

Abstract
Introduction
Arginine methylation is an abundant posttranslational modification occurring in mammalian cells and catalyzed by protein arginine methyltransferases (PRMTs). Misregulation and aberrant expression of PRMTs are associated with various disease states, notably cancer. PRMTs are prominent therapeutic targets in drug discovery.
Areas covered
The authors provide an updated review of the research on the development of chemical modulators for PRMTs. Great efforts are seen in screening and designing potent and selective PRMT inhibitors, and a number of micromolar and submicromolar inhibitors have been obtained for key PRMT enzymes such as PRMT1, CARM1, and PRMT5. The authors provide a focus on their chemical structures, mechanism of action, and pharmacological activities. Pros and cons of each type of inhibitors are also discussed.
Expert opinion
Several key challenging issues exist in PRMT inhibitor discovery. Structural mechanisms of many PRMT inhibitors remain unclear. There lacks consistency in potency data due to divergence of assay methods and conditions. Physiologically relevant cellular assays are warranted. Substantial engagements are needed to investigate pharmacodynamics and pharmacokinetics of the new PRMT inhibitors in pertinent disease models. Discovery and evaluation of potent, isoform-selective, cell-permeable and in vivo-active PRMT modulators will continue to be an active arena of research in years ahead.
Keywords: Arginine methylation, PRMT, Histone methylation, Inhibitor, Drug discovery, Chemical probe, Epigenetics
1. Overview of PRMT function in biology and disease
Arginine methylation occurs ubiquitously as one of cellular posttranslational modifications in eukaryotic organisms [1]. It is mediated by the family of N-arginine methyltransferases (PRMTs). PRMTs impact on numerous essential biological pathways (e.g. epigenetic regulation, RNA processing, DNA repair, hormone-receptor signaling, etc.) by methylating different nuclear, cytoplasmic and membrane protein substrates. Misregulation or aberrant expression of PRMTs has been found in various pathological conditions, notably cancer.
During PRMT catalysis, the methyl group of the co-substrate S-adenosylmethionine (SAM or AdoMet) is transferred to a protein substrate and replaces a hydrogen atom on the ω-NG of arginine, generating methylated arginine as a final product and leaving S-adenosyl homocysteine (SAH or AdoHcy) as the side product (Figure 1). There are mainly three types of methylated arginine products: monomethylarginine (MMA), asymmetric dimethylarginine (ADMA), and symmetric dimethylarginine (SDMA). Currently, nine members of PRMTs have been identified with established arginine methylation activity in human cells [2]. They are classified into three types: type I, II and III. Type I enzymes (EC 2.1.1.319) convert arginine to MMA and further to ADMA [2, 3], including PRMT1, -2, -3, -4 -6 and -8. PRMT4 is also known as coactivator-associated arginine methyltransferase 1, or CARM1. Type II enzyme (EC 2.1.1.320) is comprised of PRMT5 [4] and -9 [5], which produce MMA and SDMA. PRMT7 is the only type III PRMT (EC 2.1.1.321) that generates MMA [6]. The global arginine levels in the mouse embryo fibroblast (MEF) cells are found to be 1500:3:2:1 for Arg:ADMA:MMA:SDMA [7]. PRMT1 accounts for 50% of ADMA formation [8], and PRMT5 is the predominant type II enzyme, responsible for the bulk of SDMA in MEF [9].
Figure 1.

Protein arginine methylation regulated by PRMTs.
PRMTs have a wide range of protein substrates (Table 1). PRMT-1, -3, 5-, -6 and -8 prefer to methylate the glycine- and arginine-rich (GAR) region [7, 10]. There is a high degree of promiscuity in the substrate recognition by these PRMTs [1, 11]. No conserved sequences neighboring the methyl arginine sites are required [12], and the modification sites often occur in unstructured protein regions [13]. Structurally, the large binding surface for peptide substrates also supports the promiscuous nature of PRMTs [1, 11]. CARM1 tends to methylate proline-, glycine- and methionine-rich (PGM) motif [14, 15]; PRMT5 methylates GAR sequences as well as PGM regions [4]; PRMT7 was recently found to target RXR (X is a variable residue) in a lysine- and arginine-rich (KAR) motif [6] but not the previously reported GAR motif [16]; PRMT9 does not recognize GAR motif, and possesses very restricted activity on spliceosome-associated protein 145 (SAP145, also known as splicing factor 3b subunit 2, SF3B2) [5, 17].
Table 1.
Primary substrates, functions and disease relevance of PRMTs.
| PRMT | Substrate | Function | Disease relevance |
|---|---|---|---|
| PRMT1 | H4R3[188] hnRNP A1[189], BTG1[190], TIS2[190], IFNα/β[191], ILF3[192], SPT5[193], SAF-A[194], p53[195], MRE11[34], FMRP[196], Sam68[33], SLM[33], ERα[197], RUNX1[198], TAF15[199], BCR[200], CF Im59 and Im68[201], Ash2L[202], nuclear poly(A)-binding protein (PABP1)[203] | Transcriptional coactivator[134], signal transduction[197, 204], RNA splicing[205] and DNA repair[34] | Overexpressed or aberrant in breast, prostate, lung, colon, bladder cancer and leukemia.[2] Overexpressed or aberrant in pulmonary diseases: pulmonary fibrosis, pulmonary hypertension, chronic obstructive pulmonary disease (COPD) and asthma. [40, 47, 206] Play regulatory roles in cardiovascular disease[119, 207], diabetes[120, 121] and renal disease[44, 45, 122, 131]. |
| PRMT2 | H3R8[208] ERα[209], Glutathione transferase[210] | Transcriptional coactivator and androgen/estrogen receptor coactivator[211] | Overexpressed or aberrant in breast cancer [212, 213]. Overexpressed in pulmonary inflammation [206, 214]. |
| PRMT3 | FMRP[196], rpS2[128], PABP1[203] | Ribosomal homeostasis[129] | Enhanced activity in breast tumors [2]. Overexpressed in coronary heart disease [130] and chronic kidney disease [215]. |
| CARM1 | H3R2, H3R17, H3R26[216] PABP1[14]; SAP49 (CBP)/p300[217]; FMRP[196]; Sox9[218]; CA150, SmB, U1C and SF3b4[15] | Transcriptional coactivator[134], RNA splicing[219], cell proliferation[135], cell differentiation[136] | Overexpressed in breast, prostate and colorectal cancer [2]. Regulate human t-cell lymphotropic virus type 1 (HTLV1) [38]. |
| PRMT5 | H2AR3, H3R8, H4R3[138]; H3R2[220]; MBP (Myelin basic protein) [221]; LSm4, Sm D1 and Sm D3[31]; EBNA-2[222]; SPT5[193]; EBNA-1[223]; p53[200]; CBP-1[224]; CF Im68[201]; Ash2L[202]; PDCD4[225]; HoxA[41]; NFkB[226] | Transcriptional repressor[138], RNA splicing[139], signal transduction[140] and piRNA pathway[141] | Overexpression/increased activity in gastric, colorectal, lung cancer, lymphoma, and leukaemia [2]; mis-localized in prostate cancer cells [142]. Play regulatory roles in renal and cardiovascular disease [143], Huntington’s disease [144], Alzheimer’s disease [145] |
| PRMT6 | H2AR29[227], H3R2[148] PRMT6[228, 229]; HIV Tat[230]; HMGA1a[231], DNA polymerase β[149], PABP1[203] | Transcriptional repressor[150, 151] and activator[152] | Overexpressed in bladder and lung cancer [2] Suppress HIV-1 activity [39] Overexpressed or aberrant in pulmonary fibrosis, COPD and asthma [47] |
| PRMT7 | H4R3 and H2AR3[232], H3R2[220], Fibrillarin [233] | DNA damage[232], embryonic stem cell pluripotency[234], male germline gene imprinting[235, 236] | Involved in breast cancer metastasis [237] |
| PRMT8 | H2A, H4[238] MBP, PRMT8[238] EWS (Ewing sarcoma)[239] | Brain specific functions[240] | Somatic mutations were found in ovarian, skin and large intestine cancer [2] |
| PRMT9 | SAP145 (SF3B2)[9] | RNA splicing[5] | lymphoma, melanoma, testicular, and pancreatic cancers [241] |
A notable function of the PRMTs is that by depositing methyl marks onto the nucleosomal histone tails, they participate in the regulation of the dynamic transitions between transcriptionally active or silent chromatin states [18, 19]. PRMTs may act as either coactivators or repressors, depending on the modification site and status. As such, histone arginine methylation is an important component contributing to the complexity of the overall epigenetic marking system. Identified histone arginine methylation sites include H2AR3 (PRMT1, -5, -6), H2AR29/R31 (PRMT7), H3R2 (PRMT6), H3R8 (PRMT5), H3R17/R26 (CARM1), H4R3 (PRMT1, -5, -6), and H4R17/R19 (PRMT7) [3, 20–22], and there might be more to be uncovered. It is possible that a specific mark correlate to a specific chromatin state, despite the lack of understanding in their related pathways [3]. Binding of reader proteins to arginine methylation marks further recruits effectors of downstream signaling machinery, thus to mediate gene transcription, DNA replication and repair events [23, 24]. Arginine methylation can also block or promote other histone modifications or DNA methylation. The cross-talks among various epigenetic modifications further diversify the pathways of gene transcription and other fundamental DNA templated processes [21, 25, 26]. Grand challenges remain in deciphering the complex “histone codes.” Elaborating roles of PRMTs in histone modification will certainly fill in the knowledge gap.
PRMTs also have a broad spectrum of non-histone substrates, which are involved in crucial cellular regulations, such as transcription, RNA splicing, signal transduction, nuclear/cytoplasmic shuttling and DNA repair. Protein arginine methylation affects protein-protein interaction, protein-DNA or protein-RNA interaction, protein stability, subcellular localization, and enzymatic activity [3, 7, 27–29]. Selected PRMT substrates and functions are summarized in Table 1. For example, CARM1 methylates the C-terminal repeat domain (CTD) of RNA polymerase II for transcriptional control [30]. The cytoplasmic substrates of PRMT5, small nuclear ribonucleoproteins SmD1 and SmD3, are responsible for spliceosome assembly; spliceosome-associated proteins SAP49 and SAP145 are the substrates of CARM1 and PRMT9, respectively [5, 31]. PRMT1 methylates Sam68 (Src associated in mitosis, 68 kDa), an RNA-binding protein, and mediates signal transduction [32, 33]. PRMT1 also methylates the MRE11 (meiotic recombination 11) checkpoint protein for DNA damage checkpoint response and DNA repair [34]. PRMT5 takes part in a DNA damage responsive co-activator complex [35]. CARM1 methylates RNA-binding protein human antigen R (HuR) to affect its shuttling [36]. Despite the accumulating findings that reveal the molecular basis of PRMTs in the cell, our understanding of functions of this specific family of PTM enzymes remains to be further developed in depth, not to mention there is much of unknown to be discovered.
Mounting studies have pointed to multifaceted roles of PRMT misregulation in disease development. PRMT-related pathologies include virus-related diseases [37–39], inflammatory response [40, 41], cardiovascular disease [42, 43], renal disease [44, 45], diabetes [46], pulmonary disorders [47] and the most actively studied area, carcinogenesis [2, 3, 27, 48–53] (Table 1). Arginine methylation levels might be used as diagnostic biomarkers. For instance, the amount of ADMA is related to cardiovascular pathological conditions [42]; and in prostate cancer, the methylation of H4R3 predicts the risk of prostate cancer recurrence [54]. PRMTs are overexpressed in many types of cancers and some tumor types may overexpress several members of PRMTs, as listed in Table 1. Additionally, alternative splicing of PRMT isoforms may also play a critical role [55]: for example, seven PRMT1 isoforms (PRMT1v1-v7) have been found by alternative splicing in the 5′ region of its pre-mRNA [56], and PRMT1v1 is the most abundant variant. The overexpression of PRMT1v1 was observed colon cancer [57]; PRMT1v2 had the greatest increase in breast cancer and can promote the survival and invasiveness of breast cancer cells [58]. PRMT1 is a component of a mixed lineage leukaemia (MLL) transcriptional complex [59], and it is essential for the development of acute myeloid leukaemia (AML) [60]. A recent study shows that upregulated PRMT5 activity enhances AML growth [61]. PRMT5 is critical to B-cell lymphomagenesis and the maintenance of its malignant phenotype [37]. While it has been widely known that misregulation of PRMTs is involved in carcinogenesis and tumor progression, in most cases, the specific roles of these enzymes in cancer pathology are still poorly studied [2].
Along the line with biological studies on illuminating PRMT functions in physiology and pathology, developing potent and selective PRMT inhibitors has become a well-timed research endeavor in both academic laboratories and pharmaceutical industry. A number of PRMT inhibitors have been identified in the last decade through the efforts of researchers worldwide [62, 63]. Because multiple PRMT isoforms exist in the cell and each owns distinct functions, an important goal in the campaign of PRMT drug discovery is to find out effective lead compounds highly specific for one particular isoform enzyme. Pan-MTase inhibitors, such as simple SAM analogs sinefungin, SAH, methylthioadenosine (MTA), and AzaAdoMet (Figure 2), are often used as functional tools to change global methylation levels in cells and as cofactor ligands for determination of MTase X-ray structures [64]. However, due to their notorious promiscuity, pan-MTase inhibitors are not suited for use as chemical genetic tools for elucidating function of a particular PRMT isozyme in the proteomic context. In this regard, potent and isoform-selective small-molecule inhibitors will be much more desired. Encouragingly, quite a few small-molecule PRMT inhibitors have developed from different research laboratories and exhibit prominent isoform specificity, though at varying degrees. Below, we will first discuss biochemical assays used in the PRMT drug discovery, and then elaborate on different PRMT inhibitors according to their respective PRMT targets.
Figure 2.
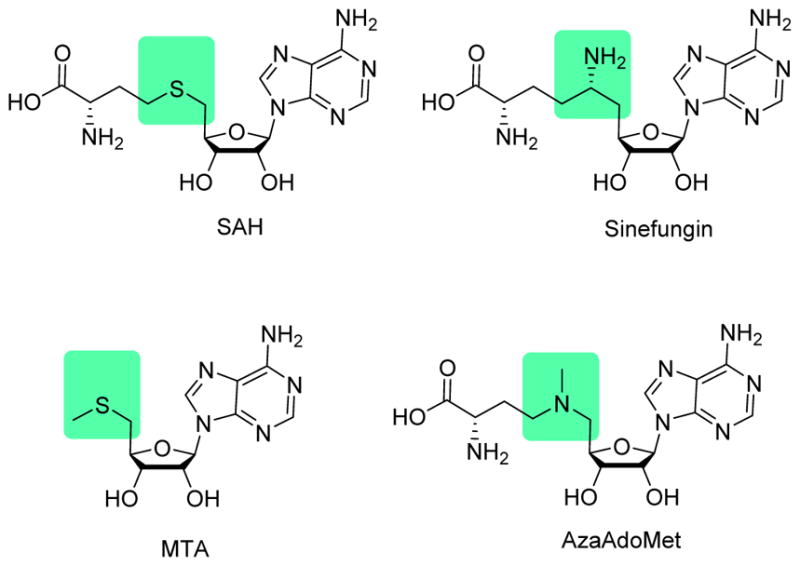
Simple SAM analogues.
2. Biochemical assays in PRMT inhibitor study
Discovery of PRMT modulators relies on effective assays to quantitatively measure PRMT activities. Quite a few biochemical assays have been developed for PRMTs and protein lysine methyltransferases (PKMTs) (see the recent reviews [65–69]). The radiometric assays have been the gold standard for quantitating in vitro methyltransferase activity of PRMTs due to their high sensitivity. In a general procedure, the radioisotope-labeled methyl group from [3H]-SAM or [14C]-SAM is first enzymatically transferred into a peptide or protein substrate of PRMTs. Prior to quantitation by autoradiography or liquid scintillation counting, the methylated substrates have to be separated from unreacted SAM by using different approaches such as polyacrylamide gel electrophoresis (radiometric gel assay, RGA) [70–76], pipette chromatography (ZipTip assay) [77], and filtering on glass fiber or phosphocellulose paper discs (radiometric filter assay, RFA) [37, 73, 78–90]. To eliminate the washing step required for the above described radiometric assays, scintillation proximity assay (SPA) has been implemented [80, 91–101], in which the scintillation signals depend on the micrometer proximity between biotinylated substrates and streptavidin-coated scintillants (either FlashPlates or streptavidin-coated microscopic beads). As such, the SAM molecules present in the bulk solution fall off the SPA distance and thus do not produce scintillation signals.
Antibody-based immunosorbent assays represent another category of widely used methods for detection of PRMT activities, in which a methylated substrate is recognized by methyl-arginine specific antibody. A typical format of these assays is enzyme-linked immunosorbent assay (ELISA) [70, 102, 103], in which methylated substrates are adsorbed onto microplate wells and incubated with primary antibody that is further probed with horseradish peroxidase (HRP)-labeled secondary antibody. After wash-out of the unbound antibodies, chemiluminescence is developed from the HRP. Dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) [71, 104–106] is similar to ELISA, except that the antibody is labeled with a lanthanide probe instead of HRP. The lanthanide dissociates from the antibody by addition of an enhancement cocktail and gains amplified fluorescence under excitation at 340 nm. Lanthanide as fluorophore features large stoke shifts, long decay time and narrow emission spectrum, which together minimize the interference of background fluorescence. Other assay methods involving lanthanide labels are homogeneous (no-wash) technologies that include time-resolved fluorescence resonance energy transfer (TR-FRET) [107–110] and AlphaLISA [107, 111]. In TR-FRET assay, energy is transferred from the donor europium chelate to the acceptor fluorophore within FRET distance (~10 nm). For AlphaLISA, donor beads convert ambient oxygen to singlet oxygen upon excitation at 680 nm, which can diffuse about 200 nm in solution. Then, the europium in the acceptor bead within this range receives energy and emits fluorescence (Figure 3).
