

Abstract
The cellular quality control system degrades abnormal or misfolded proteins and consists of three different mechanisms: the ubiquitin proteasomal system (UPS), autophagy and molecular chaperones. Any disturbance in this system causes proteins to accumulate, resulting in neurodegenerative diseases such as amyotrophic lateral sclerosis, Alzheimer's disease (AD), Parkinson's disease, Huntington's disease and prion or polyglutamine diseases. Alzheimer's disease is currently one of the most common age‐related neurodegenerative diseases. However, its exact cause and pathogenesis are unknown. Currently approved medications for AD provide symptomatic relief; however, they fail to influence disease progression. Moreover, the components of the cellular quality control system represent an important focus for the development of targeted and potent therapies for managing AD. This review aims to evaluate whether existing evidence supports the hypothesis that UPS impairment causes the early pathogenesis of neurodegenerative disorders. The first part presents basic information about the UPS and its molecular components. The next part explains how the UPS is involved in neurodegenerative disorders. Finally, we emphasize how the UPS influences the management of AD. This review may help in the design of future UPS‐related therapies for AD.
Keywords: ubiquitin, Alzheimer's disease, amyloid β, chaperones
Introduction
Biosynthesized proteins often misfold because of destabilizing mutations, stress or metabolic changes 1 and cannot perform their biological functions. Protein misfolding is prevented by the cellular quality control system, which consists of the ubiquitin proteasomal pathway (26S pathway or UPP), autophagy and molecular chaperones 2, 3. These mechanisms, which are often referred to as the cellular proteostasis network, prevent the accumulation of toxic levels of unfolded or misfolded proteins 4 by breaking the proteins down into polypeptide chains 5, 6. Any disturbance in this system causes proteins in different parts of the body to accumulate and promotes severe neurodegenerative disorders, such as Alzheimer's disease (AD), Parkinson's disease (PD) and prion or polyglutamine diseases 2, 7.
Presently, AD is one of the best described age‐related neurodegenerative diseases. However, its potential cause is not fully understood. Alzheimer's disease is characterized by a progressive loss of memory and object recognition, dementia and changes in behavior and speech. It is associated with the synthesis of defective proteins, errors in their transport to membranes and oxidative damage. Researchers have suggested that the aggregation of amyloid beta (Aβ) initiates this disease. Amyloid beta is formed when amyloid precursor protein (APP) 4, 6 undergoes successive enzymatic cleavages and is transported into the extracellular space. Overproduction or defective clearance of Aβ causes it to progressively accumulate. Amyloid beta self‐aggregates into oligomers, which obstruct proteasome function and disturb neuronal processes. Furthermore, Aβ inhibits the proteasomal pathway and promotes the formation and accumulation of hyperphosphorylated tau, a highly soluble microtubule‐binding protein 8. Normally, tau stabilizes the microtubule network within axons and facilitates the axonal transport of organelles, neurotransmitters and other cellular constituents that act as trophic factors 9, 10 Hyperphosphorylated tau self‐aggregates into neurofibrillary tangles (NFTs) and impairs axonal transport and neuronal function 8.
Protein degradation is primarily performed by proteasomes in the cytosolic and nuclear compartments 11. Changes in UPP‐based protein degradation lead to pleiotropic effects, such as neurodegeneration, synaptic dysfunction and neuron death 12. Recently, many studies have suggested that UPP affects the pathogenesis of AD 12, 13. First, ubiquitinated proteins are present in AD. In addition, misfolded proteins within all tissues are normally degraded by the UPP 8, 9. However, the exact underlying mechanism of this process continues to be investigated.
The ubiquitin proteasomal system
Intracellular protein degradation is a selective process 14 that, in eukaryotic cells, is catalyzed by proteasome, the major component of the ubiquitin proteasomal system (UPS) 8, 15. Ubiquitin proteasomal system‐mediated degradation is the primary proteolytic mechanism for normal and abnormal synaptic proteins. The UPS includes five steps: activation, conjugation, recognition, ubiquitin removal by specific deubiquitinating enzymes (DUBs) and substrate degradation by the proteasome 16, 17.
The UPS controls protein degradation by proteasome‐mediated proteolysis and regulates protein function via multiple types of ubiquitination. The UPS degrades more than 80% of normal and abnormal intracellular proteins 18. Within tissues, most intracellular proteins are degraded by the UPP, whereas extracellular proteins and several proteins on the cell surface are endocytosed and degraded via lysosomes 19. The UPP controls many cellular processes: the cell cycle, DNA transcription and repair, apoptosis and quality control 20, 21. It also maintains proteostasis during aging and disease and prevents protein misfolding and aggregation 22.
Ubiquitination and deubiquitination
Ubiquitin is a 76‐amino‐acid protein with a molecular weight of 8.5 kD that is present in all tissues in a free or covalently conjugated form. Ubiquitination is a reversible process 23 that consists of the covalent attachment of the glycine residue of ubiquitin to a lysine residue of the target protein via an isopeptide bond through the actions of ubiquitin activating (E1), conjugating (E2) and ligating (E3) enzymes 2, 24. Recently, the activity of an E4 enzyme was described. E1 and E2 enzymes prepare ubiquitin for conjugation. E3 enzymes recognize the specific substrate and catalyze the transfer of activated ubiquitin to the substrate 19, 25. E4 enzymes catalyze the conjugation of additional ubiquitin monomers to form a polyubiquitin chain, usually through lysine 48 (K48) linkages 12 (Fig. 1). For 26S proteasome degradation, a polyubiquitin chain is made of four or more ubiquitin proteins that target a single substrate 8. There are two types of E3 ligases: HECT (homologous to E6‐associated protein C‐terminus) and RING‐finger/adaptor. Only the HECT domain E3 ligase forms a covalent bond with ubiquitin through a thioester intermediate during polyubiquitination (the transfer of ubiquitin to the substrate) of abnormal proteins. The RING‐finger E3 ligase directly transfers ubiquitin from its associated E2 enzyme to the substrate 16. The fate of ubiquitinated proteins depends on the type of linkage with ubiquitin (K48, K63, K6, K11, K27, K29 and K33). K48‐linked polyubiquitinated proteins are generally degraded through the proteasomal pathway, whereas K63‐linked polyubiquitinated proteins are degraded via the lysosomal pathway 26. The different types of ubiquitin linkage exhibit the following associations: K6 with DNA repair, K11 with endoplasmic reticulum (ER)‐associated protein degradation and cell cycle regulation, K27 with ubiquitin fusion and degradation, K29 with lysosomal degradation and K33 with kinase modification 22. After the proteasome recognizes the polyubiquitinated substrate, DUBs deubiquitinate the polyubiquitin chain at different levels of the UPP. Deubiquitination may occur before or after DUBs recognize the substrate on the 26S proteasome 14. After they recognize the substrate, the catalytic core degrades the substrate, and the DUBs recycle ubiquitin and preserve monoubiquitin for additional ubiquitination 14. Deubiquitination is necessary when newly translated ubiquitin binds with the C‐terminal end of amino acids or is cleaved by ubiquitin C‐terminal hydrolase 1 (UCHL1). Approximately, 100 DUBs are found in eukaryotes 27. Of them, 27 are in the nervous system, and seven are in the proteasomal system 14 (Fig. 2). Deubiquitinating enzymes are grouped into five different classes: (i) ubiquitin C‐terminal hydrolases (UCHs), e.g. UCHL1, UCHL3, and UCHL5/UCH37; (ii) ubiquitin‐specific proteases (USPs), e.g. USP7, USP9x, and USP14; (iii) Machado‐Joseph disease protease, e.g. ataxin‐3; (iv) otubain proteases (OTUs), e.g. otubain 1 and otubain 2 and (v) metallo‐enzymes (JAMMs), e.g. PSMD14/Rpn11 and JAB1/MPN/Mov34 14, 28.
Figure 1.
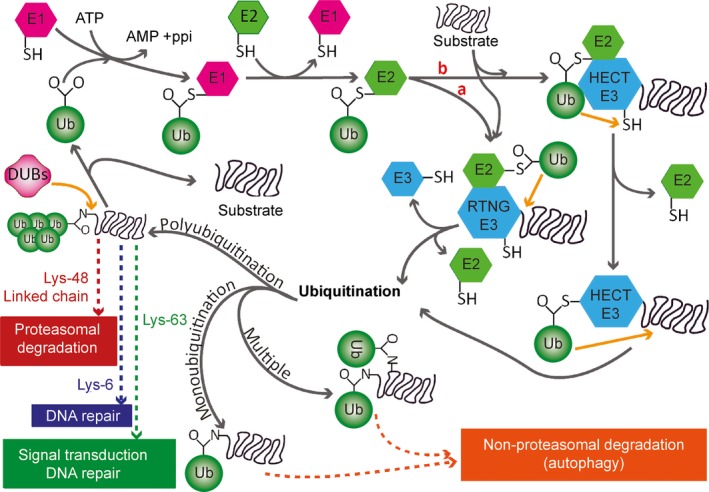
General outline of ubiquitination. Ubiquitination is the covalent attachment of the glycine residue of ubiquitin to the lysine residue of the target protein via an isopeptide bond. This process is mediated by E1, E2 and E3 enzymes. E1 ligases forms thioester bonds with ubiquitin, which is subsequently transferred to E2 ligases through a thioester linkage. E3 ligases are grouped into two types: HECT type and RING‐finger/adaptor type. Only the HECT domain E3 ligase forms a covalent bond with ubiquitin during polyubiquitination of abnormal proteins, whereas the RING‐finger E3 ligases directly transfer ubiquitin from the associated E2 enzyme to the substrate protein. Proteins can be mono‐, multiple‐ or poly‐ubiquitinated. Mono‐ and multiple‐ubiquitinated proteins are degraded by lysosomes (autophagy), whereas the degradation of polyubiquitinated proteins depends on the type of ubiquitin linkage (K48, K63, K6, K11, K27, K29 and K33). K48‐linked polyubiquitinated proteins are generally degraded by the proteasomal pathway. Polyubiquitination via K6 is associated with DNA repair; via K11, with endoplasmic reticulum‐associated protein degradation and cell cycle regulation; via K27, with ubiquitin fusion degradation; via K29, with lysosomal degradation; and via K33, with kinase modification. DUBs remove ubiquitin from the substrate protein, and the monomers are recycled.
