

Abstract
Nerve injury triggers the conversion of myelin and non‐myelin (Remak) Schwann cells to a cell phenotype specialized to promote repair. Distal to damage, these repair Schwann cells provide the necessary signals and spatial cues for the survival of injured neurons, axonal regeneration and target reinnervation. The conversion to repair Schwann cells involves de‐differentiation together with alternative differentiation, or activation, a combination that is typical of cell type conversions often referred to as (direct or lineage) reprogramming. Thus, injury‐induced Schwann cell reprogramming involves down‐regulation of myelin genes combined with activation of a set of repair‐supportive features, including up‐regulation of trophic factors, elevation of cytokines as part of the innate immune response, myelin clearance by activation of myelin autophagy in Schwann cells and macrophage recruitment, and the formation of regeneration tracks, Bungner's bands, for directing axons to their targets. This repair programme is controlled transcriptionally by mechanisms involving the transcription factor c‐Jun, which is rapidly up‐regulated in Schwann cells after injury. In the absence of c‐Jun, damage results in the formation of a dysfunctional repair cell, neuronal death and failure of functional recovery. c‐Jun, although not required for Schwann cell development, is therefore central to the reprogramming of myelin and non‐myelin (Remak) Schwann cells to repair cells after injury. In future, the signalling that specifies this cell requires further analysis so that pharmacological tools that boost and maintain the repair Schwann cell phenotype can be developed.
Introduction
The striking regeneration potential of the peripheral nervous system is clearly illustrated by comparing the outcome of a blunt injury of the spinal cord (contusion or crush) with a similar injury to the sciatic nerve in rodents. Crushing the spinal cord is followed by the formation of a fluid‐ or matrix‐filled lesion, axonal retraction, retention of myelin debris distal to the injury, and absence of any significant axonal regeneration (reviewed in Beattie et al. 1997; Vargas & Barres, 2007; Plemel et al. 2008). After sciatic nerve crush, on the other hand, axons grow readily back to their targets, redundant myelin is removed and new myelin formed around regenerated axons, with the result that nerve tissue that is broadly normal in structure and function is restored in a surprisingly short time, 3–4 weeks (reviewed in Glenn & Talbot, 2013; Scheib & Höke, 2013; Brosius Lutz & Barres, 2014).
What is the cellular basis for the fundamental difference between these two tissues, similar though they are with respect to key components: axons, myelin and glial cells? In particular, what happens in the injured peripheral nerve to make repair so straightforward in this type of experiment?
Ultimately the answer relates to a general realization emerging from studies on cell differentiation, namely that the differentiated state of mammalian cells is less stable than previously thought. It is now clear that many cell types can be induced to change their differentiation state by experimental manipulation. Most often this has involved enforced expression of transcription factors in the culture dish to convert one cell into another (reviewed in Eberhard & Tosh, 2008; Yamanaka & Blau, 2010; Sisakhtnezhad & Matin, 2012). But there is also increasing evidence for comparable events in vivo, where the cell type conversions typically take place as helpful, or adaptive, responses to injury. A classic example is the conversion of pigment epithelium of the eye into lens epithelium, which helps restore the lens after eye injury in some newts and frogs (reviewed in Tsonis et al. 2004). This adaptive cellular reprogramming, induced by injury and promoting repair, can also be seen in mammalian tissues, including skin, liver, ear (reviewed in Jessen et al. 2015 a) and, as it turns out, peripheral nerve.
Damage to a peripheral nerve, whether crush or cut, triggers extensive changes in the differentiation state both of the injured neurons and of the Schwann cells distal to the injury. The neurons change expression of hundreds of genes, including a large number of transcription factors. This response (often referred to as the cell body reaction or signalling to growth mode switch) allows the neuron to shift its function from cell–cell signalling to that of building a new axon (Blesch et al. 2012; reviewed in Fu & Gordon, 1997; Doron‐Mandel et al. 2015). Equally strikingly, the myelin and non‐myelin (Remak) Schwann cells distal to nerve injury undergo a large scale change in gene expression, probably involving some thousands of genes, and change function from maintenance of axonal ensheathment and myelin to that of supporting regeneration (Nagarajan et al. 2002; Bosse et al. 2006; Barrette et al. 2010; Arthur‐Farraj et al. 2012). Because these cells are specialized for repair and differ from other cells in the Schwann cell lineage, as described below, we refer to these cells as repair Schwann cells (or Bungner cells since they form guidance tracks for regenerating axons called Bungner's bands) (Arthur‐Farraj et al. 2012; reviewed in Jessen et al. 2015 b).
In sum, peripheral nerves owe their regenerative potential to the flexible differentiation state of PNS neurons and Schwann cells, and their ability to convert to cells devoted to repair after injury. In the CNS, this adaptive injury response is generally subdued in neurons (reviewed in Bradke et al. 2012; Doron‐Mandel et al. 2015), and oligodendrocytes, the myelin forming cells of the CNS, are not reprogrammed to repair cells and serve no helpful function for regeneration distal to injury (reviewed in Vargas & Barres, 2007).
This article will focus on the repair (Bungner) Schwann cell in nerves distal to crush or cut injury (Figs 1 and 2). We will describe the properties, generation and maintenance of this cell, and its recently discovered function in myelinophagy, the autophagic breakdown of redundant myelin in injured nerves.
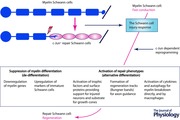
Figure 1. The repair (Bungner) Schwann cell in a developmental context .
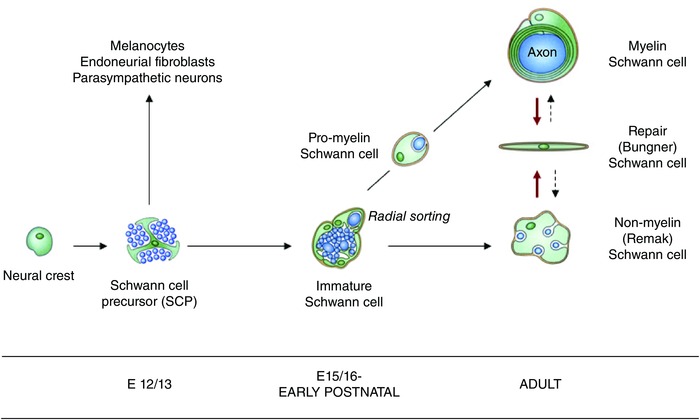
The diagram shows the repair (Bungner) Schwann cell, and the key stages of Schwann cell development, in addition to other developmental options for the Schwann cell precursor (Jessen & Mirsky, 2005). Arrows indicate developmental and injury‐related transitions. Black continuous arrows: normal development. Red arrows: the Schwann cell injury response. Dashed arrows: post‐repair re‐formation of myelin and Remak cells. Embryonic dates (E) refer to mouse development (from Jessen et al. 2015 b).
