

Abstract
Key points
In this study, we describe a new knock‐in (KI) mouse model that allows the study of the H191‐dependent regulation of T‐type Cav3.2 channels. Sensitivity to zinc, nickel and ascorbate of native Cav3.2 channels is significantly impeded in the dorsal root ganglion (DRG) neurons of this KI mouse. Importantly, we describe that this H191‐dependent regulation has discrete but significant effects on the excitability properties of D‐hair (down‐hair) cells, a sub‐population of DRG neurons in which Cav3.2 currents prominently regulate excitability.
Overall, this study reveals that the native H191‐dependent regulation of Cav3.2 channels plays a role in the excitability of Cav3.2‐expressing neurons. This animal model will be valuable in addressing the potential in vivo roles of the trace metal and redox modulation of Cav3.2 T‐type channels in a wide range of physiological and pathological conditions.
Abstract
Cav3.2 channels are T‐type voltage‐gated calcium channels that play important roles in controlling neuronal excitability, particularly in dorsal root ganglion (DRG) neurons where they are involved in touch and pain signalling. Cav3.2 channels are modulated by low concentrations of metal ions (nickel, zinc) and redox agents, which involves the histidine 191 (H191) in the channel's extracellular IS3–IS4 loop. It is hypothesized that this metal/redox modulation would contribute to the tuning of the excitability properties of DRG neurons. However, the precise role of this H191‐dependent modulation of Cav3.2 channel remains unresolved. Towards this goal, we have generated a knock‐in (KI) mouse carrying the mutation H191Q in the Cav3.2 protein. Electrophysiological studies were performed on a subpopulation of DRG neurons, the D‐hair cells, which express large Cav3.2 currents. We describe an impaired sensitivity to zinc, nickel and ascorbate of the T‐type current in D‐hair neurons from KI mice. Analysis of the action potential and low‐threshold calcium spike (LTCS) properties revealed that, contrary to that observed in WT D‐hair neurons, a low concentration of zinc and nickel is unable to modulate (1) the rheobase threshold current, (2) the afterdepolarization amplitude, (3) the threshold potential necessary to trigger an LTCS or (4) the LTCS amplitude in D‐hair neurons from KI mice. Together, our data demonstrate that this H191‐dependent metal/redox regulation of Cav3.2 channels can tune neuronal excitability. This study validates the use of this Cav3.2‐H191Q mouse model for further investigations of the physiological roles thought to rely on this Cav3.2 modulation.
Key points
In this study, we describe a new knock‐in (KI) mouse model that allows the study of the H191‐dependent regulation of T‐type Cav3.2 channels. Sensitivity to zinc, nickel and ascorbate of native Cav3.2 channels is significantly impeded in the dorsal root ganglion (DRG) neurons of this KI mouse. Importantly, we describe that this H191‐dependent regulation has discrete but significant effects on the excitability properties of D‐hair (down‐hair) cells, a sub‐population of DRG neurons in which Cav3.2 currents prominently regulate excitability.
Overall, this study reveals that the native H191‐dependent regulation of Cav3.2 channels plays a role in the excitability of Cav3.2‐expressing neurons. This animal model will be valuable in addressing the potential in vivo roles of the trace metal and redox modulation of Cav3.2 T‐type channels in a wide range of physiological and pathological conditions.
Abbreviations
- ADP
afterdepolarization potential
- AP
action potential
- Cav
voltage‐gated calcium channel
- D‐hair
down hair
- DRG
dorsal root ganglion
- KI
knock‐in
- LTCS
low threshold calcium spike
- LTMR
low‐threshold mechanoreceptor
- LVA
low‐voltage activated
- NT4
neurotrophin 4
- TPEN
N,N,N′,N′‐Tetrakis(2‐pyridylmethyl)ethylenediamine
Introduction
Low‐voltage activated (LVA) T‐type calcium channels are critically involved in neuronal excitability, especially in burst firing, in low‐threshold calcium spikes (LTCS) and rebound action potential (AP), as reviewed by Cain & Snutch (2010). They contribute to network rhythms in normal situations, exemplified by their role in thalamocortical circuits during sleep‐related oscillations, as well as in diseases associated with hyperexcitability disorders, such as in epilepsy and pain (Zamponi et al. 2010; Bourinet et al. 2014). Three genes, CACNA1G, CACNA1H and CACNA1I, encode the T‐type calcium channel isoforms, Cav3.1, Cav3.2 and Cav3.3, respectively (reviewed by Perez‐Reyes, 2003). The corresponding Cav3 currents display most properties of the native T‐type channels, although each Cav3 isoform shows some unique electrophysiological and/or pharmacological properties. The Cav3.2 channels correspond to the original ‘nickel‐sensitive’ LVA/T‐type channels that were characterized first from the dorsal root ganglion (DRG) in the pioneering studies describing these channels (Carbone & Lux, 1984; Lee et al. 1999).
In DRGs, T‐type channels play a crucial function in tuning cell excitability. They serve as amplifiers in peripheral pain transmission in a subset of low‐threshold mechanoreceptor (LTMR) sensory neurons. In these neurons, Cav3.2 channels are present in the peripheral and central terminals, as well as within the axons (Talley et al. 1999; Rose et al. 2013; François et al. 2015; Reynders et al. 2015; Usoskin et al. 2015). Cav3.2 channels in DRG neurons are involved in the patterning of afterdepolarization potentials (ADPs) and repetitive burst firing (White et al. 1989). Activity of these T‐type/Cav3.2 channels is implicated in the firing threshold and/or neurotransmitter release in DRGs and ultimately in pain processing (Todorovic & Jevtovic‐Todorovic, 2013; Bourinet et al. 2014). In a consistent manner, knocking‐down Cav3.2 expression with antisense oligonucleotides (Bourinet et al. 2005), using a knock‐out mouse (Choi et al. 2007), using selective T‐type channel blockers (Todorovic et al. 2002; Francois et al. 2013) or promoting Cav3.2 ubiquitination (Garcia‐Caballero et al. 2014) have shown antinociceptive effects in acute and/or chronic pain models.
The molecular mechanisms regulating T‐type channel activity are still poorly understood. However, studies using recombinant T‐type channels have established that the Cav3.2 isoform is modulated by redox agents (Todorovic et al. 2001) and it is much more potently inhibited, in the micromolar range, by the divalent metals zinc, nickel and copper, than the Cav3.1 and Cav3.3 isoforms (Lee et al. 1999; Jeong et al. 2003; Traboulsie et al. 2006). The histidine 191 (H191) located in the outer IS3–IS4 loop of the Cav3.2 protein is involved in the metal binding site (Kang et al. 2006). This binding site also mediates the inhibition of the Cav3.2 channel by oxidizing agents and its sensitization by reducing agents, such as l‐cysteine (Nelson et al. 2007 a,b). When H191 is mutated into glutamine (H191Q), the channel's affinity for zinc and nickel is greatly reduced (Kang et al. 2006).