Figure 2.
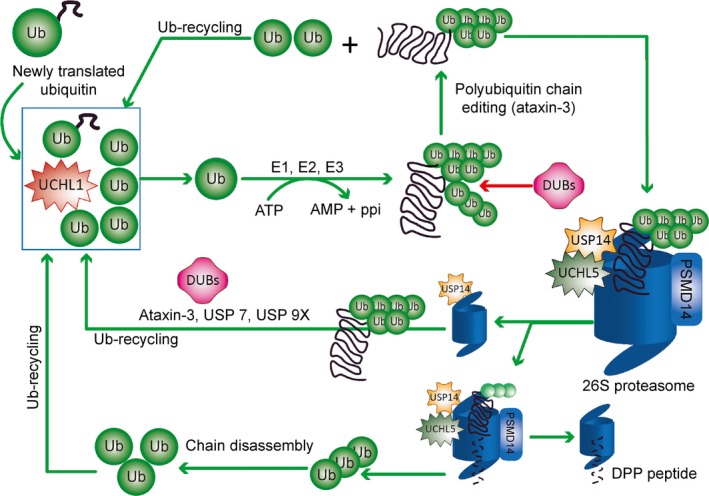
The deubiquitination process. Different types of deubiquitinase enzymes (DUBs) mediate the degradation of misfolded proteins. Newly translated ubiquitin (Ub) that is fused with the C‐terminal end of amino acids is cleaved by DUBs, such as UCHL1. UCHL1 maintains the pool of mono‐Ub and supplies ubiquitin for misfolded protein ubiquitination. Ubiquitin enzymes (E1, E2, and E3 enzymes) form the polyubiquitin chain on substrate proteins by adding mono‐UB. The polyubiquitinated chain is modified by DUBs, such as ataxin‐3, to ensure correct recognition of the substrate proteins by the 26S proteasome. The mono‐Ubs are again recycled back into the pool of mono‐Ub. DUBs, including USP14, work in two ways: (i) USP14 rescues polyubiquitinated proteins from degradation by the 26S proteasome, whereas DUBs such as USP7, ataxin‐3 and USP9X facilitate the deubiquitination of poly‐Ub chains and the recycling of mono‐Ubs back into the pool of mono‐Ub; and (ii) USP14 may also work in combination with other DUBs including PSMD14 and UCHL5, on the 26S proteasome to degrade misfolded proteins into small peptides. The poly‐Ub chain is disassembled to mono‐Ubs that are recycled back into the pool of mono‐Ub.
Ubiquitin chain trimming promotes or inhibits proteasomal degradation. The enzymes involved in this process (especially DUBs) are potential therapeutic targets for the regulation of proteasomal degradation and the treatment of AD 14, 27.
The 26S proteasome
The proteasome is a multisubunit protein complex that includes parts of the cytoplasm and the nucleus. There are several thousands of proteasomes, which are responsible for intracellular protein degradation 29 of normal and misfolded cytoplasmic proteins 30. Within the cytoplasm, the proteasome is associated with the centrosome, the cytoskeletal network and the outer surface of the ER, whereas in the nucleus, it is present throughout the nucleoplasm 31. Two types of proteasomes exist in eukaryotes: a smaller 20S proteasome that degrades unfolded proteins and a larger 26S proteasome that degrades folded proteins in an ATP‐dependent or ‐independent manner 30, 32.
A modification of the 26S proteasome, the immunoproteasome, is also involved in the pathogenesis of AD via neuroinflammation 33, 34. The 26S proteasome is a 2.5‐MDa multicatalytic protease complex that recognizes and degrades ubiquitin‐tagged substrates 19. It consists of a 20S core particle (CP) (700 kD) that is associated with one or two 19S regulatory particles (RPs) 18, 20. After the substrates are ubiquitinated, the 26S proteasome recognizes them in the presence of the lid subunits of the 19S RP, Rpn10/S5a and Rpn13/Adrm1 27. Then, the ubiquitinated substrates are deubiquitinated by Rpn11 in the presence of the DUBs USP14, UCHL5, PSMD14, UCHL1, Ataxin‐3, USP7 and USP9x 8, 14. The proteins are then unfolded through ATP hydrolysis in the 19S RP and translocated into the 20S CP for degradation 29.
The 19S RP is composed of two subcomplexes (lid and base), both of which contain at least 19 individual subunits. These subunits are responsible for specific substrate recognition, deubiquitination, and unfolding and translocation of the substrate into the proteolytic chamber for degradation 29. The lid consists of nine non‐ATPase subunits (Rpn3, 5‐9, 11, 12, and 15), and the base consists of six ATPase (Rpt1‐6) and four non‐ATPase (Rpn1, 2, 13, and 10) subunits 20 (Fig. 3). The primary function of the lid is to deubiquitinate the substrate: Rpn11 in the lid displays deubiquitinating activity 20 and aids ubiquitin recycling 24. The base subcomplex is composed of six homologous ATPase subunits (Rpt1‐6) and 4 non‐ATPase subunits (Rpn1, 2, 10 and 13). The base recognizes ubiquitin, binds to the substrate, promotes its unfolding and opens the channel of the 20S CP 35. The Rpn10/S5a and Rpn13/Adrm1 subunits of the base act as two integral receptors and coordinate binding of the proteasome to polyubiquitin chains 27. Rpn10 also stabilizes the bond between the lid and the base 20. Rpn2 is the largest subunit of the proteasome and stabilizes the 19S complex 27.
Figure 3.
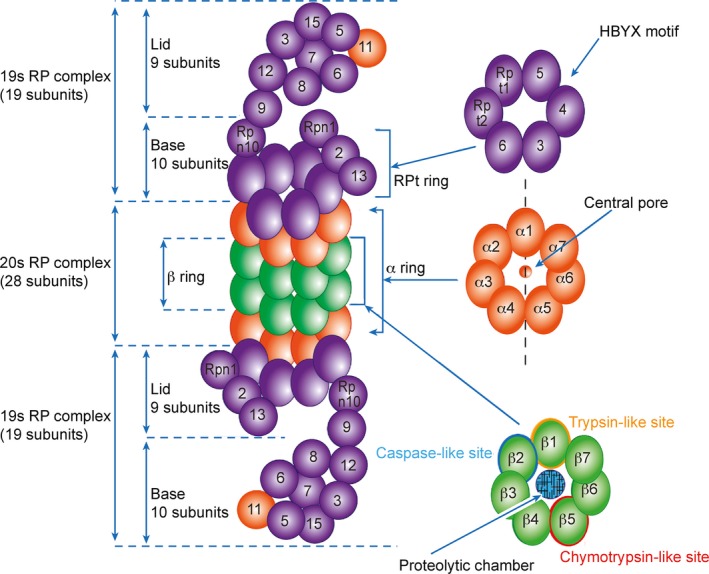
Basic molecular construction of the 26S proteasome. The proteasome consists of the 19S regulatory particle (19S‐RP) and the 20S core particle (20S‐CP). The 19S‐RP consists of 19 subunits that are divided into lid and base and is responsible for specific polyubiquitinated substrate recognition, deubiquitination, unfolding, and translocation to a proteolytic chamber. The lid consists of nine subunits (Rpn3, 5‐9, 11, 12 and 15), whereas the base consists of 10 subunits (Rpt1‐6, Rpn10, and Rpn1, 2 and 13). Rpn10/S5a and Rpn13 (hRpn1, ADRM1 or ARM1) act as ubiquitin receptors and are important for ubiquitinated protein degradation. The Rpt ring (Rpt1‐6) is associated with three HbYX motifs. In the presence of HbYX motifs, RP associates with the α‐ring of the 20S‐CP and open its N‐terminal gate to enable substrate entry. The 20S‐CP complex consists of two outer α‐rings and two inner β‐rings that together include 28 subunits. The two outer α‐rings (α1‐α7) contain a total of 14 subunits that create a central pore/gate to enable the entry of unfolded proteins into the proteolytic chamber. The two inner β‐rings (β1‐β7) also contain a total of 14 subunits and form the proteolytic chamber where protein degradation occurs. Each β‐ring contains six proteolytic sites, two β1 (trypsin‐like site), two β2 (caspase‐like site) and two β5 (chymotrypsin‐like site); these sites are responsible for the degradation of the substrate protein.
In several cases, the 19S RP can be replaced by the PA28/11S regulatory complex, which contains a single ring of seven subunits 36. Replacing three catalytic subunits in a standard proteasome forms an immunoproteasome 37. The immunoproteasome utilizes the 11S RP instead of the 19S RP to facilitate 20S gate opening and to stimulate substrate unfolding and translocation. In addition, the immunoproteasome has increased chymotrypsin‐like and trypsin‐like activities because of the six different ATPases (Rpt1‐6) at the base of the 19S RP that interact with the 20S catalytic chamber 38.