Figure 3.

Biochemical assays for PRMT activity detection. Top blocks denote radiometric assays, bottom blocks denote antibody-based assays, and right blocks denote enzyme-coupled assays. RFA: radiometric filter assay. RGA: radiometric gel assay. SPA: scintillation proximity assay. SAHN: SAH nucleosidase. ADA: adenine deaminase. ELISA: enzyme-linked immunosorbent assay. HRP: horseradish peroxidase. DELFIA: dissociation-enhanced lanthanide fluorescent immunoassay. TR-FRET: time-resolved fluorescence resonance energy transfer. SAHH: SAH hydrolase.
Quite a few assays have also been developed to detect SAH, the side product of the methylation reaction. These strategies are to convert SAH into derivatives with colorimetric, fluorescent, or luminescent properties by coupled enzymatic reactions. In the SAHH (SAH hydrolase)-coupled assay, SAH is hydrolyzed into adenosine and homocysteine, the latter of which subsequently reacts with ThioGlo or CPM, yielding strong fluorescence [112, 113] [96, 114]. In another approach, SAH is converted into adenine by SAH nucleosidase (SAHN) and then to hypoxanthine by adenine deaminase (ADA). The methylation process is monitored by the absorbance difference of adenine and hypoxanthine [115]. Additionally, strategies are also reported to convert adenine to ATP which can be monitored by luciferase-linked bioluminescence [116, 117]. Interested readers are referred to reviews [67, 68] for more discussions of methyltransferase assays.
3. PRMT1-specific inhibitors
PRMT1 is the major enzyme responsible for asymmetric dimethylation of protein arginine residues [118]. PRMT1 methylates histone H4 and many other non-histone proteins. Studies have linked PRMT to many crucial biological processes such as transcription, cell signaling, RNA splicing and DNA repair [50]. Overexpression of PRMT1 has been observed in breast, prostate, lung, colon, bladder cancer and leukemia [2]. It is also found to relate with pulmonary diseases [47], cardiovascular diseases [119], diabetes [120, 121] and renal diseases [122].
The recombinant PRMT1 is well expressed in bacteria and demonstrates robust enzymatic activity. Thus, it is no surprise that PRMT1 is the most widely used PRMT model to test arginine methylation inhibitors. In 2004, Bedford and coworkers identified AMI series of compounds, represented by compounds 1 (AMI-1) and 2 (AMI-8) (Table 2), as the first set of small molecules to target PRMTs [70]. Details about this work and related studies are discussed in Section 8.1.
Table 2.
PRMT1-specific inhibitors.
| Structure and numbering | Activity | year[ref] | |
|---|---|---|---|
| Biochemical | Cellular | ||
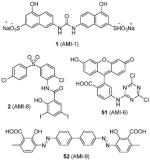
|
Assay: ELISA; RGA. Compound 1, 2, 51 and 52: IC50 0.3–8 μM (PRMT1); 100% inhibition (@ 5 μg) for PRMT-3, -4 and -6; Compound 1: inactive on PRMT5 Compound 1 and 51 (@ 5 μg): inactive for Suv39H1, Suv39H2, SET7/9 and DOT1 |
Compound 1:
|
2004[70], 2010[74], 2015[40] |
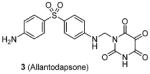
|
Assay: DELFIA IC50 on PRMT1: 1.7 μM, compared to 1.2 μM of AMI-1 |
In HepG2 cells, inhibition on H4R3 methylation level > 70% (@ 50 μM) determined by DELFIA, the same as AMI-1. | 2007[104] |
|
|
Assay: DELFIA IC50 13–17 μM (PRMT1) |
— | 2009[105] |
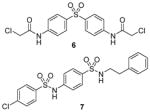
|
Assay: RGA; DELFIA Both: reversible inhibition Compound 6: IC50 1.5 μM (PRMT1); inactive on CARM1 and SET7/9 Compound 7: IC50 5 μM (PRMT1); inactive on CARM1; 60% inhibition (@ 50 μM) on SET7/9 |
Compound 6:
|
2011[71] |
|
Assay: DELFIA IC50 55 μM (PRMT1), compared with 1.2 μM of AMI-1 |
150 μM to achieve 50% inhibition of arginine methylation in HepG2 cells determined by DELFIA. | 2007[106] |
|
Assay: TR-FRET, AlphaLISA Compound 9: IC50 18 μM (PRMT1); Inhibition < 15% on CARM1 and PRMT6 (@ 100 μM) Compound 10: IC50 26 μM (PRMT1); 45% inhibition on CARM1 (@ 50 μM); 18% inhibition on PRMT6 (@ 100 μM) Note: IC50 55 μM of AMI-1 (PRMT1) |
Both: More than 40% of the proliferation of three cancer cell lines (HepG2, MCF7 and THP1), with compound 9 showing better activity. |
2014[107] |
|
Assay: RFA IC50 4.1 μM (PRMT1), 8–13 μM (PRMT3, -5, and -6), 100 μM (CARM1) |
|
2012[78] |
|
Assay: SPA; RFA More than 6-, 10-, and 25-fold selectivity for PRMT1 (3.38 μM) over CARM1 (21.5 μM), PRMT5 (35.4 μM), and PRMT8 (84.9 μM) Note: all the methylation assays were done under balanced condition |
|
2015[95] |
|
Assay: DELFIA IC50 57 μM (PRMT1), compared to 1.2 μM of AMI-1 |
|
2007[104] |
|
Assay: AlphaLISA IC50 2.0 μM (PRMT1), 10 μM (CARM1), >100 μM (PRMT5 and -6) |
|
2015[111] |
|
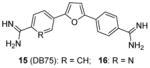
Assay: RFA IC50 on PRMT1: 15, 9.4 μM; 16, 7.2 μM; Stilbamidine (13), 15 μM. Selectivity: Both preferred PRMT1 over CARM1 (>42-fold), PRMT5 (>18-fold), and PRMT6 (>30-fold) Note: all the methylation assays were done under balanced condition |
Compound 15:
Inhibit PRMT1-mediated Smad6 methylation, revealing that the methylase activity of PRMT1 is was essential for BMP-induced Smad1/Smad5 phosphorylation and downstream signaling activation |
2013[123], 2014[79], 2015[124], 2015[125] |
|
— | — | 2015[126, 127] |
Compound 3 (allantodapsone) is found from a virtual screening on a library of 1990 compounds based on a homology-modeled PRMT1 structure. It shows IC50 of 1.7 μM on PRMT1 in DELFIA assay [104]. This compound inhibits cellular H4R3 methylation to the same level as AMI-1 at the same concentration in hepatocellular carcinoma cell line HepG2 while the H3K4 methylation level is barely impacted. Based on the pharmacophore of 3, compounds 4 and 5 are discovered by the same group from another round of virtual screening and show about 1/10 of the potency of 3 [105]. Later, a series of compounds are explored based on the structure of 3 and 4 [71]. Among these, 6 and 7 exhibit similar activity to their parental compounds in RGA assay and are demonstrated with reversible mode of inhibition. 6 is inactive to CARM1 and SET7/9 determined by DELFIA, while 7 shows 60% inhibition on SET7/9 at 50 μM and is inactive to CARM1. 6 inhibits the growth of breast cancer cell line MCF7a and prostate cancer cell line LNCaP with GI50 (i.e. concentration that produces 50% inhibition of growth) of 2 μM and 5 μM, respectively. 6 at 10 μM specifically reduces the level of androgen-dependent transcription to a significant degree. Intriguingly, compounds 3 – 7 structurally resemble the previously found AMI compound (AMI-8, compound 2) and they possess comparable inhibitory activity toward PRMT1, indicating they may share very similar pharmacophore. Notably, the selectivity profile of compound 6 is improved compared with 2.
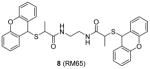
Compound 8 (RM65) is another early-stage PRMT1 inhibitor discovered from virtual screening, with IC50 around 55 μM [106]. It is less active than AMI-1 (1.2 μM) and is inactive on SET7/9 at 50 μM. High dose (150 μM) of 8 is needed to achieve 50% inhibition of arginine methylation in HepG2 cells. It is of note that selectivity profile among PRMTs has not been reported for compounds 3 – 5 and 8, so they cannot be considered as PRMT1-specific inhibitors. Besides, it is unlikely that compound 8 is useful in dissection of cellular PRMT1 function considering its relatively weak inhibitory activity.
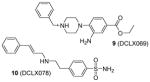
Discovered from a structure-based virtual screening, compounds 9 and 10 were subsequently validated with better inhibitory activity on PRMT1 (IC50 = 18 μM and 26 μM, respectively) than AMI-1 (55 μM) using a TR-FRET assay. Attractively, 9 is inactive to CARM1 and PRMT6 at 6-fold of the IC50 on PRMT1, but 10 shows lower selectivity. At 100 μM, both compounds inhibit more than 40% of the proliferation of HepG2, MCF7 and leukemic monocyte cell line THP1, with 9 showing better activity [107].
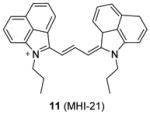
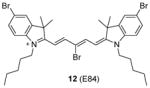
We recently screened a serial carbocyanine compounds against PRMT1 using the RFA assay [78], among which compound 11 was discovered with promising potency (IC50 = 4.1 μM) compared to AMI-1 and compound 13 (stilbamidine) (137 μM and 105 μM, respectively, determined under the same assay condition [89]). This compound also inhibits PRMT3, -5 and -6 with slightly weaker potency but is totally inactive to CARM1. The GI50 of compound 11 on cervical cancer cell line HeLa is around 1 μM at 48 h of incubation. Fluorescent imaging shows that 11 is concentrated in the nucleus and arrests the cell growth in the S phase, suggesting that it may interfere with the cell growth by causing chromatin dysfunction. In the following study [95], an extensive SAR study was performed on structurally diversified cyanine compounds, which demonstrates that symmetric structure, halogen-substituted bulky aromatic groups caging the charged N atom and hydrophobic side chain on the N atom are all essential to the activity. Among the investigated compounds, compound 12 exhibits decent potency (IC50 = 3.4 μM) on PRMT1 compared to SAH (IC50 = 0.4 μM) by the SPA measurement. Tested under the balanced condition, the compound exhibits more than 6-, 10-, and 25-fold selectivity for PRMT1 over CARM1 (IC50 = 21.5 μM), PRMT5 (IC50 = 35.4 μM), and PRMT8 (IC50 = 84.9 μM). The fluorescence intensity of 12 increases about 6-fold upon binding to PRMT1, indicative of direct interaction between PRMT1 and 12. The cellular activity of 12 was tested against chronic myelogenous leukemia cell line Meg01, acute myeloid leukemia cell line MOLM13 and erythroleukemia cell line HEL. Remarkably, 12 suppresses the cell proliferation at submicromolar level and is less sensitive in the HEL cells in which Janus kinase 2 (JAK2) has V617F mutation and is hence less dependent on PRMT1-mediated signaling pathway, indicating the anti-proliferation effect is achieved by interfering with PRMT-dependent pathway. Consistently, the global cellular methylation declines with 24h-treatment of 12 and the effect is significant at 100 nM of the compound for Meg01 and MOLM13 cells. One unique advantage of carbocyanine inhibitors is their strong fluorescent property, which can be utilized for subcellular tracking using fluorescence imaging microscopy [78].
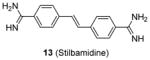
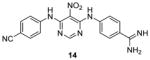
Quite a few PRMT1 inhibitors found so far contain one or two amidine groups. The amidine moiety is a structural mimic of the side-chain guanidino moiety of substrate arginine: both of which are rigidly planar, highly basic, with two ω-nitrogen atoms. Although structurally not yet confirmed, the amidine could possibly act as a substrate-competitive functional group for PRMT binding in the active site. The first such inhibitor is compound 13 (stilbamidine), identified during a screening campaign of disclosing allantodapsone 3 [71]. It inhibits PRMT1 activity with an IC50 of 57 μM in the enzymatic assay. The inhibitory effect on the H4R3 methylation by stilbamidine is the same as AMI-1 and allantodapsone in HepG2 cells. Besides, stilbamidine shows better activity than AMI-1 on reducing the transcriptional activation of an estrogen-dependent gene in MCF-7-2a cells. Yu et al. designed a serial amidines based on an amidine hit from initial screening [111]. The SAR indicates at least one amidine functionality is required for the activity, with other substitutions dictating the activity as well. Tested by AlphaLISA assay, compound 14 exhibits a 10-fold better potency than AMI-1 (2.0 μM vs 22 μM) and promising selectivity for PRMT1 over CARM1, PRMT5 and PRMT6. The docking study suggests this compound occupies the substrate arginine binding site. The cancer cell viability tests show the colon cancer cell line DLD-1 is the most sensitive (GI50 = 4.4 μM), followed by bladder cancer cell line T24 and neuroblastoma cell line SH-SY-5Y. In contrast, AMI-1 is inactive for all the tested cell lines. In DLD-1 cells, 14 reduce the H4R3 methylation level significantly starting from 3 μM.
We screened a focused library of diamidine compounds against PRMT1 under the balanced assay condition [79]. The leading hit, compound 15 (furamidine, also named DB75), shows inhibitory activity for PRMT1 at low micromolar potency. This study unravels some interesting SAR that is useful to guide further structure optimization. First, substitution by bulky group compromises the activity, which is probably caused by increased steric hindrance in the binding pocket assuming the amidino group mimics the structure of guanidine group in substrate arginine. Second, reducing the two amidino groups to one tampers the activity; perhaps the diamidine structure increases the probability of effective binding position of the amidino group. Third, the activity is reduced when the O atom in the furan is replaced by NH, especially by substituted NH, but replacing the O atom by S or Se atom was tolerated. DB75 is an old drug, originally developed for antiparasite treatment by targeting the minor groove of AT-rich DNAs. Its activity on PRMT inhibition, however, was unknown prior to our work. The IC50 of DB75 on PRMT1 is 2-fold lower than stilbamidine. DB75 shows a very favorable selectivity attribute for PRMT1 over the other PRMT members: 42-fold over CARM1, 30-fold over PRMT6, >15-fold over PRMT5. The selectivity property is also better than stilbamidine. The steady-state kinetic results show that DB75 is noncompetitive against SAM and has a strong nature of competitive inhibition with respect to the H4 peptide substrate. The competitive inhibition of DB75 versus the H4 substrate is also supported by competitive fluorescence anisotropy assay. Besides, surface plasmon resonance (SPR) assay has excluded any possibility of interaction between DB75 and H4 peptide, confirming the direct interaction between the inhibitor and enzyme. This diamidine molecule has a rigid, crescent, planar scaffold. Molecular modeling showed that the planar curving shape enables DB75 to fit the shape of the catalytic cavity of PRMT1, occupying partially the co-factor site and also spanning into the substrate arginine binding site. The computed binding free energy ΔGb arises from the better shape complementarity of compounds DB75 for the PRMT1 pocket. By contrast, PRMT5 exhibits a larger, partially solvent exposed pocket and binds the ligands less tightly in agreement with the smaller computed affinity ΔGb.
DB75 has been used as a tool compound to study PRMT1 function in different biological systems. Compound DB75 penetrated the plasma membrane of 293T cells (human embryonic kidney cell line) and inhibited cellular PRMT1 activity on ALY protein (also known as THO complex subunit 4, or THOC4) methylation, thereby regulating the turnover rate of ALY [79]. Treatment with DB75 (at 20 μM) for 72 hours led to significant inhibition of the growth of several leukemia cells. Intriguingly, cell lines derived from Down’s syndrome patients and MLL-AF9 (a fusion oncogene seen in leukemia cells) patient seems more sensitive. Compound 16, a close structural analogue of DB75, was used as a tool compound to inhibit PRMT1-mediated Smad6 methylation, revealing that the methyltransferase activity of PRMT1 is essential for bone morphogenetic proteins (BMP)-induced Smad1/Smad5 phosphorylation and downstream signaling activation [123]. Recently, we found that DB75 inhibits PRMT1-mediated putative RNA-binding protein 15 (RBM15) methylation at residue R578 [124]. Arginine methylation of RBM15 leads to its ubiquitination by the E3 ligase CNOT4 and subsequent degradation. Down-regulation of RBM15 changes alternative splicing of the downstream genes such as GATA1 (GATA-binding factor 1), C-MPL (MPL proto-oncogene) and RUNX1 (runt-related transcription factor 1 gene), which are critical for megakaryocyte differentiation. RBM15 binds to the SF3B1-containing complex in methylation-dependent manner. SF3B1 has been shown to be mutated in the myelodysplastic syndrome and other forms of leukemia. Thus, PRMT1-mediated methylation of RBM15 directly links the protein arginine methyltransferase to RNA splicing pathways. Interestingly, Siboni et al. [125] recently found that DB75 impacts on RNA splicing in a myotonic dystrophy type 1 (DM1) model with equal efficacy and low toxicity compared to another diamidine compound pentamidine. Given our observation that DB75 modulates PRMT1-mediated RBM15-SF3B1 splicing regulators, it could be possible that DB75 also regulates RNA splicing in DM1 through PRMT1 inhibition. This hypothesis has to await future experimental verification.
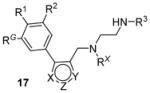
Recently, inhibitors with core structure of 17 (Table 2) were disclosed as effective inhibitors for PRMT1 (as well as PRMT6 and PRMT8) in two patents filed by Epizyme [126, 127]. It is not clear how selective and how potent they are. However, out of this series, compounds with extended side chains at para-position (R1) of phenyl group were subsequently reported as selective PRMT6 inhibitors (see Section 7).