In vivo studies have revealed that many of these H191‐dependent modulators of Cav3.2 channels have marked effects on pain (Evans & Todorovic, 2015). For example, zinc was shown to have antinociceptive properties (Safieh‐Garabedian et al. 1996; Liu et al. 1999; Nozaki et al. 2011), while reducing agents l‐cysteine and dithiothreitol (DTT), as well as the zinc chelator N,N,N′,N′‐tetrakis(2‐pyridylmethyl)ethylenediamine (TPEN) display pronociceptive properties (Nelson et al. 2005, 2007 b; Kawabata et al. 2007; Matsunami et al. 2011). The possibility that these effects are mediated through Cav3.2 channel modulation has been explored using knock‐out animals (Choi et al. 2007; Nelson et al. 2007 b). It was tentatively hypothesized that these agents would exert their modulatory role through their ability to directly modulate Cav3.2 channel activity and, consequently the electrical activity of the neurons expressing these channels. However, such demonstration clearly requires the development and characterization of a site‐specific knock‐in animal to investigate the physiological relevance of this H191 regulatory site in Cav3.2 channels. Toward this goal, we have created a mouse carrying the H191Q mutation and provide here the first characterization of the electrophysiological properties of the down hair (D‐hair) neurons of the DRG from this knock‐in mouse.
Methods
Ethical approval
The authors certify that the present work complies with The Journal of Physiology's animal checklist (http://www.physoc.org/animal‐experiments). All animal use procedures were done in accordance with the directives of the French Ministry of Agriculture (A 34‐172‐41).
Generation of Cav3.2‐H191Q knock‐in mice
Mice carrying the H191Q mutation in Cav3.2 were generated by the ‘Genetic engineering and Mouse transgenesis’ (GEMTis) facility at CIPHE (http://www.celphedia.eu/fr/centers/ciphe). An 11‐kb genomic clone containing exon 4 of the cacna1h gene was isolated from C57BL6/N mouse genomic DNA and cloned upon pACN vector. This clone was engineered to introduce the histidine‐to‐glutamine mutation (CAC to CAG) at position 191, a floxed neo cassette and a thymidine kinase encoding cassette. The targeting vector was linearized and electroporated into C57BL6/N embryonic stem cells followed by neomycin selection. Selected colonies were screened for homologous recombination by Southern blotting with Drdl digests, using 5′ and 3′ external probes or neo probe. The floxed neo cassette contained a Cre recombinase under the control of a testicular promoter to achieve its auto‐removal. Indeed, when chimeric males had gametes carrying the mutated allele in their testis, the testicular promoter was activated to trigger the Cre‐mediated recombination and the deletion of the floxed sequence. ES clones with the correct genotype were micro‐injected into Balb/CN blastocysts, and the resulting chimeric males were bred with C57BL6/N females to obtain heterozygous F1 animals (cacna1h +/H191Q) and these F1 were intercrossed to obtain homozygous mutant mice (cacna1h H191Q/H191Q), named here knock‐in (KI) mice. The genotypes were determined by PCR analysis, with forward primer 5′‐GAGAGAGGCTCAGGTTTGACA‐3′ and reverse primer 5′TGAGGCCCTCACTGGACACT‐3′. The 85‐bp difference between KI and WT alleles resulted from the remaining loxP site in the KI allele.
Western blots
Western blots were performed as described previously (Blesneac et al. 2015). Briefly, mice were anaesthetized with sodium pentobarbital and decapitated. Whole brains were dissected, frozen in liquid nitrogen and ground with a hand‐held homogenizer. The samples were then placed in a lysis buffer on ice with NP‐40 buffer containing 50 mm Tris‐HCl, pH 7.5, 125 mm NaCl, 1% NP‐40, 0.1% SDS, 2 mm EDTA, 2 mm EGTA and protease inhibitors (Roche Applied Science, Penzberg, Germany). Lysates were spun at 1000 g for 10 min at 4°C. The supernatants were retained and spun again at 13,000 g for 40 min at 4°C. The same amount of protein (60 μg) was loaded in each lane on 4–6% SDS‐PAGE gels. Proteins were then transferred onto nitrocellulose membranes and blocked with 5% powdered non‐fat milk. Primary antibodies [anti‐Cav3.2 (1:100, Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), anti‐GAPDH (1:20,000, Sigma, St Louis, MO, USA)] were incubated overnight in PBS‐T (Tween 0.05%, milk 5%) at 4°C. After three washes in PBS‐T, secondary HRP‐coupled antibodies were incubated for 1 h in PBS‐T. The signal was detected using the Super Signal West Pico Chemiluminescent system (Pierce Chemical, Rockford, IL, USA). GAPDH was used as a loading control.
DRG neuron preparation
Detailed procedures to isolate DRG neurons were described elsewhere (Francois et al. 2013, 2015). Briefly, adult male mice were anaesthetized with pentobarbital injection and transcardially perfused with Hank's balanced salt solution (HBSS, pH 7.4, 4°C). DRGs were dissected and collected in cold HBSS with 1 m Hepes, 2.5 m d‐glucose and penicillin/streptomycin. DRGs were treated with a mix of 2 mg ml−1 collagenase II and 5 mg ml−1 dispase for 40 min at 37°C, washed in HBSS and resuspended in 1 ml of neurobasal A medium (Invitrogen, Carlsbad, CA, USA) supplemented with B27, 2 mm l‐glutamine and penicillin/streptomycin. Single‐cell suspensions were obtained by five passages through three needle tips of decreasing diameter (gauge 18, 21 and 26). Cells were plated onto polyornithine/laminin‐coated dishes. After 2 h, the medium was removed and replaced with neurobasal B27 supplemented with 10 ng ml−1 neurotrophin 4 (NT4) and 2 ng ml−1 glial derived neurotrophic factor (GDNF).
Patch clamp recordings
Patch clamp recordings were performed 6–24 h after plating, on neurons with a ‘rosette’ morphology that is promoted by the presence of NT4 in the culture media, as described by Dubreuil et al. (2004). For calcium current recordings, the extracellular solution contained (in mm): 130 choline chloride, 10 TEACl, 2 NaCl, 2 CaCl2, 1 MgCl2, 10 Hepes, 5 4‐aminopyridine, 10 glucose (pH to 7.3 with TEAOH). Pipettes with a resistance of 1–2 MΩ were filled with an internal solution containing (in mm): 130 CsCl, 10 Hepes, 10 EGTA, 1 MgCl2, 2 CaCl2, 4 Mg‐ATP, 0.3 Na‐GTP (pH to 7.3 with NaOH). Whole‐cell transmembrane potentials were recorded in current‐clamp mode at room temperature. The bath solution contained (in mm): 140 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2, 10 glucose, 10 Hepes (pH to 7.4 with NaOH). Pipettes with a resistance of 1–2 MΩ were filled with an internal solution containing (in mm): 130 KCl, 40 Hepes, 5 MgCl2, 2 Mg‐ATP, 0.1 Na‐GTP (pH to 7.2 with KOH). LTCS were evoked by 2 s hyperpolarizing current injections of increasing amplitude. The presence of the T‐type current was assessed after completion of current‐clamp experiments. The amplifier was switched back to voltage‐clamp mode and the external solution was replaced by the one used for the calcium current recordings (see above). All drugs (purchased from Sigma‐Aldrich), NiCl2, ZnCl2, ascorbate, l‐cysteine and TPEN, were applied using a gravity‐driven perfusion system.