The 20S CP, also known as the 20S ‘core’ proteasome complex or 20S proteasome 39, contains peptidases that function through a threonine active site 40. Unlike the 26S proteasome, the 20S CP actively recognizes and degrades substrate proteins and is not reversibly inactivated by mild oxidative stress 41. In addition, the 20S CP can only degrade proteins that are already unfolded, whereas the 26S proteasome can degrade folded and functional proteins 42. The 20S CP is a barrel‐like structure and consists of four stacked heptameric rings (α, β, β, and α) 29. The outer two α‐rings consist of seven homologous subunits (α1‐α7), and the inner two β‐rings consist of seven homologous subunits (β1‐β7) 29. The β‐ring contains a wide pore at its center in which proteolysis occurs, and the α‐ring contains a narrow pore that allows the translocation of only unfolded proteins into the 20S CP 29. The two β‐rings in the 20S CP perform six proteolytic activities, two caspase‐like, two trypsin‐like and two chymotrypsin‐like, which are performed by the β1, β2 and β5 subunits respectively 43. A docking study 29 demonstrated that the binding of the 19S RP to the 20S CP occurs through an L‐shaped link between the base of the 19S RP and the α subunit of the 20S CP. This link forms between the putative ATPase densities and the side of the α2 subunit of the 20S CP 9, 44. Three additional links form between the 19S RP and the 20S CP. They are parallel to the long axis of the proteasome and extend from the putative ATPase region of the 19S RP to A5, A6 and the interface between A7 and A1 of the 20S CP. When the 19S RP binds to the 20S CP, the gate to the proteolytic chamber is opened through the displacement of the subunit within the α‐ring of the 20S CP 29, 45. The 20S core complex performs various catalytic activities, including cleaving the carboxy‐terminal side of hydrophobic, basic or acidic residues 46. 20S CP is activated by the ATPases and archaeal proteasome‐activating nucleotide (PAN), which consists of peptides with a C‐terminal hydrophobic tyrosine‐x (HbYX) motif 24. The N‐terminal tails of the α‐subunits of the 20S CP form a gate that opens when the HbYX peptide of the ATPase binds to the pockets of the 20S α‐ring 47. The C‐terminal peptides of the ATPases Rpt2 and Rpt5 induce and regulate gate opening 24, 48. The regulatory peptides of the PAN C‐terminus bind to the inner subunit pockets and open the gate of the 20S CP so that the substrate can enter the proteolytic chamber 24.
The 26S proteasome binds polyubiquitinated misfolded proteins, deubiquitinates them, unfolds misfolded proteins, opens the α‐gate of the 20S CP and subsequently degrades the misfolded proteins 35. Ubiquitin affects substrate susceptibility to proteasomal degradation in two ways: the ubiquitination level of the substrate promotes proteasome‐substrate interactions, and the ubiquitin chain structure influences substrate translocation to the 20S CP 49.
The ubiquitin proteasomal pathway in neurodegenerative disorders
Neurodegenerative diseases are characterized by abnormal deposition of insoluble protein aggregates or inclusion bodies within neurons 50. Many of them are caused by the accumulation of abnormal, misfolded or aberrant proteins 51; protofibril formation 52; UPS dysfunction 53; excitotoxic insult 54, 55; oxidative and nitrosative stress 56; mitochondrial injury 57; synaptic failure 58 or altered metal homeostasis 59. The UPP controls many of these processes in the nervous system. Several of its components are linked to synaptic dysfunction and genetic mutations that lead to neuronal disorders 50: UPS impairment has been associated with synaptic dysfunction in AD, PD, Huntington's disease (HD), schizophrenia, amyotrophic lateral sclerosis, ataxia, Angelman syndrome, Wallerian degeneration and gracile axonal dystrophy 50, 60. Ubiquitinated protein aggregates cause UPS dysfunction or structural changes in protein substrates that prevent their recognition and degradation. This loss of degradation has pleiotropic effects on neurons, such as cell death, degeneration and synaptic malfunction 50. Synaptic plasticity impairment and its connection to the UPP have been more extensively studied in AD than in PD and HD 60.
Alzheimer's disease is a progressive neurodegenerative disorder that is also the most frequent cause of dementia. It is characterized by the intracellular accumulation of Aβ oligomers and hyperphosphorylated tau and by the extracellular accumulation of high‐molecular‐weight deposits of Aβ peptides in fibrillar forms. These accumulations results in memory loss and severe cognitive decline 22, 50. Via the 26S proteasome, the UPP regulates Aβ metabolism and tau degradation 22. Its impairment in the neurons of AD patients causes ubiquitinated proteins to accumulate and the combinations of proteasome subunits to change 22, 60, which decreases proteasomal activity, decreases α‐secretase activity and produces Aβ 22 (Fig. 4).
Figure 4.
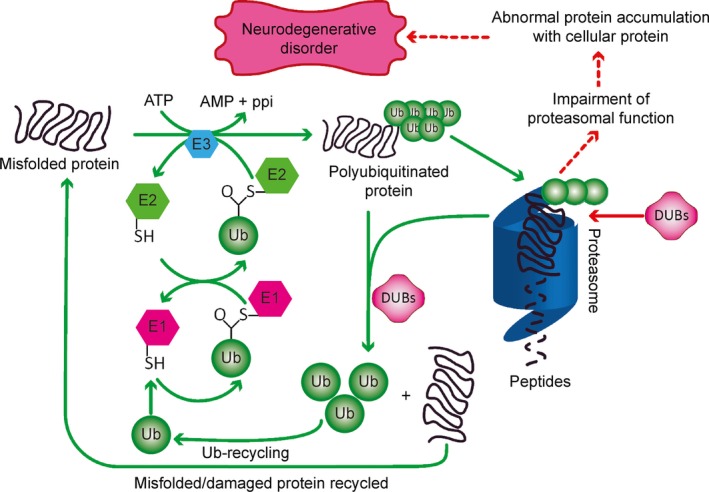
Delineation of the ubiquitin proteasome pathway in the pathogenesis of Alzheimer's disease. Improperly folded proteins are initially polyubiquitinated by the ubiquitin (Ub) enzymes E1 (Ub activating), E2 (Ub ligating) and E3 (Ub conjugating). These polyubiquitinated substrates are easily recognized by the 19S regulatory particle of the 26S proteasome. The polyubiquitinated protein is deubiquitinated by deubiquitinase enzymes (DUBs) and degraded into small peptides through 20S core particle. The polyubiquitinated proteins are deubiquitinated before they are recognized by the 26S proteasome. After they are recognized, they are polyubiquitinated again by the same mechanism and degraded by the 26S proteasome. DUBs convert poly‐Ub chains to Ub monomers, which are then recycled. Proteasome impairment decreases the degradation of amyloid beta (Aβ) and tau and promotes their oligomerization. The oligomers accumulate in neurons, resulting in neuronal death and Alzheimer's disease.
Alzheimer's disease is characterized by progressive degeneration and loss of cortical and limbic neurons. Amyloid beta peptides form senile plaques in the extracellular space, and NFTs are present intracellularly 61. Proteasomal activity is decreased in the parahippocampal gyrus, the superior and middle temporal gyri and the inferior parietal lobe, suggesting that the UPP may be involved in the pathogenesis of AD 62.
Treatment strategies for Alzheimer's disease
Protein misfolding and changes in cellular protein homeostasis are the primary causes of AD. Currently approved medications for treating AD provide symptomatic relief but fail to influence disease progression 63. In the last 10 years, drug discovery has been directed towards developing disease‐modifying agents. In fact, more than 200 medications are currently in phase 2 and 3 clinical trials 64. These compounds can be largely categorized into anti‐amyloid agents and medications that target other pathophysiological pathways (Tables 1, 2, 3, 4, 5, 6, 7). Developments in AD prevention and treatment include anti‐amyloid drugs; methods to hyperphosphorylate tau; and neuroprotective and neurotrophic approaches, such as those involving neurotrophins, neurotrophic factors and stem cells 65. Neurotrophic agents such as cerebrolysin modulate the apoptosis rate of neurons in patients affected by neurodegenerative diseases and are a novel treatment option for AD 66. The accumulation of toxic amyloid is regulated by the quality control systems of the cell (molecular chaperones, autophagy and the UPS) 22. Targeting different components of these pathways will lead to potential therapies for AD (Fig. 5).
Table 1.
Agents that target Aβ production or aggregation
| Agents | Details | Agents under investigation | Status | Ref. |
|---|---|---|---|---|
| Agents that decrease production of Aβ | β‐secretase inhibitor |
MK8931 NIC5‐15 |
(III)a
(II)a |
72 |
| γ‐secretase inhibitor |
EVP0962 CHF5074 |
(II) (II) |
||
| α‐secretase activator |
Etazolate EGCg |
(IIA) (II/III)a | ||
| Agents that decrease aggregation or oligomerization of Aβ | Anti‐aggregation nanopeptide | Tramiprosate | (III) | 72, 73 |
| Fluorinated nanoparticles | – | – |
The status of a drug in an ongoing clinical trial.
Table 2.
Other agents that target Aβ metabolism
| Targets of the agents | Details | Agents under investigation | Status | Ref. |
|---|---|---|---|---|
| Apolipoprotein (ApoE) and Aβ clearance | Nuclear receptor modulator and ApoE activator | Bexarotene | – | 74 |
| Enzymes that degrade Aβ (NEP, IDE, ECE, ACE) | Increases NEP activity |
Valproic acid Oestrogen Green tea |
– | 75 |
| Aβ transport | Antibodies against LRP | – | – | 76 |
| Intraneuronal Aβ transport | Cerebrolysin | – | 77 | |
| Aβ signaling | Blocks Fyn kinase or cellular prion proteins | Saracatinib | (II) | 76 |
| Intravenous immunoglobulin | Clearance of Aβ | – | – | 78 |
| Medications that influence Aβ blood–brain barrier transport | Inhibition of RAGE | – | – | 64 |
| Activation of LRP‐1 |
Table 3.
Immunotherapy‐based agents
| Agents | Details | Agents under investigation | Status | Ref. |
|---|---|---|---|---|
| Active immunotherapy agents (Vaccine) | DNA vaccine | pCMVE/MDC‐3Aβ11 PADRE‐C3d | – | 103 |
| Epitope‐/protein‐/VLP‐based vaccine | Adeno‐10× Aβ3 – 10+ CpG | – | ||
| Prime‐boost approach | Aβ1‐42 peptide prime/Aβ1‐42 DNA + QuilA | – | ||
| Passive immunotherapy agents | Monoclonal antibody |
Bapineuzumab Solanezumab |
(III) | 64 |
Table 4.