4. PRMT3-specific inhibitors
There are only a few non-histone substrates found for PRMT3 (Table 1). A major PRMT3 substrate is rpS2 (ribosomal protein S2), which interacts with the zinc finger of PRMT3 [128]. It has been found to play a role in ribosomal homeostasis [129]. Overexpression or enhanced activity of PRMT3 has been recognized in breast cancer [2], coronary heart disease [130] and chronic kidney disease [131].
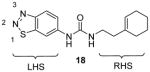
Compound 18 (Table 3) is the first reported specific inhibitor for PRMT3 [96]. From a library of 16000 diverse compounds, 18 is identified as the most potent using SAHH-coupled assay. The IC50 is determined around 2 μM with histone H4 peptide or 40S ribosomal protein S2 (rpS2) protein as substrate. The Kd is determined as 9 μM by SPR. The crystal structure of 18 and PRMT3 reveals that 18 is an allosteric inhibitor bound into the dimer interface. Binding of 18 probably causes the disorder of αX-helix, which is essential for cofactor binding. Out of a panel of five PKMTs and four other PRMTs, only PRMT5 is weakly inhibited by 18. Because the compound is metabolically unstable as tested from the in vitro assay with human and mouse liver microsomes, no cellular activity is reported for this compound. It is worthy to note that all the methylation assays were done under balanced condition, and hence the selectivity profile should be enzymatically meaningful. In addition, the inhibition mechanism was well characterized by a series of biophysics, enzymology, chemical biology and crystallography methods. All of these support that 18 is a bona fide PRMT3-specific inhibitor which is an ideal lead for further inhibitor development.
Table 3.
PRMT3-specific inhibitors.
| Structure and numbering | Activity | year[ref] | |
|---|---|---|---|
| Biochemical | |||
|
Assay: SAHH-coupled assay; SPA; RFA
Note: all the methylation assays were done under balanced condition. |
2012[96] | |
|
Assay: SPA; RFA 19a:
|
2013[80] | |
|
Biochemical | Cellular & Animal model | |
Assay: SPA
|
|
2015[97] | |
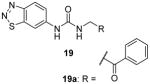
Later, a number of derivatives of compound 18 with diversified structures were subjected to SAR study [80]. The left-hand side region and the urea of 18 are crucial: replacing the two moieties by other ones causes total loss of the activity. However, the modifications of the right-hand side region are more tolerable. As a matter of fact, several compounds sharing the structure 19 have gained activity improvement by about 5 – 10 fold compared to 18. Then, 19a was picked out for the following selectivity profiling. Like 18, 19a also shows remarkable selectivity for PRMT3 against tested PKMTs, PRMTs and DNMT1, but no cellular activity is reported. The crystal structure of PRMT3-19a has confirmed the allosteric binding mode.
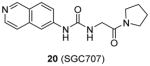
Afterwards, a great deal of efforts has been made by the same group to explore PRMT3 inhibitor with better cellular stability. Consequently, compound 20 (SGC707) was characterized as a promising lead [97]. The binding model and inhibition mechanism of SGC707 is the same as 18. The Kd is determined as 0.053 μM and 0.085 μM respectively from SPR and ITC (isothermal titration calorimetry). It is much smaller than that of compound 18, further validating that the intrinsic potency is enhanced by the structural optimization. Furthermore, it exhibits not only improved potency for PRMT3 (IC50 = 0.03 μM) but also outstanding selectivity against 31 PKMTs, PRMTs, DNMTs and RNA-methyltransferase as well as 250 non-epigenetic proteins. More importantly, SGC707 is proven to bind with PRMT3 overexpressed in HEK293 (human embryonic kidney cell line) and A549 (lung cancer cell line) cells by InCELL Hunter assay and inhibits the methylation of both endogenous and exogenous H4R3 in HEK293 cells (IC50 = 0.225 μM and 0.091 μM, respectively). No toxicity was observed after 24h-treatment of the compound. In addition, the pharmacokinetic study suggests the compound is feasible for in vivo in the future.
The structural basis of this set of inhibitors is the allosteric site binding, but this pocket can also be observed in other PRMTs (see Section 9 and review [132]). The high specificity is probably achieved by the variability of the sequence lining the pocket, implicating that the underlying rationale could also be applied to design specific inhibitors for other PRMTs. Of note is that compared with the SAH-bound PRMT3 [133], the residue R396 of the 18-bound PRMT3 adopts a distinct orientation which creates an open space to allow the inhibitor binding. Therefore, if one attempts to perform structure-based virtual screening for inhibitors targeting this allosteric site of other PRMTs, it is better to use a structural model based on the compound 18-bound PRMT3 structure instead of the SAH-bound crystal structure. In addition, the structure of PRMT dimer rather than monomer should be used since compounds 18 thru 20 are all shown to interact with both monomers.
5. CARM1-specific inhibitors
CARM1 is a transcriptional co-activator [134], and also involved in RNA splicing [15], cell proliferation [135] and cell differentiation [136]. It has a wide range of substrates including histones, transcriptional regulators, and splicing factors, as summarized in Table 1. CARM1 is overexpressed in breast cancer, prostate cancer and colorectal cancer [2]; and it also regulates pathology of human T-cell lymphotropic virus type 1 (HTLV1) [38].
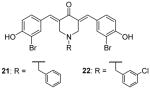
During the screening for multivalent epigenetic inhibitors, a set of curcumin derivatives were found with inclination to inhibit CARM1 in the radiometric assay [137]. Later, one of the hits (21, Table 4) was chosen as a lead to develop inhibitor to selectively target CARM1. Various substitutions on the benzyl group in R group were explored. With RGA assay, 21 inhibited CARM1 with IC50 of 8.6 μM and is over 80-fold less sensitive to PRMT1 and SET7/9, and all the compounds show similar activity and selectivity to 21, with 22 as the most potent one. Tested against a broader panel of PRMTs and PKMTs, 21 and 22 (at 100 μM) exhibit inhibition on PRMT3, -5 and -6 at different extents, but weaker than on CARM1. In HEK293 cells, 21 but not 22 suppressed the methylation of the transfected PABP1 (poly(A)-binding protein 1) protein. To investigate the inhibitory effect of the modulation of CARM1 on biological events, 21 and 22 were applied to treat LNCaP cells transfected with prostate-specific antigen promoter. The dose-dependent inhibition of the transcription level was observed starting from 4 μM for both compounds. At this concentration, 21 did not affect the cell viability [72].
Table 4.
CARM1-specific inhibitors.
| Structure and numbering | Activity | year[ref] | |
|---|---|---|---|
| Biochemical | Cellular | ||
|
Assay: RGA
|
|
2011[72] |
|
Assay: RFA Both:
|
Compound 34:
|
2009[81] |
| Biochemical | |||
|
|
Assay: RFA IC50 (CARM1): Compound 23, 1.8 μM; Compound 24–27, 0.08–0.23 μM; Compound 26: inactive to PRMT1 and -3 (IC50 > 25 μM); moderate permeability against PAMPA. |
2008[82], 2009[83] | |
|
|
Assay: RFA IC50 (CARM1): compound 28–32, 0.04–0.06 μM; Compound 31: inactive to PRMT1 and -3 (IC50 > 25 μM); improved permeability. |
2009[83] | |
|
|
Assay: RFA
|
2009[84] | |
|
Assay: RFA Compound 41: IC50 0.07 μM (CARM1); Inactive to PRMT1 and -3 (IC50 > 25 μM); Compound 42: IC50 0.12μM (CARM1); |
2009[85] | |
|
Assay: RFA
|
2011[86] | |
|
Assay: SPA IC50 0.80 μM for SETD2, 2.2 μM for SET7/9, 3.0 μM for CARM1, 9.5 μM for PRMT1, >100 μM for SET8, G9a, and GLP. |
2015[98] | |
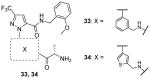
Another class of extensively studied compounds for CARM1 is the Pyrazole inhibitors. The hit 23 was found with IC50 of 1.8 μM for CARM1. Modification on the R part (Table 4) can only be well tolerated if there is an aryl substitute in the structure. Besides, any analogues replacing the south end glycine decrease the activity except (S)-alanine which slightly improved the potency. From the optimization, compound 24 – 27 display IC50 at submicromolar level with 26 showing the best potency (IC50 = 0.08 μM) [82]. 26 is inactive on PRMT1 and -3 (IC50 > 25 μM). Because 26 showed only modest permeability by PAMPA (parallel artificial membrane permeability assay), the SAR is further explored [83]: 1) the amide was replaced with 1,3,4-oxadiazole and 1,2,4-oxadiazole and the former one (compound 28) retained the activity and improved permeability against PAMPA; 2) the activity also declines by replacing the 3-trifluoremethyl pyrazole with other five-member heterocycle; 3) aryl groups are still tolerated for the R group. Finally, compounds 28 – 32 is found to show the nanomolar-leveled IC50. In the following assays, the representative compound 31 showed satisfactory selectivity for CARM1 over PRMT1 and -3.
Based on 23, structural optimization is performed by replacing the phenyl group with other aromatic ring and exploring more aryl groups for the R group by another lab [81]. From them, 33 and 34 are determined with similar IC50. The selectivity of 33 and 34 are also found promising for CARM1 against PRMT1 and SET7/9 (both IC50 > 100 μM). However, no inhibition was observed for cellular methylation of H3R26 by the treatment of 34 at 5 μM for 48 h. In addition, 34 failed to show any significant activity in the hormone-dependent assays (IC50 > 10 μM), which is probably due to the poor cell permeability considering their structural resemblance to 23. Envisaging that the alanine group (south end of 23) might be the cause, the lab then sought to improve the quality by replacing the alanine with other groups which generated compound 35 with a N2-methyl-1,2-diamine group in south end showing IC50 of 0.2 μM [84]. Further modification of the R group yielded compound 36 – 39 (IC50 = 0.32 – 0.9 μM). However, substitution on the phenyl group of the core of 23 decreased the activity to different extent. Besides, compound 40, a “N2-methyl-1,2-diamine” version of 29, exhibits similar activity (IC50 = 0.59 μM). 35 – 40 has indeed acquired enhanced pharmacokinetic profile (lower clearance, longer half-life and smaller volumes of distribution) compared with 33 while still retaining decent potency. Though selectivity property is yet to know for 35 – 40, it could be deduced that they might be still selective for CARM1 over PRMT1 and -3 since they basically share the same type of structure with 23 – 34.
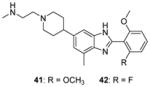
Another structural class of CARM1 inhibitors (Benzo[d]imidazole inhibitors) represented by compounds 41 and 42 is identified [85] from screening campaign by the same lab who discovered 23 – 32. Removing or modification of the 2-methylaminoethyl group of 41 results complete loss of the activity while substitution of the middle benzimidazole seems tolerable. The potency and selectivity of 41 and 42 are as good as 24 – 27, but no further cellular activity is reported.
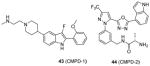
In the following work [86], the co-crystal structures with CARM1 are determined for the Pyrazole (43) and Benzo[d]imidazole (44) inhibitors, of which the potency and selectivity are comparable to 31 and 41. They both bind to the substrate arginine binding pocket as seen in the ternary structures of CARM1, SAH (or sinefungin), and the inhibitor. In addition, cofactor binding is a prerequisite for the binding of the inhibitors according to the ITC experiment. This work not only verifies the binding pattern between inhibitors and CARM1 but also provides substantial information for rational tailoring the compound with better pharmacokinetic property without compromising the activity in the future. Of caution that the selectivity of all inhibitors (23 – 44) discussed under this category can only be considered as apparent selectivity because whether the assays were done under balanced condition is not clear.
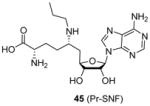
In a recent study [98] releasing sinefungin derivatives as inhibitors for the PKMT enzyme SETD2 (SET domain containing 2), compound 45 (Pr-SNF) exhibited low micromolar inhibition on CARM1 (IC50 = 3 μM) though it was demonstrated a much better inhibitor for SETD2. The fact that Pr-SNF and sinefungin (Figure 2) possess very close potency for CARM1 indicates the attached propyl group on sinefungin is well tolerated by CARM1 and has the potential to be further tailored to generate CARM1-specific inhibitors.
6. PRMT5-specific inhibitors
PRMT5 is the predominant type II arginine methyltransferase [4], which participates in transcriptional repression [138], RNA splicing [139], signal transduction [140] and piRNA pathway [141]. PRMT5 is upregulated in gastric cancer, colorectal cancer, lung cancer, lymphoma, and leukaemia [2]. It is also overexpressed and dyslocalized in prostate cancer cells [142]. Furthermore, PRMT5 has found to be involved in renal and cardiovascular disease [143], Huntington’s disease [144] and Alzheimer’s disease [145].
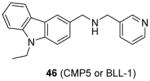
Very recently, a few studies have reported inhibitors targeting PRMT5. Firstly, compound 46 (CMP5, or BLL-1 according to the patent [146], Table 5) is reported in delineating the role of overexpressed PRMT5 in Epstein-Barr virus (EBV)-associated B-cell lymphomagenesis [37]. 46 is discovered from a virtual screening based on a modeled PRMT5 structure. The inhibition on symmetric methylation of the H3R8 and H4R3 was tested in JeKo cells (a mantle cell lymphoma cell line). The study confirms that treatment with 46 reduces histone methylation by specifically inhibiting PRMT5 rather than PRMT1, CARM1 or PRMT7 through RFA assay. The selective inhibition of 46 (50 μM) on the symmetric arginine methylation is also confirmed by an immunofluorescence staining against D-9 cells (lymphoma cell line). The inhibition on viability of lymphoma cells treated with 46 for 24h became significant when concentration of compound went up to 20 – 40 μM. In contrast, the normal resting B cells was only mildly affected even treated for 72h at 100 μM of 46, indicative of nontoxicity towards normal B cells. Further detailed work unravels the underlying mechanism of the cytotoxicity of 46. Using 46 as a molecular tool, the study demonstrates that PRMT5 and p65 and HDAC3 constitute a transcriptional repressive complex on miR96 (microRNA 96) promoter which in turn enhances the expression of PRMT5 itself and hence the overexpression of PRMT5 is maintained. The overexpressed PRMT5 is then utilized by EBV to silence tumor suppressor gene ST7 (suppression of tumorigenicity 7) and PTPROt (protein tyrosine phosphatase receptor-type O truncated), which causes the B-cell immortalization.
Table 5.
PRMT5-specific inhibitors.
| Structure and numbering | Activity | year[ref] | |
|---|---|---|---|
| Biochemical | Cellular & Animal model | ||
 Structure is drawn according to patent WO2011079236 A1 |
Assay: RFA 10–100 μM of compound 46 decreased histone methylation by inhibiting PRMT5 rather than PRMT1, CARM1 or PRMT7 |
|
2011[146], 2015[37] |
|
Assay: RFA 10 μM compound exhibited about 80% inhibition on PRMT5 but no obvious inhibition on PRMT1, CARM1 and PRMT7. |
|
2014[87], 2015[147] |
|
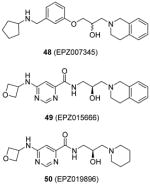
Assay: SPA; TR-FRET Compound 48: IC50 0.4 μM (PRMT5 : MEP50) Compound 49:
Note: all the methylation assays were done under balanced condition |
Compound 49: Cellular activity on MCL cell lines (Z-138, Maver-1, Mino, Granta-519 and Jeko-1): IC50 4 – 340 nM for inhibition on SmD3 dimethylation; IC50 60 – 900 nM for anti-cell-growth effect. In vivo activity:
|
2015[108–110] |
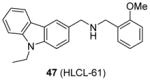
Compound 46 is subjected to further structural optimization with multiple ligand simultaneous docking method [147]. Compound 47 (HLCL-61), in which the pyridine of 46 is replaced with an ortho-methoxylphenyl group, gains improved activity, perhaps due to an addition of hydrogen bond with enzyme. This compound (10 μM) exhibits about 80% inhibition on PRMT5 but no obvious inhibition on PRMT1, CARM1 and PRMT7. The cellular activity of 47 is tested against AML samples. It decreases the symmetric dimethylation level of H3R8 and H4R3 though the concentration used was not clear. Decreased cell viability is also detected after treatment of 47, with the GI50 ranging 7 – 21 μM at 48h treatment. Further assays suggest compound 47 can promote apoptosis and induce differentiation. Next, 47 is used to dissect the function of PRMT5 in the myeloid leukemia growth and the authors find PRMT5 are critical to AML by silencing tumor suppressor microRNA miR29b while inducing transcription of a receptor tyrosine kinase gene, FLT3 (Fms-like tyrosine kinase 3).
