

Continuous multiple sulfur isotope profiles from South African rocks pinpoint the Great Oxygenation Event in the geologic record.
Keywords: Oxygen, atmosphere, Great Oxidation Event, GOE, Paleoproterozoic, sulfur isotopes
Abstract
Molecular oxygen (O2) is, and has been, a primary driver of biological evolution and shapes the contemporary landscape of Earth’s biogeochemical cycles. Although “whiffs” of oxygen have been documented in the Archean atmosphere, substantial O2 did not accumulate irreversibly until the Early Paleoproterozoic, during what has been termed the Great Oxygenation Event (GOE). The timing of the GOE and the rate at which this oxygenation took place have been poorly constrained until now. We report the transition (that is, from being mass-independent to becoming mass-dependent) in multiple sulfur isotope signals of diagenetic pyrite in a continuous sedimentary sequence in three coeval drill cores in the Transvaal Supergroup, South Africa. These data precisely constrain the GOE to 2.33 billion years ago. The new data suggest that the oxygenation occurred rapidly—within 1 to 10 million years—and was followed by a slower rise in the ocean sulfate inventory. Our data indicate that a climate perturbation predated the GOE, whereas the relationships among GOE, “Snowball Earth” glaciation, and biogeochemical cycling will require further stratigraphic correlation supported with precise chronologies and paleolatitude reconstructions.
INTRODUCTION
Earth’s present atmosphere is notable for its remarkably high concentration of ~21% by volume of molecular oxygen (O2) upon which all complex life depends, whereas geologic and geochemical constraints suggest that before ~2.45 billion years ago (Ga), the atmosphere was essentially devoid of this gas (1–4). The rise of the atmospheric O2 partial pressure (pO2) is proposed to have occurred in two main episodes, one at the beginning and the other at the end of the Proterozoic Eon (3, 5, 6). More recent studies, most of which were driven by the emergence of redox-sensitive trace element proxies, suggest a more complex and dynamic evolution scenario for pO2 (7–11). Therefore, continuous records and precise geochronological constraints are absolutely critical to defining and understanding these patterns in relation to the myriad of underlying biological and geological drivers (12).
The initial episode of demonstrable pO2 increase is generally termed the Great Oxygenation Event (GOE) and was first identified by the disappearance of oxygen-sensitive detrital minerals such as pyrite, uraninite, and siderite from sedimentary rocks together with the widespread appearance of Fe-rich paleosols in geological sequences (3, 13). A paucity of unambiguously continuous sedimentary records around this time meant that the GOE was only roughly constrained to the interval of 2.45 to 2.2 Ga (3, 14). Geochemical data, including redox-sensitive trace element concentrations and isotope compositions, as well as the isotope composition of sulfide minerals that may have higher stratigraphic resolutions, help unravel the history of atmospheric pO2 and the timing of the GOE (1, 7, 8, 15–17). However, caution is warranted because these geochemical proxies likely have different sensitivities to changes in pO2 and may not respond uniformly (1, 7, 8, 18, 19).
Mass-independent fractionation of sulfur isotopes (S-MIF) is thought to originate from the photolysis and/or photoexcitation of volcanogenic SO2 by ultraviolet light in an anoxic atmosphere (1, 20–23). Although the specific fractionation mechanisms are still under investigation (20–22, 24), the atmospheric origin of S-MIF signals, such as the Δ33S proxy (see definition in Materials and Methods), is supported by the widespread occurrence of anomalous sulfur isotope fractionation in sulfate and sulfide minerals in Archean and early Paleoproterozoic sedimentary rocks and their absence from younger strata (25–30). Furthermore, results of one-dimensional (1D) photochemical modeling suggested that atmospheric pO2 must have been less than 10−5 times the present atmospheric level (PAL) for the production and preservation of S-MIF signals (31). Although a previous study by Zahnle et al. (32) suggested a slightly lower pO2 (~10−7 PAL), the link between S-MIF and the absence of atmospheric O2 remains strong. By accepting the original value of ~10−5 PAL as the nominal threshold above which S-MIF is no longer preserved, we apply it here as a robust tool for tracing the first rise in pO2 to >10−5 PAL because of its potential for high stratigraphic resolution (1, 31, 33).
On the basis of the transition from S-MIF to S-MDF (mass-dependent fractionation of sulfur isotopes), dating the GOE stems mainly from work carried out on sedimentary rocks from South Africa (18, 34, 35). Although data from the Huronian Supergroup in Canada and the Hamersley and Turee Creek groups in Western Australia also provide information on the timing of the GOE (36, 37), the age constraints from these sedimentary sections are less precise, roughly between ~2.2 and ~2.4 Ga. However, there have been large gaps in sedimentary sulfur isotope records in South Africa (Fig. 1). The original work by Bekker et al. (18) was based on S-MDF signatures in pyrite from the uppermost Rooihoogte and the lowermost Timeball Hill formations (~4-m interval straddling the Rooihoogte–Timeball Hill boundary) in the EBA-2 core (Figs. 1 and 2) and suggested that the GOE should be older than 2316 ± 7 million years ago (Ma). There is a gap of ~150 million years (My) between these S-MDF signatures and the youngest S-MIF signals found in pyrite in the Brockman Iron Formation in Western Australia (1, 38) and a gap of ~100 My between S-MIF in the Koegas Formation in the Griqualand West Basin in South Africa (39). Subsequently, Guo et al. (34) reduced this time and information gap from sulfur isotope measurements of carbonate-associated sulfate (CAS) and pyrite in the Duitschland Formation (Fig. 1). However, there remained a large uncertainty about the timing and exact nature of the GOE as a major sequence boundary and a gap of about 300 m for the shale succession of which no unweathered materials are available in outcrop, which separates the S-MIF signals obtained in the lower Duitschland Formation from the S-MDF signatures obtained in the uppermost part of the upper Duitschland Formation (Fig. 1). Thus, there is a pressing need and an opportunity to gather continuous S-MIF records to provide a more complete picture of the timing, tempo, and consequences of the GOE.
Fig. 1. Geologic sequence in South Africa between 2.5 and 2.2 Ga.
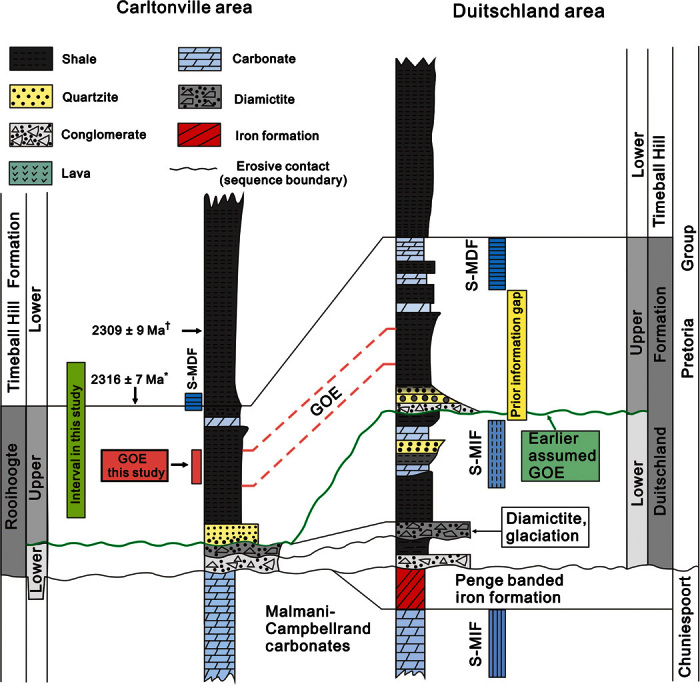
Stratigraphic correlation of the Rooihoogte and Duitschland Formation between the Carltonville and Duitschland area in the Transvaal Basin, South Africa. Note that the diagram is not drawn according to the real stratal thicknesses. The coarse green wavy line represents the sequence boundary in the middle Rooihoogte/Duitschland Formation that was previously proposed as the time interval in which the GOE occurred (34). Previously, the transition from S-MIF to S-MDF was obscured by an apparent hiatus in the Duitschland Formation. The pink interval represents the GOE identified in this study. The dashed vertical lines in the S-MIF box represent small Δ33S values (<2‰), whereas the solid lines represent the presence of large Δ33S values (>2‰). *From Hannah et al. (40). †From Rasmussen et al. (41).