Agents that target tau
| Targets of the agents | Details | Agents under investigation | Status | Ref. |
|---|---|---|---|---|
| Hyperphosphorylation |
Kinase inhibitor GSK‐3α GSK‐3β CDK5 |
Lithium AZD‐1080 Minocycline Cerebrolysin |
– | 64, 104 |
| Inhibits histone deacetylase | HDAC6 | – | 73 | |
| Microtubule stabilizers | Improves axonal transport, microtubule density |
Epothilone NAP (NAPVSIPQ) D‐SAL (SALLRSIPA) |
– | 76 |
| Blocking tau oligomerization | Reduce tau–tau interactions |
Astemizole Lansoprazole Methylene blue |
– | |
| Enhancing tau degradation | Inhibit heat shock protein 90 | Curcumin | – | |
| Vaccination therapy | Promote immunological clearance of tau | – | – |
Table 5.
Agents that target neurotransmitter signaling
Table 6.
Agents that target cellular metabolic pathways
| Agents | Details | Agents under investigation | Status | Ref. |
|---|---|---|---|---|
| Agents that target intracellular signaling cascades | Phosphodiesterase inhibitor |
AVE‐8112 BCA‐909 THPP‐1 |
– | 76 |
| Lipoprotein inhibitor |
Rilapladib (NCT01428453 |
(II) | ||
| Agents that target mitochondrial dysfunction | Mitochondrial‐targeted ROS scavenger therapy | Szeto‐schiller peptide (SS‐31) | – | |
| Lipoic acid omega‐3 fatty acid combination therapy |
NCT01780974 NCT01058941 |
(I/II) | ||
| Agents that target epigenetics | DNA methylation and histone modification | Vitamin B | – | 115 |
| Agents that target oxidative stress | Vitamin E + selenium | NCT00040378 | (III) | |
| Melatonin | NCT00940589 | (II) | ||
| Caspase inhibitors | Neuroprotective benefits | – | – | |
| Metal chelators | Inhibit zinc and copper from binding to Aβ | Clioquinol | (II) | 116 |
| Metal protein attenuation | PBT2 | (II) |
Table 7.
Agents with other targets
| Agents | Details | Agents under investigation | Status | Ref. |
|---|---|---|---|---|
| Glial cell modulators | Direct glial target | ONO‐2506 | – | 64 |
| RAGE receptor antagonist | TTP 488 | – | ||
| TNFα agonist | Enbrel | – | ||
| Anti‐inflammatory agents | NSAID | CHF5074 and SC560 | – | |
| Cholesterol‐lowering agents | Pleiotropic effect |
Simvastatin Atorvastatin |
(III) | 116 |
| Neurogenesis | Pleiotropic effect | Cerebrolysin | – | 76 |
| Multi‐target‐directed ligands | Multifunctional compound | Ladostigil (TV3326) | – | |
| Neurotrophins | NGF brain delivery via adeno‐associated virus vector system |
CERE‐110 NCT00876863 NCT00087789 |
(I)a | 124 |
| Based on stereotactic implantation containing NGF‐producing cell | NsG0202 | (I) | ||
| Nutritional supplements | Combination medical food | Souvenaid® | – | 125 |
| Octanoic acid or caprylic acid | Axona® | – | ||
| Natural products, e.g. quercetin | 96, 126 | |||
| Neuroprotective gonadotropin hormone | 76 | |||
| NOS modulators | ||||
| Nucleic acid agents | ||||
| Agents that modulate synaptic plasticity and nerve growth | ||||
| Stem cells | 127 | |||
The status of a drug in an ongoing clinical trial.
Figure 5.
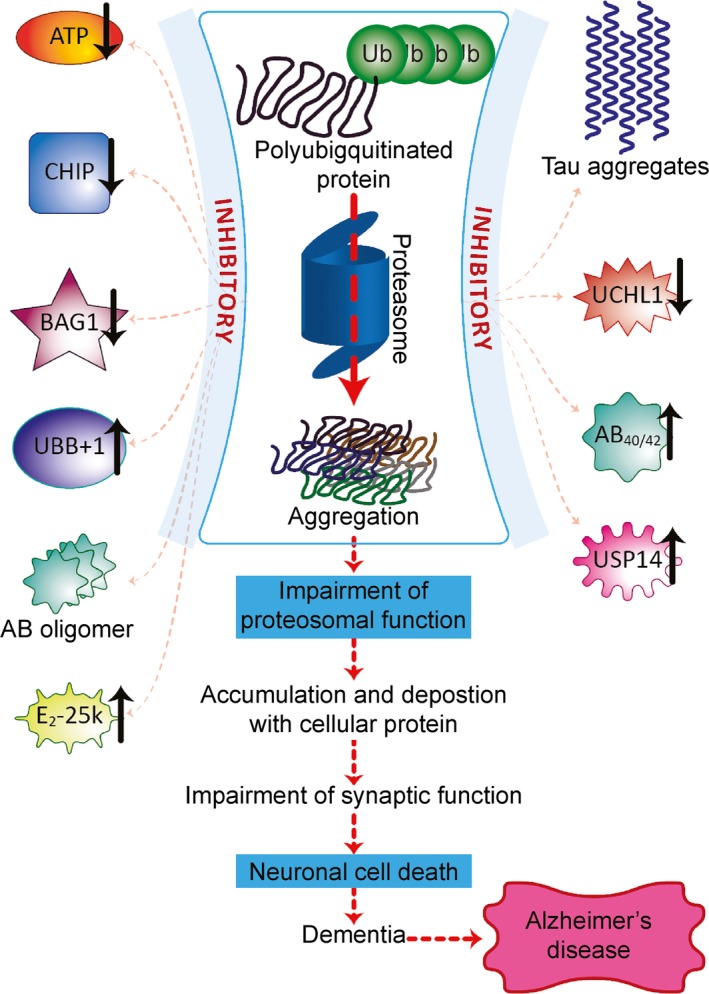
Approaches for the management of Alzheimer's disease. Inhibitors of the 26S proteasome cause Aβ and tau aggregation, which impairs proteasomal function and promotes the accumulation of Aβ and tau in neurons, resulting in neuronal death and Alzheimer's disease. ATP hydrolysis is necessary for both ubiquitination and 26S proteasomal degradation of ubiquitinated proteins. UBB +1 is selectively expressed in neurons and competes with normal ubiquitin for binding to target proteins. UBB +1 blocks ubiquitin‐dependent proteolysis by interacting with E2‐25k/Hip‐2 and might induce neurotoxicity and the abnormal accumulation of proteins. BAG1 and Hsp70 regulate the proteasomal degradation of tau. UCHL1 accelerates β‐secretase degradation and affects APP processing and Aβ production. USP14 inhibits proteasome activity. Aβ40 and Aβ42 oligomers decrease the trypsin‐like, chymotrypsin‐like and peptidylglutamyl‐like activity of the proteasome and promote intraneuronal accumulation of Aβ conjugates. Low levels of CHIP promote the accumulation of phosphorylated tau species, inhibiting the proteasome and decreasing protein degradation.
Therapies targeting misfolding
Neurodegenerative diseases share common cellular and molecular mechanisms, such as protein aggregation and inclusion body formation. The protein aggregates consist of fibrils of misfolded amyloid proteins that have taken on a β‐sheet conformation. The formation of these aggregates can be prevented by two pathways: the Hsp40/Hsp60/Hsp70 pathway mediates protein aggregation, while the proteolytic system removes misfolded proteins 67. Several agents may be able to target the protein misfolding pathway. For example, Congo red binds to proteins with β‐sheet structures, alters the protein misfolding pathway and reduces toxicity in vivo 24, 68, 69, 70, 71. Recent therapeutic developments include peptides that interfere with the post‐translational modifications that are responsible for the misfolding and aggregation of Aβ and tau. They also include the up‐regulation of molecular chaperones to increase the clearance of protein aggregates 69. Of the ATP‐dependent endogenous proteasome activators (PA700, PA200 and PA28), targeting PA700 may be the most successful therapeutic approach for increasing misfolded protein degradation 79.
Therapies targeting the different quality control systems of the cell
Ubiquitin‐dependent protein degradation occurs via two major catabolic systems: the endosomal/lysosomal system and the ATP‐dependent, non‐lysosomal proteolysis system, which is known as the UPS.
Autophagy (the endosomal/lysosomal system)
Through autophagy, lysosomes degrade normal and aggregated proteins that are usually found under conditions of stress or injury. Independent alterations in the endocytic pathway activate the lysosomal system and increase the number and density of lysosomes as well as gene expression 80. The latter effect results in the synthesis of all classes of lysosomal hydrolases, including cathepsin 81. Lysosomal cathepsin B is up‐regulated either by the accumulation of proteins or by the modulator 2‐Phe‐Ala‐diazomethyl ketone (PADK). Systemic administration of PADK increases cathepsin B activity, which increases the clearance of intracellular Aβ and decreases its extracellular deposition. PADK inhibits protein accumulation and promotes synaptic integrity in vivo and in vitro 82. Thus, modulators of lysosomal activity show great promise for the treatment of neurodegenerative diseases.
Autophagy reduces the accumulation and facilitates the clearance of normal or mutant Aβ 80. The Aβ that is generated in endosomes and autophagic vacuoles is delivered to lysosomes, where it is cleared through lysosomal proteolysis under normal conditions 83. Enhancing lysosomal proteolysis in TgCRND8 transgenic mice increased the clearance of autophagy substrates, which reduced intracellular and extracellular Aβ levels and reversed multiple cognitive deficits 84. Rapamycin could be effective in preventing or reversing AD pathology, because it inhibits motor activity and enhances autophagy. Rapamycin also inhibits NFT formation and tau phosphorylation and reduces cognitive impairment 80. Hence, drugs that induce autophagy could reduce or eliminate protein aggregates 85. Impaired clearance and accumulation of autophagic vacuoles in AD‐affected neurons causes abnormal autophagy 86, 87; this effect might block the neuroprotective effect of autophagy and limit the accumulation of toxic proteins (including tau), thus promoting neuronal cell death 88.
Molecular chaperones (Hsps)
Molecular chaperones and the UPS are the first and second lines of defense against protein misfolding and aggregation. Chaperones regulate the folding of newly synthesized proteins and the refolding or transfer of misfolded proteins to protein degradation systems 22. Lower molecular weight Hsps (12–43 kD) are ATP independent, whereas higher molecular weight Hsps (>43 kD) are ATP dependent. Several studies 89, 90, 91, 92, 93 have demonstrated that the chaperone network can be targeted to develop therapeutic strategies for AD 94. Chaperones bind to Aβ and tau and regulate their degradation. Both Hsp90 and Hsp70 participate in APP metabolism 95.