Figure 2. The structure of repair Schwann cells .
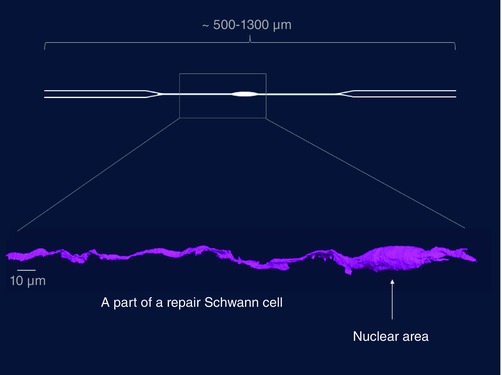
A repair Schwann cell in the distal stump (4‐week transected tibial nerve without re‐innervation), as shown by serial block face scanning electron microscopy. Only a part of the cell is shown, as indicated by the box superimposed on a schematic diagram of a repair cell (R. Mirsky, K. R. Jessen H. Armer and P. Munro, unpublished).
The Schwann cell injury response: de‐differentiation, activation or reprogramming
Although it is obvious that the axonal death distal to nerve crush (or cut) is accompanied by a radical phenotypic change in Schwann cells, there has long been disagreement in the literature about how this change is best understood. Some groups view the Schwann cell injury response as de‐differentiation (e.g. Chen et al. 2007; discussed in Jessen & Mirsky, 2008), while others see it as activation (Armstrong et al. 2007; Campana, 2007; Webber & Zochodne, 2010; Allodi et al. 2012). The dual nomenclature is confusing, because these terms appear to contradict each other by implying loss or gain of phenotypes respectively. But it is likely to have a simple explanation, which is that that the Schwann cell injury response in fact involves both of these processes. Thus after injury, myelin Schwann cells lose their characteristic gene expression pattern, which represents loss of differentiated features, de‐differentiation, but simultaneously they activate a set of repair‐related phenotypes, a repair programme, as detailed in the following section.
Previously we pointed out that the generation of repair Schwann cells could be compared to injury responses in other tissues, where cells also change phenotype to promote healing (adaptive reprogramming) (Jessen et al. 2015 a). Notably, these cell type conversions also involve the combination of de‐differentiation and activation. Thus, the injury‐induced conversion of pigment‐to‐lens epithelium in the newt involves the depigmentation and inhibition of melanogenesis (de‐differentiaton), but also the gain (activation) of crystallin synthesis to form the transparent lens (reviewed in Shen et al. 2004; Tsonis et al. 2004). Similarly in mammals, the conversion of α‐cells to β‐cells in the pancreatic islets following destruction of β‐cells involves loss of glucagon expression (de‐differentiation) and gain (activation) of insulin expression (Thorel et al. 2010; Chera et al. 2014). Such cell type conversions are generally referred to as transdifferentiation or direct (or lineage) reprogramming (Graf & Enver, 2009; Sisakhtnezhad & Matin, 2012). These concepts and terminology appear equally applicable to the Schwann cell injury response, reflecting a more accurate understanding than de‐differentiation or activation, words that refer only to a part of the underlying process (reviewed in Jessen et al. 2015 a).
Key events in Schwann cell reprogramming
Axons can be interrupted in two ways, by nerve cut or crush. After crush, the basal lamina tubes around individual axon/Schwann cell units are intact, and an axon remains within its native basal lamina sheath as it regenerates into the distal stump. After cut, the connective tissue and basal lamina sheaths are interrupted. If cut nerves are not repaired by re‐attaching the proximal and distal stumps, a tissue bridge forms between the two ends of the nerve, through which axons accompanied by Schwann cells, regeneration units, grow towards the distal stump guided by fibroblasts and blood vessels, meeting Schwann cell outgrowth from the cut end of the distal stump (Morris et al. 1972; Friede & Bischhausen, 1980; Meller, 1987; Barrette et al. 2008; Parrinello et al. 2010; Cattin et al. 2015). The Schwann cells of the regeneration units that project from the proximal stump are the daughter cells of the cells associated with cut axons just proximal to the injury. Although they have abandoned myelin differentiation, they may never have lost contact with axons, and the exact differentiation state of these cells is not clear. Nerve repair by re‐attaching the cut ends, a standard clinical treatment, leaves only a microscopic gap to be filled by a bridge and regeneration units.
Irrespective of whether axons are severed by crush or cut, the Schwann cell injury response in the nerve stump distal to the injury is similar. For simplicity this response will be discussed here in terms of myelin Schwann cells only; similar principles are likely to hold for non‐myelin (Remak) cells. The injury response can be viewed as having two principal components.
One of these is the reversal of myelin differentiation. Genes coding for the key myelin transcription factor Egr2 (Krox20), the enzymes of cholesterol synthesis, structural proteins such as P0, myelin basic protein (MBP), and membrane associated proteins like myelin associated glycoprotein (MAG) and periaxin are all rapidly down‐regulated (reviewed in Chen et al. 2007; Jessen & Mirsky, 2008). Conversely molecules that characterize pre‐myelinating Schwann cells in developing nerves (immature Schwann cells), including L1, neural cell adhesion molecule (NCAM), p75 neurotrophin receptor (p75NTR) and glial fibrillary acidic protein (GFAP), are up‐regulated (Chen et al. 2007; Jessen & Mirsky, 2008).