Fig. 2. The δ34S and Δ33S profiles of pyrite in the three cores.

All isotope data are reported relative to VCDT (Vienna Cañon Diablo Troilite). The dashed lines represent three-point moving averages, whereas the shaded regions represent 1 SEM on the three points. The transitional interval highlighted in pink is defined on the basis of variation of Δ33S records. The stars adjacent to the KEA-4 lithologic column correspond to samples for which thin-section photomicrographs are shown in fig. S2. The Re-Os date of 2316 ± 7 Ma on syngenetic pyrite from the boundary between the Timeball Hill and Rooihoogte formations in the EBA-2 core is from Hannah et al. (40).
One underlying problem is that there has been no consensus about the definition of GOE despite the term having been used in the literature for decades. Here, we define the GOE as the time interval during which the atmosphere changed from an anoxic state to one with sufficient pO2 to prevent S-MIF signal preservation in sedimentary rocks. Therefore, the key questions we aimed to address in this study relate to when the atmosphere first irreversibly passed the pO2 > 10−5 PAL threshold (the conclusion of the GOE) and the rate at which the transition occurred (the duration of the GOE).
Because the oldest presently known S-MDF signals are found around the Rooihoogte–Timeball Hill boundary, we measured, at high stratigraphic resolutions, multiple sulfur isotopic ratios in diagenetic pyrite in a continuous sequence from the lower Timeball Hill down to the upper Rooihoogte formations in South Africa (fig. S1). The samples in this study were collected from three diamond drill cores (KEA-4, EBA-2, and EBA-4) that penetrate the sequence in the western Transvaal Basin (see the Supplementary Materials). The Rooihoogte–Timeball Hill boundary has been dated to be 2316 ± 7 My old on the basis of a Re-Os isochron in spatially close synsedimentary to early diagenetic pyrite having the same initial 187Os/188Os ratios in one of our cores (EBA-2) (40). This is supported by a tuff zircon U-Pb age of 2309 ± 9 My for the lowermost Timeball Hill Formation in the nearby UD49 core (41) and the youngest detrital zircon U-Pb age of 2324 ± 17 My for the Timeball Hill Formation (42) (Fig. 1). Therefore, these new data fill the previous gap in data and pinpoint the transition from S-MIF to S-MDF or the GOE as being present in a sedimentary rock sequence with compelling indications for continuous deposition.
RESULTS
Evidence for diagenetic pyrite
Multiple lines of evidence show that the pyrite grains analyzed in this study are authigenic. First, the sizes of the pyrite grains are much larger than coexisting terrestrial particles, mainly comprising quartz and clay minerals (fig. S2, A to I). A detrital origin for the pyrite is not feasible on the basis of hydraulic equivalence because the density of pyrite is higher than that of other minerals. Second, the size range of the pyrite grains does not change according to that of the matrix (fig. S2F). Third, some pyrite grains distinctly disturb sedimentary laminae (fig. S2, B to E and G to I), suggesting that the crystals grew in place before compaction and, therefore, are early diagenetic in origin. Furthermore, pyrite morphology and size do not change across the rock sequence studied here (fig. S2, A to C versus D to I).
Multiple sulfur isotope records
Both the pyrite in bulk rock and that drilled from layered or granular pyrite show similar sulfur isotopic compositions (see Materials and Methods, Fig. 2, and table S1). On the basis of the time series variation trends of Δ33S and the relationships between Δ33S and Δ36S values (see Materials and Methods for the definition of Δ36S), we have divided the sequence in the three cores into three intervals, termed S-MIF, transitional, and S-MDF (Fig. 2). The pyrite in the S-MIF interval (below the pink band in Fig. 2) carries distinct S-MIF signals, with Δ33S reaching as high as +8‰ and the Δ36S/Δ33S ratios centering around −0.98, which is characteristic of the Neoarchean array (Fig. 3) (30, 43). In contrast, in the S-MDF interval (above the pink band in Fig. 2), all pyrite is characterized by near-zero Δ33S values (|Δ33S| < 0.3‰) and Δ36S/Δ33S ratios close to the theoretical MDF value (approximately –6.7) (44). The intervening transitional interval (the pink band in Fig. 2) contains pyrite with both small (|Δ33S| < 0.5‰ in the lower part) and large (0.5 to 2.0‰ in the upper part) Δ33S values and a Δ36S/Δ33S slope that is higher (less negative) than both the underlying S-MIF interval and the overlying S-MDF interval. Thus, the evolution from the S-MIF to the transitional interval is characterized by a sharp negative shift in Δ33S and a significant increase in the slope of Δ36S/Δ33S, whereas the changes from the transitional to the S-MDF interval are characterized by the disappearance of S-MIF signals and a decrease in the slope of Δ36S/Δ33S.
Fig. 3. Triple sulfur isotope cross-plots of the three intervals of the study sequence.

(A) δ34S versus δ33S. (B) Δ36S versus Δ33S. The green dashed lines in (A) represent the MDF array. The blue and purple dashed lines in (B) represent the mass-independent and the theoretical mass-dependent fractionation slope, respectively (1, 44). The red dashed lines in both panels represent the regression line of samples in the transitional interval.
DISCUSSION
The Δ33S values of pyrite in the S-MIF interval (up to +8‰) are among the highest values in the entire geologic record [for example, Johnston (27)]. These data are significantly larger than published data in the interval between ~2.5 and ~2.3 Ga that are commonly located in the range of −1 to 3‰ (25, 27). This may partially reflect sample bias because only a few geologic formations that contain rock types suitable for sulfur isotope analysis are currently known. As a result, data are nearly exclusively from the Transvaal Supergroup in South Africa (34), the Hamersley and Turee Creek groups in Western Australia (36), and the Huronian Supergroup in Canada (37). The large Δ33S values might be due to some particular facet of the atmospheric chemistry at that time (22, 45). The high, spatially heterogeneous, and temporally variable Δ33S values and the large area of the Transvaal Basin where these rocks were deposited suggest that the photochemical products of SO2 in an anoxic atmosphere were the primary origin of these signals. Recycling of continental sulfides with S-MIF signals, which has previously been used to interpret the small Δ33S values in this time interval (25, 46), cannot explain these new data (see the Supplementary Materials). This is also supported by the Δ36S/Δ33S slope of −0.9, which is widespread in Neoarchean sedimentary rocks and interpreted to be of atmospheric origin (9, 25, 30, 43). Accordingly, our data indicate that the atmosphere was effectively devoid of oxygen (nominally <10−5 PAL) when the lower part of the upper Rooihoogte Formation was being deposited (Figs. 2 and 3).
