Gao H., Bousamra M., Miller D. M., Altered regulation of metabolic pathways in human lung cancer discerned by 13C stable isotope-resolved metabolomics (SIRM). Mol. Cancer 8, 41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Hensley C. T., Faubert B., Yuan Q., Lev-Cohain N., Jin E., Kim J., Jiang L., Ko B., Skelton R., Loudat L., Wodzak M., Klimko C., McMillan E., Butt Y., Ni M., Oliver D., Torrealba J., Malloy C. R., Kernstine K., Lenkinski R. E., DeBerardinis R. J., Metabolic heterogeneity in human lung tumors. Cell 164, 681–694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Davidson S. M., Papagiannakopoulos T., Olenchock B. A., Heyman J. E., Keibler M. A., Luengo A., Bauer M. R., Jha A. K., O’Brien J. P., Pierce K. A., Gui D. Y., Sullivan L. B., Wasylenko T. M., Subbaraj L., Chin C. R., Stephanopolous G., Mott B. T., Jacks T., Clish C. B., Vander Heiden M. G., Environment impacts the metabolic dependencies of ras-driven non-small cell lung cancer. Cell Metab. 23, 517–528 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Birsoy K., Wang T., Chen W. W., Freikman E., Abu-Remaileh M., Sabatini D. M., An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 162, 540–551 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Sullivan L. B., Gui D. Y., Hosios A. M., Bush L. N., Freinkman E., Vander Hediden M. G., Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 162, 552–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]