Another report of PRMT5 inhibitors is from the research group in Epizyme [108, 110]. From several rounds of screening by a combination of various biochemical assays (TR-FRET and SPA) against a diversity library with 370000 small molecules, compound 48 (EPZ007345) was identified as a desirable inhibitor for PRMT5:MEP50 (methylosome protein 50) complex with IC50 around 0.3 μM. Iterative structural optimization was further performed to improve potency. The activity of 48 as well as its analogues was established for the inhibition on PRMT5 via the radiometric assay, decline of cellular methylation and suppression of cell proliferation (Z-138 cells). Any attempted modification on the tetrahydroisoquinoline part diminishes the potency due to a deleterious effect on the cation–π interaction between the ligand and enzyme, which is further confirmed by co-crystal structure of 48 and PRMT5:MEP50. Besides, amide replacing the alkoxy phenyl ring improves the activity by providing additional hydrogen bonds. After further optimization on the physiochemical property, compound 49 (EPZ015666) is obtained with desired clearance in human and mouse liver microsomes while retaining promising potency (IC50 = 0.022 μM). EPZ015666 shows outstanding selectivity against the other PRMTs (PRMT1, -3, -4, -6, -7 and -8; no inhibition at 50 μM) and a panel comprising 14 PKMTs, with all the assays being performed under the balanced condition. EPZ015666 acts as a substrate-competitor based on mechanism-of-inhibition study, which is also confirmed by the co-crystal structure of PRMT5:MEP50–SAM–EPZ015666. Interestingly, the binding of the cofactor SAM is prerequisite for the activity of this compound, as seen in the CARM1 inhibitors aforementioned. The global symmetric dimethylation (including the methylation on SmD3 protein) in Z-138 cells is found to be inhibited by EPZ015666, but not by an inactive analogue 50 (EPZ019896), which together support the cellular on-target effect. In a panel of five MCL cell lines, dose-dependent inhibition was observed for the symmetric dimethylation of SmD3 with IC50 ranging 4 – 340 nM. The cellular on-target binding of EPZ015666 was further confirmed by the cellular thermal shift assay. Longer period anti-proliferation (12 days) by EPZ015666 is observed for all the tested cell lines with IC50 ranging 60 – 900 nM. Excitingly, EPZ015666 shows antitumor effect in mice bearing Z-138 and Maver-1 xenografts. After 21 days of continuous oral dosing, 200 mg/kg of EPZ015666 resulted in significant tumor growth inhibition (> 90%, > 70% and > 45% for Z-138, Maver-1 and Granta-519 cell lines, respectively) with limited effect on the bodyweight. Accordingly, the symmetric methylation level in the xenograft tumors was decreased in a dose-dependent manner. A following study [109] reports that EPZ015666 exhibits low clearance in human, mouse and rat liver microsomes and higher clearance in dog liver microsomes as well as hepatocytes. This study provides important information about similarity of the metabolism profiles between different organisms and human, which may facilitate the transition from preclinical models to human test.
7. PRMT6-specific inhibitors
PRMT6 is a nuclear enzyme that primarily targets histone H3R2[148] and DNA polymerase β152 [149] (additional substrates are listed in Table 1). Studies show that PRMT6 can act as transcriptional repressor and activator through different pathways [150–152]. It is overexpressed in bladder and lung cancer cells, involved in pulmonary disorders [47] and can suppress HIV-1 activity [39].
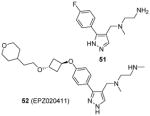
As mentioned in Section 2, compounds with the core structure of 17, represented by compound 51 (Table 6), were found to show promising potency to inhibit PRMT1, -6 and -8 via the SPA measurement [99, 126, 127, 153]. Extension of the para-position of the phenyl group of 51 with alkoxy group retains the activity. Further extension of the length of the group at para-position of the phenyl group results retained activity to PRMT6 and decreased activity to PRMT1 and -8, while large substitution on the meta-position yields opposite effect: the activity is diminished toward PRMT6 but not PRMT1 and -8. During the structural optimization, some analogues even show low nanomolar level of potency (IC50 ranging 5 – 20 nM) and remarkable selectivity for PRMT6 [99]. Out of them is compound 52 (EPZ020411, IC50 = 10 nM) that exhibits desired pharmacokinetic property as well as excellent selectivity (> 100 fold) for PRMT6 over other tested PRMTs (PRMT3, -4, -5 and -7) and PKMTs. The crystal structure demonstrates EPZ020411 inhibits PRMT6 by occupying the putative substrate arginine pocket. In A375 cells with transiently overexpressed PRMT6, inhibition on H3R2 methylation by EPZ020411 was observed does-dependently with IC50 at 0.64 μM. In contrast, the inhibition on PRMT1-specific methylation mark was seen at the 10 fold of the IC50 for PRMT6. In addition, the pharmacokinetic study suggests the bioavailability of EPZ020411 in rat by subcutaneous dosing (5 mg/kg), but not by oral dosing, is satisfactory for further in vivo efficacy profiling.
Table 6.
PRMT6-specific inhibitors.
| Structure and numbering | Activity | year[ref] | |
|---|---|---|---|
| Biochemical | Cellular | ||
|
Assay: SPA Compound 52:
Note: all the methylation assays were done under balanced condition |
Compound 52:
|
2015[99] |
|
Assay: SPA Compound 53:
Note: all the methylation assays were done under balanced condition |
Compound 53:
|
2015[100] |
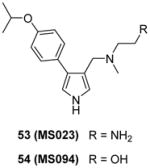
Inspired by the discovery of EPZ020411, a serial derivatives were designed to target type I PRMTs lately, in which compounds were tested using SPA method under the balanced condition [100]. Modifications are introduced on the pyrazole ring as well as the substitution of the phenyl group and ethylenediamino of EPZ020411. As a replacement of the pyrazole core, pyrrole produces better activity towards all the type I PRMTs than 1,2,3-triazole ring. Besides, para-isopropoxy group improves the potency compared with meta-trifluoromethyl group. Slight modification on the ethylenediamino group, such as altering the distal NH2 to OH or addition of a carbonyl group, totally abolishes the activity, supporting the authors’ hypothesis that the ethylenediamino is critical for the activity. Among others, compound 53 (MS023) has been extensively characterized at biochemical, biophysical and cellular levels. It shows nanomolar-leveled activity for tested type I PRMTs with best inhibition on PRMT6 and -8 (IC50 = 4 and 5 nM, respectively) but is inactive to type II and III PRMTs as well as a total of 28 PKMTs, DNMTs and histone lysine demethylases at up to 10 μM. The binding between PRMT6 and compound MS023 is confirmed by ITC (Kd = 6 nM) and differential scanning fluorimetry (DSF). In addition there is no binding between MS023 and nine tested methyllysine or methylarginine reader proteins as evaluated by DSF or differential static light scattering (DSLS). The co-crystal structure of PRMT6-SAH-MS023 reveals similar ligand-enzyme interactions as observed for EPZ020411, with the inhibitor occupying the arginine binding site. However, the mechanism of action (MOA) studies suggest compound MS023 is a noncompetitive inhibitor to both SAM and peptide substrate, which reminds that the active site-binding inhibitor does not warrant a competitive MOA [154]. After 48h-treatment, MS023 showed dose-dependent inhibition on endogenous PRMT1 activity on H4R3 in MCF7 cells (IC50 = 9 nM) and on overexpression PRMT6 activity on H3R2 in HEK293 cells (IC50 = 56 nM). In contrast, a structurally resembled but completely inactive analog, compound 54 (MS094), did not change the methylation status. The effect of MS023 on proliferation depends on exposure time and cell type: 4-day treatment of the inhibitor on HEK293 cells resulted great decrease of the cell growth at 1 μM while it required 10 μM and 10-day treatment of the inhibitor to affect cell growth significantly for the least sensitive MCF7 cells.
8. Other PRMT inhibitors
8.1 Promiscuous PRMT inhibitors
As aforementioned, AMI series are the first reported small molecule PRMT inhibitors. From an ELISA-based high throughput screening campaign against a library containing 9000 diverse chemicals, nine compounds (including AMI-1) stand out with low micromolar level inhibition for all tested PRMTs (PRMT-1, -3, -4 and -6) [70], but are inactive for PRMT5 [74]. The poor selectivity for PRMT1 may be related to the mechanism of inhibition (see Section 8.3). Among them, only AMI-1 (1) and AMI-8 (2) (Table 2) are inactive against the tested PKMTs. AMI-1 was able to inhibit the methylation level of exogenous nucleolar protein 3 (Npl3) and endogenous Sam68 protein in HeLa cells and suppress the effect of PRMT1 and CARM1 on nuclear receptor dependent transcriptional activation in MCF7 cells. Lately, AMI-1 was used to investigate the function of PRMT1 in chronic Ag-induced pulmonary inflammation (AIPI) in the E3 rat asthma model [40]. Similar to observation in PRMT1-knockdown rats, AMI-1-treated AIPI rats showed reduced COX2 (cyclooxygenase-2) production and humoral immune response, and abrogated mucus secretion and collagen generation.
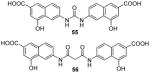
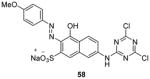
Enlightened by AMI-1, 51 (AMI-6) and 52 (AMI-9), compounds 55 – 58 (Table 7) were designed and exhibited similar activities to their progenitors on inhibiting the PRMT1-mediated methylation. Though they are inactive to SET7/9, 55, 56 [73] and 58 [74] still do not show selectivity among PRMTs (PRMT-1, 3, 4 or -6) while 57 [102] is only tested against PRMT1. Western blot analysis shows the arginine methylation level of exogenous Npl3p protein in HeLa cells was inhibited by 55 at 50 μM [73]. In VCaP cells (a prostate cancer cell line), dose-dependent inhibitory effect of 57 was observed on the expression level of two androgen-dependent genes [102]. In 293T cells, the methylation level of H3 protein Arg17 (H3R17, a major methylation site of CARM1 [155]) was decreased to 55% by 58 at 100 μM, in contrast to around 80% by AMI-1 of the same concentration [74]. Furthermore, 58 is demonstrated to reduce the secretion of interferon γ (IFNγ) and interleukin-4 (IL-4) by Type 1 T-helper (Th1) cells and Type 2 T-helper (Th2) cells, respectively, possibly due to interrupted association between PRMT1 and NIP45 (45 kDa NF-AT-interacting protein, a protein involved in transcription of IL-4 gene when methylated by PRMT1).
Table 7.
Other PRMT inhibitors.
| Structure and numbering | Activity | year[ref] | |
|---|---|---|---|
| Biochemical | Cellular | ||
| 1) Promiscuous PRMT inhibitors | |||
|
Assay: RFA; western blot; RGA Compound 55: IC50 112μM (PRMT1); similar activity to AMI-1 against PRMT-1,-3 and -4; Compound 56: IC50 272 μM (PRMT1) Note: IC50 of AMI-1 was 92 μM (PRMT1) |
Compound 55: Inhibition on methylation of transiently transfected Npl3p start to show at 50 μM | 2010[73] |
|
|
Assay: ELISA IC50 6.1 μM (PRMT1); 15% – 20% inhibition on PRMT5 (@ 100 μM) |
Dose-dependent inhibitory effect on the expression level of androgen-dependent genes (TMPRSS2 and FKBP5); 50% of the maximum inhibition was achieved at 25μM. | 2013[102] |
|
Assay: RGA Better activity than AMI-1 on PRMT1, -3, -4, -5, -6 and -8; IC50 4.2 μM (PRMT1); IC50 2.7 μM (CARM1); Inactive on SET7/9 |
|
2010[74] |
| 2) Bisubstrate inhibitors | |||
|
Assay: RGA IC50 3–6 μM (PRMT1); IC50 > 100 μM (CARM1); IC50 > 100 μM (SET7/9) |
— | 2010[75], 2011[76] |
|
Assay: ELISA IC50 of compound 61 and 62: IC50 0.1–0.2 μM (CARM1); > 5 μM (PRMT1, PRMT6); IC50 of compound 63: 3.2 μM (PRMT6); > 50 μM (PRMT1, CARM1) |
— | 2015[103] |
|
Assay: SPA IC50 6 μM (PRMT5/PRMT7); inactive to other PRMTs Note: all the methylation assays were done under balanced condition |
Start to decrease the methylation level of SmD and SmB proteins in MBMDA231 cells at 10 μM. | 2015[101] |
| 3) Inhibitors with substrate-targeting property | |||
|
Assay: RFA; western blot Bind histone H3 instead of CARM1 as determined by ITC, targeting residue sequence KAPRK Specifically inhibit methylation of H3R17 by CARM1 (IC50 around 25 μM); no effect to acetylation of H3K14 and H3K18 by p300. |
Affect H3R17 methylation but not H3K14 and H3K18 acetylation in HeLa cells; Repress the expression of p53-responsive gene, p21, by decreasing H3R17 methylation |
2010[88] |
|
Assay: RFA Compound 66: IC50 12 μM (PRMT1 with H4(1-20) peptide), 7.1 μM (PRMT3 with R4 peptide), 2 mM (CARM1 with H3(1-31) peptide), 39 μM (PRMT6 with H3(1-31) peptide), 21 μM on (p300 with H4(1-20) peptide) Bind H4(1-20) peptide as validated by uv, fluorescence and mass spectra . Note: : IC50 of AMI-1 is 137 μM on PRMT1 with H4(1-20) peptide as substrate |
— | 2010[139] |
|
Assay: RFA Compound 68: IC50 41 μM (PRMT1 with H4(1-20) peptide) Compound 69:
Note: : IC50 of AMI-1 is 77 μM on PRMT1 with H4(1-20) peptide as substrate |
Compound 68: 10 μM achieve 70% inhibition on proliferation of LNCaP C-81 Cells at in regular medium and 30% inhibition in steroid-reduced condition |
2012[242] |
Since these inhibitors exhibit relatively low selectivity, more selective inhibitors are highly desired when it comes to the study of specific PRMT subtype. It is worth noting that the methylation assays done in these studies are not performed under balanced condition (see Section 9 for detailed discussion) [156]. Further studies are needed to validate their selectivity profiles. As a matter of fact, structurally related compounds (3 – 7, Table 2) are proved with improved selectivity.
8.2 Substrate-based inhibitors
Inhibitors based on the amino acid sequence of PRMT substrates presumably possess high affinity and specificity via extensive interaction between peptide and the targeted enzyme. Several sets of peptide inhibitors have been designed and studied, with some showing promising potency and selectivity [157–161]. Interested readers are recommended to refer to a recent review on PRMT peptide inhibitors for more detailed information [162]. Nonetheless, the substrate sequences of PRMT are usually highly charged (rich in Arg and Lys) in physiological pH, so the corresponding peptide inhibitors by themselves are neither orally available nor metabolically stable. From this point of view, the peptide inhibitors are more useful as chemical probes to study function and mechanism of PRMTs at biochemical and cellular levels.
Exploration of small bisubstrate analogue as inhibitor is a strategy that has been justified successful in achieving specificity between enzyme isoforms in the field of kinase [163, 164]. The cofactor SAM is the primary methyl donor for methyltransferase [165]. It is not surprising that simple SAM analogues, such as SAH, sinefungin, MTA (Figure 2) act as pan-inhibitors for methyltransferase and can result in global decrease of methylation [64]. However, structural studies shows that active sites of methyltransferases bear different degrees of plasticity and structural uniqueness [132]. Thus, it is possible to design SAM analogs for selective inhibition of a target methyltransferase, a successful example being DOT1L selective inhibitors [166, 167].
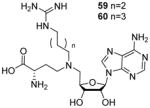
The SAM analogue AzaAdoMet (Figure 2) is a promiscuous methyltransferase inhibitor[168]. Dowden et al. [75, 76] synthesized several AzaAdoMet derivatives, and found that compound 59 and 60 (Table 7) inhibited PRMT1 with an IC50 value of 2.9 and 5.6 μM, respectively, and was inactive against CARM1 and SET7/9. The docking result suggests a bisubstrate binding mode and the four or five methylene linker is favored by PRMT1 but not by CARM1. The work has justified the strategy that combination of structural features from both substrate and cofactor could achieve selectivity not only between different methyltransferases families but also for different members within the PRMT family.
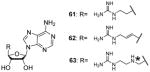
Following this methodology, compounds 61 and 62, with shorter linkers between the two feature groups, are developed with IC50 at sub-micromolar level (0.12 and 0.15 μM, respectively) for CARM1 and the selectivity is at least more than 50-fold over PRMT1 and PRMT6. One of the compound (63) in the same series is found to be a selective PRMT6 inhibitor [103]. Interestingly, replacing the asterisked NH in R group (Table 7) with O or S atom totally abolished the activity to all the tested PRMTs. However, compound 61 and 62 did not show significant effect on the proliferation of two cancer cell lines even at the concentration of 100 μM, possibly due to the high polarity and poor membrane permeability.
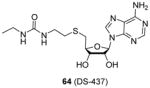
A similar structure (compound 64), rationally designed based on the ligand bound crystal structure of hPRMT5 and xlPRMT5, is found to be a potent PRMT5/PRMT7 dual inhibitor (IC50 = 6 μM for both enzyme), but inactive against a panel of 29 other human protein-, DNA-, and RNA-methyltransferases [101]. Being less polar than the compounds above, 64 exhibits dose-dependent inhibition on the methylation level of endogenic SmD and SmB proteins in breast carcinoma MB-MDA231 cells. The inhibition of 64 becomes significant when the concentration is larger than 10 μM. This study is encouraging because it suggests small bisubstrate inhibitors, in contrast to the peptide inhibitors, are capable to be tailored with appropriate hydrophobicity for penetrating cell membrane.
8.3 Histone-interacting inhibitors
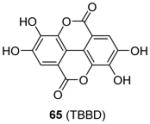
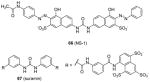
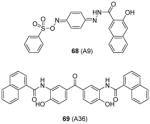
Some PRMT inhibitors are found to possess capability of binding to the histone tail peptides, which include compound AMI-1 (1) and 65 – 69 (Table 7). Compound 65 (TBBD, also known as ellagic acid) was found to be a specific CARM1 inhibitor (IC50 = 25 μM), which specifically inhibits methylation of H3R17 [88]. The ITC assay shows that 65 binds to a peptide sequence KAPRK of histone H3, highlighting it is a substrate-targeting inhibitor. Compound 66 (NS-1) was found to show more favorable IC50 (12 μM) than AMI-1 (137 μM) in PRMT1 inhibition. However, the interaction between 66 (as well as AMI-1) and the histone substrate was substantiated by mass spectrometry, UV, and fluorescence, suggesting the compound inhibits PRMT1 activity largely by binding with the substrate [89]. Unlike the recognition of TBBD for short peptide sequence, 66 has higher activity on H4(1-20) peptide than H4(1-11) peptide, suggesting the distal residues in the peptide sequence participate in the interaction of 66 with H4. 66 shows similar IC50 (21 μM) to inhibit the acetylation of H4(1-20) by p300 (an acetyltransferase), further supporting its substrate-targeting property. The same study also suggests that suramin (compound 67), which is structurally similar to 66 and AMI-1, also acts by binding with the H4 substrate [169]. In addition, compound 68 (A9) and 69 (A36), discovered from pharmacophore-based virtual screening, are validated as histone-H4-tail-targeting inhibitors as well (IC50 = 41 μM and 12 μM) [90]. Because of their nature of interacting with the histone tails (and possibly other histone-like sequences), careful caution should be taken on interpreting the biological effects of these histone-targeting compounds in cells. This type of unique inhibitors could find use as complementary chemical probes together with enzyme inhibitors to study mechanism and function of nucleosome modifications.