The near-zero Δ33S values and mass-dependent Δ36S/Δ33S signature slope (approximately −6.7) in the S-MDF interval indicate that mass-independent fractionation did not take place or was not preservable during the deposition of the uppermost part of the Rooihoogte and lower Timeball Hill formations. This is entirely consistent with previous research that only investigated a narrow stratigraphic interval straddling the Rooihoogte–Timeball Hill boundary and the top Duitschland Formation (18, 34). Thus, our data indicate that the atmosphere had a substantial pO2 (>10−5 PAL) following the deposition of the uppermost part of the Rooihoogte Formation. Although mechanisms other than pO2, such as decrease of pCH4, have been proposed to explain the disappearance of mass-independent fractionation signals (32), geochemical cycles of CH4 and O2 are intimately linked, as we show below. A significant increase in marine sulfate levels in this interval (see below), widespread hematite-rich oolitic ironstone, and a high enrichment of chromium in the middle part of the Timeball Hill Formation (16, 47) all suggest that a substantial rise in pO2 was at play.
Meanwhile, our data also indicate that before the substantial rise in pO2, the atmosphere underwent significant compositional changes indicated by the sharp decline in the magnitude of Δ33S values and dynamic fluctuations in the Δ36S/Δ33S slope observed in the pyrites of the transitional interval (Figs. 2 and 3). Because the Δ36S/Δ33S ratio in the transitional interval cannot be explained by the mixing of signals from MIF and MDF intervals (see the Supplementary Materials), we suggest that the decrease in Δ33S values in the transitional interval was caused by an increase of pO2, which weakened the preservation of Δ33S signals from photochemistry through mixing different atmospheric sulfur pools; however, this homogenization was incomplete (9, 25). Some samples in this interval in all three cores that have near-zero Δ33S values lend support to this hypothesis. This suggests that pO2 started to increase slightly in the transitional interval. Decreases in the magnitude of Δ33S values have also been ascribed to the shielding effect of organic haze, such as the small Δ33S values present in the Mesoarchean (9, 37, 48). This effect would also decrease the Δ36S/Δ33S slope (9). For example, the Mesoarchean Δ36S/Δ33S slope (approximately −1.5) is lower (more negative) than that of the Neoarchean (approximately −0.9). The higher Δ36S/Δ33S slope in the transitional interval suggests that the effect of organic haze might not be the key factor for the decline in Δ33S values (Fig. 3). Therefore, we hypothesize that this change in atmospheric composition was the prelude to the substantial increase in pO2, representing the GOE as defined above.
The high stratigraphic resolution and continuous record of the transition from S-MIF to S-MDF signals in these three cores speak to dynamic processes as underpinning the substantial rise in pO2 and provide an opportunity to precisely constrain the time of pO2 rise at the GOE to >10−5 PAL, as well as the duration of the transition. The package of sediment between where S-MIF signals cease to be discernible (that is, an oxidized atmosphere) and the Rooihoogte–Timeball Hill boundary is approximately 5 m thick in all three cores (Fig. 2). Sequence stratigraphic analysis indicates continuous deposition from the upper Rooihoogte Formation to the lower Timeball Hill Formation (49) and is supported by uniformly fine sedimentary rock (mudstone, including black shale and siltstone, and few carbonates) throughout (see the Supplementary Materials). Although with some uncertainty, a conservative sedimentation rate can be assumed on the basis of previously reported sedimentation rates in slightly older parts of the same basin—rates that are also similar to modern values (50). The decompacted sedimentation rate of modern siliciclastic sediments, except for deep-sea clay, is >5 m/My (51). Assuming 80% compaction for shale and 50% of time represented by deposition, the duration of the 5-m package is less than 10 My. Applying this estimate to the age of the Rooihoogte–Timeball Hill boundary (2316 ± 7 Ma), our data constrain the termination of the GOE to be within 2316 to 2326 Ma (±7 My).
The sedimentary package corresponding to the transitional interval, which represents the duration of the GOE, is thin. It spans about 5 m of strata in all three cores (Fig. 2), which, according to the conservative sedimentation rate assumed above, represents <10 My. If we assume a more rapid sedimentation rate (about 50 m/My) of siliciclastic sediments in a foreland basin as the Rooihoogte Formation deposited (52) and a lower compacted ratio of the siltstone/mudstone relative to black shale, the transitional interval would represent less than 1 My in time. In either case, the GOE took place on a geologically rapid time scale. Such a fast rise of pO2 is consistent with atmospheric modeling results that predict a rapid and irreversible transition from stable anoxic to oxic atmospheres (53, 54) and may provide constraints on the controversial problem of whether the rise of O2 during the GOE was primarily a consequence of an increase in the flux of oxygen sources or a decrease in the flux of oxygen sinks (34, 55–57).
The GOE not only ended the ultraviolet photoreaction of SO2 in the troposphere but also had effects on exogenic sulfur cycling. A large magnitude (about 45‰) of negative shift in δ34S occurred in the lower part of the S-MDF interval (Fig. 2). The pyrite around the Rooihoogte–Timeball Hill boundary in these three cores is depleted in 34S, with δ34S values as low as −35‰. Although minor occurrences of low δ34S pyrite have recently been reported in late Archean strata by in situ analysis (58), samples studied here are the oldest samples that exhibit low δ34S values in bulk rock analyses (18, 59). The extremely 34S-depleted pyrite suggests that microbial sulfate reduction occurred under sulfate-unlimited conditions in the upper portion of the MDF interval (60). In contrast, high δ34S values of +2.4 to +24.4‰ further downcore in the S-MIF and transitional intervals are consistent with quantitative sulfate reduction under low oceanic sulfate levels (60) before the GOE. Therefore, these data suggest that marine sulfate levels increased considerably following the GOE. An increase in the marine sulfate reservoir at about 2.3 Ga is also supported by the large sulfur isotope separation between CAS and pyrite (attributed to microbial sulfate reduction) recorded in the topmost Duitschland Formation (34), as well as the recent finding of gypsum in sabkha-facies evaporites at ~2.31 Ga (41).
The negative shift in δ34S values occurred above the disappearance of S-MIF in all three cores by ~3 m of strata (Fig. 2), representing as much as ~6 My in time on the basis of the sedimentation rate assumed above. Thus, the increase in seawater sulfate lagged the rise in pO2 by ~6 My. An elevated sulfate flux into the ocean through oxidative pyrite weathering is an expected consequence of higher pO2 after the GOE and can partly contribute to the rise in the oceanic sulfate concentration. It has been suggested that the threshold of pO2 required to induce high pyrite weathering rates is the same as or a little lower than that required for the preservation of S-MIF signals (7, 43, 61). Therefore, the rise in seawater sulfate levels should coincide with or predate the disappearance of MIF. The observed time lag and opposite sequence of events suggest that other factors, not only the changes in sources (for example, sulfide weathering), contributed to the expansion of the oceanic sulfate reservoir, possibly including variation in the sinks of the seawater sulfate.