The second and vital constituent of the injury response involves the novel appearance of a set of phenotypes which are not active in Schwann cells in normal mature nerves or in Schwann cells in developing nerves. This repair programme includes a number of components. First, the up‐regulation of neurotrophic factors and surface proteins that promote axonal elongation and the survival of injured neurons, including glial cell line‐derived neurotrophic factor (GDNF), artemin, brain‐derived neurotrophic factor (BDNF), neurotrophin‐3 (NT3), nerve growth factor (NGF), vascular endothelial growth factor (VEGF), erythropoietin, pleiotrophin, p75NTR and N‐cadherin (Fontana et al. 2012; Brushart et al. 2013; reviewed in Boyd & Gordon, 2003; Chen et al. 2007; Scheib & Höke, 2013; Wood & Mackinnon, 2015). Second, it involves the activation of an innate immune response, including the upregulation of cytokines including tumour necrosis factor α (TNFα), interleukin‐1α (Il‐1α), Il‐1β, leukaemia inhibitory factor (LIF) and monocyte chemotactic protein 1 (MCP‐1) by the Schwann cells in the distal stump (reviewed in Martini et al. 2008; Rotshenker, 2011). This allows repair Schwann cells to interact with immune cells and, in particular, recruit macrophages to the nerve. This immune response likely promotes nerve regeneration in numerous ways. Cytokines such as Il‐6 and LIF not only attract macrophages to the injured nerve but can also act on neurons to promote axonal regeneration (Hirota et al. 1996; Cafferty et al. 2001; reviewed in Bauer et al. 2007). In addition, macrophages that invade nerves and ganglia provide an additional sustained source of cytokines, promote vascularisation of the distal nerve (Barrette et al. 2008; Niemi et al. 2013; Cattin et al. 2015), and co‐operate with Schwann cells to degrade myelin debris that potentially inhibits axon growth during the second phase of myelin clearance (see further below) (reviewed in Hirata & Kawabuchi, 2002; Rotshenker, 2011). Third, injury prompts the formation of regeneration tracks to help guide the growth of axons. The repair Schwann cells adopt an elongated bipolar morphology (Fig. 2) and align in columns (bands of Bungner) inside the basal lamina tubes that previously enclosed either myelin or Remak cells and their associated axons prior to injury (Stoll & Müller, 1999). This structure provides essential substrate and guidance cues to enable regenerating axons to reconnect with their target tissues. The fourth component of the repair programme is the activation of autophagy for myelin breakdown, which will be discussed in the following section.
Together the activation of the de‐differentiation and repair programmes recasts Schwann cells of intact nerves as cells that are equipped in a number of ways to promote regeneration, namely as repair (Bungner) Schwann cells. These cells ensheath axons and transform back to myelin and Remak cells in regenerated nerves. The repair Schwann cell is therefore a transient cell state that meets the particular demands that arise in injured tissue.
Schwann cells clear myelin by activation of myelinophagy
Surprisingly, during the first 5–7 days after injury Schwann cells themselves take a major part in breaking down their own redundant myelin sheaths. It has been estimated that about 50% of the myelin is degraded during this first phase of myelin clearance (Perry et al. 1995; see also Niemi et al. 2013). The second phase of myelin clearance is dominated by macrophages, which gradually accumulate in damaged nerves and phagocytose myelin debris (Ramaglia et al. 2008; Vargas et al. 2010; reviewed in Hirata & Kawabuchi, 2002; Dubový et al. 2013). During injury‐induced Schwann cell reprogramming, therefore, myelin cells not only switch off myelin maintenance, but also switch on an intracellular pathway for myelin destruction. Myelin breakdown by Schwann cells is often described as phagocytosis (e.g. Stoll & Müller, 1999; Hirata & Kawabuchi, 2002), although earlier authors were more cautious (Holtzman & Novikoff, 1965). But this notion is problematic because phagocytosis is primarily a mechanism for ingestion of extracellular material, while myelin is from the start a Schwann cell (membrane) component, and there is no evidence that myelin separates from Schwann cells to a significant extent during the first, Schwann cell‐dependent, phase of myelin clearance.
This problem has been revisited in recent work. This shows that the first step in myelin breakdown is the activation of an actin‐dependent process for dividing the myelin sheath into discrete oval‐shaped intracellular segments, which gradually break up into smaller myelin remains (Jung et al. 2011). Our own work indicates that these myelin fragments are delivered to lysosomes for digestion by a form of selective, mechanistic target of rapamycin (mTOR)‐independent autophagy, myelinophagy, which is distinct from the classical, mTOR‐dependent starvation autophagy mechanisms (Gomez‐Sanchez et al. 2015). Autophagy is strongly activated in injured Schwann cells, and myelin debris can be directly seen in double membrane autophagosomes, the distinctive intracellular ferries that deliver cargo to lysosomes during the autophagy process. Pharmacological or genetic autophagy inhibition also inhibits myelin breakdown (Fig. 3). Autophagy of myelin has also been seen in Schwann cells during axonal degeneration in the dental pulp (Suzuki et al. 2015).
Figure 3. Outline of myelinophagy .
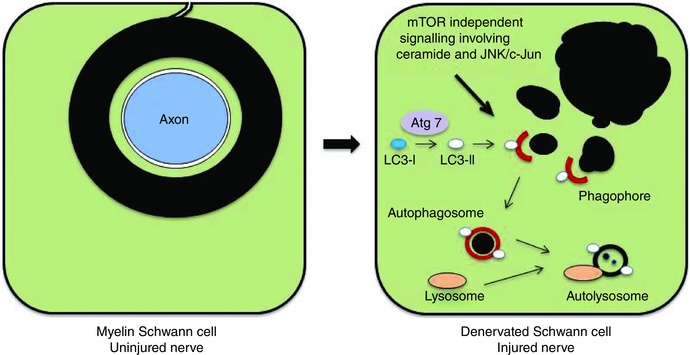
Left diagram: a transverse section through a myelin Schwann cell in an uninjured nerve. The myelin sheath is in direct continuity with the Schwann cell membrane and a component of the Schwann cell. Right diagram: a myelin Schwann cell after nerve injury and axonal degeneration. The myelin sheath has broken up into myelin ovoid and smaller fragments lying in the Schwann cell cytoplasm. The proposed role of autophagy in digesting these fragments is illustrated (from Gomez‐Sanchez et al. 2015).
The identification of autophagy as the key process by which Schwann cells do away with their myelin sheaths as they convert to repair cells makes biological sense, because autophagy is the typical mechanism by which cells digest their own components, organelles and large macromolecular complexes. Schwann cell phagocytosis of extracellular myelin debris may well play a part during the second macrophage‐dominated phase of myelin clearance.
It would appear to be potentially hazardous for the Schwann cell to possess a pathway that can effectively destroy its own myelin sheath. It remains to be seen whether this self‐eating of myelin is liable to be inappropriately activated, and whether this plays a part in any of the large number of myelin disorders. The possibility that this may be the case is raised by our observation that in a mouse model of CMT1A, the most common hereditary demyelinating neuropathy in man, uninjured nerves show evidence of enhanced autophagy activation (Gomez‐Sanchez et al. 2015). Classical, mTOR‐dependent starvation autophagy can also be activated in Schwann cells where it can serve the beneficial function of degrading protein aggregates that form in some myelin mutants (Rangaraju et al. 2010).
It is intriguing that in the CNS the transected optic nerve, where myelin is not degraded after injury, shows little evidence of the activation of autophagy (Gomez‐Sanchez et al. 2015).