Taken together, considerable success has been achieved in the discovery of PRMT inhibitors. The discovered inhibitors contain different structural scaffolds and act through different modalities, providing powerful and versatile molecular tools to understand PRMT mechanism and function. PRMTs are found to methylate hundreds to thousands of cellular proteins, symbolic of their diverse roles in myriad pathways. Inhibition of PRMTs via small molecule inhibitors is a powerful chemical genetic strategy for interrogating the pathophysiological function of PRMTs in various disease pathways. Validated leads will serve as potential drug candidates for pharmacological treatment of arginine methylation-involved diseases.
9. Expert opinion
Since the first report of small molecule inhibitors for arginine methylation by Bedford and coworkers in 2004, the field of PRMT drug discovery has been productive, which is evidenced by the appearance of several micromolar and submicromolar small molecule inhibitors targeting different PRMT members, such as DB75 (15) for PRMT1 [79], SGC707 (20) for PRMT3 [97] and EPZ015666 (49) for PRMT5 [108]. Such appreciable progress is made possible owing to multiple factors. A fundamental impetus lies in the biomedical community’s enthusiastic quest for novel disease targets and the improved understanding of PRMT functions in disease progression pathways. Although PRMT inhibitor discovery research until now still lags behind the area of PKMT inhibitor discovery [170–174], the scenario is constantly changing. During year 2014–2015, the investment on PRMT inhibitor discovery increased steadily and several patents were filed pertaining to PRMT inhibitors [87, 126, 127, 146, 153]. That said, great strides are in imperative demand in this field. We discuss three key challenging issues that need to be addressed for the field to leap forward.
First, structural mechanisms of the majority of PRMT inhibitors remain unclear. Although dozens of structurally diverse PRMT inhibitors have been available, co-crystal structures of PRMT-inhibitor complexes are significantly needed at this stage. Only a few PRMT-inhibitor complex structures were available [80, 86, 96, 97, 99, 108]. Co-crystal structures of PRMT-inhibitor complexes are of value to drug discovery for several reasons: structural determination is a straightforward means for target validation of inhibitors for PRMT binding and inhibition; structural information is useful to understand the MOA of PRMT inhibitors; structural insights can be obtained for inhibitor optimization and rational design to make more potent leads. For example, both sulphone inhibitors and furan inhibitors (represented by compounds 6 and DB75, respectively) have a similar crescent-shaped backbone scaffold and have terminal heteroatomtic groups. It would be tempting to postulate that these two types of compounds target PRMT1 activity with similar binding structural mode. Crystal or NMR structures of PRMT1 complexed with these inhibitors will be the key to approving or disapproving the proposition.
It is a tremendous advance in the recent years that X-ray crystal structures of the majority of PRMTs have been resolved by research groups in America, Asia and Europe. These high-resolution structures undoubtedly deliver a thrust to speed up PRMT inhibitor discovery either by virtual screening or structure-based rational design. From the structural point of view, small molecule ligand targeting PRMT enzyme can bind to either the active site or an allosteric site. The deeply buried SAM binding pocket of PRMT is conserved among PRMTs and also share certain structural similarity with other methyltransferases, such as small molecule-, RNA-, and DNA-methyltransferases [22, 132, 175]. With regard to the substrate binding region, the surface grooves of PRMTs that likely accommodate substrate peptide backbones are shallow and wide. Besides, the substrate promiscuity of PRMTs suggests that the substrate binding grooves lack specificity regarding molecular recognition. Therefore, designing small molecules to target the substrate binding grooves would be highly challenging. On the other hand, exploration of distinct allosteric sites in PRMTs might be recommended as an alternative and effective strategy to identify small molecule inhibitors for specific PRMT members.
We analyzed the crystal structures of PRMTs using the Discovery Studio software to locate possible exosite binding pockets (Figure 4). Two major allosteric cavities are identified in the PRMT structures. Cavity 1 is located near the “double-E” loop that contacts the incoming arginine residue and is essential for catalytic activity. Cavity 1 of PRMT1, PRMT6 and PRMT8 have a volume in the range of 150 – 220 Å3 and is located at almost the same location, with the loop between β13 and β14 sheets possessing the largest sequence diversity (Figure 4H, Cavity 1). The cavity 1 of PRMT3 [96] is located toward the β-barrel and can only be seen in the “inactive” form (PDB: 3SMQ) but not in the “active” form (PDB: 1F3L) [133], indicating that locking this cavity might interrupt the enzymatic activity though its volume is relatively smaller (64 Å3). No such cavity is found in the CARM1 structure [86].
Figure 4. Illustration of possible allosteric cavities in PRMTs.

The pink colored mesh represents the catalytic pocket. The allosteric cavities are identified using the Discovery Studio software. Numbering of α-helices and β-sheets follows the orders of secondary structures in PRMT1. Panel H shows the alignment of the sequences in PRMT1, -3, -6 and -8 lining the two allosteric cavities.
Cavity 2 is located underneath the dimerization arm (formed by αE, β7 and αH). Targeting this region could influence the dimerization and hence the enzyme activity. This conclusion is supported by recent molecular simulation study showing that the dimerization arm allosterically influences SAM binding and catalytic activity of PRMT1 [176]. Cavity 2 is shared by PRMT1, -3, -4, -6, -7 (C-terminal domain) and -8 (volume ranging 100 – 160 Å3) but with varied sequence of αE-helix (Figure 4H, Cavity 2) and cavity shape. For PRMT8 [177], the sequence corresponding αE and αF helices in other PRMTs exists as coils (denoted as “αE”- and “αF”-coil, respectively), leaving a large gap between this motif and αG helix to form cavity 3. PRMT7 has two tandem PRMT core domains (denoted as N-terminal domain and C-terminal domain), possibly mimicking the canonical PRMT dimer architecture [178]. Cavities 2 and 3 are found in the C-terminal domain of mPRMT7 with a summed area of 263 Å3 and the sequence of hPRMT7 lining this pocket is quite distinct from the other human PRMTs (not shown). There are additional unique cavities, such as a surface area (442 Å3) near the middle of the C-terminal domain of PRMT7 (whether binding to this area is useful for interference of enzymatic activity is yet to discover) and a large area (2148 Å3) between TIM-barrel (Figure 4D, green) and MEP50 (Figure 4D, yellow) in the PRMT5 complex structure [179]. The differences of the residue sequences lining the sidewalls of the allosteric sites between different PRMTs might render the specificity of inhibitor recognition for particular isoforms.
Second, caution should be taken on appropriate interpretation of reported potency values of PRMT inhibitors. The most common metric used in potency characterization is IC50, i.e. the concentration of inhibitor that produces 50% inhibition of enzyme activity. The choices of assay methods and conditions for IC50 measurements often vary in one study from another, which sometimes yield large divergence in reported IC50. For example, literature reported IC50 of AMI-1 (1) in PRMT1 activity inhibition vary substantially, from 1.2 μM [104], 8.8 μM [70], 22 μM [111], 67 μM [73], 77 μM [90], 92 μM [169], 137 μM [89], to 376 μM [89]. As a result of such divergence, the definition of “strong inhibition” could be interpreted differently by individual researchers. One should keep in mind that IC50 values directly depend on assay methods, reagent sources and concentrations, and other experimental conditions. IC50 of a single inhibitor can vary largely under different experimental conditions. Depending upon its working mechanism, IC50 of an inhibitor can change in different manners as the concentration of substrate increases: it can either increase (for competitive inhibitors), decrease (for uncompetitive inhibitor), or remain unaffected (for classic noncompetitive inhibitors). In contrast, inhibition constant (Ki) or binding affinity constant (Kd,), which represents the intrinsic affinity of a ligand for a specific enzyme, is independent of substrate concentration. Thus, Ki is more stable in comparison to IC50 with regard to experimental condition changes. Measurement for Ki or Kd, however, is neither necessary nor simple for every hit in the initial round of screening. Practically, inclusion of a standard PRMT inhibitor (e.g. sinefungin or SAH) as a proper control is highly recommended in PRMT inhibition assays in order to achieve meaningful potency comparison for the IC50 data obtained by different laboratories. Because the exact IC50—[S] relationship is determined by the action mechanism of an inhibitor, we recommend that PRMT inhibitor screening and IC50 measurement be carried out in the initial velocity phase and under the balanced assay condition, in which the concentration of the selected substrate is equal or close to its Michaelis constant, Km. The balanced assay condition is an experimental choice to identify structurally diversified inhibitors of enzyme that may act through different mechanisms, which can minimize the pitfall of missing hits in library screening. The balanced assay condition is also important in evaluating the selectivity profile of an inhibitor against one PRMT versus the other PRMT isoforms. In selectivity profiling, if inappropriate substrate concentrations are chosen for different PRMTs, the drawn conclusion about inhibitor selectivity could be inaccurate and biased. The reason is that individual PRMT isoforms have different Km for their substrates. For instance, for a substrate-competitive inhibitor, relationship of IC50 to substrate concentration follows the equation of . As such, IC50 is affected by both Km and substrate concentration. Under the balanced assay condition ([S] = Km and IC50 = 2Ki), the ratio of IC50 is a faithful representation of the inhibition constant Ki, thus can be compared between different PRMTs. Interested readers can read more details from the reference [156].
Third, extensive studies should be invested on target validation of new PRMT inhibitors in both in vitro and in vivo models. A good lesson is learnt from the mechanism study of PRMT1 inhibition by AMI-1 (1): although originally expected to be a PRMT1-binding inhibitor, later biophysical experiments show strong interaction existing between the inhibitor and the histone substrate, which likely is the main reason for observed PRMT1 inhibition [89]. Amongst the reported PRMT inhibitors, dozens may have chemical susceptibility of being pan-assay interference compounds (PAINS). PAINS refer to those compounds which are reactive to amino acids in protein and therefore inhibit enzyme non-specifically. Increasing advocacy has been made to filter off PAINS in the early stage of drug discovery [180–182]. Besides, overly hydrophobic compounds that are prone to form aggregates could cause non-specific enzyme inhibition [183].
Target validation of PRMT inhibitors in cellulo and in vivo is particularly important and needed. In a cellular setting in which the inhibitor is exposed to the entire proteome and complex metabolic networks, its effective binding to the protein target becomes much more challenging than in biochemical assays. Since it is hardly possible for a drug candidate to have a sole molecular target, the selectivity is critical for confirming the correlation between the inhibition of targeted PRMT and corresponding phenotype [184, 185]. Many PRMT inhibitors demonstrate anti-tumor effects in cellular assays. Whether such phenotypic effects are a direct result of PRMT inhibition is an important issue. Therefore, following discovery of PRMT inhibitors, extensive pharmacological examination of those inhibitors in cellulo and in animal models should be conducted. It would be necessary to develop effective and physiologically relevant cellular assays for individual PRMTs and their chemical modulators. However, this is quite challenging because different PRMTs have different function and substrate profile, and different cellular systems may respond differently to a single PRMT inhibitor. Specific cellular reporters will be useful for assaying PRMT inhibitors. Wei et al. implemented a GFP-PABP1 reporter to study CARM1 modulators [186]. In this approach, GFP-PABP1 substrate protein is transiently expressed in MCF7 cells, and the methylation event is captured by methyl-PABP1-specific primary antibody and terbium-labeled secondary antibody in the cell lysate, leading to the formation of the antibody/substrate complex. Upon excitation of terbium by light at 340 nm, the proximity between terbium and GFP enables TR-FRET to occur, which reflects the methyltransferase activity of CARM1. In principle, similar assay platform can be created to study activities of other PRMT enzymes. Of further notion is that chemical proteomics in combination with mass spectrometry has gained increased popularity in inhibitor selectivity profiling and in vivo study of drug-proteome interactions [187]. It remains to see if chemoproteomic assays can be designed and applied for target validation of new PRMT inhibitors.
The majority of PRMT enzymes still lack nanomolar potent inhibitors. There is no PRMT2, -7, -8 and -9 specific inhibitors reported yet. Functions of PRMT7 and PRMT9 are studied only recently. PRMT inhibitors thus far are largely evaluated in biochemical and cellular models. A pressing issue is the inadequate study of pharmacodynamics and pharmacokinetic attributes of PRMT inhibitors in animal disease models. Most importantly, a big gap exists for PRMT inhibitors to translate into clinical drug leads. Thus authentication of PRMT inhibitors as an effective therapeutic strategy warrants further investigation. We foresee that generation and evaluation of potent, isoform-selective, pharmacokinetically amenable, and in vivo-active molecules will continue to be an active arena of research in years ahead.
Article highlights.
PRMTs are crucial players in the regulation of fundamental cellular processes and disease pathways.
PRMTs are emerging disease targets for drug discovery.
PRMT modulators are powerful tools to elucidate PRMT function in physiology and pathology, and are potential therapeutic agents for the treatment of various diseases, notably cancer.
Specific inhibitors have been developed for PRMT1-, -3, -4, -5 and -6.
Caution should be taken on possible off-target effects of new PRMT inhibitors.
Breakthroughs are needed to translate PRMT inhibitors into clinical candidates.