Geochemical data and mass balance analysis suggest that sulfate sinks must have been relatively much stronger before the GOE than in the modern ocean. As shown in table S2, in the modern sulfur cycle, volcanic sulfur input can comprise as much as 10% of total sulfur in the ocean-atmosphere system, which is dominantly from pyrite and gypsum weathering. Very low pO2 before the GOE would severely limit the amount of sulfur input from weathering processes. Therefore, sulfur inputs into the pre-GOE ocean-atmosphere system are dominated by volcanic fluxes, which can be one to three times higher than the modern value, considering the likely higher heat flux and the associated higher volcanic gas fluxes (53). Thus, assuming that the sulfate reduction sink is first order with respect to sulfate concentration and that residence times were similar, expected sulfate levels before the GOE would be at least 10% (and perhaps as much as 30%) of today’s values (2.8 to 8.4 mM). This is not consistent with models that suggest extremely low seawater sulfate levels before the GOE, possibly as low as 2.5 μM (34, 62). Therefore, a much higher sink of seawater sulfate is required to maintain the very low sulfate levels previously suggested (8, 34, 60). We describe below how pyrite burial through microbial sulfate reduction modulated by pO2 can simultaneously account for the low seawater sulfate levels before the GOE and the delayed rise to high sulfate levels after the GOE.
To discern the sequence of carbon, sulfur, and oxygen cycle perturbations, we constructed a model of biogeochemical cycles. This model integrates the sulfur cycle model to a coupled atmospheric-biogeochemical model of CH4 and O2 (53, 54). In the sulfur cycle model, continental sulfide weathering and volcanic sulfur were the two main sources of seawater sulfate, whereas pyrite burial and sulfate burial were the two main sinks (see Materials and Methods and table S2). We assumed that sulfide weathering was coupled to pO2, and volcanic sulfur fluxes were scaled to the outgassing rate of reducing gases (63). Sulfate burial is proportional to seawater sulfate content. Pyrite burial is assumed to be governed by microbial sulfate reduction dependent on sulfate content and on pO2 to account for the influence of quality and availability of organic matter to sulfate-reducing microbes.
Our model both reproduces the rapid rise of oxygen (53, 54) (consistent with the rapid disappearance of S-MIF in the cores we analyzed here) and predicts a time lag of ~10 My between the increase in pO2 and the subsequent rise in sulfate from ~10 μM to >1 mM (Fig. 4); this is comparable to the lag in isotope signals estimated above. Although the elevated pO2 enhances the oxidative weathering of sulfide minerals on land, thereby increasing sulfate flux into the oceans, the modeled increase in sulfide weathering flux alone cannot explain the increase in sulfate levels by approximately two orders of magnitude (Fig. 4). Rather, the required increase in seawater sulfate can be simulated if the flux of pyrite burial decreases in response to higher pO2 (Fig. 4). This behavior can be accounted for by a reduced supply of labile organic matter due to enhanced aerobic remineralization of organic matter as a consequence of the rise of O2 (see “Model description” in Materials and Methods and the Supplementary Materials).
Fig. 4. Model results showing the increase in pO2 and seawater sulfate level across the GOE.
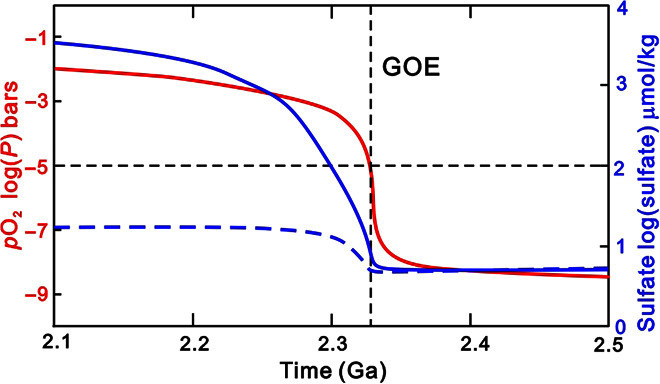
The red line represents the atmospheric pO2. The blue solid and dashed lines represent seawater sulfate levels modeled with the pyrite burial flux coupled or decoupled with pO2, respectively.
In addition to the influence on sulfur biogeochemical cycles, the rapid oxygenation of the atmosphere may have had a pronounced impact on Earth’s climate. Multiple layers of diamictite have been observed in Canada (Huronian Supergroup), South Africa (Transvaal Supergroup), and Australia (Turee Creek Group), suggesting that a series of glaciations took place between 2.45 and 2.2 Ga (56, 64), the timeframe in which the GOE also occurred. Because the rise of pO2 would consume CH4 through a series of photochemical reactions, any atmospheric CH4 would have been lost during the GOE. Because CH4 is a strong greenhouse gas, the substantial decrease of pCH4 could have led to the Paleoproterozoic “Snowball Earth” glaciation (56, 65). In one of the cores analyzed here (EBA-4), a layer of diamictite occurs below the GOE, suggesting that glaciation (though of unknown magnitude and at unknown latitude) predated the GOE. Such a relationship is consistent with biogeochemical modeling that suggests the collapse of a methane greenhouse (immediately followed by ice ages) before the rise of atmospheric O2 (32). However, neither these models nor available age constraints from the sections we studied provide any information on the duration of the lag between these two postulated events. In addition, the relative timing of the GOE and the Snowball Earth glaciation represented by the Makganyene diamictite in the Griqualand West Basin in South Africa (56) also remains uncertain. In particular, the age of the Makganyene diamictite is controversial; it could be either older than 2.3 Gy according to the Pb-Pb age (2394 ± 26 Ma) of the Mooidraai dolomite overlying the Makganyene diamictite and Ongeluk lava (66) and the youngest age (2436.2 ± 6.6 Ma) of detrital zircons on top of the Makganyene diamictite (67), or younger than 2.3 Gy on the basis of the Pb-Pb age (2222 ± 13 Ma) of the Ongeluk lava overlying the Makganyene diamictite (68). These large chronological uncertainties mean that the Makganyene may be older, younger, or contemporaneous with the sections studied in our cores. Additional high-resolution dating and paleolatitude constraints are therefore required to shed light on this outstanding problem. Notwithstanding the remaining geochronological gaps, our work identifies a discrete sequence of geological events during the GOE and thus provides a new, robust framework for future research on the coevolution of Earth’s life and the surface environment.
MATERIALS AND METHODS
Sample preparation
Two types of pyrite, disseminated fine-grain pyrite and macroscopic layered or granular pyrite, were analyzed in this study. For the fine-grain pyrite, we used the portion of samples without macroscopic pyrite (those that can be observed with the naked eye) (fig. S2). The samples with interbedded black mudstone and gray siltstone were divided into two subsamples according to their lithology (mudstone and siltstone) to examine potential relationships to lithology. The rock was cut into small chips, washed with deionized water, and dried at 60°C in an oven. The chips were then ground to fine powder (<100 mesh) using a stainless steel puck mill, which was cleaned between samples by grinding baked quartz sand, followed by sequential washing with tap and deionized water. For the samples containing macroscopic pyrite, either layered pyrite or granular pyrite, we used a microdrill (~1 mm in diameter) to obtain pyrite powder to compare sulfur isotope compositions for disseminated and macroscopic pyrite.