Statistical analysis
Data are presented as means ± SEMs. Statistical analyses were performed using either Mann–Whitney's test for two‐group comparison, or two‐way ANOVA combined with Sidak's post hoc analysis for multiple comparisons, using GraphPad Prism software (GraphPad Software Inc., La Jolla, CA, USA). P values were considered statistically significant at P < 0.05 (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
Generation of the mutant mouse carrying the mutation H191Q on Cav3.2
We have generated a mouse line carrying a histidine‐to‐glutamine point mutation at position 191 of the Cav3.2 subunit (Cav3.2‐H191Q). As described in Fig. 1 A, a C‐to‐G mutation was introduced using homologous recombination into the cacna1h gene encoding the Cav3.2 protein of mouse (C57BL6) embryonic stem cells. The resulting Cav3.2‐H191Q knock‐in mouse line (hereafter KI mouse) was genotyped using PCR analysis with mutation‐specific primers to reveal WT (280 bp) and mutant (315 bp) alleles (Fig. 1 B). Homozygous KI mice are viable, fertile and indistinguishable from their control littermates. The amount of Cav3.2 protein, as determined by Western blot analysis, was unchanged in the whole brain of KI mice compared to WT mice (Fig. 1 C).
Figure 1. Generation of a knock‐in (KI) mouse line carrying the mutation H191Q on the T‐type Cav3.2 channel .
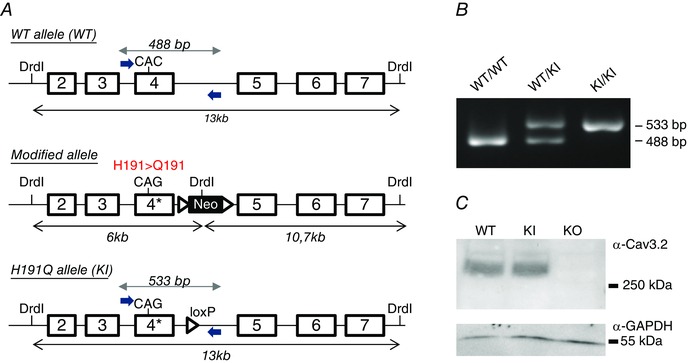
A, the Cav3.2 allele was created by homologous recombination (see Materials and Methods). The CAC codon encoding histidine 191 (H191) in exon 4 of the WT allele (upper panel) was replaced by the codon CAG encoding glutamine (Q), and a loxP‐flanked neo cassette was introduced 3′ from exon 4 to select for embryonic stem cells harbouring the knock‐in (KI) allele (middle panel). The final mutant allele was obtained after excision of the neo cassette by a Cre recombinase treatment of embryonic stem cells (lower panel). B, henotyping of the Cav3.2‐H191Q KI mice. PCR analysis using specific primers positioned in A revealed the WT allele (488 bp) in both WT and heterozygous animals as well as the mutant allele (533 bp) in both KI and heterozygous animals. The 45 bp difference between mutant and WT alleles results from the remaining loxP site in the mutant allele. C, representative Western‐blot analysis of the expression level of the Cav3.2 protein in the whole brain of WT and KI mice; a whole brain extract from knock‐out mice for Cav3.2 (KO) was used as a negative control in these experiments. The expression level of α‐GAPDH was used as loading control in these experiments (lower panel).
Altered sensitivity to zinc, nickel and ascorbate of the Cav3.2 current of KI mouse DRG neurons
To determine the electrophysiological properties and sensitivity of the Cav3.2 channel to its H191‐dependent modulators, i.e. zinc, nickel and ascorbate, we recorded whole cell T‐type currents from a population of medium‐sized dissociated DRG neurons, the D‐hair cells. D‐hair cells could be unambiguously identified from their typical ‘rosette‐like’ morphology in short‐term culture conditions (see inset to Fig. 2 A), allowing us to study a homogeneous neuron population among the various DRG neuron subtypes (Dubreuil et al. 2004; Francois et al. 2013). As shown in Fig. 2 A, no difference in calcium current density could be identified between the D‐hair neurons from WT and KI mice and this current was efficiently blocked (>90%) in both conditions in the presence of 10 μm TTA‐A2, a selective T‐type channel blocker (Francois et al. 2013). D‐hair cells are mechanoreceptor neurons that express Cav3.2 current of large amplitude with classical Cav3.2‐like properties, including half‐potential (V 0.5) for steady‐state activation in the range of −60 mV (Fig. 2 B, Table 1), V 0.5 for steady‐state inactivation in the range of −72 mV (Fig. 2 C, Table 1), and typical activation, inactivation and deactivation kinetics (Table 1, Fig. 2 D). Importantly, no difference could be identified regarding these T‐type current electrophysiological properties recorded between D‐hair neurons from WT and KI mice (Fig. 2, Table 1). Also, the properties of the high voltage activated current were found to be similar (see Fig. 2 B). Another hallmark property of the T‐type/Cav3.2 channel isoform is its inhibition by low concentration of nickel (Shin et al. 2003; Dubreuil et al. 2004) and zinc (Traboulsie et al. 2006). To perform a pharmacological analysis of the T‐type channels in WT and KI D‐hair neurons, T‐type currents were recorded in the presence of 2 mm external calcium concentration during a depolarizing test pulse at −30 mV (90 ms duration) from a holding potential at −80 mV (see Fig. 3 A). Figure 3 B shows the dose–response of zinc inhibition of Cav3.2 current in D‐hair neurons from WT mice and from KI mice. The IC50 for zinc inhibition of the WT channel was 1.7 ± 0.4 μm (n = 7), contrasting with the IC50 determined for the KI channel which was 10 ± 2 μm (n = 5). We also assessed the effect of other H191‐dependent modulators on neuronal Cav3.2 current from the KI mouse. Application of the oxidizing agent ascorbate at 300 μm blocked 27.9 ± 1.9% (n = 7) of WT current and only 8.1 ± 1.5% (n = 9) of KI current (Fig. 3 C). Also, the blocking effect of nickel ions was significantly reduced for the Cav3.2 current recorded on D‐hair neurons from KI mice. Nickel blocked 29.5 ± 2.7% of WT current at 1 μm and 79.8 ± 0.6% at 30 μm, whereas it blocked respectively 12.9 ± 1.8 and 28 ± 2.3% of KI current (n = 8) (Fig. 3 C). In good agreement with the absence of free zinc in our standard experimental conditions, neither the zinc chelator TPEN (10 μm) nor the reducing agent l‐cysteine (100 μm) could modulate the T‐type current density in both WT neurons (93.5 ± 1.5%, n = 7, and 91.9 ± 1.2%, n = 6, for TPEN and l‐cysteine, respectively) and KI neurons (95.4 ± 1.8 %, n = 8, and 96.0 ± 2.0%, n = 7, for TPEN and l‐cysteine, respectively; Fig. 3 D). Importantly, similar experiments performed in the presence of 1 μm zinc revealed the ability of both TPEN and l‐cysteine to unmask a basal inhibition of WT Cav3.2 channels by free extracellular zinc (121.5 ± 2.1%, n = 7, for TPEN and 122.4 ± 2,3%, n = 6, for l‐cysteine in WT neurons and 104 ± 1.4%, n = 7, for TPEN and 97.9 ± 2.4%, n = 6, for l‐cysteine in KI neurons; Fig. 3 E). Collectively, these data demonstrate that the Cav3.2 channels present in neurons from mice carrying the H191Q mutation exhibit a reduced sensitivity to the main representative pharmacological agents known to modulate Cav3.2 channels through the H191 residue.