Hsp70 is ATP dependent and the major target for AD treatment. Increased Hsp70 levels inhibit its ATPase activity and might be an effective treatment strategy for AD 94. In 5XFAD transgenic mice, recombinant human Hsp70 reduces Aβ plaque formation 96. At very high concentrations, methylene blue inhibits Hsp70 and increases tau clearance 68. Curcumin was also shown to increase Hsp70 and Hsp90 activity, which could inhibit or delay amyloid formation and reduce neuronal death 97, 98. The co‐chaperone protein Bcl2‐associated athanogene (BAG‐1) forms a complex with Hsp70 and tau and could inhibit tau degradation 99 in cell cultures, increasing both tau and APP levels 99, 100.
Hsp90 regulates the misfolding and stabilization of neurotoxic proteins and facilitates tau pathology in AD 101. Inhibiting Hsp90 reduces levels of hyperphosphorylated and Ser/Thr‐mutated tau as well as of the kinases that are involved in persistent hyperphosphorylation 102. Moreover, Hsp90 inhibition induces heat shock factor, Hsp70 and small heat shock proteins (sHsps). This inhibition also facilitates Hsp70 binding to abnormal proteins to form a complex that is ubiquitinated by CHIP (carboxyl C‐terminus of Hsp70‐interacting protein) and degraded through proteolysis. Thus, Hsp90 inhibition increases tau degradation and might be a potential therapeutic target for tau‐based neurodegeneration in AD 101. The antibiotic geldanamycin was the first Hsp90 inhibitor to be identified; geldanamycin was shown to compete with ATP and stop neurotoxic protein folding and stabilization 86, 87, 105, 106. Although geldanamycin is toxic and cannot penetrate the blood–brain barrier, other agents have been developed. 17‐AAG and PU‐DZ8 are synthetic purine scaffold proteins that cross the blood–brain barrier, inhibit Hsp90, and decrease levels of phosphorylated tau 107.
The ubiquitin proteasomal system
The role of the UPS in AD has been demonstrated through observations of the downregulation of E1 and E2 enzymes, the oxidation of DUBs, the accumulation of mutated ubiquitin, the detection of proteasome subunits and decreased proteasomal activity 43. Toxic Aβ is formed when β‐secretase cleaves APP and generates a C‐terminal fragment (APPCTFB or C99). This fragment is subsequently cleaved by γ‐secretase to produce toxic Aβ40 and Aβ42. Proteasome inhibitors up‐regulated APP C99, suggesting that they may decrease β‐secretase activity 62.
UPS components as potential therapeutic targets in AD management
In the pathogenesis of AD, it remains unclear whether Aβ accumulation causes UPS dysfunction or vice versa 13, 92.
The 26S proteasome ubiquitinates and degrades proteins other than β‐ and γ‐secretase that are involved in APP metabolism 20. The overexpression of an APP mutant isoform has been correlated with increased oxidative stress and decreased proteasome activity 84. Moreover, proteins associated with AD (most notably AHNAK, gelsolin and tenascin‐R) were found to exhibit strong interactions with ubiquitin C. This change in AD associated proteins was observed in regions that are frequently affected in AD (the hippocampus, parietal cortex and cerebellum), suggesting that ubiquitin C promotes age‐related neurodegeneration 108. Aβ40 and Aβ42 oligomers, but not their monomers, also inhibit proteasomal activity in cell‐free assays 22.
The UPS processes and degrades presenilin‐1 (ps‐1), presenilin‐2 (ps‐2) and mutated presenilin, which are key proteins in the pathogenesis of AD. Therefore, proteasomes and mutated presenilin might indirectly influence Aβ production 30. Ubiquilin regulates UPS activity and presenilin metabolism. It decreases γ‐secretase activity and downregulates presenilin and its N‐terminal and C‐terminal fragments 13. Ubiquilin‐1 is functionally associated with AD and interacts with both ps‐1 and ps‐2 13.
The UPS ubiquitinates and degrades β‐secretase 109. Up‐regulating proteasomal degradation of β‐secretase and γ‐secretase decreases Aβ levels. Proteasome activation can also be used to clear tau 85. Hence, the UPS may provide an alternative target for new therapies for AD 13. An age‐dependent decrease in proteasomal activity may cause both Aβ and tau to accumulate. Once Aβ and tau aggregates are formed, they decrease proteasome activity and create a vicious cycle that increases Aβ and tau accumulation 110. Normal tau in the normal brain and hyperphosphorylated and misfolded tau in the AD brain are present at both pre‐ and postsynaptic terminals 111. Abnormally folded and hyperphosphorylated tau accumulates in dendrites, axons and somas 112. In addition, changes in proteasomal subunit composition alter proteasomal activity in AD 33. Thus, tau oligomers might accumulate in AD synapses because of increased proteasomal components and UPS dysfunction 111. BAG‐1 and Hsp70 regulate the proteasomal degradation of tau 22. Kinases (such as glycogen synthase kinase‐3 and casein kinase‐1) phosphorylate tau 26 and might be candidate therapeutic targets for AD. Tau co‐immunoprecipitates with CHIP, an E3 ubiquitin ligase that ubiquitinates tau before the proteasome degrades it. Phosphorylated tau accumulates in the neurons of CHIP knockout mice and inhibits the interaction between tau and CHIP in AD 113. In vitro studies of AD brains showed that high CHIP levels were associated with less tau aggregation and fewer NFTs 114. In addition, the CHIP knockout mice showed the accumulation of species of phosphorylated tau 113. Thus, the proteasome is directly involved in NFT accumulation, and reduced proteasomal activity might cause tangle formation in patients with AD 43.
Ubiquitin C‐terminal hydrolase 1 is oxidized and downregulated in AD 110, and UCHL1 inhibition up‐regulates β‐secretase. UCHL1 also accelerates β‐secretase degradation, impairs APP processing and decreases Aβ 109. Furthermore, the synaptic dysfunction that is induced by Aβ can be reduced by increasing UCHL1 expression 110. Hence, UCHL1 is a promising target for the development of therapeutic agents for AD.
Importance of UPP in the management of Alzheimer's disease
Amyloid beta neurotoxicity inhibits the proteasome in association with increased brain levels of E2‐25k/Hip‐2. E2‐25k/Hip‐2‐mediated proteasome inhibition can directly induce neuronal death and indirectly increase Aβ production 45. In vitro, E2‐25k/Hip‐2‐mediated ubiquitination of a mutated form of ubiquitin (UBB+1) at Lys48 was suggested to inhibit proteasome activity 87. In vitro, cultures exposed to Aβ42 up‐regulate E2‐25K/Hip‐2 expression in neurons 117. E2‐25k/Hip‐2 induction is required for neuronal cell death under Aβ toxicity. It inhibits proteasome activity and promotes the accumulation of ubiquitin conjugates, which increases the levels of apoptosis signal regulating kinase‐1/C‐Jun N‐terminal kinase and leads to Aβ neurotoxicity. Therefore, E2‐25k/Hip‐2 might be a potential therapeutic target in AD 118.
Ubiquitin is an endogenous reporter of proteasome activity in human AD pathology and can inhibit proteasomal degradation 43. Current studies have shown that UBB+1 accumulates in the brains of AD patients 43. UBB+1 is selectively expressed in neurons and competes with normal ubiquitin for binding to proteasomal target proteins. It also interacts with E2‐25k/Hip‐2 to inhibit the 26S proteasome and block ubiquitin‐dependent proteasomal proteolysis 61, 119. Polyubiquitin chains that contain UBB+1 are refractory to DUB lysis and alter proteasomal degradation of the substrate 120. UBB+1 also impairs mitochondrial movement and promotes neurodegeneration in primary neurons 121. Proteasome‐associated proteins contribute to the proteasome's proteolytic function and regulate its activity through calcium signaling. The function of these proteasome‐associated proteins was also impaired in AD brains 30.
The loss of proteasome activity with age leads to decreased subunit expression, alterations and replacement of proteasome subunits and the formation of inhibitory cross‐linked proteins 122. In transgenic (Tg2576) mice, ubiquitin and proteasome activity decreased with age, whereas Aβ42 levels increased. In addition, in cultured neurons, extracellular administration of Aβ markedly decreased proteasome activity 123.
Recent therapeutic developments
Enhancing chaperone function, chaperone up‐regulation and aggregate clearance or targeting post‐translational modifications (inhibiting oxidation and phosphorylation) could inhibit protein misfolding and aggregation. For example, current AD therapeutic strategies that target Hsp70 include the induction of endogenous Hsp70, exogenous Hsp70 utilization and Hsp70 inhibition. The combination of agents that affect more than one of these therapeutic targets will be the most effective treatment strategy for AD 69.
E3 enzymes determine the specificity of UPS‐mediated proteolysis. Hence, identifying their specific allosteric modulators could provide an effective therapeutic target 21, 69. PROTACS (proteolysis‐targeting chimeric molecules) bind to E3 ubiquitin ligases. They promote the synthesis and attachment of the poly‐ubiquitin chain to the target protein, which is followed by protein degradation by the 26S proteasome. Therefore, developing PROTACS could prevent the accumulation and aggregation of tau proteins 128.
Resveratrol is a phytoalexin polyphenolic compound (trans‐3, 4, 5‐trihydroxystilbene) from grapes and wines. As a result of its anti‐amyloidogenic activity, resveratrol might show therapeutic potential in AD 129. Resveratrol promotes neuroprotection via multiple mechanisms 13: it inhibits Aβ aggregation, scavenges oxidants, reduces inflammation 13 and removes Aβ via the proteasomal pathway 129.
Betulinic acid, a lupine‐type pentacyclic triterpene derived from birch trees, activates the chymotrypsin‐like activity of the proteasome 130. Overexpressing arsenite‐inducible RNA‐associated protein, which stabilizes proteasome activity in the absence of ATP 131, is another therapeutic approach to maintain proteasome function.