Footnotes
Financial and Competing Interests Disclosure:
The authors are supported by the National Institutes of Health through a National Institute of General Medical Sciences grant R01GM086717. The authors are also financially supported by the National Science Foundation and American Heart Association. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1•.Fuhrmann J, Clancy KW, Thompson PR. Chemical biology of protein arginine modifications in epigenetic regulation. Chem Rev. 2015;115:5413–61. doi: 10.1021/acs.chemrev.5b00003. An updated review on protein arginine methylation and deimination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2••.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. An outstanding review on PRMT function in carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 3•.Di Lorenzo A, Bedford MT. Histone arginine methylation. FEBS Lett. 2011;585:2024–31. doi: 10.1016/j.febslet.2010.11.010. An excellent review on histone arginine methylation and its biological function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Branscombe TL, Frankel A, Lee J-H, et al. PRMT5 (janus kinase-binding protein 1) catalyzes the formation of symmetric dimethylarginine residues in proteins. J Biol Chem. 2001;276:32971–76. doi: 10.1074/jbc.M105412200. [DOI] [PubMed] [Google Scholar]
- 5•.Yang Y, Hadjikyriacou A, Xia Z, et al. P RMT9 is a Type II methyltransferase that methylates the splicing factor SAP145. Nat Commun. 2015;6:6428. doi: 10.1038/ncomms7428. An important article about function and substrate of PRMT9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6•.Feng Y, Maity R, Whitelegge JP, et al. Mammalian protein arginine methyltransferase 7 (PRMT7) specifically targets RXR sites in lysine- and arginine-rich regions. J Biol Chem. 2013;288:37010–25. doi: 10.1074/jbc.M113.525345. An important article about the substrate recognition of PRMT7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bedford MT. Arginine methylation at a glance. Journal of Cell Science. 2007;120:4243–46. doi: 10.1242/jcs.019885. [DOI] [PubMed] [Google Scholar]
- 8.Dhar S, Vemulapalli V, Patananan AN, et al. Loss of the major Type I arginine methyltransferase PRMT1 causes substrate scavenging by other PRMTs. Sci Rep. 2013;3:1311. doi: 10.1038/srep01311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hadjikyriacou A, Yang Y, Espejo A, et al. Unique features of human protein arginine methyltransferase 9 (PRMT9) and its substrate rna splicing factor SF3B2. J Biol Chem. 2015;290:16723–43. doi: 10.1074/jbc.M115.659433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gary JD, Clarke S. RNA and protein interactions modulated by protein arginine methylation. Prog Nucleic Acid Res Mol Biol. 1998;61:65–131. doi: 10.1016/s0079-6603(08)60825-9. [DOI] [PubMed] [Google Scholar]
- 11•.Morales Y, Cáceres T, May K, et al. Biochemistry and regulation of the protein arginine methyltransferases (PRMTs) Arch Biochem Biophys. 2016;590:138–52. doi: 10.1016/j.abb.2015.11.030. An excellent review about PRMT biochemistry and regulation. [DOI] [PubMed] [Google Scholar]
- 12.Shien DM, Lee TY, Chang WC, et al. Incorporating structural characteristics for identification of protein methylation sites. J Comput Chem. 2009;30:1532–43. doi: 10.1002/jcc.21232. [DOI] [PubMed] [Google Scholar]
- 13.Kölbel K, Ihling C, Kühn U, et al. Peptide backbone conformation affects the substrate preference of protein arginine methyltransferase I. Biochemistry. 2012;51:5463–75. doi: 10.1021/bi300373b. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Bedford MT. PABP1 identified as an arginine methyltransferase substrate using high-density protein arrays. EMBO Rep. 2002;3:268–73. doi: 10.1093/embo-reports/kvf052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng D, Cote J, Shaaban S, et al. The Arginine Methyltransferase CARM1 regulates the coupling of transcription and mrna processing. Mol Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Lee J-H, Cook JR, Yang Z-H, et al. PRMT7, a new protein arginine methyltransferase that synthesizes symmetric dimethylarginine. J Biol Chem. 2005;280:3656–64. doi: 10.1074/jbc.M405295200. [DOI] [PubMed] [Google Scholar]
- 17.Hadjikyriacou A, Yang Y, Bedford M, et al. Characterization of the activity and biological function of human protein arginine methyltransferase 9 (PRMT9) FASEB J. 2015;29 doi: 10.1074/jbc.M115.659433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 19.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 20.Lee DY, Teyssier C, Strahl BD, et al. Role of protein methylation in regulation of transcription. Endocr Rev. 2005;26:147–70. doi: 10.1210/er.2004-0008. [DOI] [PubMed] [Google Scholar]
- 21.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–60. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 22••.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. An excellent review on protein arginine methyltransferase. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yun M, Wu J, Workman JL, et al. Readers of histone modifications. Cell Res. 2011;21:564–78. doi: 10.1038/cr.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Musselman CA, Lalonde M-E, Cote J, et al. Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol. 2012;19:1218–27. doi: 10.1038/nsmb.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molina-Serrano D, Schiza V, Kirmizis A. Cross-talk among epigenetic modifications: lessons from histone arginine methylation. Biochem Soc Trans. 2013;41:751–59. doi: 10.1042/BST20130003. [DOI] [PubMed] [Google Scholar]
- 26.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–12. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 27.Wei H, Mundade R, Lange KC, et al. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle. 2014;13:32–41. doi: 10.4161/cc.27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee Y-H, Stallcup MR. Minireview: Protein arginine methylation of nonhistone proteins in transcriptional regulation. Mol Endocrinol. 2009;23:425–33. doi: 10.1210/me.2008-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Blanchet F, Schurter BT, Acuto O. Protein arginine methylation in lymphocyte signaling. Curr Opin Immunol. 2006;18:321–28. doi: 10.1016/j.coi.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 30.Sims RJ, Rojas LA, Beck D, et al. The C-Terminal Domain of RNA Polymerase II is modified by site-specific methylation. Science. 2011;332:99–103. doi: 10.1126/science.1202663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brahms H, Raymackers J, Union A, et al. The C-terminal RG dipeptide repeats of the spliceosomal Sm proteins D1 and D3 contain symmetrical dimethylarginines, which form a major B-cell epitope for anti-Sm autoantibodies. J Biol Chem. 2000;275:17122–29. doi: 10.1074/jbc.M000300200. [DOI] [PubMed] [Google Scholar]
- 32.Côté J, Boulanger M-C, Bedford MT, et al. Sam68 RNA binding protein is an in vivo substrate for protein arginine N-methyltransferase 1. Mol Biol Cell. 2003;14:274–87. doi: 10.1091/mbc.E02-08-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rho J, Choi S, Jung C-R, et al. Arginine methylation of Sam68 and SLM proteins negatively regulates their poly (U) RNA binding activity. Arch Biochem Biophys. 2007;466:49–57. doi: 10.1016/j.abb.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 34.Boisvert F-M, Déry U, Masson J-Y, et al. Arginine methylation of MRE11 by PRMT1 is required for DNA damage checkpoint control. Genes Dev. 2005;19:671–76. doi: 10.1101/gad.1279805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jansson M, Durant ST, Cho E-C, et al. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431–39. doi: 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- 36.Doller A, Pfeilschifter J, Eberhardt W. Signalling pathways regulating nucleo-cytoplasmic shuttling of the mRNA-binding protein HuR. Cell Signal. 2008;20:2165–73. doi: 10.1016/j.cellsig.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 37.Alinari L, Mahasenan KV, Yan F, et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood. 2015;125:2530–43. doi: 10.1182/blood-2014-12-619783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jeong S-J, Lu H, Cho W-K, et al. Coactivator-associated arginine methyltransferase 1 enhances transcriptional activity of the human T-cell lymphotropic virus type 1 long terminal repeat through direct interaction with Tax. J Virol. 2006;80:10036–44. doi: 10.1128/JVI.00186-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie B, Invernizzi CF, Richard S, et al. Arginine methylation of the human immunodeficiency virus type 1 tat protein by PRMT6 negatively affects tat interactions with both cyclin T1 and the Tat transactivation region. J Virol. 2007;81:4226–34. doi: 10.1128/JVI.01888-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun Q, Liu L, Roth M, et al. PRMT1 Upregulated by epithelial proinflammatory cytokines participates in COX2 expression in fibroblasts and chronic antigen-induced pulmonary inflammation. J Immunol. 2015;195:298–306. doi: 10.4049/jimmunol.1402465. [DOI] [PubMed] [Google Scholar]
- 41.Bandyopadhyay S, Harris DP, Adams GN, et al. HOXA9 methylation by PRMT5 is essential for endothelial cell expression of leukocyte adhesion molecules. Mol Cell Biol. 2012;32:1202–13. doi: 10.1128/MCB.05977-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouras G, Deftereos S, Tousoulis D, et al. Asymmetric dimethylarginine (ADMA): a promising biomarker for cardiovascular disease? Curr Top Med Chem. 2013;13:180–200. doi: 10.2174/1568026611313020007. [DOI] [PubMed] [Google Scholar]
- 43.Teerlink T, Luo Z, Palm F, et al. Cellular ADMA: Regulation and action. Pharmacol Res. 2009;60:448–60. doi: 10.1016/j.phrs.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cvetković T, Pavlović R, Đorđević V, et al. Dimethylarginine – biomarkers in progression of kidney disease. J Med Biochem. 2012;31:301–08. [Google Scholar]
- 45.Raptis V, Kapoulas S, Grekas D. Role of asymmetrical dimethylarginine in the progression of renal disease. Nephrology. 2013;18:11–21. doi: 10.1111/j.1440-1797.2012.01659.x. [DOI] [PubMed] [Google Scholar]
- 46.Krzyzanowska K, Mittermayer F, Wolzt M, et al. ADMA, cardiovascular disease and diabetes. Diabetes Res Clin Pract. 2008;82(Supplement 2):S122–S26. doi: 10.1016/j.diabres.2008.09.024. [DOI] [PubMed] [Google Scholar]
- 47.Zakrzewicz D, Zakrzewicz A, Preissner KT, et al. Protein arginine methyltransferases (PRMTs): promising targets for the treatment of pulmonary disorders. Int J Mol Sci. 2012;13:12383–400. doi: 10.3390/ijms131012383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cha B, Jho E-H. Protein arginine methyltransferases (PRMTs) as therapeutic targets. Expert Opin Ther Targets. 2012;16:651–64. doi: 10.1517/14728222.2012.688030. [DOI] [PubMed] [Google Scholar]
- 49.Teyssier C, Le Romancer M, Sentis S, et al. Protein arginine methylation in estrogen signaling and estrogen-related cancers. Trends Endocrinol Metab. 2010;21:181–89. doi: 10.1016/j.tem.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 50.Nicholson TB, Chen T, Richard S. The physiological and pathophysiological role of PRMT1-mediated protein arginine methylation. Pharmacological Research. 2009;60:466–74. doi: 10.1016/j.phrs.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 51.Scoumanne A, Chen X. Protein methylation: a new mechanism of p53 tumor suppressor regulation. Histol Histopathol. 2008;23:1143–9. doi: 10.14670/hh-23.1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng YG, Wu J, Chen Z, et al. Chemical regulation of epigenetic modifications: Opportunities for new cancer therapy. Med Res Rev. 2008;28:645–87. doi: 10.1002/med.20120. [DOI] [PubMed] [Google Scholar]
- 53.Park JH, Szemes M, Vieira GC, et al. Protein arginine methyltransferase 5 is a key regulator of the MYCN oncoprotein in neuroblastoma cells. Mol Oncol. 2015;9:617–27. doi: 10.1016/j.molonc.2014.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–66. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 55.Baldwin RM, Morettin A, Cote J. Role of PRMTs in cancer: Could minor isoforms be leaving a mark? World J Biol Chem. 2014;5:115–29. doi: 10.4331/wjbc.v5.i2.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Goulet I, Gauvin G, Boisvenue S, et al. Alternative splicing yields protein arginine methyltransferase 1 isoforms with distinct activity, substrate specificity, and subcellular localization. J Biol Chem. 2007;282:33009–21. doi: 10.1074/jbc.M704349200. [DOI] [PubMed] [Google Scholar]
- 57.Mathioudaki K, Papadokostopoulou A, Scorilas A, et al. The PRMT1 gene expression pattern in colon cancer. Br J Cancer. 2008;99:2094–9. doi: 10.1038/sj.bjc.6604807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baldwin RM, Morettin A, Paris G, et al. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle. 2012;11:4597–612. doi: 10.4161/cc.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cheung N, Chan LC, Thompson A, et al. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol. 2007;9:1208–15. doi: 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- 60.Shia W-J, Okumura AJ, Yan M, et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood. 2012;119:4953–62. doi: 10.1182/blood-2011-04-347476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tarighat S, Santhanam R, Frankhouser D, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2015 doi: 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaniskan HU, Konze KD, Jin J. Selective inhibitors of protein methyltransferases. J Med Chem. 2015;58:1596–629. doi: 10.1021/jm501234a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Medina-Franco J. Epi-Informatics: Discovery and development of small molecule epigenetic drugs and probes. Elsevier Science; 2016. [Google Scholar]
- 64.Zhang J, Zheng YG. SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chem Biol. 2015 doi: 10.1021/acschembio.5b00812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li KK, Luo C, Wang D, et al. Chemical and biochemical approaches in the study of histone methylation and demethylation. Med Res Rev. 2012;32:815–67. doi: 10.1002/mrr.20228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wooderchak WL, Zhou ZS, Hevel J. Assays for S-adenosylmethionine (AdoMet/SAM)-dependent methyltransferases. Curr Protoc Toxicol. 2008;Chapter 4(Unit 4):26. doi: 10.1002/0471140856.tx0426s38. [DOI] [PubMed] [Google Scholar]
- 67.Zeng H, Xu W. Enzymatic assays of histone methyltransferase enzymes. In: Zheng YG, editor. Epigenetic technological applications. Academic Press; San Diego: 2015. pp. 333–61. [Google Scholar]
- 68.Luo M. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem Biol. 2012;7:443–63. doi: 10.1021/cb200519y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Spannhoff A, Hauser A-T, Heinke R, et al. The emerging therapeutic potential of histone methyltransferase and demethylase inhibitors. ChemMedChem. 2009;4:1568–82. doi: 10.1002/cmdc.200900301. [DOI] [PubMed] [Google Scholar]
- 70.Cheng D, Yadav N, King RW, et al. Small molecule regulators of protein arginine methyltransferases. J Biol Chem. 2004;279:23892–99. doi: 10.1074/jbc.M401853200. [DOI] [PubMed] [Google Scholar]
- 71.Bissinger E-M, Heinke R, Spannhoff A, et al. Acyl derivatives of p-aminosulfonamides and dapsone as new inhibitors of the arginine methyltransferase hPRMT1. Biorg Med Chem. 2011;19:3717–31. doi: 10.1016/j.bmc.2011.02.032. [DOI] [PubMed] [Google Scholar]
- 72.Cheng D, Valente S, Castellano S, et al. Novel 3,5-bis(bromohydroxybenzylidene)piperidin-4-ones as coactivator-associated arginine methyltransferase 1 Inhibitors: Enzyme selectivity and cellular activity. J Med Chem. 2011;54:4928–32. doi: 10.1021/jm200453n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Castellano S, Milite C, Ragno R, et al. Design, synthesis and biological evaluation of carboxy analogues of arginine methyltransferase inhibitor 1 (AMI-1) ChemMedChem. 2010;5:398–414. doi: 10.1002/cmdc.200900459. [DOI] [PubMed] [Google Scholar]
- 74.Bonham K, Hemmers S, Lim Y-H, et al. Effects of a novel arginine methyltransferase inhibitor on T-helper cell cytokine production. FEBS J. 2010;277:2096–108. doi: 10.1111/j.1742-4658.2010.07623.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dowden J, Hong W, Parry RV, et al. Toward the development of potent and selective bisubstrate inhibitors of protein arginine methyltransferases. Bioorg Med Chem Lett. 2010;20:2103–05. doi: 10.1016/j.bmcl.2010.02.069. [DOI] [PubMed] [Google Scholar]
- 76.Dowden J, Pike RA, Parry RV, et al. Small molecule inhibitors that discriminate between protein arginine N-methyltransferases PRMT1 and CARM1. Org Biomol Chem. 2011;9:7814–21. doi: 10.1039/c1ob06100c. [DOI] [PubMed] [Google Scholar]
- 77.Suh-Lailam BB, Hevel JM. A fast and efficient method for quantitative measurement of S-adenosyl-l-methionine-dependent methyltransferase activity with protein substrates. Anal Biochem. 2010;398:218–24. doi: 10.1016/j.ab.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 78.Sinha SH, Owens EA, Feng Y, et al. Synthesis and evaluation of carbocyanine dyes as PRMT inhibitors and imaging agents. Eur J Med Chem. 2012;54:647–59. doi: 10.1016/j.ejmech.2012.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79•.Yan L, Yan C, Qian K, et al. Diamidine Compounds for selective inhibition of protein arginine methyltransferase 1. J Med Chem. 2014;57:2611–22. doi: 10.1021/jm401884z. This article reported a class of diamidine compounds for PRMT inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liu F, Li F, Ma A, et al. Exploiting an allosteric binding site of prmt3 yields potent and selective inhibitors. J Med Chem. 2013;56:2110–24. doi: 10.1021/jm3018332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Allan M, Manku S, Therrien E, et al. N-Benzyl-1-heteroaryl-3-(trifluoromethyl)-1H-pyrazole-5-carboxamides as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:1218–23. doi: 10.1016/j.bmcl.2008.12.075. [DOI] [PubMed] [Google Scholar]
- 82.Purandare AV, Chen Z, Huynh T, et al. Pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2008;18:4438–41. doi: 10.1016/j.bmcl.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 83.Huynh T, Chen Z, Pang S, et al. Optimization of pyrazole inhibitors of coactivator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:2924–27. doi: 10.1016/j.bmcl.2009.04.075. [DOI] [PubMed] [Google Scholar]