Figure 2. Electrophysiological properties of T‐type currents in DRG neurons from WT and KI mice .
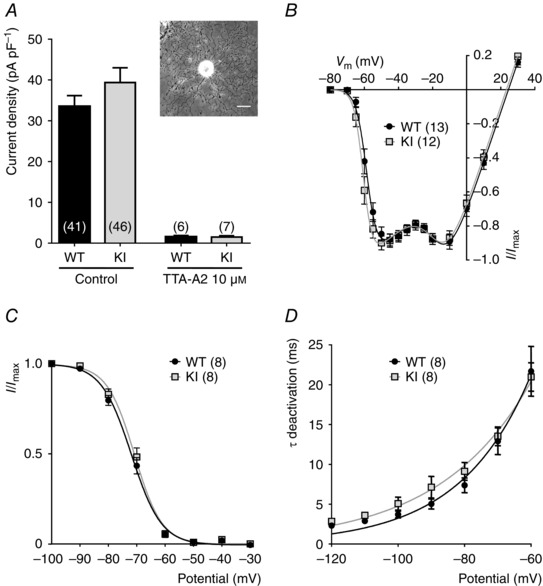
A, T‐type calcium current density in D‐hair DRG neurons from wild‐type (WT) and knock‐in (KI) mice, in control conditions (left) and in the presence of 10 μm of the T‐type channel inhibitor TTA‐A2 (right). For each cell, the current amplitude (in pA) was measured for a depolarization step from −80 to −30 mV and normalized for the cell capacitance (pF) in current density (pA pF–1). Values are expressed as mean ± SEM. Inset: bright field image of a typical D‐hair neuron with its ‘rosette’ morphology. Scale bar = 30 μm. B, normalized I–V relationship for the WT (grey square) and the KI (black circle) current evoked by 90 ms depolarizing step pulses from a holding potential at −80 mV. C, steady‐state inactivation curves obtained by stepping the membrane potential at −30 mV from conditioning depolarizing pulses ranging from −100 to −30 mV. D, deactivation kinetics as a function of the voltage.
Table 1.
Electrophysiological properties of T‐type currents in D‐hair neurons from WT and KI mice
| WT | KI | |
|---|---|---|
| Activation | ||
| V 0.5 (mV) | −58.1 ± 0.77 (13) | −60.27 ± 0.73 (12) |
| Slope (mV) | 2.68 ± 0.21 (13) | 2.3 ± 0.27 (12) |
| Inactivation | ||
| V 0.5 (mV) | −72 ± 1.02 (8) | −71.06 ± 1.05 (8) |
| Slope (mV) | 4.88 ± 0.25 (8) | 4.5 ± 0.3 (8) |
| Activation kinetics | ||
| τ @ −50 mV (ms) | 8.44 ± 0.43 (13) | 7.2 ± 0.56 (12) |
| Inactivation kinetics | ||
| τ @ −50 mV (ms) | 17.73 ± 0.62 (13) | 16.8 ± 0.53 (12) |
Data are presented as mean ± SEM with (n) the number of cells tested. Note that none of the values was statistically different between WT and KI conditions.
Figure 3. Metal/redox sensitivity of T‐type currents in DRG neurons from WT and KI mice .
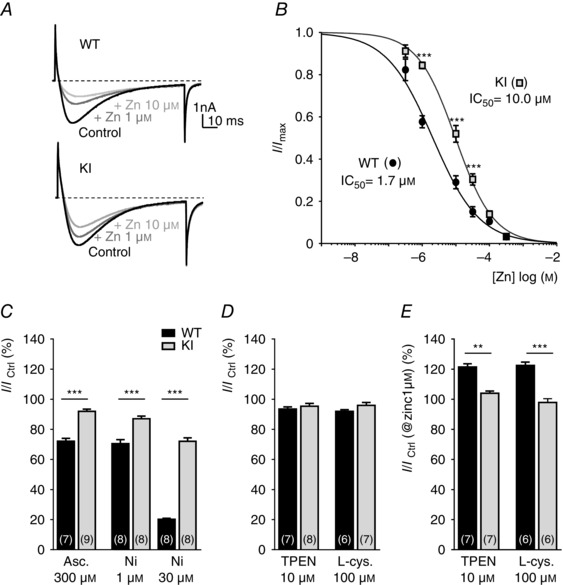
A, representative T‐type current traces recorded from D‐hair cells isolated from a WT mouse (upper trace) and a KI mouse (lower trace) evoked by 90 ms depolarization steps from −80 to −30 mV, before and after exposure to 1 and 10 μm zinc. B, dose–response curves obtained for zinc inhibition of T‐type currents in neurons from WT mice and KI mice. Current amplitude was normalized to the peak current in the absence of zinc, and the remaining current was plotted against zinc concentration. The IC50 values were obtained from fitted data using a sigmoidal dose–response with variable Hill slope equation. Values are mean ± SEM (***P < 0.001; two‐way ANOVA). C, the ability of ascorbate (300 μm) and nickel ions (1 and 30 μm) to inhibit T‐type currents in D‐hair neurons from WT (black bars) and KI (grey bars) mice is represented as the percentage of remaining current following drug application. D, effect of zinc chelator TPEN (10 μm) and l‐cysteine (100 μm) on T‐type current. Note that TPEN and l‐cysteine in control conditions fail to modulate either the WT or the KI current. WT TPEN vs. KI TPEN: P = 0.52; WT l‐cysteine vs. KI l‐cysteine: P = 0.0734; Mann–Whitney). E, effect of TPEN (10 μm) and l‐cysteine (100 μm) on the T‐type current from WT and KI mice in the presence of 1 μm zinc. Values are expressed as mean ± SEM (**P < 0.01; ***P < 0.001; Mann–Whitney).