Future prospects
Emerging evidence suggests that the substrate‐specific components of the UPS, such as the E3 ligase, may impair ubiquitination, which results in UPS aberrations, defective proteasome‐mediated protein degradation, the accumulation of toxic proteins and neuronal death. Studies of the interaction between Aβ and the proteasomal system have shown that Aβ40 binds directly to the inside of the 20S proteasome, blocks the peptide channel and inhibits the chymotrypsin‐like activity of proteasome 132. Tau accumulation also inhibits the proteasome and reduces protein degradation 18, 133. Several proteases break down tau, including calpain; caspases 3, 6 and 9; cathepsin D and L; and puromycin‐sensitive aminopeptidase. These proteases limit tau proteolysis and may contribute to AD pathology. However, they may also be potential therapeutic targets for this disease 134.
The development of proteasome activators is another potential approach to prevent Aβ accumulation in AD 135. Proteasome‐activating peptide 1 increases the chymotrypsin‐like activity of the proteasome in fibroblast cell cultures. It also down‐regulates and prevents the aggregation of oxidized proteins in amyotrophic lateral sclerosis 135.
USP14, a proteasome‐associated DUB, inhibits the degradation of ubiquitin‐protein conjugates both in vivo and in vitro. USP14 inhibits proteasome activity by trimming the ubiquitin chain on the substrate. Inhibition of USP14 accelerates the degradation of oxidized proteins and increases cell resistance to oxidative stress 136. IU1 is a chain‐trimming enzyme that inhibits USP14 and prevents the rescue of ubiquitinated forms of the neurotoxic proteins tau and ataxin. IU1 acts on the proteasome and was found to have a therapeutic benefit in AD 14. IU1 was also shown to promote the degradation of several overexpressed proteins (tau, TDP‐43 and ATXN3) in various neurodegenerative diseases 27. As ubiquitin chain trimming can promote or inhibit proteasomal degradation, future studies should identify whether IU1 inhibits other DUBs or proteases in human cells 27.
Conclusion
The UPS and its components are key factors in AD treatment. The UPS mediates the pathogenesis of AD through various mechanisms. Full understanding of the mechanisms of proteasomal activity is critical for the development of improved therapeutic and diagnostic strategies for managing AD. The development of effective medications also requires comprehensive understanding of the role of proteasome inhibition and how neuronal death occurs in patients with AD. Amyloid beta has been shown to inhibit proteasomes in vitro, even though it remains unclear whether AD patients show the same pattern. Future AD therapies should reduce protein aggregation by targeting specific components of the UPS (e.g. the 26S proteasome, ubiquitin and DUBs). Another strategy could be to enhance proteasome activity by inhibiting USP14. To develop new therapies for AD, we need to fully understand how the components of the UPS interact in protein degradation.
Conflicts of interest
All of the authors declare that they have no potential conflicts of interest to disclose.
Acknowledgements
The authors are grateful to Dr. Mangala Lahkar, Coordinator of Institutional Level Biotech hub (IBT hub), NIPER Guwahati, for her support and to her superb staff for their helpful discussions. We would also like to acknowledge Dr. Maria Balea from the ‘RoNeuro’ Institute for Neurological Research and Diagnostic for English knowledge expertise.
References
- 1. Brockwell DJ, Radford SE. Intermediates: ubiquitous species on folding energy landscapes? Curr Opin Struct Biol. 2007; 17: 30–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ciechanover A, Kwon YT. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp Mol Med. 2015; 47: e147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bukau B, Weissman J, Horwich A. Molecular chaperones and protein quality control. Cell. 2006; 125: 443–51. [DOI] [PubMed] [Google Scholar]
- 4. Zhao Y, Macgurn JA, Liu M, et al The ART‐Rsp5 ubiquitin ligase network comprises a plasma membrane quality control system that protects yeast cells from proteotoxic stress. Elife. 2013; 2: e00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. David CR. The roles of intracellular protein‐degradation pathways in neurodegeneration. Nature. 2006; 443: 780–6. [DOI] [PubMed] [Google Scholar]
- 6. Lim J, Yue Z. Neuronal aggregates: formation, clearance, and spreading. Dev Cell. 2015; 32: 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng C, Geetha T, Babu JR. Failure of ubiquitin proteasome system: risk for neurodegenerative diseases. Neurodegener Dis. 2014; 14: 161–75. [DOI] [PubMed] [Google Scholar]
- 8. Koulich E, Li X, DeMartino GN. Relative structural and functional roles of multiple deubiquitylating proteins associated with mammalian 26S proteasome. Mol Biol Cell. 2008; 19: 1072–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Köhler A, Bajorek M, Groll M, et al The substrate translocation channel of the proteasome. Biochimie. 2001; 83: 325–32. [DOI] [PubMed] [Google Scholar]
- 10. Alexandra E, Luis S, Manuela LP. The amyloid stretch hypothesis: recruiting proteins toward the dark side. PANAS. 2005; 102: 16672–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stadtmueller BM, Hill CP. Proteasome activators. Mol Cell. 2011; 41: 8–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Upadhya SC, Hegde AN. Role of the ubiquitin proteasome system in Alzheimer's disease. BMC Biochem. 2007; 8 (Suppl. 1): S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hong L, Huang HC, Jiang ZF. Relationship between amyloid‐beta and the ubiquitin‐proteasome system in Alzheimer's disease. Neurol Res. 2014; 36: 276–82. [DOI] [PubMed] [Google Scholar]
- 14. Ristic G, Tsou WL, Todi SV. An optimal ubiquitin‐proteasome pathway in the nervous system: the role of deubiquitinating enzymes. Front Mol Neurosci. 2014; 7: 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dantuma NP, Bott LC. The ubiquitin‐proteasome system in neurodegenerative diseases: precipitating factor, yet part of the solution. Front Mol Neurosci. 2014; 7: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nalepa G, Rolfe M, Harper JW. Drug discovery in the ubiquitin‐proteasome system. Nat Rev Drug Discov. 2006; 5: 596–613. [DOI] [PubMed] [Google Scholar]
- 17. Tai H‐C, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008; 9: 826–38. [DOI] [PubMed] [Google Scholar]
- 18. Wang J, Maldonado MA. The ubiquitin‐proteasome system and its role in inflammatory and autoimmune diseases. Cell Mol Immunol. 2006; 3: 255–61. [PubMed] [Google Scholar]
- 19. Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin‐proteasome pathway in normal and disease states. J Am Soc Nephrol. 2006; 17: 1807–19. [DOI] [PubMed] [Google Scholar]
- 20. Xie Y. Structure, assembly and homeostatic regulation of the 26S proteasome. J Mol Cell Biol. 2010; 2: 308–17. [DOI] [PubMed] [Google Scholar]
- 21. Eldridge AG, O'Brien T. Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 2010; 17: 4–13. [DOI] [PubMed] [Google Scholar]
- 22. Morawe T, Hiebel C, Kern A, et al Protein homeostasis, aging and Alzheimer's disease. Mol Neurobiol. 2012; 46: 41–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Komander D, Clague MJ, Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol. 2009; 10: 550–63. [DOI] [PubMed] [Google Scholar]
- 24. Goldberg AL. Protein degradation and protection against misfolded or damaged proteins. Nature. 2003; 426: 895–9. [DOI] [PubMed] [Google Scholar]
- 25. Nguyen LK, Dobrzyński M, Fey D, et al Polyubiquitin chain assembly and organization determine the dynamics of protein activation and degradation. Front Physiol. 2014; 5: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lehman NL. The ubiquitin proteasome system in neuropathology. Acta Neuropathol. 2009; 118: 329–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu CW, Jacobson AD. Functions of the 19S complex in proteasomal degradation. Trends Biochem Sci. 2013; 38: 103–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Balakirev MY, Tcherniuk SO, Jaquinod M, et al Otubains: a new family of cysteine proteases in the ubiquitin pathway. EMBO Rep. 2003; 4: 517–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. da Fonseca PC, Morris EP. Structure of the human 26S proteasome: subunit radial displacements open the gate into the proteolytic core. J Biol Chem. 2008; 283: 23305–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Keller JN, Hanni KB, Markesbery WR. Impaired proteasome function in Alzheimer's disease. J Neurochem. 2000; 75: 436–9. [DOI] [PubMed] [Google Scholar]
- 31. Wojcik C, DeMartino GN. Intracellular localization of proteasomes. Int J Biochem Cell Biol. 2003; 35: 579–89. [DOI] [PubMed] [Google Scholar]
- 32. Liu C‐W, Li X, Thompson D, et al ATP binding and ATP hydrolysis play distinct roles in the function of 26S proteasome. Mol Cell. 2006; 24: 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mishto M, Bellavista E, Santoro A, et al Immunoproteasome and LMP2 polymorphism in aged and Alzheimer's disease brains. Neurobiol Aging. 2006; 27: 54–66. [DOI] [PubMed] [Google Scholar]
- 34. Orre M, Kamphuis W, Dooves S, et al Reactive glia show increased immunoproteasome activity in Alzheimer's disease. Brain. 2013; 136: 1415–31. [DOI] [PubMed] [Google Scholar]