A comparison of repair Schwann cells and immature Schwann cells
Myelin and Remak Schwann cells develop from immature Schwann cells in perinatal nerves (reviewed in Jessen & Mirsky, 2005). The de‐differentiation model of the Schwann cell injury response therefore predicts the generation of a cell in injured nerves that is similar to the immature Schwann cell found in developing nerves prior to myelination. Comparison of these two cells reveals, however, substantial differences.
First, they show a distinct molecular expression profile. This is exemplified by GDNF, oligodendrocyte transcription factor 1 (Olig 1), sonic hedgehog (Shh) and artemin, all of which are controlled by c‐Jun and highly up‐regulated in Schwann cells of injured nerves but absent/low in immature cells (Arthur‐Farraj et al. 2012; Fontana et al. 2012). These genes therefore serve as markers that distinguish repair Schwann cells in adult nerves from immature Schwann cells in perinatal nerves. One of these, GDNF, is detected in Schwann cell precursors of early embryonic nerves but is down‐regulated before birth (Piirsoo et al. 2010). Other genes including Olig1, Shh and artemin are absent/low both in immature cells and in earlier stages of the Schwann cell lineage (Lu et al. 2000; Zhou et al. 2000; Lin et al. 2015). Because these genes therefore appear to be expressed de novo after injury, they serve as distinctive markers of repair Schwann cells.
Further extensive differences in gene expression between immature and repair Schwann cells are indicated in a study comparing developing and regenerating nerves (Bosse et al. 2006). This work identified over a hundred genes that were regulated distal to nerve crush, although these genes were not developmentally regulated, being expressed at similar levels in newborn and adult uninjured nerves. Most of these genes were up‐regulated after injury, suggesting that induction of a large number of injury‐specific genes is one of the features that differentiate repair Schwann cells from cells in developing nerves prior to myelination.
Second, denervated cells, unlike immature cells, are engaged in a local, innate immune response, involving expression of a number of cytokines, and the attraction and activation of macrophages (reviewed in Martini et al. 2008; Gaudet et al. 2011; Rotshenker, 2011).
Third, an important function of denervated cells, but not immature cells, is to organize the clearance of myelin, indirectly by macrophage recruitment and directly by myelin autophagy (myelinophagy) (reviewed in Hirata & Kawabuchi, 2002; Gomez‐Sanchez et al. 2015; Suzuki et al. 2015).
Fourth, another key function of denervated cells is to guide growing axons to their target areas, which they accomplish by forming the Bungner regeneration tracks (Stoll & Müller, 1999). Perhaps surprisingly, a comparable guidance function is not shared by developing Schwann cells, because in embryos, peripheral axons can find their way to their target fields essentially normally in the absence of glial cells (Schwann cell precursors and immature Schwann cells) (Grim et al. 1992; Riethmacher et al. 1997).
Lastly, transcriptional controls differ in these two cell types, since c‐Jun is essential for the specification of the denervated cell, but this transcription factor appears to be dispensable for the generation of other cells in the Schwann cell lineage including functional immature Schwann cells (Arthur‐Farraj et al. 2012).
This comparison reveals two distinct Schwann cell phenotypes with separate functions and transcriptional controls. The essential role of immature Schwann cells is to act as a pool from which myelin and non‐myelin (Remak) cells develop, while Schwann cells in damaged nerves carry out specific functions related to regeneration and wound repair. This is consistent with the idea that the Schwann cell injury response represents a reprogramming process, which converts myelin and Remak Schwann cells to a Schwann cell specialized for repair (reviewed in Jessen et al. 2015 a,b).
The generation of the repair Schwann cell: the role of c‐Jun
The transcription factor c‐Jun is low or absent in Schwann cell precursors, up‐regulated in immature Schwann cells but suppressed during postnatal development, although it remains detectable in many non‐myelin (Remak) cells, and to a lesser extent in myelin cells, in adult nerves (Parkinson et al. 2004; 2008; Arthur‐Farraj et al. 2012; Hantke et al. 2014; Klein et al. 2014). It has long been known that c‐Jun is rapidly induced to high levels in the Schwann cells of injured nerves (De Felipe & Hunt, 1994; Shy et al. 1996). The functional significance of this was demonstrated with the generation of mice in which c‐Jun is selectively inactivated in Schwann cells (c‐Jun cKO mice). Uninjured nerves are essentially normal in these mutants, indicating that c‐Jun is not essential for Schwann cell development. Axonal regeneration and functional recovery after injury are, however, strikingly compromised or absent. The regeneration failure in c‐Jun cKO mice is due to the central function of c‐Jun in Schwann cell reprogramming, since this factor controls both components of the Schwann cell injury response, de‐differentiation of myelin cells and activation of the repair programme (Arthur‐Farraj et al. 2012).
c‐Jun promotes de‐differentiation, being required for the normal suppression of myelin genes after injury, including the Pzero and myelin basic protein genes and the gene encoding the pro‐myelin transcription factor Egr2 (Krox20). This suppressive function of c‐Jun and its cross‐antagonistic relationship with Egr2 (Krox20) was uncovered before its role in regeneration and helped give rise to the notion that c‐Jun, together with a number of other transcriptional regulators, such as Notch, Sox2, Pax3 and Id2, acted as a negative regulator of myelination (Parkinson et al. 2004; 2008; reviewed in Jessen & Mirsky, 2008). While many of these genes may be important for regulating the rate or onset of myelination during development, the key in vivo role for c‐Jun‐mediated suppression of myelin genes appears to be that of helping to supress myelin gene expression after injury.
c‐Jun is also essential for the normal activation of the repair programme, as seen from the following observations (Arthur‐Farraj et al. 2012; Fontana et al. 2012). First, in c‐Jun cKO mice the Schwann cells distal to injury fail to normally up‐regulate important trophic factors and cell surface proteins that support survival and axon growth, including GDNF, artemin and BDNF, p75NTR and N‐cadherin. Of these, GDNF and artemin have been shown to be direct targets of c‐Jun. Substantial numbers of dorsal root ganglion (DRG) sensory neurons and facial motoneurons die after sciatic and facial nerve injury, respectively, in c‐Jun cKO mice, revealing a key function for repair Schwann cells, and c‐Jun signalling, in support of neuronal survival. Second, because c‐Jun promotes myelinophagy, c‐Jun cKO nerves show long term delay in myelin clearance. Third, the regeneration tracks (Bungner bands) that denervated Schwann cells attempt to form without c‐Jun are structurally disorganized. In culture, c‐Jun in necessary for what has become known as the ‘typical’ narrow, bi/tripolar Schwann cell morphology, with c‐Jun‐negative cells tending to be flattened and sheet‐forming. Similarly in vivo, c‐Jun appears to be required for the conversion of the more complex and flattened structure of the myelin Schwann cell to the narrow and rod‐like morphology of repair cells which is required for the formation of normal regeneration columns.