- 84.Therrien E, Larouche G, Manku S, et al. 1,2-Diamines as inhibitors of co-activator associated arginine methyltransferase 1 (CARM1) Bioorg Med Chem Lett. 2009;19:6725–32. doi: 10.1016/j.bmcl.2009.09.110. [DOI] [PubMed] [Google Scholar]
- 85.Wan H, Huynh T, Pang S, et al. Benzo[d]imidazole inhibitors of Coactivator Associated Arginine Methyltransferase 1 (CARM1)—Hit to Lead studies. Bioorg Med Chem Lett. 2009;19:5063–66. doi: 10.1016/j.bmcl.2009.07.040. [DOI] [PubMed] [Google Scholar]
- 86.Sack JS, Thieffine S, Bandiera T, et al. Structural basis for CARM1 inhibition by indole and pyrazole inhibitors. Biochem J. 2011;436:331–39. doi: 10.1042/BJ20102161. [DOI] [PubMed] [Google Scholar]
- 87.Baiocchi RA, Li C, LAI H, et al. WO2014145214 A2. Inhibitors of prmt5 and methods of their use. 2014
- 88.Selvi BR, Batta K, Kishore AH, et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17. J Biol Chem. 2010;285:7143–52. doi: 10.1074/jbc.M109.063933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89•.Feng Y, Li M, Wang B, et al. Discovery and mechanistic study of a class of protein arginine methylation inhibitors. J Med Chem. 2010;53:6028–39. doi: 10.1021/jm100416n. This article showed that some compounds inhibit PRMT activity due to their interaction with histone substrate. [DOI] [PubMed] [Google Scholar]
- 90.Wang J, Chen L, Sinha SH, et al. Pharmacophore-based virtual screening and biological evaluation of small molecule inhibitors for protein arginine methylation. J Med Chem. 2012;55:7978–87. doi: 10.1021/jm300521m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rathert P, Cheng X, Jeltsch A. Continuous enzymatic assay for histone lysine methyltransferases. BioTechniques. 2007;43:602. doi: 10.2144/000112623. 04, 06 passim. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dhayalan A, Dimitrova E, Rathert P, et al. A continuous protein methyltransferase (G9a) assay for enzyme activity measurement and inhibitor screening. J Biomol Screen. 2009;14:1129–33. doi: 10.1177/1087057109345528. [DOI] [PubMed] [Google Scholar]
- 93.Wu J, Xie N, Feng Y, et al. Scintillation proximity assay of arginine methylation. J Biomol Screen. 2012;17:237–44. doi: 10.1177/1087057111414903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ibanez G, Shum D, Blum G, et al. A high throughput scintillation proximity imaging assay for protein methyltransferases. Comb Chem High Throughput Screen. 2012;15:359–71. doi: 10.2174/138620712800194468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hu H, Owens EA, Su H, et al. Exploration of cyanine compounds as selective inhibitors of protein arginine methyltransferases: synthesis and biological evaluation. J Med Chem. 2015;58:1228–43. doi: 10.1021/jm501452j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96•.Siarheyeva A, Senisterra G, Allali-Hassani A, et al. An allosteric inhibitor of protein arginine methyltransferase 3. Structure. 2012;20:1425–35. doi: 10.1016/j.str.2012.06.001. The first report on allosteric PRMT3 inhibitor. [DOI] [PubMed] [Google Scholar]
- 97.Kaniskan HÜ, Szewczyk MM, Yu Z, et al. A Potent, Selective and cell-active allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3) Angew Chem Int Ed. 2015;54:5166–70. doi: 10.1002/anie.201412154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zheng W, Ibáñez G, Wu H, et al. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J Am Chem Soc. 2012;134:18004–14. doi: 10.1021/ja307060p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mitchell LH, Drew AE, Ribich SA, et al. Aryl Pyrazoles as potent inhibitors of arginine methyltransferases: identification of the first PRMT6 tool compound. ACS Med Chem Lett. 2015;6:655–59. doi: 10.1021/acsmedchemlett.5b00071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Eram MS, Shen Y, Szewczyk MM, et al. A potent, selective, and cell-active inhibitor of human type i protein arginine methyltransferases. ACS Chem Biol. 2015 doi: 10.1021/acschembio.5b00839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Smil D, Eram MS, Li F, et al. Discovery of a dual PRMT5–PRMT7 inhibitor. ACS Med Chem Lett. 2015;6:408–12. doi: 10.1021/ml500467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fontán N, García-Domínguez P, Álvarez R, et al. Novel symmetrical ureas as modulators of protein arginine methyl transferases. Biorg Med Chem. 2013;21:2056–67. doi: 10.1016/j.bmc.2013.01.017. [DOI] [PubMed] [Google Scholar]
- 103.van Haren M, van Ufford LQ, Moret EE, et al. Synthesis and evaluation of protein arginine N-methyltransferase inhibitors designed to simultaneously occupy both substrate binding sites. Org Biomol Chem. 2015;13:549–60. doi: 10.1039/c4ob01734j. [DOI] [PubMed] [Google Scholar]
- 104.Spannhoff A, Heinke R, Bauer I, et al. Target-based approach to inhibitors of histone arginine methyltransferases. J Med Chem. 2007;50:2319–25. doi: 10.1021/jm061250e. [DOI] [PubMed] [Google Scholar]
- 105.Heinke R, Spannhoff A, Meier R, et al. virtual screening and biological characterization of novel histone arginine methyltransferase PRMT1 inhibitors. ChemMedChem. 2009;4:69–77. doi: 10.1002/cmdc.200800301. [DOI] [PubMed] [Google Scholar]
- 106.Spannhoff A, Machmur R, Heinke R, et al. A novel arginine methyltransferase inhibitor with cellular activity. Bioorg Med Chem Lett. 2007;17:4150–53. doi: 10.1016/j.bmcl.2007.05.088. [DOI] [PubMed] [Google Scholar]
- 107.Xie Y, Zhou R, Lian F, et al. Virtual screening and biological evaluation of novel small molecular inhibitors against protein arginine methyltransferase 1 (PRMT1) Org Biomol Chem. 2014;12:9665–73. doi: 10.1039/c4ob01591f. [DOI] [PubMed] [Google Scholar]
- 108••.Chan-Penebre E, Kuplast KG, Majer CR, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015;11:432–37. doi: 10.1038/nchembio.1810. Report of EPZ015666 as a selective PRMT5 inhibitor. [DOI] [PubMed] [Google Scholar]
- 109.Rioux N, Duncan KW, Lantz RJ, et al. Species differences in metabolism of EPZ015666, an oxetane-containing protein arginine methyltransferase-5 (PRMT5) inhibitor. Xenobiotica. 2015:1–10. doi: 10.3109/00498254.2015.1072253. [DOI] [PubMed] [Google Scholar]
- 110.Duncan KW, Rioux N, Boriack-Sjodin PA, et al. Structure and Property Guided Design in the Identification of PRMT5 tool compound EPZ015666. ACS Med Chem Lett. 2015 doi: 10.1021/acsmedchemlett.5b00380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu X-R, Tang Y, Wang W-J, et al. Discovery and structure–activity analysis of 4-((5-nitropyrimidin-4-yl)amino)benzimidamide derivatives as novel protein arginine methyltransferase 1 (PRMT1) inhibitors. Bioorg Med Chem Lett. 2015 doi: 10.1016/j.bmcl.2015.06.095. [DOI] [PubMed] [Google Scholar]
- 112.Collazo E, Couture JF, Bulfer S, et al. A coupled fluorescent assay for histone methyltransferases. Anal Biochem. 2005;342:86–92. doi: 10.1016/j.ab.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 113.Wang R, Ibanez G, Islam K, et al. Formulating a fluorogenic assay to evaluate S-adenosyl-L-methionine analogues as protein methyltransferase cofactors. Mol BioSyst. 2011;7:2970–81. doi: 10.1039/c1mb05230f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Allali-Hassani A, Wasney GA, Siarheyeva A, et al. Fluorescence-based methods for screening writers and readers of histone methyl marks. J Biomol Screen. 2012;17:71–84. doi: 10.1177/1087057111422256. [DOI] [PubMed] [Google Scholar]
- 115.Dorgan KM, Wooderchak WL, Wynn DP, et al. An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal Biochem. 2006;350:249–55. doi: 10.1016/j.ab.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 116.Hemeon I, Gutierrez JA, Ho MC, et al. Characterizing DNA methyltransferases with an ultrasensitive luciferase-linked continuous assay. Anal Chem. 2011;83:4996–5004. doi: 10.1021/ac200816m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ibanez G, McBean JL, Astudillo YM, et al. An enzyme-coupled ultrasensitive luminescence assay for protein methyltransferases. Anal Biochem. 2010;401:203–10. doi: 10.1016/j.ab.2010.03.010. [DOI] [PubMed] [Google Scholar]
- 118.Tang J, Frankel A, Cook RJ, et al. PRMT1 Is the Predominant Type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–30. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 119.Bouras G, Deftereos S, Tousoulis D, et al. Asymmetric dimethylarginine (ADMA): a promising biomarker for cardiovascular disease? Curr Top Med Chem. 2013;13:180–200. doi: 10.2174/1568026611313020007. [DOI] [PubMed] [Google Scholar]
- 120.Lajer M, Tarnow L, Jorsal A, et al. Plasma concentration of asymmetric dimethylarginine (ADMA) predicts cardiovascular morbidity and mortality in type 1 diabetic patients with diabetic nephropathy. Diabetes Care. 2008;31:747–52. doi: 10.2337/dc07-1762. [DOI] [PubMed] [Google Scholar]
- 121.Iwasaki H. Impaired PRMT1 activity in the liver and pancreas of type 2 diabetic Goto–Kakizaki rats. Life Sci. 2009;85:161–66. doi: 10.1016/j.lfs.2009.05.007. [DOI] [PubMed] [Google Scholar]
- 122.Schwedhelm E, Böger RH. The role of asymmetric and symmetric dimethylarginines in renal disease. Nat Rev Neurol. 2011;7:275–85. doi: 10.1038/nrneph.2011.31. [DOI] [PubMed] [Google Scholar]
- 123.Xu J, Wang AH, Oses-Prieto J, et al. Arginine methylation initiates BMP-induced Smad signaling. Mol Cell. 2013;51:5–19. doi: 10.1016/j.molcel.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zhang L, Tran NT, Su H, et al. Cross-talk between PRMT1-mediated methylation and ubiquitylation on RBM15 controls RNA splicing. eLife. 2015;4 doi: 10.7554/eLife.07938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Siboni RB, Bodner MJ, Khalifa MM, et al. Biological efficacy and toxicity of diamidines in myotonic dystrophy type 1 models. J Med Chem. 2015;58:5770–80. doi: 10.1021/acs.jmedchem.5b00356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mitchell LH, Shapiro G, Richard C, et al. US8952026. PRMT1 inhibitors and uses thereof. 2015
- 127.Kuntz KW, Mitchell LH, Shapiro G, et al. US9023883. PRMT1 inhibitors and uses thereof. 2015
- 128.Swiercz R, Cheng D, Kim D, et al. Ribosomal protein rpS2 is hypomethylated in PRMT3-deficient mice. J Biol Chem. 2007;282:16917–23. doi: 10.1074/jbc.M609778200. [DOI] [PubMed] [Google Scholar]
- 129.Bachand F, Silver PA. PRMT3 is a ribosomal protein methyltransferase that affects the cellular levels of ribosomal subunits. EMBO J. 2004;23:2641–50. doi: 10.1038/sj.emboj.7600265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen X, Niroomand F, Liu Z, et al. Expression of nitric oxide related enzymes in coronary heart disease. Basic Res Cardiol. 2006;101:346–53. doi: 10.1007/s00395-006-0592-5. [DOI] [PubMed] [Google Scholar]
- 131.Baylis C. Arginine, arginine analogs and nitric oxide production in chronic kidney disease. Nat Clin Pract Neurol. 2006;2:209–20. doi: 10.1038/ncpneph0143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132•.Schapira M, Ferreira de Freitas R. Structural biology and chemistry of protein arginine methyltransferases. MedChemComm. 2014;5:1779–88. doi: 10.1039/c4md00269e. An excellent review on structural analysis of PRMTs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhang X, Zhou L, Cheng X. Crystal structure of the conserved core of protein arginine methyltransferase PRMT3. EMBO J. 2000;19:3509–19. doi: 10.1093/emboj/19.14.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell. 2004;117:735–48. doi: 10.1016/j.cell.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 135.Fujiwara T, Mori Y, Chu DL, et al. CARM1 regulates proliferation of PC12 cells by methylating HuD. Mol Cell Biol. 2006;26:2273–85. doi: 10.1128/MCB.26.6.2273-2285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chen SL, Loffler KA, Chen D, et al. The Coactivator-associated arginine methyltransferase is necessary for muscle differentiation CARM1 coactivates myocyte enhancer factor-2. J Biol Chem. 2002;277:4324–33. doi: 10.1074/jbc.M109835200. [DOI] [PubMed] [Google Scholar]
- 137.Mai A, Cheng D, Bedford MT, et al. Epigenetic multiple ligands: mixed histone/protein methyltransferase, acetyltransferase, and class III deacetylase (sirtuin) inhibitors. J Med Chem. 2008;51:2279–90. doi: 10.1021/jm701595q. [DOI] [PubMed] [Google Scholar]
- 138.Pal S, Vishwanath SN, Erdjument-Bromage H, et al. Human SWI/SNF-associated PRMT5 methylates histone H3 arginine 8 and negatively regulates expression of ST7 and NM23 tumor suppressor genes. Mol Cell Biol. 2004;24:9630–45. doi: 10.1128/MCB.24.21.9630-9645.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Deng X, Gu L, Liu C, et al. Arginine methylation mediated by the Arabidopsis homolog of PRMT5 is essential for proper pre-mRNA splicing. Proc Natl Acad Sci. 2010;107:19114–19. doi: 10.1073/pnas.1009669107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Andreu-Perez P, Esteve-Puig R, de Torre-Minguela C, et al. Protein arginine methyltransferase 5 regulates ERK1/2 signal transduction amplitude and cell fate through CRAF. Sci Signal. 2011;4:ra58–ra58. doi: 10.1126/scisignal.2001936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Nishida KM, Okada TN, Kawamura T, et al. Functional involvement of Tudor and dPRMT5 in the piRNA processing pathway in Drosophila germlines. EMBO J. 2009;28:3820–31. doi: 10.1038/emboj.2009.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Gu Z, Li Y, Lee P, et al. Protein arginine methyltransferase 5 functions in opposite ways in the cytoplasm and nucleus of prostate cancer cells. PLoS One. 2012;7:e44033. doi: 10.1371/journal.pone.0044033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bode-Böger SM, Scalera F, Kielstein JT, et al. Symmetrical dimethylarginine: a new combined parameter for renal function and extent of coronary artery disease. J Am Soc Nephrol. 2006;17:1128–34. doi: 10.1681/ASN.2005101119. [DOI] [PubMed] [Google Scholar]
- 144.Ratovitski T, Arbez N, Stewart JC, et al. PRMT5-mediated symmetric arginine dimethylation is attenuated by mutant huntingtin and is impaired in Huntington’s Disease (HD) Cell Cycle. 2015 doi: 10.1080/15384101.2015.1033595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Quan X, Yue W, Luo Y, et al. The protein arginine methyltransferase PRMT5 regulates Aβ-induced toxicity in human cells and Caenorhabditis elegans models of Alzheimer’s disease. J Neurochem. 2015;134:969–77. doi: 10.1111/jnc.13191. [DOI] [PubMed] [Google Scholar]
- 146.Baiocchi R, Li C, Li PK, et al. WO2011079236 A1. Compositions and methods for cancer detection and treatment. 2011
- 147.Tarighat SS, Santhanam R, Frankhouser D, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2015 doi: 10.1038/leu.2015.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Guccione E, Bassi C, Casadio F, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–37. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- 149.El-Andaloussi N, Valovka T, Toueille M, et al. Arginine methylation regulates DNA polymerase β. Mol Cell. 2006;22:51–62. doi: 10.1016/j.molcel.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 150.Neault M, Mallette FA, Vogel G, et al. Ablation of PRMT6 reveals a role as a negative transcriptional regulator of the p53 tumor suppressor. Nucleic Acids Res. 2012;40:9513–21. doi: 10.1093/nar/gks764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Stein C, Riedl S, Rüthnick D, et al. The arginine methyltransferase PRMT6 regulates cell proliferation and senescence through transcriptional repression of tumor suppressor genes. Nucleic Acids Res. 2012;40:9522–33. doi: 10.1093/nar/gks767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Di Lorenzo A, Yang Y, Macaluso M, et al. A gain-of-function mouse model identifies PRMT6 as a NF-κB coactivator. Nucleic Acids Res. 2014;42:8297–309. doi: 10.1093/nar/gku530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chesworth R, Mitchell LH, Shapiro G, et al. US9133189. Argnine methyltransferase inhibitors and uses thereof. 2014
- 154.Blat Y. Non-competitive inhibition by active site binders. Chem Biol Drug Des. 2010;75:535–40. doi: 10.1111/j.1747-0285.2010.00972.x. [DOI] [PubMed] [Google Scholar]
- 155.Bauer UM, Daujat S, Nielsen SJ, et al. Methylation at arginine 17 of histone H3 is linked to gene activation. EMBO Rep. 2002;3:39–44. doi: 10.1093/embo-reports/kvf013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156•.Copeland RA. In evaluation of enzyme inhibitors in drug discovery. John Wiley & Sons, Inc; 2013. Assay Considerations for Compound Library Screening; pp. 123–68. A recommended reading on enzyme inhibitor screen assays. [Google Scholar]
- 157.t Hart P, Thomas D, van Ommeren R, et al. Analogues of the HIV-Tat peptide containing Nη-modified arginines as potent inhibitors of protein arginine N-methyltransferases. MedChemComm. 2012;3:1235–44. [Google Scholar]
- 158.t Hart P, Lakowski TM, Thomas D, et al. Peptidic partial bisubstrates as inhibitors of the protein arginine n-methyltransferases. ChemBioChem. 2011;12:1427–32. doi: 10.1002/cbic.201100074. [DOI] [PubMed] [Google Scholar]
- 159.Lakowski TM, ‘t Hart P, Ahern CA, et al. Nη-Substituted arginyl peptide inhibitors of protein arginine n-methyltransferases. ACS Chem Biol. 2010;5:1053–63. doi: 10.1021/cb100161u. [DOI] [PubMed] [Google Scholar]
- 160.Obianyo O, Causey CP, Osborne TC, et al. A Chloroacetamidine-based inactivator of protein arginine methyltransferase 1: Design, synthesis, and in vitro and in vivo evaluation. ChemBioChem. 2010;11:1219–23. doi: 10.1002/cbic.201000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Osborne T, Weller Roska RL, Rajski SR, et al. In situ generation of a bisubstrate analogue for protein arginine methyltransferase 1. J Am Chem Soc. 2008;130:4574–75. doi: 10.1021/ja077104v. [DOI] [PubMed] [Google Scholar]
- 162.Vhuiyan M, Thomas D, Hossen F, et al. Targeting protein arginine N-methyltransferases with peptide-based inhibitors: opportunities and challenges. Future Med Chem. 2013;5:2199–206. doi: 10.4155/fmc.13.184. [DOI] [PubMed] [Google Scholar]
- 163.Parang K, Cole PA. Designing bisubstrate analog inhibitors for protein kinases. Pharmacol Ther. 2002;93:145–57. doi: 10.1016/s0163-7258(02)00184-5. [DOI] [PubMed] [Google Scholar]
- 164.Lavogina D, Enkvist E, Uri A. Bisubstrate inhibitors of protein kinases: from principle to practical applications. ChemMedChem. 2010;5:23–34. doi: 10.1002/cmdc.200900252. [DOI] [PubMed] [Google Scholar]