- 35. Tanaka K. The proteasome: overview of structure and functions. Proc Jpn Acad Ser B Phys Biol Sci. 2009; 85: 12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Massaly N, Frances B, Mouledous L. Roles of the ubiquitin proteasome system in the effects of drugs of abuse. Front Mol Neurosci. 2015; 7: 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function, and antigen presentation. Prog Mol Biol Transl Sci. 2012; 109: 75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bar‐Nun S, Glickman MH. Proteasomal AAA‐ATPases: structure and function. Biochim Biophys Acta. 2012; 1823: 67–82. [DOI] [PubMed] [Google Scholar]
- 39. Bedford L, Paine S, Sheppard PW, et al Assembly, structure, and function of the 26S proteasome. Trends Cell Biol. 2010; 20: 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Coux O, Tanaka K, Goldberg AL. Structure and functions of the 20S and 26S proteasomes. Annu Rev Biochem. 1996; 65: 801–47. [DOI] [PubMed] [Google Scholar]
- 41. Davies KJ. Degradation of oxidized proteins by the 20S proteasome. Biochimie. 2001; 83: 301–10. [DOI] [PubMed] [Google Scholar]
- 42. Jung T, Grune T. The proteasome and the degradation of oxidized proteins: part I—structure of proteasomes. Redox Biol. 2013; 1: 178–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hol EM, van Leeuwen FW, Fischer DF. The proteasome in Alzheimer's disease and Parkinson's disease: lessons from ubiquitin B + 1. Trends Mol Med. 2005; 11: 488–95. [DOI] [PubMed] [Google Scholar]
- 44. Voges D, Zwickl P, Baumeister W. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem. 1999; 68: 1015–68. [DOI] [PubMed] [Google Scholar]
- 45. Lee S‐H, Moon J‐H, Yoon SK, et al Stable incorporation of ATPase subunits into 19 S regulatory particle of human proteasome requires nucleotide binding and C‐terminal tails. J Biol Chem. 2012; 287: 9269–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ciechanover A. Intracellular protein degradation: from a vague idea thru the lysosome and the ubiquitin‐proteasome system and onto human diseases and drug targeting. Cell Death Differ. 2005; 12: 1178–90. [DOI] [PubMed] [Google Scholar]
- 47. Rabl J, Smith DM, Yu Y, et al Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Mol Cell. 2008; 30: 360–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Benaroudj N, Zwickl P, Seemüller E, et al ATP hydrolysis by the proteasome regulatory complex PAN serves multiple functions in protein degradation. Mol Cell. 2003; 11: 69–78. [DOI] [PubMed] [Google Scholar]
- 49. Lu Y, B‐h Lee, King RW, et al Substrate degradation by the proteasome: a single‐molecule kinetic analysis. Science. 2015; 348: 1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hegde AN, Upadhya SC. Role of ubiquitin‐proteasome‐mediated proteolysis in nervous system disease. Biochim Biophys Acta. 2011; 1809: 128–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Majd S, Power JH, Grantham HJ. Neuronal response in Alzheimer's and Parkinson's disease: the effect of toxic proteins on intracellular pathways. BMC Neurosci. 2015; 16: 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rahman M, Zetterberg H, Lendel C, et al Binding of human proteins to amyloid‐beta protofibrils. ACS Chem Biol. 2015; 10: 766–74. [DOI] [PubMed] [Google Scholar]
- 53. Ying Z, Wang H, Wang G. The ubiquitin proteasome system as a potential target for the treatment of neurodegenerative diseases. Curr Pharm Des. 2013; 19: 3305–14. [DOI] [PubMed] [Google Scholar]
- 54. Daniels RW, Miller BR, DiAntonio A. Increased vesicular glutamate transporter expression causes excitotoxic neurodegeneration. Neurobiol Dis. 2011; 41: 415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lau A, Tymianski M. Glutamate receptors, neurotoxicity and neurodegeneration. Pflugers Arch. 2010; 460: 525–42. [DOI] [PubMed] [Google Scholar]
- 56. Johnson DA, Johnson JA. Nrf2‐a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med. 2015; 88: 253–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cabezas‐Opazo FA, et al Mitochondrial dysfunction contributes to the pathogenesis of Alzheimer's disease. Oxid Med Cell Longev. 2015; 2015: 509654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Engel PA. Does metabolic failure at the synapse cause Alzheimer's disease? Med Hypotheses. 2014; 83: 802–8. [DOI] [PubMed] [Google Scholar]
- 59. Sensi S. Metal homeostasis in dementia. Free Radic Biol Med. 2014; 75 (Suppl. 1): S9. [DOI] [PubMed] [Google Scholar]
- 60. Hegde AN. The ubiquitin‐proteasome pathway and synaptic plasticity. Learn Mem. 2010; 17: 314–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Naujokat C, Hoffmann S. Role and function of the 26S proteasome in proliferation and apoptosis. Lab Invest. 2002; 82: 965–80. [DOI] [PubMed] [Google Scholar]
- 62. Qing H, Zhou W, Christensen MA, et al Degradation of BACE by the ubiquitin‐proteasome pathway. FASEB J. 2004; 18: 1571–3. [DOI] [PubMed] [Google Scholar]
- 63. Salomone S, Caraci F, Leggio GM, et al New pharmacological strategies for treatment of Alzheimer's disease: focus on disease modifying drugs. Br J Clin Pharmacol. 2012; 73: 504–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Aprahamian I, Stella F, Forlenza OV. New treatment strategies for Alzheimer's disease: is there a hope? Indian J Med Res. 2013; 138: 449–60. [PMC free article] [PubMed] [Google Scholar]
- 65. Gavrilova SI, Kolykhalov IV, Fedorova YB, et al Possibilities of preventive treatment of Alzheimer's disease: results of the 3‐year open prospective comparative study on the efficacy and safety of the course therapy with cerebrolysin and cavinton in elderly patients with the syndrome of mild cognitive impairment. Treatment of neurological and psychiatric diseases. 2010; 110: 62. [PubMed] [Google Scholar]
- 66. Fragoso YD, Dantas DC. Cerebrolysin for Alzheimer's disease. Cochrane Database of Systematic Reviews 2002, Issue 3. Art. No.: CD003801. [Google Scholar]
- 67. Paul S. Dysfunction of the ubiquitin‐proteasome system in multiple disease conditions: therapeutic approaches. BioEssays. 2008; 30: 1172–84. [DOI] [PubMed] [Google Scholar]
- 68. O'Leary JC, Li Q, Marinec P, et al Phenothiazine‐mediated rescue of cognition in tau transgenic mice requires neuroprotection and reduced soluble tau burden. Mol Neurodegener. 2010; 5: article 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rochet JC. Novel therapeutic strategies for the treatment of protein‐misfolding diseases. Expert Rev Mol Med. 2007; 9: 1–34. [DOI] [PubMed] [Google Scholar]
- 70. Ross C, Poirier M. Protein aggregation and neurodegenerative disease. Nature Med. 2004; 10: S10–7. [DOI] [PubMed] [Google Scholar]
- 71. Upadhya SC, Hegde AN. Ubiquitin‐proteasome pathway components as therapeutic targets for CNS maladies. Curr Pharm Des. 2005; 11: 3807–28. [DOI] [PubMed] [Google Scholar]
- 72. Qiutian J, Yulin D, Hong Q. Potential therapeutic strategies for Alzheimer's disease targeting or beyond β‐amyloid: insights from clinical trials. BioMed Research International. 2014; 2014: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Saraiva A, Cardoso I, Pereira MC, et al Controlling amyloid‐beta peptide (1‐42) oligomerization and toxicity by fluorinated nanoparticles. ChemBioChem. 2010; 11: 1905–13. [DOI] [PubMed] [Google Scholar]
- 74. Katherine DL, Kebreten FM, Dexter LL, et al Treatment with bexarotene, a compound that increases apolipoprotein‐E, provides no cognitive benefit in mutant APP/PS1 mice. Mol Neurodegener. 2013; 8: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Miners JS, Barua N, Kehoe PG, et al Aβ‐degrading enzymes: potential for treatment of Alzheimer disease. J Neuropathol Exp Neurol. 2011; 70: 944–59. [DOI] [PubMed] [Google Scholar]
- 76. Kumar A, Singh A, Ekavali. A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol Rep. 2015; 67: 195–203. [DOI] [PubMed] [Google Scholar]
- 77. Edward R, Magdalena T, Michael M, et al Cerebrolysin decreases amyloid‐b production by regulating amyloid protein precursor maturation in a transgenic model of Alzheimer's disease. J Neurosci Res. 2006; 83: 1252–61. [DOI] [PubMed] [Google Scholar]
- 78. Nygaard HB. Current and emerging therapies for Alzheimer's disease. Clin Ther. 2013; 35: 1480–9. [DOI] [PubMed] [Google Scholar]
- 79. Verma R, Peters NR, D'Onofrio M, et al Ubistatins inhibit proteasome‐dependent degradation by binding the ubiquitin chain. Science. 2004; 306: 117–20. [DOI] [PubMed] [Google Scholar]
- 80. Cai Z, Yan LJ. Rapamycin, autophagy, and Alzheimer's disease. J Biochem Pharmacol Res. 2013; 1: 84–90. [PMC free article] [PubMed] [Google Scholar]
- 81. Nixon RA, Mathews PM, Cataldo AM. The neuronal endosomal‐lysosomal system in Alzheimer's disease. J Alzheimers Dis. 2001; 3: 97–107. [DOI] [PubMed] [Google Scholar]
- 82. Butler D, Nixon RA, Bahr BA. Potential compensatory responses through autophagic/lysosomal pathways in neurodegenerative diseases. Autophagy. 2006; 2: 234–7. [DOI] [PubMed] [Google Scholar]
- 83. Yang D‐S, Stavrides P, Mohan PS, et al Reversal of autophagy dysfunction in the TgCRND8 mouse model of Alzheimer's disease ameliorates amyloid pathologies and memory deficits. Brain. 2011; 134: 258–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cecarini V, Bonfili L, Cuccioloni M, et al Crosstalk between the ubiquitin–proteasome system and autophagy in a human cellular model of Alzheimer's disease. Biochim Biophys Acta. 2012; 1822: 1741–51. [DOI] [PubMed] [Google Scholar]
- 85. Lane RF, Shineman DW, Steele JW, et al Beyond amyloid: the future of therapeutics for Alzheimer's disease. Adv Pharmacol. 2012; 64: 213–71. [DOI] [PubMed] [Google Scholar]