Properties of the action potentials in DRG neurons from KI mice
To evaluate the role of H191‐dependent regulation of the Cav3.2 channels on neuronal excitability, we performed current‐clamp recordings on freshly cultured D‐hair neurons. Action potentials (APs) were evoked by 4 ms current injections of increasing intensity. Typical recordings of evoked APs are presented in Fig. 4 A for a WT neuron (upper panel) and for a KI neuron (lower panel), in control condition (left part) and in the presence of 30 μm nickel (right part). Under control conditions, the rheobase (threshold current) was similar in WT neurons (0.82 ± 0.06 nA, n = 15) and KI neurons (0.71 ± 0.06 nA, n = 13). After application of 30 μm nickel, the rheobase was significantly increased in WT neurons (0.9 ± 0.06 nA, n = 15), consistent with previously described findings (Dubreuil et al. 2004), whereas it remained unchanged in KI neurons (0.72 ± 0.05 nA, n = 13) (Fig. 4 B). Of interest, the AP in mouse D‐hair neurons is characterized by a spike followed by a pronounced ADP and this ADP is dependent on the activity of T‐type Cav3.2 channels (Figs 4 and 5; see also Dubreuil et al. 2004). Noticeably, the ADP amplitude was significantly larger in KI neurons as compared to WT neurons (Fig. 5 B). Application of zinc (Zn; Fig. 5 C) and nickel (Ni; Fig. 5 D) at low concentration (1 μm) was sufficient to induce a strong inhibition of the ADP amplitude on WT neurons (65 ± 35%, n = 6; 77 ± 25%, n = 7, respectively). Importantly, inhibition of the ADP amplitude in the presence of 1 μm zinc or nickel was lost on KI neurons. A higher concentration of zinc (30 μm) completely abolished the ADP in WT neurons and also significantly reduced ADP amplitude in KI neurons (Fig. 5 C). In contrast, 30 μm nickel abolishes the ADP in WT neurons but had no effect on KI neurons (Fig. 5 D). These data show that ADP properties can be modulated by H191‐dependent regulation of T‐type Cav3.2 channels.
Figure 4. Action potential (AP) properties in DRG neurons from WT and KI mice .
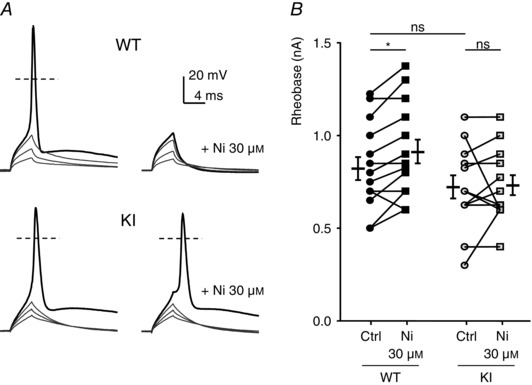
A, typical traces of evoked APs. Depolarization currents of increasing amplitude were injected to trigger an AP in the absence (left) or presence of 1 μm zinc (right) in D‐hair neurons from WT (upper panels) and KI (lower panels) mice. The resting potential of these neurons was maintained at −70 mV. B, threshold current for an AP generation (Rheobase) in control condition and with 30 μm nickel for neurons from WT mice (left) and KI mice (right). Statistical analysis was performed with repeated‐measures two‐way ANOVA (WT Ctrl vs. WT Ni 30 μm, *P < 0.05; WT Ctrl vs. KI Ctrl, P = 0.52; WT Ni 30 μm vs. KI Ni 30 μm, P = 0.08; KI Ctrl vs. KI Ni 30 μm, P = 0.95).
Figure 5. Afterdepolarization (ADP) properties in DRG neurons from WT and KI mice .
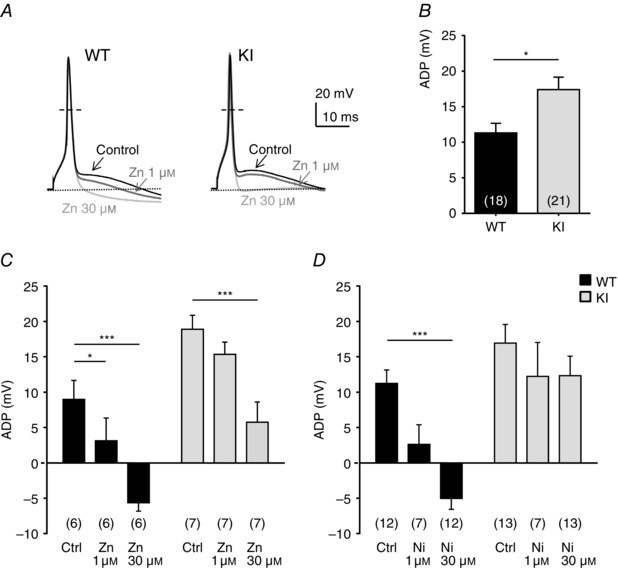
A, representative APs with ADP before and after application of zinc (1 and 30 μm) in D‐hair neurons of WT (left) and KI mice (right). The resting membrane potential of the neurons was maintained at −70 mV. B, ADP amplitude (in mV) in control condition is determined as the difference in voltage from the resting potential value (*P < 0.05; Mann‐–Whitney). C and D, effect of the application of zinc at 1 and 30 μm (C) and nickel at 30 μm (D) on ADP amplitude. Statistical analysis was performed with two‐way ANOVA followed by Sidak's test (*P < 0.05; ***P < 0.001; KI Ctrl vs. KI Zn 1 μm, P = 0.22; WT Ctrl vs. WT Ni 1 μm, P = 0.11; KI Ctrl vs. KI Ni 1 μm, P = 0.61; KI Ctrl vs. KI Ni 30 μm, P = 0.43).
Properties of the LTCS in DRG neurons of KI mice
To further examine the neuronal excitability in the D‐hair neurons from the KI mice, we investigated their ability to generate LTCS. It was previously shown that LTCS properties rely on T‐type channel properties (Dubreuil et al. 2004; Francois et al. 2013). Using 2 s hyperpolarizing steps of increasing amplitude, in the range −70 to −100 mV, led to the appearance of typical LTCS crowned with a single sodium channel‐dependent AP (Fig. 6 A). In control conditions, the membrane potential necessary to trigger an AP was not statistically different between WT and KI neurons (WT: −88.2 ± 1.8 mV, n = 7; KI: −94.2 ± 2.0 mV, n = 9, P = 0.09). Application of 1 μm zinc prevented the triggering of an AP for the same hyperpolarization level in WT neurons (Fig. 6 A, upper left panel, Fig. 6 B). This blocking effect on the threshold of LTCS was lost in KI neurons (Fig. 6 A, lower left panel, Fig. 6 B). Regarding LTCS amplitude, which was measured in the range 5–10 ms after the AP peak as the difference from the resting membrane potential, it was unchanged between WT neurons and KI neurons in control conditions (Fig. 7 A, B). However, LTCS amplitude was strongly inhibited following the application of 1 μm zinc in WT neurons but only moderately reduced in KI neurons. The LTCS inhibition in WT neurons was significantly higher than in KI neurons (Fig. 7 C). Similar experiments were conducted in the presence of nickel (Fig. 7 D). LTCS amplitude in WT neurons was significantly reduced by increasing concentrations of nickel (1 and 30 μm), in contrast to LTCS amplitude in KI neurons which was unchanged following application 30 μm nickel. Together our data show that DRG neurons from the KI mouse appear more excitable due to a loss of inhibition, which indicates that the H191‐dependent regulation of Cav3.2 channels can influence the excitability of neurons expressing these channels.