Pyrite extraction
Pyrite sulfur was extracted using the chromium reduction method of Canfield et al. (69), which is briefly described here. For microdrilled pyrite samples, ~15 mg of powder was used for extraction. For the bulk rock (disseminated pyrite) samples, before pyrite sulfur extraction, ~1 to 8 g of rock powder was reacted with 6 M HCl to remove any carbonate and acid-volatile sulfide. The carbonate contents of the samples are less than 5%. The acid residue was washed with deionized water until neutralized (pH > 6) and then dried in an oven at 60°C over 24 hours. The pyrite powder or the bulk rock acid residue was reduced with CrCl2 at boiling under nitrogen atmosphere for 2 hours. The evolved H2S was flushed with N2 flow and collected in the Zn-acetate trap as Zn sulfide (ZnS). Finally, ~1 to 3 ml of silver nitrate (AgNO3) was added to precipitate silver sulfide (Ag2S), which was then cleaned with deionized water.
Sulfur isotope analysis
Multiple sulfur isotope compositions (32S, 33S, 34S, and 36S) were measured according to the method of Ono et al. (44), which is briefly described here. Approximately 2 mg of Ag2S was reacted with elemental fluorine for >6 hours at 300°C. The evolved SF6 was purified by a gas chromatograph equipped with a column packed with a 5 Å molecular sieve, followed by a column packed with HayeSep Q. Isotope ratios of the purified SF6 were measured using a magnetic sector isotope ratio mass spectrometer (ThermoFinnigan MAT 253) operated in a dual-inlet mode. Replicate analyses (n = 28) of a reference material, IAEA-S1, yielded 2σ (2 SDs) of 0.26, 0.014, and 0.19‰ for δ34S, Δ33S and Δ36S, respectively (70).
Sulfur isotope ratios are reported using the conventional δ notation in per mil (‰)
| (1) |
where xRsample and xRVCDT are the isotope ratios (xS/32S, where x = 33, 34, or 36) of the sample and VCDT, respectively. The isotope ratios of VCDT (xRVCDT) are defined by the international reference material (IAEA-S1) to be −0.055, −0.300, and −1.14‰ for δ33S, δ34S, and δ36S, respectively (70).
The following definitions for ΔxS are used
| (2) |
| (3) |
Model description
We constructed a coupled atmosphere-ocean biogeochemical model of O2, CH4, and sulfur cycles. The O2 and CH4 part of the model follows those of Claire et al. (53) and Goldblatt et al. (54) (fig. S3). The aim of the model is to test factors contributing to the observed time lag between the loss of S-MIF signals (GOE) and the subsequent 34S-depleted sulfide (the rise of marine sulfate reservoir) and the mechanism(s) for the rise in seawater sulfate level. The model keeps track of the budget of O2, CH4, and total sulfur ([O], [M], and [S], respectively) in the atmosphere-ocean system.
The source of O2 in the model atmosphere-ocean system is oxygenic photosynthesis (FPROD). The produced organic carbon (CORG) is consumed through aerobic respiration (FRESP), net sulfate reduction that leads to pyrite burial (FBPY), fermentation to CO2 and CH4 (FFERM), or the burial of organic carbon (FBORG).
| (4) |
Aerobic respiration is assumed to follow Monod kinetics with a half-saturation constant at 0.01 PAL pO2 (3.7 × 105 Tmol). The pyrite burial term is parameterized as a function of [S] and [O] with Monod-type dependence on [S] with the half-saturation at 1.4× 104 Tmol [0.01 mM (71)]. The stoichiometry factor of 2 accounts for the stoichiometry of sulfate reduction coupled to organic carbon respiration (fig. S3). For simplicity, we did not explicitly model different kinetics for different forms of sulfur (for example, thiosulfate, S8, or sulfate) that are likely present in the Archean oceans (1, 30). Microbial sulfate reduction depends not only on sulfate but also on the availability and quality of electron donors (72), and pyrite burial depends on the reactivity and availability of iron (73). Because aerobic respiration is limited in the pre-GOE oceans, sulfate reducers are expected to have access to more labile organics compared to post-GOE oceans. To account for this, we parameterized FBPY as a power function ([O]θ, with θ = −0.05 ± 0.01). This [O] dependency is not due to the reoxidation of H2S by O2, which is a net aerobic respiration and does not lead to pyrite precipitation. The value of θ determines the steady state [S] for the model pre- and post-GOE oceans but does not significantly affect the time scale of the rise of sulfate levels (fig. S5). Following Goldblatt et al. (54), FBORG is assumed to be independent of [O]. This is because the decay rate of organic matter below oxygenated surface sediments is limited by hydrolysis and access to hydrolytic enzymes but not bottom water oxygen [for example, Betts and Holland (74)]. We assume steady state for [CORG] and solved FFERM, which produced [M] at one-half stoichiometry (fig. S3).
The continuity equations for O2, CH4, and sulfur reservoirs are
| (5) |
| (6) |
| (7) |
where FATM is the atmospheric oxidation of CH4 by O2, which follows a series of reactions but is parameterized as a pseudo–second-order reaction (katm[O][M]), where katm is obtained by fitting a 2D polynomial function of [O] and [M] from Fig. 3 of Claire et al. (53). FESC is the hydrogen escape to space (taken as the first order to [M]) and is a net sink for [O] and [M]. FVR is the flux of reductants from volcanic gas (mostly H2) and decreases with time (53). Because we set the organic burial (FBORG) constant, the GOE occurs when FVR < FBORG (53, 54). Volcanic sulfur flux (FVS) was scaled to FVR using the H2/SO2 ratio of volcanic gas (63). Oxidative weathering of organics on land (FWO) and of pyrite (FWPY) are assumed to follow a power function and Monod kinetics with respect to [O]. Equations 5 to 7 are numerically integrated with implicit Euler scheme with a variable time step. Flux terms are defined and described in table S2.
Supplementary Material
Acknowledgments
We thank B. Olszewski for assistance with sulfur isotope analysis; A. Whitehill, G. Fournier, L. Kump, and J. Kirschvink for vaulable discussions; T. Grove for microscope access; Z. She and J. Yan for advice on pyrite genesis; T. Lyons and an anonymous reviewer for constructive and helpful reviews; and B. Wing and C. Goldblatt for their comments on an earlier version of this manuscript. Funding: This work was supported by awards from the Simons Foundation (to R.E.S.), the NASA Astrobiology Institute (NNA13AA90A to R.E.S), the 973 Program (grant nos. 2011CB808800 and 2013CB955704 to G.L.), the Chinese National Natural Science Foundation (grant no. 41472170 to G.L.), the 111 Project (grant no. B08030 to G.L.), and NSF (EAR-1338810 to R.E.S. and S.O.). Author contributions: G.L., S.O., and R.E.S. designed the research. G.L., S.O., and N.J.B. collected the samples. G.L. and S.O. carried out the isotope analyses. S.O. constructed the model. All authors contributed to data analysis and the writing of the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1600134/DC1
Geological background
Potential of sulfur MIF signals originated from weathering of preexisting S-MIF
Sensitivity analysis of C-O-S biogeochemical cycling model
fig. S1. Simplified geological map of the Transvaal Supergroup in the Transvaal and Griqualand West basins, Kaapvaal Craton, South Africa.
fig. S2. Representative transmitted light photomicrographs showing pyrite (opaque particles) distribution patterns in the samples analyzed in this study.
fig. S3. Cartoon figure showing the C-S-O geochemical model used in this study.
fig. S4. Sensitivity analysis for the power coefficient (θ) of pyrite burial and pO2.
fig. S5. Sensitivity analysis for the coefficient (γ) of weathering of sulfide and pO2.
table S1. Multiple sulfur isotope composition of the pyrite analyzed in this study. All data are relative to VCDT.
table S2. Parameters used for the Archean-Proterozoic oxygen-methane-sulfur cycle model.