Evidence is emerging that epigenetic mechanisms such as histone methylation state and miRNA also take part in the activation of the repair programme, since demethylation of H3K27 and down‐regulation of key miRNAs have been implicated in the activation of important injury factors including Shh, insulin‐like growth factor binding protein 2 (Igfbp2), Olig1 and GDNF (Lin et al. 2015; Ma et al. 2015).
The innate Schwann cell immune response to injury is to some extent regulated by c‐Jun because in cut nerves of c‐Jun cKO mice macrophage invasion is reduced at the injury site, and degenerating nerves in these mice contain large numbers of bloated macrophages. Many cytokines are, however, normally up‐regulated in the mutants and macrophage numbers are not significantly altered in crushed nerves or in cut nerves away from the location of the injury (Arthur‐Farraj et al. 2012). This suggests the participation of other pathways in driving the immune response. The extracellular signal‐regulated protein kinases 1 and 2 (ERK1/2)–mitogen‐activated protein kinase (MAPK) signalling pathway is activated in injured nerves and has been implicated in the control of immune functions in Schwann cells, in particular the activation of MCP‐1 expression, a key factor in attracting monocytes/macrophages to damaged nerves, and additional aspects of Schwann cell de‐differentation (Sheu et al. 2000; Harrisingh et al. 2004; Fischer et al. 2008; Groh et al. 2010; Shin et al. 2013). The other main MAPK signalling pathways, p38 and JNK, are also activated by nerve injury, and all three MAPK pathways have been implicated in promoting de‐differentiation of myelin cells (Myers et al. 2003; Parkinson et al. 2004; Yang et al. 2012). Demyelination after injury is also accelerated by Notch signalling, which is activated in Schwann cells in distal stumps. Inappropriate activation of Notch or Raf/ERK in Schwann cells of uninjured nerves is sufficient to cause demyelination, even in uninjured nerves (Woodhoo et al. 2009; Napoli et al. 2012).
c‐Jun and nerve disease
As predicted from animal studies, in human nerves c‐Jun is also expressed at low levels in normal Schwann cells, but up‐regulated in a number of different neuropathic conditions that do not involve nerve cut or crush (Hutton et al. 2011). In line with this, c‐Jun is elevated in uninjured nerves of a mouse model of the most common human genetic demyelinating neuropathy, CMT1A (Hantke et al. 2014). If this is prevented by genetically inactivating c‐Jun, the CMT1A mice exhibit distal sensory axonopathy and deterioration in sensory–motor performance. In a mouse model of another human genetic demyelinating disease, CMT1X, c‐Jun levels are also increased in Schwann cells of uninjured nerves and at least one c‐Jun target, GDNF, is also elevated (Klein et al. 2014). In both of these mouse models, the c‐Jun elevation is seen in the nuclei of Schwann cells that retain myelin differentiation. Further, in a mouse mutant involving inactivation of the liver kinase B1 (LKB1) in Schwann cells and showing axonal damage without overt demyelination, c‐Jun protein is elevated as well as c‐Jun‐associated injury molecules including GDNF and Shh (Beirowski et al. 2014).
Taken together these observations raise two points. First, they indicate that significant c‐Jun elevation is compatible with myelin differentiation and does not cause demyelination. This is supported by our observations that a mouse engineered to show 5‐ to 8‐fold overexpression of c‐Jun protein in Schwann cells, nevertheless has relatively normal myelin sheaths. Although significant, these c‐Jun levels are about an order of magnitude lower than those seen after nerve cut (J. Gomez‐Sanchez, R. Mirsky and K. R. Jessen, unpublished). Second, they suggest that even in uninjured nerves, Schwann cells respond to adverse conditions, in these cases caused genetically, by relatively modest c‐Jun activation, which is low enough to be compatible with the myelin differentiation but high enough to activate repair‐related genes, representing a graded neuroprotective Schwann cell response to nerve distress that does not involve the overt generation of the repair phenotype associated with nerve injury.
Maintenance of the repair phenotype
In line with the wide‐ranging role of c‐Jun in the specification of repair Schwann cells, reduced activation of Schwann cell c‐Jun in damaged nerves has recently been implicated in the age‐dependent reduction in regeneration that is typical of older animals (Painter et al. 2014). Another case of reduced c‐Jun expression is seen in Schwann cells in chronically denervated distal nerve stumps. In distal stumps of cut mouse nerves (without regeneration), c‐Jun levels are significantly lower at 10 weeks after injury than they are after 1 or 4 weeks (L. Wagstaff, K. R. Jessen and R. Mirsky, unpublished). This observation suggests that the repair phenotype is not stable, but fades with time. This is likely to be relevant in regenerating nerves of humans and other larger animals where the more distal Schwann cells are without axonal contact for many months while axons slowly elongate through the nerve. It is well established that during this time the axon‐free distal nerve stump gradually loses its repair‐supportive capacity, and that this decline is one of the key reasons for regeneration failure in humans (Höke, 2006; Sulaiman & Gordon, 2009). Therefore it will be relevant to determine whether reduced growth support is caused by reduced c‐Jun expression, and whether the Bungner repair phenotype can be maintained for long periods of time, or re‐activated, by promoting c‐Jun signalling. Analysis of the cell‐extrinsic and cell‐intrinsic pathways that maintain the repair phenotype, and the identification of pharmacological tools that promote this cell state (Heinen et al. 2015), is clearly an important future research direction.
Additional information
Competing interests
None declared.
Author contributions
Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
The work from the authors' own laboratory and quoted in this article was funded by Wellcome Trust Programme grants (091119 and 074665), an MRC project grant (G0600967) and grant agreement No. HEALTH‐F2‐2008‐201535 from the European Community's Seventh Framework Program (FP7/2007‐2013).
Biographies
Kristjan R. Jessen obtained MSc and PhD degrees in Neuroscience at UCL, London. He has been a Professor of Developmental Neurobiology in the Department of Cell and Developmental Biology, UCL, London since 1993.
Rhona Mirsky has a PhD in Radiochemistry from Cambridge University. She has been a Professor of Developmental Neurobiology in the same Department since 1990. They run a joint laboratory focusing on (i) early Schwann cell development and the biology of the Schwann cell precursor (SCP), (ii) the molecular control of myelination, and (iii) the response of Schwann cells to injury and genetic disease, including the processes of demyelination, axonal regeneration and nerve repair. They are both Fellows of the Academy of Medical Sciences.