Figure 6. Rebound activity, low threshold calcium spike (LTCS) and its modulation by zinc in DRG neurons from WT and KI mice .
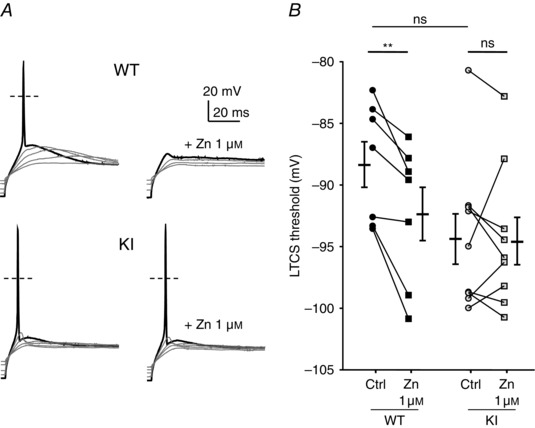
A, typical traces of the rebound potential LTCS evoked following 2 s hyperpolarizing steps of increasing amplitude in D‐hair neurons in control (left panel) and in the presence of zinc (1 μm, right panel) from WT (upper panel) and KI (lower panel) mice. B, LTCS threshold in control condition (circle) and in the presence of zinc (1 μm, square) in D‐hair neurons from WT mice (black symbols) and KI (grey symbols) mice. Statistical analysis was performed with repeated‐measures two‐way ANOVA (**P < 0.01; KI Ctrl vs. KI Zn 1 μm, P = 0.99; WT Ctrl vs. KI Ctrl, P = 0.09; WT Zn 1 μm vs. KI Zn 1 μm, P = 0.70).
Figure 7. Zinc modulation of LTCS amplitude in DRG neurons from WT and KI mice .
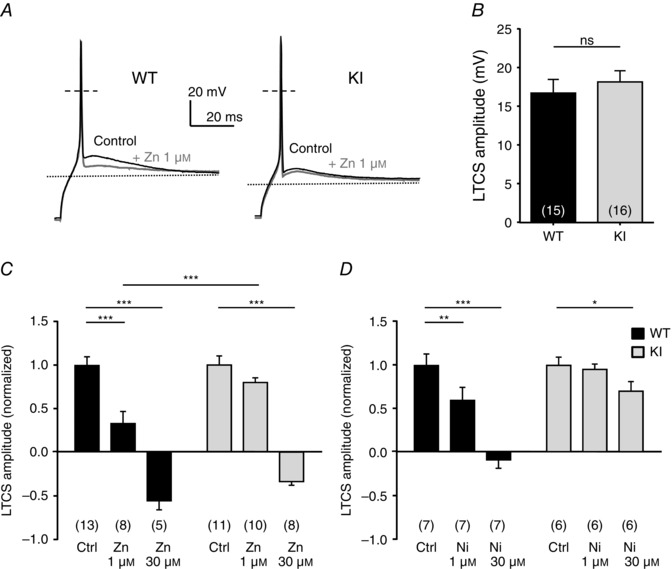
A, typical traces of LTCS evoked by a 2 s hyperpolarizing pulse in D‐hair neurons from WT (left) and KI mice (right) before and after exposure to 1 μm zinc. B, averaged amplitude of the LTCS in DRG neurons from WT and KI mice. The amplitude of the LTCS is measured as the difference from the resting membrane potential value (P = 0.27; Mann–Whitney). C, normalized amplitude of the LTCS before and after exposure to 1 and 30 μm zinc. Statistical analysis was performed with two‐way ANOVA followed by Sidak's test (***P < 0.001; KI Ctrl vs. KI Zn 1 μm, P = 0.19). D, normalized amplitude of the LTCS before and after exposure to 1 and 30 μm nickel. Statistical analysis was performed with two‐way ANOVA followed by Sidak's test (*P < 0.05; **P < 0.01; ***P < 0.001; KI Ctrl vs. KI Ni 1 μm, P = 0.96).
Discussion
In this study, we describe a Cav3.2‐H191Q knock‐in (KI) mouse model that allows the study of the H191‐dependent regulation of Cav3.2 channels. The sensitivity to zinc, nickel and ascorbate of native Cav3.2 channels is significantly impeded in the DRG neurons of this KI mouse. We report that this H191‐dependent regulation has discrete but significant effects on the excitability properties of D‐hair cells, a subpopulation of DRG neurons in which Cav3.2 currents play a prominent role in regulating excitability.
Because there was previously no means to assess directly the relevance of the H191‐dependent modulation of native Cav3.2 channels, as reviewed by Evans & Todorovic (2015), we have generated a mouse model harbouring an H191Q point mutation in its cacna1h gene. In this Cav3.2‐H191Q KI mouse model, we found no change in the Cav3.2 protein expression level, in the Cav3.2 current density and in the electrophysiological properties. Substitution of the H191 residue in the Cav3.2 isoform by glutamine (Q), the corresponding residue in the nickel/zinc‐resistant Cav3.1 isoform, has proven efficient in significantly reducing the nickel/zinc sensitivity of the Cav3.2 isoform in heterologous expression studies (Kang et al. 2006). Of importance, the H191Q mutation also disrupts the metal/redox‐dependent regulation of Cav3.2 channels in KI mice. However, compared to studies performed with recombinant Cav3.2 channels (human isoform) expressed in HEK‐293 cells (Traboulsie et al. 2006; Nelson et al. 2007 b), we found that the IC50 for zinc inhibition was 2‐fold higher in WT mouse neurons (1.7 μm) and 4‐fold lower in KI mouse neurons (10.0 μm), compared to that obtained with human recombinant WT Cav3.2 and Cav3.2‐H191Q channels (0.8 and 42 μm, respectively). This difference in zinc sensitivity between the native and recombinant Cav3.2 channels could be related to a difference in the primary sequence of the mouse Cav3.2 protein, which is not identical to its human counterpart. Indeed, zinc binds to a complex high affinity metal‐binding site at the external surface of the Cav3.2 channel. This metal‐binding site comprises not only the H191 residue in the extracellular IS3–IS4 loop (Kang et al. 2006) but also D189 and G190 in the IS3–IS4 loop as well as D140 in the outer IS2 segment (Kang et al. 2010). Although the protein sequence alignment indicates that these four residues are conserved among most species, including mouse and human (Uniprot Nos. AAK21607 and NP_001005407, respectively, not shown), one neighbouring neutral residue, alanine (A141) in the human sequence, is substituted by a negatively charged residue, aspartate (D141), in the mouse Cav3.2 protein. Of note, when expressed in Xenopus oocytes, the A‐to‐D mutation introduced into the human Cav3.2 protein (A141D) was found to increase the IC50 value for zinc inhibition from 3 to 12.5 μm (Kang et al. 2010). Also, post‐translational maturation of the Cav3.2 channel in neurons and/or association with putative Cav3.2‐interacting proteins might be different in neurons and in HEK‐293 cells, providing a distinct environment of the metal‐binding site among mammalian cell types able to modify the zinc/nickel affinity for the channel. Nevertheless, our study establishes that in the range 1–30 μm of zinc and nickel, there is a significant difference in current inhibition between neuronal WT and H191Q Cav3.2 channels which therefore validates that the H191 residue in Cav3.2 channels is an important component of the zinc/nickel binding site, as reported with recombinant Cav3.2 channels.