REFERENCES AND NOTES
- 1.Farquhar J., Bao H., Thiemens M., Atmospheric influence of earth’s earliest sulfur cycle. Science 289, 756–758 (2000). [DOI] [PubMed] [Google Scholar]
- 2.H. D. Holland, The Chemistry of the Atmosphere and Oceans (Wiley-Interscience, New York, 1978) [Google Scholar]
- 3.Holland H. D., The oxygenation of the atmosphere and oceans. Philos. Trans. R. Soc. London Ser. B 361, 903–915 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rasmussen B., Buick R., Redox state of the Archean atmosphere: Evidence from detrital heavy minerals in ca. 3250-2750 Ma sandstones from the Pilbara Craton, Australia. Geology 27, 115–118 (1999). [Google Scholar]
- 5.Canfield D. E., The early history of atmospheric oxygen: Homage to Robert M. Garrels. Annu. Rev. Earth Planet. Sci. 33, 1–36 (2005). [Google Scholar]
- 6.Kump L. R., The rise of atmospheric oxygen. Nature 451, 277–278 (2008). [DOI] [PubMed] [Google Scholar]
- 7.Anbar A. D., Duan Y., Lyons T. W., Arnold G. L., Kendall B., Creaser R. A., Kaufman A. J., Gordon G. W., Scott C., Garvin J., Buick R., A whiff of oxygen before the Great Oxidation Event? Science 317, 1903–1906 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Crowe S. A., Døssing L. N., Beukes N. J., Bau M., Kruger S. J., Frei R., Canfield D. E., Atmospheric oxygenation three billion years ago. Nature 501, 535–539 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Kurzweil F., Claire M., Thomazo C., Peters M., Hannington M., Strauss H., Atmospheric sulfur rearrangement 2.7 billion years ago: Evidence for oxygenic photosynthesis. Earth Planet. Sci. Lett. 366, 17–26 (2013). [Google Scholar]
- 10.Lyons T. W., Reinhard C. T., Planavsky N. J., The rise of oxygen in Earth’s early ocean and atmosphere. Nature 506, 307–315 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Planavsky N. J., Reinhard C. T., Wang X., Thomson D., McGoldrick P., Rainbird R. H., Johnson T., Fischer W. W., Lyons T. W., Low mid-proterozoic atmospheric oxygen levels and the delayed rise of animals. Science 346, 635–638 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Bowring S. A., Schmitz M. D., High-precision U-Pb zircon geochronology and the stratigraphic record. Rev. Mineral. Geochem. 53, 305–326 (2003). [Google Scholar]
- 13.H. D. Holland, The Chemical Evolution of the Atmosphere and Ocean (Princeton University Press, Princeton, NJ, 1984). [Google Scholar]
- 14.Kasting J. F., Ono S., Palaeoclimates: The first two billion years. Philos. Trans. R. Soc. London Ser. B 361, 917–929 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frei R., Gaucher C., Poulton S. W., Canfield D. E., Fluctuations in Precambrian atmospheric oxygenation recorded by chromium isotopes. Nature 461, 250–253 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Konhauser K. O., Lalonde S. V., Planavsky N. J., Pecoits E., Lyons T. W., Mojzsis S. J., Rouxel O., Barley M. E., Rosìere C., Fralick P. W., Kump L. R., Bekker A., Aerobic bacterial pyrite oxidation and acid rock drainage during the Great Oxidation Event. Nature 478, 369–374 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Scott C., Lyons T. W., Bekker A., Shen Y., Poulton S. W., Chu X., Anbar A. D., Tracing the stepwise oxygenation of the Proterozoic ocean. Nature 452, 456–459 (2008). [DOI] [PubMed] [Google Scholar]
- 18.Bekker A., Holland H. D., Wang P.-L., Rumble D. III, Stein H. J., Hannah J. L., Coetzee L. L., Beukes N. J., Dating the rise of atmospheric oxygen. Nature 427, 117–120 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Beukes N. J., Dorland H., Gutzmer J., Nedachi M., Ohmoto H., Tropical laterites, life on land, and the history of atmospheric oxygen in the Paleoproterozoic. Geology 30, 491–494 (2002). [Google Scholar]
- 20.Ono S., Whitehill A. R., Lyons T. W., Contribution of isotopologue self-shielding to sulfur mass-independent fractionation during sulfur dioxide photolysis. J. Geophys. Res. Atmos. 118, 2444–2454 (2013). [Google Scholar]
- 21.Whitehill A. R., Ono S., Excitation band dependence of sulfur isotope mass-independent fractionation during photochemistry of sulfur dioxide using boradband light sources. Geochim. Cosmochim. Acta 94, 238–253 (2012). [Google Scholar]
- 22.Whitehill A. R., Xie C., Hu X., Xie D., Guo H., Ono S., Vibronic origin of sulfur mass-independent isotope effect in photoexcitation of SO2 and the implications to the early earth’s atmosphere. Proc. Natl. Acad. Sci. U.S.A. 110, 17697–17702 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Farquhar J., Savarino J., Airieau S., Thiemens M. H., Observation of wavelength-sensitive mass-independent sulfur isotope effects during SO2 photolysis: Implications for the early atmosphere. J. Geophys. Res. Planets 106, 32829–32839 (2001). [Google Scholar]
- 24.Claire M. W., Kasting J. F., Domagal-Goldman S. D., Stüeken E. E., Buick R., Meadows V. S., Modeling the signature of sulfur mass-independent fractionation produced in the Archean atmosphere. Geochim. Cosmochim. Acta 141, 365–380 (2014). [Google Scholar]
- 25.Farquhar J., Wing B. A., Multiple sulfur isotopes and the evolution of the atmosphere. Earth Planet. Sci. Lett. 213, 1–13 (2003). [Google Scholar]
- 26.Izon G., Zerkle A. L., Zhelezinskaia I., Farquhar J., Newton R. J., Poulton S. W., Eigenbrode J. L., Claire M. W., Multiple oscillations in Neoarchaean atmospheric chemistry. Earth Planet. Sci. Lett. 431, 264–273 (2015). [Google Scholar]
- 27.Johnston D. T., Multiple sulfur isotopes and the evolution of Earth’s surface sulfur cycle. Earth Sci. Rev. 106, 161–183 (2011). [Google Scholar]
- 28.Ono S., Beukes N. J., Rumble D., Origin of two distinct multiple-sulfur isotope compositions of pyrite in the 2.5 Ga Klein Naute Formation, Griqualand West Basin, South Africa. Precambrian Res. 169, 48–57 (2009). [Google Scholar]
- 29.Ono S., Beukes N. J., Rumble D., Fogel M. L., Early evolution of atmospheric oxygen from multiple-sulfur and carbon isotope records of the 2.9 Ga Mozaan Group of the Pongola Supergroup, Southern Africa. S. Afr. J. Geol. 109, 97–108 (2006). [Google Scholar]