This review was presented at the symposium “Axon regeneration and remyelination in the peripheral and central nervous systems”, which took place at Physiology 2015, Cardiff, UK between 6–8 July 2015.
References
- Allodi I, Udina E & Navarro X (2012). Specificity of peripheral nerve regeneration: interactions at the axon level. Prog Neurobiol 98, 16–37. [DOI] [PubMed] [Google Scholar]
- Armstrong SJ, Wiberg M, Terenghi G & Kingham PJ (2007). ECM molecules mediate both Schwann cell proliferation and activation to enhance neurite outgrowth. Tissue Eng 13, 2863–2870. [DOI] [PubMed] [Google Scholar]
- Arthur‐Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M, Wicher GK, Mitter R, Greensmith L, Behrens A, Raivich G, Mirsky R & Jessen KR (2012). c‐Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75, 633–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrette B, Calvo E, Vallières N & Lacroix S (2010). Transcriptional profiling of the injured sciatic nerve of mice carrying the Wld(S) mutant gene: identification of genes involved in neuroprotection, neuroinflammation, and nerve regeneration. Brain Behav Immun 24, 1254–1267. [DOI] [PubMed] [Google Scholar]
- Barrette B, Hébert MA, Filali M, Lafortune K, Vallières N, Gowing G, Julien JP & Lacroix S (2008). Requirement of myeloid cells for axon regeneration. J Neurosci 28, 9363–9376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer S, Kerr BJ & Patterson PH (2007). The neuropoietic cytokine family in development, plasticity, disease and injury. Nat Rev Neurosci 8, 221–232. [DOI] [PubMed] [Google Scholar]
- Beattie MS, Bresnahan JC, Komon J, Tovar CA, Van Meter M, Anderson DK, Faden AI, Hsu CY, Noble LJ, Salzman S & Young W (1997). Endogenous repair after spinal cord contusion injuries in the rat. Exp Neurol 148, 453–463. [DOI] [PubMed] [Google Scholar]
- Beirowski B, Babetto E, Golden JP, Chen YJ, Yang K, Gross RW, Patti GJ & Milbrandt J (2014). Metabolic regulator LKB1 is crucial for Schwann cell‐mediated axon maintenance. Nat Neurosci 17, 1351–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesch A, Lu P, Tsukada S, Alto LT, Roet K, Coppola G, Geschwind D & Tuszynski MH (2012). Conditioning lesions before or after spinal cord injury recruit broad genetic mechanisms that sustain axonal regeneration: superiority to camp‐mediated effects. Exp Neurol 235, 162–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosse F, Hasenpusch‐Theil K, Küry P & Müller HW (2006). Gene expression profiling reveals that peripheral nerve regeneration is a consequence of both novel injury‐dependent and reactivated developmental processes. J Neurochem 96, 1441–1457. [DOI] [PubMed] [Google Scholar]
- Boyd JG & Gordon T (2003). Neurotrophic factors and their receptors in axonal regeneration and functional recovery after peripheral nerve injury. Mol Neurobiol 27, 277–324. [DOI] [PubMed] [Google Scholar]
- Bradke F, Fawcett JW & Spira ME (2012). Assembly of a new growth cone after axotomy: the precursor to axon regeneration. Nat Rev Neurosci 13, 183–193. [DOI] [PubMed] [Google Scholar]
- Brosius Lutz A, Barres BA (2014). Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev Cell 28, 7–17. [DOI] [PubMed] [Google Scholar]
- Brushart TM, Aspalter M, Griffin JW, Redett R, Hameed H, Zhou C, Wright M, Vyas A & Höke A (2013). Schwann cell phenotype is regulated by axon modality and central‐peripheral location, and persists in vitro. Exp Neurol 247, 272–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Gavazzi I, Powell J, McMahon SB, Heath JK, Munson J, Cohen J & Thompson SW (2001). Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci 21, 7161–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana WM (2007). Schwann cells: Activated peripheral glia and their role in neuropathic pain. Brain Behav Immun 21, 522–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattin AL, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJ, Garcia Calavia N, Guo Y, McLaughlin M, Rosenberg LH, Quereda V, Jamecna D, Napoli I, Parrinello S, Enver T, Ruhrberg C & Lloyd AC (2015). Macrophage‐induced blood vessels guide schwann cell‐mediated regeneration of peripheral nerves. Cell 162, 1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZL, Yu WM & Strickland S (2007). Peripheral regeneration. Annu Rev Neurosci 30, 209–233. [DOI] [PubMed] [Google Scholar]
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F & Herrera PL (2014). Diabetes recovery by age‐dependent conversion of pancreatic δ‐cells into insulin producers. Nature 514, 503–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe C & Hunt SP (1994). The differential control of c‐jun expression in regenerating sensory neurons and their associated glial cells. J Neurosci 14, 2911–2923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron‐Mandel E, Fainzilber M & Terenzio M (2015). Growth control mechanisms in neuronal regeneration. FEBS Lett 589, 1669–1677. [DOI] [PubMed] [Google Scholar]
- Dubový P, Jančálek R & Kubek T (2013). Role of inflammation and cytokines in peripheral nerve regeneration. Int Rev Neurobiol 108, 173–206. [DOI] [PubMed] [Google Scholar]
- Eberhard D & Tosh D (2008). Transdifferentiation and metaplasia as a paradigm for understanding development and disease. Cell Mol Life Sci 65, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer S, Weishaupt A, Troppmair J & Martini R (2008). Increase of MCP‐1 (CCL2) in myelin mutant Schwann cells is mediated by MEK‐ERK signaling pathway. Glia 56, 836–843. [DOI] [PubMed] [Google Scholar]
- Fontana X, Hristova M, Da Costa C, Patodia S, Thei L, Makwana M, Spencer‐Dene B, Latouche M, Mirsky R, Jessen KR, Klein R, Raivich G & Behrens A (2012). c‐Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J Cell Biol 198, 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friede RL & Bischhausen R (1980). The fine structure of stumps of transected nerve fibers in subserial sections. J Neurol Sci 44, 181–203. [DOI] [PubMed] [Google Scholar]
- Fu SY & Gordon T (1997). The cellular and molecular basis of peripheral nerve regeneration. Mol Neurobiol 14, 67–116. [DOI] [PubMed] [Google Scholar]