- 165.Struck A-W, Thompson ML, Wong LS, et al. S-adenosyl-methionine-dependent methyltransferases: highly versatile enzymes in biocatalysis, biosynthesis and other biotechnological applications. ChemBioChem. 2012;13:2642–55. doi: 10.1002/cbic.201200556. [DOI] [PubMed] [Google Scholar]
- 166.Daigle SR, Olhava EJ, Therkelsen CA, et al. Selective killing of mixed lineage leukemia cells by a potent small-molecule DOT1L inhibitor. Cancer Cell. 2011;20:53–65. doi: 10.1016/j.ccr.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Yu W, Chory EJ, Wernimont AK, et al. Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat Commun. 2012;3:1288. doi: 10.1038/ncomms2304. [DOI] [PubMed] [Google Scholar]
- 168.Thompson MJ, Mekhalfia A, Jakeman DL, et al. Homochiral synthesis of an aza analogue of S-adenosyl-L-methionine (AdoMet) and its binding to the E. coli methionine repressor protein (MetJ) Chem Commun. 1996:791–92. [Google Scholar]
- 169.Ragno R, Simeoni S, Castellano S, et al. Small molecule inhibitors of histone arginine methyltransferases: Homology modeling, molecular docking, binding mode analysis, and biological evaluations. J Med Chem. 2007;50:1241–53. doi: 10.1021/jm061213n. [DOI] [PubMed] [Google Scholar]
- 170.Copeland RA. Protein methyltransferase inhibitors as personalized cancer therapeutics. Drug Discov Today Ther Strateg. 2012;9:e83–e90. [Google Scholar]
- 171.Kaniskan HÜ, Jin J. Chemical probes of histone lysine methyltransferases. ACS Chem Biol. 2014 doi: 10.1021/cb500785t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Momparler RL, Côté S. Targeting of cancer stem cells by inhibitors of DNA and histone methylation. Expert Opin Investig Drugs. 2015;24:1031–43. doi: 10.1517/13543784.2015.1051220. [DOI] [PubMed] [Google Scholar]
- 173.Yost JM, Korboukh I, Liu F, et al. Targets in epigenetics: inhibiting the methyl writers of the histone code. Curr Chem Genomics. 2011;5:72–84. doi: 10.2174/1875397301005010072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Spannhoff A, Sippl W, Jung M. Cancer treatment of the future: Inhibitors of histone methyltransferases. Int J Biochem Cell Biol. 2009;41:4–11. doi: 10.1016/j.biocel.2008.07.024. [DOI] [PubMed] [Google Scholar]
- 175.Krause CD, Yang Z-H, Kim Y-S, et al. Protein arginine methyltransferases: Evolution and assessment of their pharmacological and therapeutic potential. Pharmacol Ther. 2007;113:50–87. doi: 10.1016/j.pharmthera.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 176.Zhou R, Xie Y, Hu H, et al. Molecular mechanism underlying PRMT1 dimerization for SAM binding and methylase activity. J Chem Inf Model. 2015 doi: 10.1021/acs.jcim.5b00454. [DOI] [PubMed] [Google Scholar]
- 177.Lee W-C, Lin W-L, Matsui T, et al. Protein arginine methyltransferase 8: tetrameric structure and protein substrate specificity. Biochemistry. 2015 doi: 10.1021/acs.biochem.5b00995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cura V, Troffer-Charlier N, Wurtz J-M, et al. Structural insight into arginine methylation by the mouse protein arginine methyltransferase 7: a zinc finger freezes the mimic of the dimeric state into a single active site. Acta Crystallogr Sect D. 2014;70:2401–12. doi: 10.1107/S1399004714014278. [DOI] [PubMed] [Google Scholar]
- 179.Antonysamy S, Bonday Z, Campbell RM, et al. Crystal structure of the human PRMT5:MEP50 complex. Proc Natl Acad Sci. 2012;109:17960–65. doi: 10.1073/pnas.1209814109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Dahlin JL, Nissink JWM, Strasser JM, et al. PAINS in the assay: Chemical mechanisms of assay interference and promiscuous enzymatic inhibition observed during a sulfhydryl-scavenging HTS. J Med Chem. 2015;58:2091–113. doi: 10.1021/jm5019093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181•.Baell J, Walters MA. Chemistry: Chemical con artists foil drug discovery. Nature. 2014;513:481–83. doi: 10.1038/513481a. This article discusses the issue of PAINS in drug discovery. [DOI] [PubMed] [Google Scholar]
- 182.Whitty A. Growing PAINS in academic drug discovery. Future Med Chem. 2011;3:797–801. doi: 10.4155/fmc.11.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.McGovern SL, Caselli E, Grigorieff N, et al. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45:1712–22. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 184.Bunnage ME, Chekler ELP, Jones LH. Target validation using chemical probes. Nat Chem Biol. 2013;9:195–99. doi: 10.1038/nchembio.1197. [DOI] [PubMed] [Google Scholar]
- 185.Arrowsmith CH, Audia JE, Austin C, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11:536–41. doi: 10.1038/nchembio.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Zeng H, Wu J, Bedford MT, et al. A TR-FRET-based functional assay for screening activators of CARM1. ChemBioChem. 2013;14:827–35. doi: 10.1002/cbic.201300029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Bantscheff M, Lemeer S, Savitski MM, et al. Quantitative mass spectrometry in proteomics: critical review update from 2007 to the present. Anal Bioanal Chem. 2012;404:939–65. doi: 10.1007/s00216-012-6203-4. [DOI] [PubMed] [Google Scholar]
- 188.Strahl BD, Briggs SD, Brame CJ, et al. Methylation of histone H4 at arginine 3 occurs in vivo and is mediated by the nuclear receptor coactivator PRMT1. Curr Biol. 2001;11:996–1000. doi: 10.1016/s0960-9822(01)00294-9. [DOI] [PubMed] [Google Scholar]
- 189.Rajpurohit R, Lee SO, Park JO, et al. Enzymatic methylation of recombinant heterogeneous nuclear RNP protein A1. Dual substrate specificity for S-adenosylmethionine: histone-arginine N-methyltransferase. J Biol Chem. 1994;269:1075–82. [PubMed] [Google Scholar]
- 190.Lin W-J, Gary JD, Yang MC, et al. The mammalian immediate-early TIS21 protein and the leukemia-associated BTG1 protein interact with a protein-arginine N-methyltransferase. J Biol Chem. 1996;271:15034–44. doi: 10.1074/jbc.271.25.15034. [DOI] [PubMed] [Google Scholar]
- 191.Abramovich C, Yakobson B, Chebath J, et al. A protein-arginine methyltransferase binds to the intracytoplasmic domain of the IFNAR1 chain in the type I interferon receptor. EMBO J. 1997;16:260–66. doi: 10.1093/emboj/16.2.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Tang J, Kao PN, Herschman HR. Protein-arginine methyltransferase I, the predominant protein-arginine methyltransferase in cells, interacts with and is regulated by interleukin enhancer-binding factor 3. J Biol Chem. 2000;275:19866–76. doi: 10.1074/jbc.M000023200. [DOI] [PubMed] [Google Scholar]
- 193.Kwak YT, Guo J, Prajapati S, et al. Methylation of SPT5 regulates its interaction with RNA polymerase II and transcriptional elongation properties. Molecular cell. 2003;11:1055–66. doi: 10.1016/s1097-2765(03)00101-1. [DOI] [PubMed] [Google Scholar]
- 194.Herrmann F, Bossert M, Schwander A, et al. Arginine methylation of scaffold attachment factor A by heterogeneous nuclear ribonucleoprotein particle-associated PRMT1. J Biol Chem. 2004;279:48774–79. doi: 10.1074/jbc.M407332200. [DOI] [PubMed] [Google Scholar]
- 195.Boisvert F-M, Rhie A, Richard S, et al. The GAR motif of 53BP1 is arginine methylated by PRMT1 and is necessary for 53BP1 DNA binding activity. Cell Cycle. 2005;4:1834–41. doi: 10.4161/cc.4.12.2250. [DOI] [PubMed] [Google Scholar]
- 196.Dolzhanskaya N, Merz G, Denman RB. Alternative splicing modulates protein arginine methyltransferase-dependent methylation of fragile X syndrome mental retardation protein. Biochemistry. 2006;45:10385–93. doi: 10.1021/bi0525019. [DOI] [PubMed] [Google Scholar]
- 197.Le Romancer M, Treilleux I, Leconte N, et al. Regulation of estrogen rapid signaling through arginine methylation by PRMT1. Mol Cell. 2008;31:212–21. doi: 10.1016/j.molcel.2008.05.025. [DOI] [PubMed] [Google Scholar]
- 198.Zhao X, Jankovic V, Gural A, et al. Methylation of RUNX1 by PRMT1 abrogates SIN3A binding and potentiates its transcriptional activity. Genes Dev. 2008;22:640–53. doi: 10.1101/gad.1632608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Jobert L, Argentini M, Tora L. PRMT1 mediated methylation of TAF15 is required for its positive gene regulatory function. Exp Cell Res. 2009;315:1273–86. doi: 10.1016/j.yexcr.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 200.Jansson M, Durant ST, Cho EC, et al. Arginine methylation regulates the p53 response. Nat Cell Biol. 2008;10:1431–9. doi: 10.1038/ncb1802. [DOI] [PubMed] [Google Scholar]
- 201.Martin G, Ostareck-Lederer A, Chari A, et al. Arginine methylation in subunits of mammalian pre-mRNA cleavage factor I. RNA. 2010;16:1646–59. doi: 10.1261/rna.2164210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Butler JS, Zurita-Lopez CI, Clarke SG, et al. Protein-arginine methyltransferase 1 (PRMT1) methylates Ash2L, a shared component of mammalian histone H3K4 methyltransferase complexes. J Biol Chem. 2011;286:12234–44. doi: 10.1074/jbc.M110.202416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Perreault A, Lemieux C, Bachand F. Regulation of the nuclear poly (A)-binding protein by arginine methylation in fission yeast. J Biol Chem. 2007;282:7552–62. doi: 10.1074/jbc.M610512200. [DOI] [PubMed] [Google Scholar]
- 204.Aletta JM, Cimato TR, Ettinger MJ. Protein methylation: a signal event in post-translational modification. Trends Biochem Sci. 1998;23:89–91. doi: 10.1016/s0968-0004(98)01185-2. [DOI] [PubMed] [Google Scholar]
- 205.Zhang L, Wang R, Su H, et al. Arginine methylation of RBM15 by PRMT1 controls alternative splicing of genes involved in megakaryocyte differentiation. Blood. 2013;122:2443–43. [Google Scholar]
- 206.Sun Q, Yang X, Zhong B, et al. Upregulated protein arginine methyltransferase 1 by IL-4 increases eotaxin-1 expression in airway epithelial cells and participates in antigen-induced pulmonary inflammation in rats. J Immunol. 2012;188:3506–12. doi: 10.4049/jimmunol.1102635. [DOI] [PubMed] [Google Scholar]
- 207.Vallance P, Leiper J. Cardiovascular biology of the asymmetric dimethylarginine: dimethylarginine dimethylaminohydrolase pathway. Atertio Thromb Vasc Biol. 2004;24:1023–30. doi: 10.1161/01.ATV.0000128897.54893.26. [DOI] [PubMed] [Google Scholar]
- 208.Lakowski T, Frankel A. Kinetic analysis of human protein arginine N-methyltransferase 2: formation of monomethyl-and asymmetric dimethyl-arginine residues on histone H4. Biochem J. 2009;421:253–61. doi: 10.1042/BJ20090268. [DOI] [PubMed] [Google Scholar]
- 209.Qi C, Chang J, Zhu Y, et al. Identification of protein arginine methyltransferase 2 as a coactivator for estrogen receptor α. J Biol Chem. 2002;277:28624–30. doi: 10.1074/jbc.M201053200. [DOI] [PubMed] [Google Scholar]
- 210.Buss H, Dörrie A, Schmitz ML, et al. Phosphorylation of serine 468 by GSK-3β negatively regulates basal p65 NF-κB activity. J Biol Chem. 2004;279:49571–74. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- 211.Meyer R, Wolf SS, Obendorf M. PRMT2, a member of the protein arginine methyltransferase family, is a coactivator of the androgen receptor. J Steroid Biochem Mol Biol. 2007;107:1–14. doi: 10.1016/j.jsbmb.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 212.Zhong J, Cao R-X, Hong T, et al. Identification and expression analysis of a novel transcript of the human PRMT2 gene resulted from alternative polyadenylation in breast cancer. Gene. 2011;487:1–9. doi: 10.1016/j.gene.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 213.Zhong J, Cao RX, Zu XY, et al. Identification and characterization of novel spliced variants of PRMT2 in breast carcinoma. FEBS J. 2012;279:316–35. doi: 10.1111/j.1742-4658.2011.08426.x. [DOI] [PubMed] [Google Scholar]
- 214.Yildirim AO, Bulau P, Zakrzewicz D, et al. Increased protein arginine methylation in chronic hypoxia: role of protein arginine methyltransferases. Am J Respir Cell Mol Biol. 2006;35:436–43. doi: 10.1165/rcmb.2006-0097OC. [DOI] [PubMed] [Google Scholar]
- 215.Matsuguma K, Ueda S, Yamagishi S-i, et al. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J Am Soc Nephrol. 2006;17:2176–83. doi: 10.1681/ASN.2005121379. [DOI] [PubMed] [Google Scholar]
- 216.Schurter BT, Koh SS, Chen D, et al. Methylation of histone H3 by coactivator-associated arginine methyltransferase 1. Biochemistry. 2001;40:5747–56. doi: 10.1021/bi002631b. [DOI] [PubMed] [Google Scholar]
- 217.Lee Y-H, Coonrod SA, Kraus WL, et al. Regulation of coactivator complex assembly and function by protein arginine methylation and demethylimination. Proc Natl Acad Sci U S A. 2005;102:3611–16. doi: 10.1073/pnas.0407159102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ito T, Yadav N, Lee J, et al. Arginine methyltransferase CARM1/PRMT4 regulates endochondral ossification. BMC Dev Biol. 2009;9:47. doi: 10.1186/1471-213X-9-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Cheng D, Cote J, Shaaban S, et al. The arginine methyltransferase CARM1 regulates the coupling of transcription and mRNA processing. Mol Cell. 2007;25:71–83. doi: 10.1016/j.molcel.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 220.Migliori V, Müller J, Phalke S, et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat Struct Mol Biol. 2012;19:136–44. doi: 10.1038/nsmb.2209. [DOI] [PubMed] [Google Scholar]
- 221.Baldwin G, Carnegie P. Specific enzymic methylation of an arginine in the experimental allergic encephalomyelitis protein from human myelin. Science. 1971;171:579–81. doi: 10.1126/science.171.3971.579. [DOI] [PubMed] [Google Scholar]
- 222.Barth S, Liss M, Voss MD, et al. Epstein-Barr virus nuclear antigen 2 binds via its methylated arginine-glycine repeat to the survival motor neuron protein. J Virol. 2003;77:5008–13. doi: 10.1128/JVI.77.8.5008-5013.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Shire K, Kapoor P, Jiang K, et al. Regulation of the EBNA1 Epstein-Barr virus protein by serine phosphorylation and arginine methylation. J Virol. 2006;80:5261–72. doi: 10.1128/JVI.02682-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Yang M, Sun J, Sun X, et al. Caenorhabditis elegans protein arginine methyltransferase PRMT-5 negatively regulates DNA damage-induced apoptosis. PLoS Genet. 2009;5:e1000514. doi: 10.1371/journal.pgen.1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Powers MA, Fay MM, Factor RE, et al. Protein arginine methyltransferase 5 accelerates tumor growth by arginine methylation of the tumor suppressor programmed cell death 4. Cancer Res. 2011;71:5579–87. doi: 10.1158/0008-5472.CAN-11-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Wei H, Wang B, Miyagi M, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc Natl Acad Sci. 2013;110:13516–21. doi: 10.1073/pnas.1311784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Waldmann T, Izzo A, Kamieniarz K, et al. Methylation of H2AR29 is a novel repressive PRMT6 target. Epigenetics and Chromatin. 2011;4:11. doi: 10.1186/1756-8935-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Frankel A, Yadav N, Lee J, et al. The novel human protein arginine n-methyltransferase PRMT6 is a nuclear enzyme displaying unique substrate specificity. J Biol Chem. 2002;277:3537–43. doi: 10.1074/jbc.M108786200. [DOI] [PubMed] [Google Scholar]
- 229.Singhroy DN, Mesplède T, Sabbah A, et al. Automethylation of protein arginine methyltransferase 6 (PRMT6) regulates its stability and its anti-HIV-1 activity. Retrovirology. 2013;10:73. doi: 10.1186/1742-4690-10-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Boulanger M-C, Liang C, Russell RS, et al. Methylation of Tat by PRMT6 regulates human immunodeficiency virus type 1 gene expression. J Virol. 2005;79:124–31. doi: 10.1128/JVI.79.1.124-131.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Sgarra R, Lee J, Tessari MA, et al. The AT-hook of the chromatin architectural transcription factor high mobility group A1a is arginine-methylated by protein arginine methyltransferase 6. J Biol Chem. 2006;281:3764–72. doi: 10.1074/jbc.M510231200. [DOI] [PubMed] [Google Scholar]
- 232.Karkhanis V, Wang L, Tae S, et al. Protein arginine methyltransferase 7 regulates cellular response to DNA damage by methylating promoter histones H2A and H4 of the polymerase δ catalytic subunit gene, POLD1. J Biol Chem. 2012;287:29801–14. doi: 10.1074/jbc.M112.378281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zurita-Lopez CI, Sandberg T, Kelly R, et al. Human protein arginine methyltransferase 7 (PRMT7) is a type III enzyme forming ω-NG-monomethylated arginine residues. J Biol Chem. 2012;287:7859–70. doi: 10.1074/jbc.M111.336271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Wang Y-C, Peterson SE, Loring JF. Protein post-translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24:143–60. doi: 10.1038/cr.2013.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Biermann K, Steger K. Epigenetics in male germ cells. J Androl. 2007;28:466–80. doi: 10.2164/jandrol.106.002048. [DOI] [PubMed] [Google Scholar]
- 236.Jelinic P, Stehle J-C, Shaw P. The testis-specific factor CTCFL cooperates with the protein methyltransferase PRMT7 in H19 imprinting control region methylation. PLoS Biol. 2006;4:e355. doi: 10.1371/journal.pbio.0040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Yao R, Jiang H, Ma Y, et al. PRMT7 induces epithelial-to-mesenchymal transition and promotes metastasis in breast cancer. Cancer Res. 2014;74:5656–67. doi: 10.1158/0008-5472.CAN-14-0800. [DOI] [PubMed] [Google Scholar]
- 238.Sayegh J, Webb K, Cheng D, et al. Regulation of protein arginine methyltransferase 8 (PRMT8) activity by its n-terminal domain. J Biol Chem. 2007;282:36444–53. doi: 10.1074/jbc.M704650200. [DOI] [PubMed] [Google Scholar]
- 239.Kim J-D, Kako K, Kakiuchi M, et al. EWS is a substrate of type I protein arginine methyltransferase, PRMT8. Int J Mol Med. 2008;22:309–15. [PubMed] [Google Scholar]
- 240.Lee J, Sayegh J, Daniel J, et al. PRMT8, a new membrane-bound tissue-specific member of the protein arginine methyltransferase family. J Biol Chem. 2005;280:32890–6. doi: 10.1074/jbc.M506944200. [DOI] [PubMed] [Google Scholar]
- 241.Uhlén M, Fagerberg L, Hallström BM, et al. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 242.Wang JCL, Sinha SH, et al. Pharmacophore-based virtual screening and biological evaluation of small molecule inhibitors for protein arginine methylation. J Med Chem. 2012;55:7978–87. doi: 10.1021/jm300521m. [DOI] [PMC free article] [PubMed] [Google Scholar]