- 30.Ono S., Eigenbrode J. L., Pavlov A. A., Kharecha P., Rumble D. III, Kasting J. F., Freeman K. H., New insights into Archean sulfur cycle from mass-independent sulfur isotope records from the Hamersley Basin, Australia. Earth Planet. Sci. Lett. 213, 15–30 (2003). [Google Scholar]
- 31.Pavlov A. A., Kasting J. F., Mass-independent fractionation of sulfur isotopes in Archean sediments: Strong evidence for an anoxic Archean atmosphere. Astrobiology 2, 27–41 (2004). [DOI] [PubMed] [Google Scholar]
- 32.Zahnle K., Claire M., Catling D., The loss of mass-independent fractionation in sulfur due to a Palaeoproterozoic collapse of atmospheric methane. Geobiology 4, 271–283 (2006). [Google Scholar]
- 33.Farquhar J., Johnston D. T., Wing B. A., Habicht K. S., Canfield D. E., Airieau S., Thiemens M. H., Multiple sulphur isotopic interpretations of biosynthetic pathways: Implications for biological signatures in the sulphur isotope record. Geobiology 1, 27–36 (2003). [Google Scholar]
- 34.Guo Q., Strauss H., Kaufman A. J., Schröder S., Gutzmer J., Wing B., Baker M. A., Bekker A., Jin Q., Kim S.-T., Farquhar J., Reconstructing earth’s surface oxidation across the Archean-Proterozoic transition. Geology 37, 399–402 (2009). [Google Scholar]
- 35.Johnson J. E., Gerpheide A., Lamb M. P., Fischer W. W., O2 constraints from Paleoproterozoic detrital pyrite and uraninite. Geol. Soc. Am. Bull. 126, 813 (2014). [Google Scholar]
- 36.Williford K. H., Van Kranendonk M. J., Ushikubo T., Kozdon R., Valley J. W., Constraining atmospheric oxygen and seawater sulfate concentrations during Paleoproterozoic glaciation: In situ sulfur three-isotope microanalysis of pyrite from the Turee Creek Group, Western Australia. Geochim. Cosmochim. Acta 75, 5686–5705 (2011). [Google Scholar]
- 37.Papineau D., Mojzsis S. J., Schmitt A. K., Multiple sulfur isotopes from Paleoproterozoic Huronian interglacial sediments and the rise of atmospheric oxygen. Earth Planet. Sci. Lett. 255, 188–212 (2007). [Google Scholar]
- 38.Mojzsis S. J., Coath C. D., Greenwood J. P., McKeegan K. D., Harrison T. M., Mass-independent isotope effects in Archean (2.5 to 3.8 Ga) sedimentary sulfides determined by ion microprobe analysis. Geochem. Cosmochim. Acta 67, 1635–1658 (2003). [Google Scholar]
- 39.Johnson J. E., Webb S. M., Thomas K., Ono S., Kirschvink J. L., Fischer W. W., Manganese-oxidizing photosynthesis before the rise of cyanobacteria. Proc. Natl. Acad. Sci. U.S.A. 110, 11238–11243 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hannah J. L., Bekker A., Stein H. J., Markey R. J., Holland H. D., Primitive Os and 2316 Ma age for marine shale: Implications for Paleoproterozoic glacial events and the rise of atmospheric oxygen. Earth Planet. Sci. Lett. 225, 43–52 (2004). [Google Scholar]
- 41.Rasmussen B., Bekker A., Fletcher I. R., Correlation of Paleoproterozoic glaciations based on U–Pb zircon ages for tuff beds in the Transvaal and Huronian Supergroups. Earth Planet. Sci. Lett. 382, 173–180 (2013). [Google Scholar]
- 42.H. C. Dorland, Provenance ages and timing of sedimentation of selected Neoarchean and Paleoproterozoic successions on the Kaapvaal craton, thesis, Rand Afrikaans University (2004). [Google Scholar]
- 43.Kaufman A. J., Johnston D. T., Farquhar J., Masterson A., Lyons T. W., Bates S., Anbar A. D., Arnold G. L., Garvin J., Buick R., Late Archean biospheric oxygenation and atmospheric evolution. Science 317, 1900–1903 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Ono S., Wing B., Johnston D., Farquhar J., Rumble D., Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles. Geochim. Cosmochim. Acta 70, 2238–2252 (2006). [Google Scholar]
- 45.Halevy I., Johnston D. T., Schrag D. P., Explaining the structure of the archean mass-independent sulfur isotope record. Science 329, 204–207 (2010). [DOI] [PubMed] [Google Scholar]
- 46.Reinhard C. T., Planavsky N. J., Lyons T. W., Long-term sedimentary recycling of rare sulphur isotope anomalies. Nature 497, 100–103 (2013). [DOI] [PubMed] [Google Scholar]
- 47.N. J. Beukes, H. C. Dorland, J. Gutzmer, paper presented at the Geological Society of America, Denver, CO, 2002. [Google Scholar]
- 48.Zerkle A. L., Claire M. W., Domagal-Goldman S. D., Farquhar J., Poulton S. W., A bistable organic-rich atmosphere on the Neoarchaean Earth. Nat. Geosci. 5, 359–363 (2012). [Google Scholar]
- 49.Catuneanu O., Eriksson P. G., Sequence stratigraphy of the Precambrian Rooihoogte–Timeball Hill rift succession, Transvaal Basin, South Africa. Sediment. Geol. 147, 71–88 (2002). [Google Scholar]
- 50.Altermann W., Nelson D. R., Sedimentation rates, basin analysis and regional correlations of three Neoarchaean and Palaeoproterozoic sub-basins of the Kaapvaal craton as inferred from precise U–Pb zircon ages from volcaniclastic sediments. Sediment. Geol. 120, 225–256 (1998). [Google Scholar]
- 51.G. Einsele, Sedimentary Basins Evolution, Facies and Sediment Budget (Springer, Berlin, Germany, 1992), 628 pp. [Google Scholar]
- 52.L. L. Coetzee, Genetic stratigraphy of the Paleoproterozoic Pretoria Group in the western Transvaal, thesis, Rand Afrikaans University (2001). [Google Scholar]
- 53.Claire M. W., Catling D. C., Zahnle K. J., Biogeochemical modelling of the rise in atmospheric oxygen. Geobiology 4, 239–269 (2006). [Google Scholar]
- 54.Goldblatt C., Lenton T. M., Watson A. J., Bistability of atmospheric oxygen and the great oxidation. Nature 443, 683–686 (2006). [DOI] [PubMed] [Google Scholar]
- 55.Konhauser K. O., Pecoits E., Lalonde S. V., Papineau D., Nisbet E. G., Barley M. E., Arndt N. T., Zahnle K., Kamber B. S., Oceanic nickel depletion and a methanogen famine before the Great Oxidation Event. Nature 458, 750–753 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Kopp R. E., Kirschvink J. L., Hiburn I. A., Nash C. Z., The Paleoproterozoic snowball earth: A climate disaster triggered by the evolution of oxygenic photosynthesis. Proc. Natl. Acad. Sci. U.S.A. 102, 11131–11136 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kump L. R., Barley M. E., Increased subaerial volcanism and the rise of atmospheric oxygen 2.5 billion years ago. Nature 448, 1033–1036 (2007). [DOI] [PubMed] [Google Scholar]