- Gaudet AD, Popovich PG & Ramer MS (2011). Wallerian degeneration: gaining perspective on inflammatory events after peripheral nerve injury. J Neuroinflammation 8, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenn TD & Talbot WS (2013). Signals regulating myelination in peripheral nerves and the Schwann cell response to injury. Curr Opin Neurobiol 23, 1041–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Sanchez JA, Carty L, Iruarrizaga‐Lejarreta M, Palomo‐Irigoyen M, Varela‐Rey M, Griffith M, Hantke J, Macias‐Camara N, Azkargorta M, Aurrekoetxea I, De Juan VG, Jefferies HB, Aspichueta P, Elortza F, Aransay AM, Martínez‐Chantar ML, Baas F, Mato JM, Mirsky R, Woodhoo A & Jessen KR (2015). Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 210, 153–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf T & Enver T (2009). Forcing cells to change lineages. Nature 426, 587–594. [DOI] [PubMed] [Google Scholar]
- Grim M, Halata Z & Franz T (1992). Schwann cells are not required for guidance of motor nerves in the hindlimb in Splotch mutant mouse embryos. Anat Embryol (Berl) 186, 311–318. [DOI] [PubMed] [Google Scholar]
- Groh J, Heinl K, Kohl B, Wessig C, Greeske J, Fischer S & Martini R (2010). Attenuation of MCP‐1/CCL2 expression ameliorates neuropathy in a mouse model for Charcot‐Marie‐Tooth 1X. Hum Mol Genet 19, 3530–3543. [DOI] [PubMed] [Google Scholar]
- Hantke J, Carty L, Wagstaff LJ, Turmaine M, Wilton DK, Quintes S, Koltzenburg M, Baas F, Mirsky R & Jessen KR (2014). c‐Jun activation in Schwann cells protects against loss of sensory axons in inherited neuropathy. Brain 137, 2922–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrisingh MC, Perez‐Nadales E, Parkinson DB, Malcolm DS, Mudge AW & Lloyd AC (2004). The Ras/Raf/ERK signalling pathway drives Schwann cell dedifferentiation. EMBO J 23, 3061–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen A, Beyer F, Tzekova N, Hartung HP & Küry P (2015). Fingolimod induces the transition to a nerve regeneration promoting Schwann cell phenotype. Exp Neurol 271, 25–35. [DOI] [PubMed] [Google Scholar]
- Hirata K & Kawabuchi M (2002). Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc Res Tech 57, 541–547. [DOI] [PubMed] [Google Scholar]
- Hirota H, Kiyama H, Kishimoto T & Taga T (1996). Accelerated nerve regeneration in mice by upregulated expression of interleukin (IL) 6 and IL‐6 receptor after trauma. J Exp Med 183, 2627–2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höke A (2006). Neuroprotection in the peripheral nervous system: rationale for more effective therapies. Arch Neurol 63, 1681–1685. [DOI] [PubMed] [Google Scholar]
- Holtzman E & Novikoff AB (1965). Lysozomes in the rat sciatic nerve following crush. J Cell Biol 27, 651–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton EJ, Carty L, Laurá M, Houlden H, Lunn MP, Brandner S, Mirsky R, Jessen K & Reilly MM (2011). c‐Jun expression in human neuropathies: a pilot study. J Peripher Nerv Syst. 16, 295–303. [DOI] [PubMed] [Google Scholar]
- Jessen KR & Mirsky R (2005). The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci 6, 671–682. [DOI] [PubMed] [Google Scholar]
- Jessen KR & Mirsky R (2008). Negative regulation of myelination: relevance for development, injury, and demyelinating disease. Glia 56, 1552–1565. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R & Arthur‐Farraj P (2015. a). The role of cell plasticity in tissue repair: adaptive cellular reprogramming. Developmental Cell 34, 613–620. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R & Lloyd AC (2015. b). Schwann cells: development and role in nerve repair. Cold Spring Harb Perspect Biol 7, a020487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Cai W, Lee HK, Pellegatta M, Shin YK, Jang SY, Suh DJ, Wrabetz L, Feltri ML & Park HT (2011). Actin polymerization is essential for myelin sheath fragmentation during Wallerian degeneration. J Neurosci 31, 2009–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein D, Groh J, Wettmarshausen J & Martini R (2014). Nonuniform molecular features of myelinating Schwann cells in models for CMT1: distinct disease patterns are associated with NCAM and c‐Jun upregulation. Glia 62, 736–750. [DOI] [PubMed] [Google Scholar]
- Lin HP, Oksuz I, Hurley E, Wrabetz L & Awatramani R (2015). Microprocessor complex subunit DiGeorge syndrome critical region gene 8 (Dgcr8) is required for Schwann cell myelination and myelin maintenance. J Biol Chem 290, 24294–24307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu QR, Yuk D, Alberta JA, Zhu Z, Pawlitzky I, Chan J, McMahon AP, Stiles CD & Rowitch DH (2000). Sonic hedgehog‐regulated oligodendrocyte lineage genes encoding bHLH proteins in the mammalian central nervous system. Neuron 25, 317–332. [DOI] [PubMed] [Google Scholar]
- Ma KH, Hung HA, Srinivasan R, Xie H, Orkin SH & Svaren J (2015). Regulation of peripheral nerve myelin maintenance by gene repression through polycomb repressive complex 2. J Neurosci 35, 8640–8652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini R, Fischer S, López‐Vales R & David S (2008). Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 56, 1566–1577. [DOI] [PubMed] [Google Scholar]
- Meller K (1987). Early structural changes in the axoplasmic cytoskeleton after axotomy studied by cryofixation. Cell Tissue Res 250, 663–672. [DOI] [PubMed] [Google Scholar]
- Morris JH, Hudson AR & Weddell G (1972). A study of degeneration and regeneration in the divided rat sciatic nerve based on electron microscopy. II. The development of the “regenerating unit”. Z Zellforsch Mikrosk Anat 124, 103–130. [PubMed] [Google Scholar]