D‐hair neurons are a population of mechanoreceptor DRG neurons that express a large density of Cav3.2 current (Shin et al. 2003; Dubreuil et al. 2004; Wang & Lewin, 2011; Hilaire et al. 2012). These channels represent a main route for calcium entry and are associated with AP generation and electrical activity. In Cav3.2 knock‐out mice, D‐hair neurons showed lower firing frequency and reduced mechanosensitivity, compared to WT mice (Wang & Lewin, 2011). Establishing that the T‐type current in D‐hair cells was significantly less sensitive to zinc and nickel in the KI mouse allowed us to probe the role of the H191‐dependent modulation of the Cav3.2 channel in shaping the AP of D‐hair neurons. Using current‐clamp experiments combined with nickel/zinc pharmacological analysis we found that the rheobase threshold current was significantly increased in the presence of 30 μm nickel in WT D‐hair neurons, an effect that was lost in KI D‐hair neurons. A particularity of the AP waveform in D‐hair neurons is the presence of ADP that is dependent on the T‐type channels (Dubreuil et al. 2004). Here we describe that in the presence of 1–30 μm nickel, the ADP was unchanged in the KI D‐hair neurons, contrary to that observed in WT neurons. A similar differential effect was found following application of 1 μm zinc, although the ADP was markedly inhibited using a higher concentration (30 μm). The contribution of H191‐dependent regulation of Cav3.2 channels to the LTCS was also identified: (1) in the presence of low zinc or nickel concentration, the hyperpolarization potential necessary to trigger an LTCS crowned by a single spike as well as the LTCS amplitude were unchanged in KI D‐hair neurons, while significantly increased and reduced, respectively, in WT neurons. Together, these data confirmed previous studies on the important role of Cav3.2 channels on the AP and LTCS properties of D‐hair neurons and demonstrate that the H191‐dependent regulation of these channels can contribute to the D‐hair neuron excitability.
In our current‐clamp experiments, we have identified a differential ability of nickel and zinc ions to modulate D‐hair cell excitability. We found that AP and LTCS of D‐hair KI neurons were not, or poorly, affected by low concentrations of nickel but partly altered with corresponding concentrations of zinc. Indeed, zinc is an important trace metal that is involved in many biochemical and physiological processes in mammalian tissues (Mathie et al. 2006) and free zinc modulates many membrane receptors and channels, such as NMDA receptors (Paoletti et al. 1997; Nozaki et al. 2011), glycine receptors (Nevin et al. 2003), TRPA1 (Hu et al. 2009), ASIC channels (Baron et al. 2001), potassium channels (Easaw et al. 1999; Zhang et al. 2001; Clarke et al. 2004; Prost et al. 2004), sodium channels (Sheets & Hanck, 1992; Kuo et al. 2004) and other calcium channels (Magistretti et al. 2003) especially Cav2.3 (Shcheglovitov et al. 2012).
There is a need for such a site‐specific knock‐in mouse model to investigate the physiological relevance of H191‐dependent regulation of Cav3.2 channels and we believe that this new animal model will be instrumental to explore the role of this modulation in physiological situations. Indeed, a few other site‐specific knock‐in animal models dedicated to the study of the physiological relevance of zinc modulation have been developed recently, including (1) a Glra1‐D80A knock‐in mouse line lacking a zinc binding site in the glycine receptor (GlyR) α1 subunit gene (Hirzel et al. 2006) and (2) an NR2A‐H128S knock‐in mouse line lacking the high‐affinity zinc‐binding site in the NR2A subunit of NMDA receptor (Nozaki et al. 2011). Using Glra1‐D80A animals, Hirzel et al. (2006) found a significant reduction in the potentiation of glycine currents by low concentrations of zinc in Glra1‐D80A neurons as compared to WT neurons. This study also provides evidence that the Glra1‐D80A knock‐in mice exhibit severe motor deficits and a hyperekplexia phenotype mimicking the hereditary Startle disease. Using acute hippocampus and spinal cord slices from NR2A‐H128S mice, Nozaki et al. (2011) reported that NMDA excitatory postsynaptic currents (EPSCs) were barely affected by a submicromolar zinc concentration, contrary to that observed with WT mouse slices in which NMDA EPSCs were markedly inhibited. They also found that, using pain models of inflammatory pain (complete Freund's adjuvant) and neuropathic pain (sciatic nerve ligation), NR2A‐H128S animals developed enhanced mechanical allodynia with loss of zinc‐induced analgesia. Taken together, these landmark studies emphasize how important is the development of dedicated animal models to unravel the modulation properties and physiological relevance of trace metal regulation of channels and receptors and how it critically impacts neuronal excitability.
A current challenge is to identify the ability of endogenous trace metals, especially zinc, to modulate tonically and/or dynamically native channels and receptors. Using hippocampal slices from NR2A‐H128S animals, it was established recently that an ambient zinc level at mossy fibre–CA3 synapses was not high enough to tonically occupy a high‐affinity zinc site on NMDA receptors in the presence of physiological extracellular magnesium concentrations (Vergnano et al. 2014). Regarding Cav3.2 channels, evidence for tonic inhibition by zinc was found in peripheral C‐type nociceptors that also express large Cav3.2 T‐type currents (Nelson et al. 2007 b). In their study, Nelson et al. (2007 b) showed that reducing agents, such as l‐cysteine, as well as synthetic and endogenous zinc chelators, diethylenetriaminepenta‐acetic acid (DTPA), TPEN, DTT and BSA, could sensitize Cav3.2 current and favour AP firing in C‐type nociceptors from WT mice but not from Cav3.2 knock‐out mice. These experiments suggested that in WT animals, zinc chelation could effectively remove Cav3.2 channel tonic inhibition, favouring thermal hyperalgesia (Evans & Todorovic, 2015). However, because most reducing agents used have non‐specific chelating abilities, channel modification could occur by chelation of trace metals or through alternative mechanisms, such as reduction of cysteine residues. In addition, other studies have suggested that the endogenous gasotransmitters, such as hydrogen sulfide (H2S) and carbon monoxide (CO), could also play a role in Cav3.2 channel regulation through redox‐related mechanisms involving H191 (Boycott et al. 2013; Elies et al. 2014; Sekiguchi et al. 2014). This redox modulation of Cav3.2 channels could also be involved in the GABAB receptor inhibition of T‐type channels in DRG neurons (Huang et al. 2015). Of interest, it was recently reported that chelating zinc with TPEN could facilitate modulation of GABAergic inhibitory post‐synaptic currents in hippocampal granule cells, an effect that was occluded when blocking T‐type channels (Grauert et al. 2014). Increased Cav3.2 channel activity due to the removal of a chelatable zinc fraction would enhance cellular excitability in fast‐spiking interneurons without affecting the properties of granule cells, supporting that zinc modulation of GABAergic transmission to granule cells would involve H191‐dependent regulation of Cav3.2 channels.