- 86. Boland B, Kumar A, Lee S, et al Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer's disease. J Neurosci. 2008; 28: 6926–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Eskelinen E‐L, Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochim Biophys Acta. 2009; 1793: 664–73. [DOI] [PubMed] [Google Scholar]
- 88. Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci. 2007; 120: 4081–91. [DOI] [PubMed] [Google Scholar]
- 89. Blair LJ, Sabbagh JJ, Dickey CA. Targeting Hsp90 and its co‐chaperones to treat Alzheimer's disease. Expert Opin Ther Tar. 2014; 18: 1219–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Ou J‐R, Tan M‐S, Xie A‐M, et al Heat shock protein 90 in Alzheimer's disease. Biomed Res Int. 2014; 2014: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Salminen A, Ojala J, Kaarniranta K, et al Hsp90 regulates tau pathology through co‐chaperone complexes in Alzheimer's disease. Prog Neurobiol. 2011; 93: 99–110. [DOI] [PubMed] [Google Scholar]
- 92. Takalo M, Haapasalo A, Natunen T, et al Targeting ubiquilin‐1 in Alzheimer's disease. Expert Opin Ther Tar. 2013; 17: 795–810. [DOI] [PubMed] [Google Scholar]
- 93. Wilhelmus MM, De Waal RM, Verbeek MM. Heat shock proteins and amateur chaperones in amyloid‐Beta accumulation and clearance in Alzheimer's disease. Mol Neurobiol. 2007; 35: 203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Jinwal UK, Koren J, O'Leary JC, et al Hsp70 ATPase modulators as therapeutics for Alzheimer's and other neurodegenerative diseases. Mol Cell Pharmacol. 2010; 2: 43–6. [PMC free article] [PubMed] [Google Scholar]
- 95. Gao X, Hu H. Quality control of the proteins associated with neurodegenerative diseases. Acta Biochim Biophys Sin (Shanghai). 2008; 40: 612–8. [DOI] [PubMed] [Google Scholar]
- 96. Bobkova NV, Garbuz DG, Nesterova I, et al Therapeutic effect of exogenous hsp70 in mouse models of Alzheimer's disease. J Alzheimers Dis. 2014; 38: 425–35. [DOI] [PubMed] [Google Scholar]
- 97. Maiti P, Manna J, Veleri S, et al Molecular chaperone dysfunction in neurodegenerative diseases and effects of curcumin. Biomed Res Int. 2014; 2014: 495091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Mishra S, Palanivelu K. The effect of curcumin (turmeric) on Alzheimer's disease: an overview. Ann Indian Acad Neurol. 2008; 11: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Elliott E, Laufer O, Ginzburg I. BAG‐1M is up‐regulated in hippocampus of Alzheimers disease patients and associates with tau and APP proteins. J Neurochem. 2009; 109: 1168–78. [DOI] [PubMed] [Google Scholar]
- 100. Elliott E, Tsvetkov P, Ginzburg I. BAG‐1 associates with Hsc70· Tau complex and regulates the proteasomal degradation of Tau protein. J Biol Chem. 2007; 282: 37276–84. [DOI] [PubMed] [Google Scholar]
- 101. Carman A, Kishinevsky S, Koren J 3rd, et al Chaperone‐dependent neurodegeneration: a molecular perspective on therapeutic intervention. J Alzheimers Dis Parkinsonism. 2013; 2013: (suppl10). pii:007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Jinwal UK, Trotter JH, Abisambra JF, et al The Hsp90 kinase co‐chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J Biol Chem. 2011; 286: 16976–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Alves RPS, Yang MJ, Batista MT, et al Alzheimer's disease: is a vaccine possible? Braz J Med Biol Res. 2014; 47: 438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Ubhi K, Rockenstein E, Doppler E, et al Neurofibrillary and neurodegenerative pathology in APP‐transgenic mice injected with AAV2‐mutant TAU: neuroprotective effects of Cerebrolysin. Acta Neuropathol. 2009; 117: 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chouraki V, Seshadri S. Genetics of Alzheimer's disease. Adv Genet. 2014; 87: 245–94. [DOI] [PubMed] [Google Scholar]
- 106. Schulte TW, Neckers LM. The benzoquinone ansamycin 17‐allylamino‐17‐demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemoth Pharm. 1998; 42: 273–9. [DOI] [PubMed] [Google Scholar]
- 107. Luo W, Dou F, Rodina A, et al Roles of heat‐shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. PNAS. 2007; 104: 9511–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Manavalan A, Mishra M, Feng L, et al Brain site‐specific proteome changes in aging‐related dementia. Exp Mol Med. 2013; 45: e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Zhang M, Deng Y, Luo Y, et al Control of BACE1 degradation and APP processing by ubiquitin carboxyl‐terminal hydrolase L1. J Neurochem. 2012; 120: 1129–38. [DOI] [PubMed] [Google Scholar]
- 110. Oddo S. The ubiquitin‐proteasome system in Alzheimer's disease. J Cell Mol Med. 2008; 12: 363–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Tai H‐C, Serrano‐Pozo A, Hashimoto T, et al The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin‐proteasome system. Am J Pathol. 2012; 181: 1426–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Avila J, Lucas JJ, Perez M, et al Role of tau protein in both physiological and pathological conditions. Physiol Rev. 2004; 84: 361–84. [DOI] [PubMed] [Google Scholar]
- 113. Dickey CA, Yue M, Lin W‐L, et al Deletion of the ubiquitin ligase CHIP leads to the accumulation, but not the aggregation, of both endogenous phospho‐and caspase‐3‐cleaved tau species. J Neurosci. 2006; 26: 6985–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Sahara N, Murayama M, Mizoroki T, et al In vivo evidence of CHIP up‐regulation attenuating tau aggregation. J Neurochem. 2005; 94: 1254–63. [DOI] [PubMed] [Google Scholar]
- 115. Casey C, Jeannette NS, Yari C, et al Acetylation: a new key to unlock tau's role in neurodegeneration. Alzheimers Res Ther. 2014; 6: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Galimberti D, Scarpini E. Disease‐modifying treatments for Alzheimer's disease. Ther Adv Neurol Disord. 2011; 4: 203–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Salon ML, Pasquini L, Moreno MB, et al Relationship between β‐amyloid degradation and the 26S proteasome in neural cells. Exp Neurol. 2003; 180: 131–43. [DOI] [PubMed] [Google Scholar]
- 118. Song S, Kim S‐Y, Hong Y‐M, et al Essential role of E2‐25K/Hip‐2 in mediating amyloid‐β neurotoxicity. Mol Cell. 2003; 12: 553–63. [DOI] [PubMed] [Google Scholar]
- 119. De Vrij FM, Sluijs JA, Gregori L, et al Mutant ubiquitin expressed in Alzheimer's disease causes neuronal death1. FASEB J. 2001; 15: 2680–8. [DOI] [PubMed] [Google Scholar]
- 120. Huang Q, Figueiredo‐Pereira ME. Ubiquitin/proteasome pathway impairment in neurodegeneration: therapeutic implications. Apoptosis. 2010; 15: 1292–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tan Z, Sun X, F‐y Hou, et al Mutant ubiquitin found in Alzheimer's disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 2007; 14: 1721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Carrard G, Bulteau AL, Petropoulos I, et al Impairment of proteasome structure and function in aging. Int J Biochem Cell Biol. 2002; 34: 1461–74. [DOI] [PubMed] [Google Scholar]
- 123. Oh S, Hong HS, Hwang E, et al Amyloid peptide attenuates the proteasome activity in neuronal cells. Mech Ageing Dev. 2005; 126: 1292–9. [DOI] [PubMed] [Google Scholar]
- 124. Mangialasche F, Solomon A, Winblad B, et al Alzheimer's disease: clinical trials and drug development. Lancet Neurol. 2010; 9: 702–16. [DOI] [PubMed] [Google Scholar]
- 125. Swaminathan A, Jicha GA. Nutrition and prevention of Alzheimer's dementia. Front Aging Neurosci. 2014; 6: 282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Regitz C, Dubling LM, Wenzel U. Amyloid‐beta (Aβ (1‐42))‐induced paralysis in Caenorhabditis elegans is inhibited by the polyphenol quercetin through activation of protein degradation pathways. Mol Nutr Food Res. 2014; 58: 1931–40. [DOI] [PubMed] [Google Scholar]
- 127. Abdel‐Salam OM. Stem cell therapy for Alzheimer's disease. CNS Neurol Disord Drug Targets. 2011; 10: 459–85. [DOI] [PubMed] [Google Scholar]
- 128. Schneekloth AR, Pucheault M, Tae HS, et al Targeted intracellular protein degradation induced by a small molecule: en route to chemical proteomics. Bioorg Med Chem Lett. 2008; 18: 5904–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Marambaud P, Zhao H, Davies P. Resveratrol promotes clearance of Alzheimer's disease amyloid‐β peptides. J Biol Chem. 2005; 280: 37377–82. [DOI] [PubMed] [Google Scholar]
- 130. Huang L, Ho P, Chen CH. Activation and inhibition of the proteasome by betulinic acid and its derivatives. FEBS Lett. 2007; 581: 4955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Stanhill A, Haynes CM, Zhang Y, et al An arsenite‐inducible 19S regulatory particle‐associated protein adapts proteasomes to proteotoxicity. Mol Cell. 2006; 23: 875–85. [DOI] [PubMed] [Google Scholar]
- 132. Gregori L, Hainfeld JF, Simon MN, et al Binding of amyloid beta protein to the 20 S proteasome. J Biol Chem. 1997; 272: 58–62. [DOI] [PubMed] [Google Scholar]
- 133. Tseng BP, Green KN, Chan JL, et al Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging. 2008; 29: 1607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Wang Y, Mandelkow E. Degradation of tau protein by autophagy and proteasomal pathways. Biochem Soc Trans. 2012; 40: 644–52. [DOI] [PubMed] [Google Scholar]
- 135. Dal Vechio FH, Cerqueira F, Augusto O, et al Peptides that activate the 20S proteasome by gate opening increased oxidized protein removal and reduced protein aggregation. Free Radic Biol Med. 2014; 67: 304–13. [DOI] [PubMed] [Google Scholar]
- 136. Lee BH, Lee MJ, Park S, et al Enhancement of proteasome activity by a small‐molecule inhibitor of USP14. Nature. 2010; 467: 179–84. [DOI] [PMC free article] [PubMed] [Google Scholar]