- Myers RR, Sekiguchi Y, Kikuchi S, Scott B, Medicherla S, Protter A & Campana WM (2003). Inhibition of p38 MAP kinase activity enhances axonal regeneration. Exp Neurol 184, 606–614. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Le N, Mahoney H, Araki T & Milbrandt J (2002). Deciphering peripheral nerve myelination by using Schwann cell expression profiling. Proc Natl Acad Sci USA 99, 8998–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh MC, White IJ, Woodhoo A & Lloyd AC (2012). A central role for the ERK‐signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73, 729–742. [DOI] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco‐Lisowitz A, Roldán‐Hernández L, Lindborg JA, Mandell D & Zigmond RE (2013). A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci 33, 16236–16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MW, Brosius Lutz A, Cheng YC, Latremoliere A, Duong K, Miller CM, Posada S, Cobos EJ, Zhang AX, Wagers AJ, Havton LA, Barres B, Omura T & Woolf CJ (2014). Diminished Schwann cell repair responses underlie age‐associated impaired axonal regeneration. Neuron 83, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur‐Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R & Jessen KR (2008). c‐Jun is a negative regulator of myelination. J Cell Biol 181, 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Droggiti A, Dickinson S, D'Antonio M, Mirsky R & Jessen KR (2004). Krox‐20 inhibits Jun‐NH2‐terminal kinase/c‐Jun to control Schwann cell proliferation and death. J Cell Biol 164, 385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrinello S, Napoli I, Ribeiro S, Wingfield Digby P, Fedorova M, Parkinson DB, Doddrell RD, Nakayama M, Adams RH & Lloyd AC (2010). EphB signaling directs peripheral nerve regeneration through Sox2‐dependent Schwann cell sorting. Cell 143, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, Tsao JW, Fearn S & Brown MC (1995). Radiation‐induced reductions in macrophage recruitment have only slight effects on myelin degeneration in sectioned peripheral nerves of mice. Eur J Neurosci 7, 271–280. [DOI] [PubMed] [Google Scholar]
- Piirsoo M, Kaljas A, Tamm K & Timmusk T (2010). Expression of NGF and GDNF family members and their receptors during peripheral nerve development and differentiation of Schwann cells in vitro. Neurosci Lett 469, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plemel JR, Duncan G, Chen K‐W, Shannon C, Park S, Sparling JS & Tetzlaff W (2008). A graded forceps crush spinal cord injury model in mice. J Neurotrauma 25, 350–370. [DOI] [PubMed] [Google Scholar]
- Ramaglia V, Wolterman R, de Kok M, Vigar MA, Wagenaar‐Bos I, King RH, Morgan BP & Baas F (2008). Soluble complement receptor 1 protects the peripheral nerve from early axon loss after injury. Am J Pathol 172, 1043–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Verrier JD, Madorsky I, Nicks J, Dunn WA Jr & Notterpek L (2010). Rapamycin activates autophagy and improves myelination in explant cultures from neuropathic mice. J Neurosci 30, 11388–11397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riethmacher D, Sonnenberg‐Riethmacher E, Brinkmann V, Yamaai T, Lewin GR & Birchmeier C (1997). Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature 389, 725–730. [DOI] [PubMed] [Google Scholar]
- Rotshenker S (2011). Wallerian degeneration: the innate‐immune response to traumatic nerve injury. J Neuroinflammation 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheib J & Höke A (2013). Advances in peripheral nerve regeneration. Nat Rev Neurol 9, 668–676. [DOI] [PubMed] [Google Scholar]
- Shen C‐N, Burke ZD & Tosh D (2004). Transdifferentiation, metaplasia and tissue regeneration. Organogenesis 1, 36–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu JY, Kulhanek DJ & Eckenstein FP (2000). Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp Neurol 166, 392–402. [DOI] [PubMed] [Google Scholar]
- Shin YK, Jang SY, Park JY, Park SY, Lee HJ, Suh DJ & Park HT (2013). The Neuregulin‐Rac‐MKK7 pathway regulates antagonistic c‐jun/Krox20 expression in Schwann cell dedifferentiation. Glia 61, 892–904. [DOI] [PubMed] [Google Scholar]
- Shy ME, Shi Y, Wrabetz L, Kamholz J & Scherer SS (1996). Axon‐Schwann cell interactions regulate the expression of c‐jun in Schwann cells. J Neurosci Res 43, 511–525. [DOI] [PubMed] [Google Scholar]
- Sisakhtnezhad S & Matin MM (2012). Transdifferentiation: a cell and molecular reprogramming process. Cell Tissue Res 348, 379–396. [DOI] [PubMed] [Google Scholar]
- Stoll G & Müller HW (1999). Nerve injury, axonal degeneration and neural regeneration: basic insights. Brain Pathol 9, 313–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulaiman OA & Gordon T (2009). Role of chronic Schwann cell denervation in poor functional recovery after nerve injuries and experimental strategies to combat it. Neurosurgery 65(4 Suppl), A105–A114. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Lovera M, Schmachtenberg O & Couve E (2015). Axonal degeneration in dental pulp precedes human primary teeth exfoliation. J Dent Res 94, 1446–1453. [DOI] [PubMed] [Google Scholar]
- Thorel F, Népote V, Avril I, Kohno K, Desgraz R, Chera S & Herrera PL (2010). Conversion of adult pancreatic α‐cells to β‐cells after extreme β‐cell loss. Nature 464, 1149–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsonis PA, Madhavan M, Tancous EE & Del‐Roi‐Tsonis K (2004). A newt's eye view of lens regeneration. Int J Dev Biol 48, 975–980. [DOI] [PubMed] [Google Scholar]
- Vargas ME & Barres BA (2007). Why is Wallerian degeneration in the CNS so slow? Annu Rev Neurosci 30, 153–179. [DOI] [PubMed] [Google Scholar]
- Vargas ME, Watanabe J, Singh SJ, Robinson WH & Barres BA (2010). Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proc Natl Acad Sci USA 107, 11993–11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber C & Zochodne D (2010). The nerve regenerative microenvironment: early behavior and partnership of axons and Schwann cells. Exp Neurol 223, 51–59. [DOI] [PubMed] [Google Scholar]
- Wood MD & Mackinnon SE (2015). Pathways regulating modality‐specific axonal regeneration in peripheral nerve. Exp Neurol 265, 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhoo A, Alonso MB, Droggiti A, Turmaine M, D'Antonio M, Parkinson DB, Wilton DK, Al‐Shawi R, Simons P, Shen J, Guillemot F, Radtke F, Meijer D, Feltri ML, Wrabetz L, Mirsky R & Jessen KR (2009). Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat Neurosci 12, 839–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S & Blau HM (2010). Nuclear reprogramming to a pluripotent state by three approaches. Nature 465, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang DP, Kim J, Syed N, Tung YJ, Bhaskaran A, Mindos T, Mirsky R, Jessen KR, Maurel P, Parkinson DB & Kim HA (2012). p38 MAPK activation promotes denervated Schwann cell phenotype and functions as a negative regulator of Schwann cell differentiation and myelination. J Neurosci 32, 7158–7168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Wang S & Anderson DJ (2000). Identification of a novel family of oligodendrocyte lineage‐specific basic helix‐loop‐helix transcription factors. Neuron 25, 331–343. [DOI] [PubMed] [Google Scholar]