- 58.Zhelezinskaia I., Kaufman A. J., Farquhar J., Cliff J., Large sulfur isotope fractionations associated with Neoarchean microbial sulfate reduction. Science 346, 742–744 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Cameron E. M., Sulphate and sulphate reduction in early Precambrian oceans. Nature 296, 145–148 (1982). [Google Scholar]
- 60.Habicht K. S., Gade M., Thamdrup B., Berg P., Canfield D. E., Calibration of sulfate levels in the Archean ocean. Science 298, 2372–2374 (2002). [DOI] [PubMed] [Google Scholar]
- 61.Reinhard C. T., Raiswell R., Scott C., Anbar A. D., Lyons T. W., A late Archean sulfidic sea stimulated by early oxidative weathering of the continents. Science 326, 713–716 (2009). [DOI] [PubMed] [Google Scholar]
- 62.Crowe S. A., Paris G., Katsev S., Jones C., Kim S.-T., Zerkle A. L., Nomosatryo S., Fowle D. A., Adkins J. F., Sessions A. L., Farquhar J., Canfield D. E., Sulfate was a trace constituent of Archean seawater. Science 346, 735–739 (2014). [DOI] [PubMed] [Google Scholar]
- 63.Holland H. D., Why the atmosphere became oxygenated: A proposal. Geochim. Cosmochim. Acta 73, 5241–5255 (2009). [Google Scholar]
- 64.Martin D. M., Depositional setting and implications of paleoproterozoic glaciomarine sedimentation in the Hamersley Province, Western Australia. Geol. Soc. Am. Bull. 111, 189–203 (1999). [Google Scholar]
- 65.Haqq-Misra J. D., Domagal-Goldman S. D., Kasting P. J., Kasting J. F., A revised, hazy methane greenhouse for the Archean Earth. Astrobiology 8, 1127–1141 (2009). [DOI] [PubMed] [Google Scholar]
- 66.Bau M., Romer R. L., Lüders V., Beukes N. J., Pb, O, and C isotopes in silicified Mooidraai dolomite (Transvaal Supergroup, South Africa): Implications for the composition of Paleoproterozoic seawater and ‘dating’ the increase of oxygen in the Precambrian atmosphere. Earth Planet. Sci. Lett. 174, 43–57 (1999). [Google Scholar]
- 67.Moore J. M., Polteau S., Armstrong R. A., Corfu F., Tsikos H., The age and correlation of the Postmasburg Group, southern Africa: Constraints from detrital zircon grains. J. Afr. Earth Sci. 64, 9–19 (2012). [Google Scholar]
- 68.Cornell D. H., Schütte S. S., Eglington B. L., The Ongeluk basaltic andesite formation in Griqualand West, South Africa: Submarine alteration in a 2222 Ma proterozoic sea. Precambrian Res. 79, 101–123 (1996). [Google Scholar]
- 69.Canfield D. E., Raiswell R., Westrich J. T., Reaves C. M., Berner R. A., The use of chromium reduction in the analysis of reduced inorganic sulfur in sediments and shales. Chem. Geol. 54, 149–155 (1986). [Google Scholar]
- 70.Ono S., Keller N., Rouxel O., Alt J., Sulfur-33 constraints on the origin of secondary pyrite in altered ocean basement. Geochim. Cosmochim. Acta 87, 323–340 (2012). [Google Scholar]
- 71.Bradley A. S., Leavitt W. D., Schmidt M., Knoll A. H., Girguis P. R., Johnston D. T., Patterns of sulfur isotope fractionation during microbial sulfate reduction. Geobiology 14, 91–101 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Sim M. S., Ono S., Donovan K., Templer S. P., Bosak T., Effect of electron donors on the fractionation of sulfur isotopes by a marine Desulfovibrio sp. Geochim. Cosmochim. Acta 75, 4244–4259 (2011). [Google Scholar]
- 73.Canfield D. E., Reactive iron in marine sediments. Geochim. Cosmochim. Acta 53, 619–632 (1989). [DOI] [PubMed] [Google Scholar]
- 74.Betts J. N., Holland H. D., The oxygen content of ocean bottom waters, the burial efficiency of organic carbon, and the regulation of atmospheric oxygen. Palaeogeogr. Palaeoclimtol. Palaeoecol. 97, 5–18 (1991). [PubMed] [Google Scholar]
- 75.Bekker A., Kaufman A. J., Karhu J., Beukes N. J., Swart Q. D., Coetzee L. L., Eriksson K. A., Chemostratigraphy of the Paleoproterozoic Duitschland Formation, South Africa: Implications for coupled climate change and carbon cycling. Am. J. Sci. 301, 261–285 (2001). [Google Scholar]
- 76.Sumner D. Y., Beukes N. J., Sequence stratigraphic development of the Neoarchean Transvaal carbonate platform, Kaapvaal Craton, South Africa. S. Afr. J. Geol. 109, 11–22 (2006). [Google Scholar]
- 77.Hoffman P. F., The great oxidation event and a Siderian snowball earth: MIF-S based correlation of Paleoproterozoic glacial epochs. Chem. Geol. 362, 143–156 (2013). [Google Scholar]
- 78.Cheney E. S., Sequence stratigraphy and plate tectonic significance of the Transvaal succession of southern Africa and its equivalent in Western Australia. Precambrian Res. 79, 3–24 (1996). [Google Scholar]
- 79.Beukes N. J., Gutzmer J., Origin and paleoenvironmental significance of major iron formations at the Archean-Paleoproterozoic boundary. Soc. Econ. Geol. Rev. 15, 5–47 (2008). [Google Scholar]
- 80.Berner R. A., Raiswell R., Burial of organic carbon and pyrite sulfur in sediments over Phanerozoic time: A new theory. Geochim. Cosmochim. Acta 47, 855–862 (1983). [Google Scholar]
- 81.Garrels R. M., Lerman A., Phanerozoic cycles of sedimentary carbon and sulfur. Proc. Natl. Acad. Sci. U.S.A. 78, 4652–4656 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/2/5/e1600134/DC1
Geological background
Potential of sulfur MIF signals originated from weathering of preexisting S-MIF
Sensitivity analysis of C-O-S biogeochemical cycling model
fig. S1. Simplified geological map of the Transvaal Supergroup in the Transvaal and Griqualand West basins, Kaapvaal Craton, South Africa.
fig. S2. Representative transmitted light photomicrographs showing pyrite (opaque particles) distribution patterns in the samples analyzed in this study.
fig. S3. Cartoon figure showing the C-S-O geochemical model used in this study.
fig. S4. Sensitivity analysis for the power coefficient (θ) of pyrite burial and pO2.
fig. S5. Sensitivity analysis for the coefficient (γ) of weathering of sulfide and pO2.
table S1. Multiple sulfur isotope composition of the pyrite analyzed in this study. All data are relative to VCDT.
table S2. Parameters used for the Archean-Proterozoic oxygen-methane-sulfur cycle model.