The major finding of this study is that impairment of the native H191‐dependent regulation of T‐type Cav3.2 channels reveals it potential role in the excitability of Cav3.2‐expressing neurons. This study opens new avenues for precise investigation of the role of the H191‐dependent regulation of T‐type Cav3.2 channels. Over the last decade, a role of Cav3.2 channels in a variety of physiological functions has been proposed (Huc et al. 2009). This includes pain detection and processing (Todorovic & Jevtovic‐Todorovic, 2007; Bourinet et al. 2014), neuronal excitability and pacemaker activities (Liao et al. 2011), hormone secretion (Maturana et al. 2009; Enyeart & Enyeart, 2015), neurotransmitter release (Weiss et al. 2012; Carbone et al. 2014), vascular tone (Hansen, 2015) and fertilization (Bernhardt et al. 2015). The Cav3.2‐H191Q mouse model will be helpful to decipher a putative role of this Cav3.2 modulation, not only in the physiological processes listed above, but also in a variety of disease states that are thought to rely on altered Cav3.2 activity, such as neuropathic pain, epilepsy, primary aldosteronism and cardiac hypertrophy (Evans & Todorovic, 2015). Future studies should benefit from the use of these transgenic animals lacking H191 to address in vivo the potential roles of the trace metal and redox modulation of T‐type channels in a wide range of physiological and pathological conditions, as well as to support the development of selective Cav3.2 channel modulators targeting the H191‐dependent regulation.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
P.L. conceived the study. P.L., E.B. and T.V. designed experiments. T.V. carried out all experimental work and data analysis. P.L. and T.V. wrote the paper. P.L., E.B. and T.V. revised and approved the final version.
Funding
T.V. was supported by a fellowship from the French Ministry of Research and Education. This work was supported by Laboratory of Excellence ‘Ion Channel Science and Therapeutics’ (ANR‐11‐LABX‐0015‐01), by AFM grant (AFM‐PainT) to E.B., and by grants from Agence Nationale de la Recherche to E.B. (ANR‐08‐NMPS‐025, ANR‐09‐MNPS‐037) and to P.L. (ANR‐09‐MNPS‐035 and ANR‐10‐BLAN‐1601).
Acknowledgements
We are grateful to Iulia Blesneac, Amaury François, Luc Forichon, Sophie Laffray and Anne Pizzoccaro for technical support, as well as to Jean Chemin and Nathalie Guérineau for helpful comments.
References
- Baron A, Schaefer L, Lingueglia E, Champigny G & Lazdunski M (2001). Zn2+ and H+ are coactivators of acid‐sensing ion channels. J Biol Chem 276, 35361–35367. [DOI] [PubMed] [Google Scholar]
- Bernhardt ML, Zhang Y, Erxleben CF, Padilla‐Banks E, McDonough CE, Miao YL, Armstrong DL & Williams CJ (2015). CaV3.2 T‐type channels mediate Ca2+ entry during oocyte maturation and following fertilization. J Cell Sci 128, 4442–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blesneac I, Chemin J, Bidaud I, Huc‐Brandt S, Vandermoere F & Lory P (2015). Phosphorylation of the Cav3.2 T‐type calcium channel directly regulates its gating properties. Proc Natl Acad Sci USA 112, 13705–13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrere C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A & Nargeot J (2005). Silencing of the Cav3.2 T‐type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J 24, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourinet E, Altier C, Hildebrand ME, Trang T, Salter MW & Zamponi GW (2014). Calcium‐permeable ion channels in pain signaling. Physiol Rev 94, 81–140. [DOI] [PubMed] [Google Scholar]
- Boycott HE, Dallas ML, Elies J, Pettinger L, Boyle JP, Scragg JL, Gamper N & Peers C (2013). Carbon monoxide inhibition of Cav3.2 T‐type Ca2+ channels reveals tonic modulation by thioredoxin. FASEB J 27, 3395–3407. [DOI] [PubMed] [Google Scholar]
- Cain SM & Snutch TP (2010). Contributions of T‐type calcium channel isoforms to neuronal firing. Channels 4, 44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone E, Calorio C & Vandael DH (2014). T‐type channel‐mediated neurotransmitter release. Pflugers Arch 466, 677–687. [DOI] [PubMed] [Google Scholar]
- Carbone E & Lux HD (1984). A low voltage‐activated, fully inactivating Ca channel in vertebrate sensory neurones. Nature 310, 501–502. [DOI] [PubMed] [Google Scholar]
- Choi S, Na HS, Kim J, Lee J, Lee S, Kim D, Park J, Chen CC, Campbell KP & Shin HS (2007). Attenuated pain responses in mice lacking CaV3.2 T‐type channels. Genes Brain Behav 6, 425–431. [DOI] [PubMed] [Google Scholar]
- Clarke CE, Veale EL, Green PJ, Meadows HJ & Mathie A (2004). Selective block of the human 2‐P domain potassium channel, TASK‐3, and the native leak potassium current, IKSO, by zinc. J Physiol 560, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubreuil AS, Boukhaddaoui H, Desmadryl G, Martinez‐Salgado C, Moshourab R, Lewin GR, Carroll P, Valmier J & Scamps F (2004). Role of T‐type calcium current in identified D‐hair mechanoreceptor neurons studied in vitro. J Neurosci 24, 8480–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easaw JC, Jassar BS, Harris KH & Jhamandas JH (1999). Zinc modulation of ionic currents in the horizontal limb of the diagonal band of Broca. Neuroscience 94, 785–795. [DOI] [PubMed] [Google Scholar]
- Elies J, Scragg JL, Huang S, Dallas ML, Huang D, MacDougall D, Boyle JP, Gamper N & Peers C (2014). Hydrogen sulfide inhibits Cav3.2 T‐type Ca2+ channels. FASEB J 28, 5376–5387. [DOI] [PubMed] [Google Scholar]
- Enyeart JJ & Enyeart JA (2015). Adrenal fasciculata cells express T‐type and rapidly and slowly activating L‐type Ca2+ channels that regulate cortisol secretion. Am J Physiol Cell Physiol 308, C899–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JG & Todorovic SM (2015). Redox and trace metal regulation of ion channels in the pain pathway. Biochem J 470, 275–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois A, Kerckhove N, Meleine M, Alloui A, Barrere C, Gelot A, Uebele VN, Renger JJ, Eschalier A, Ardid D & Bourinet E (2013). State‐dependent properties of a new T‐type calcium channel blocker enhance CaV3.2 selectivity and support analgesic effects. Pain 154, 283–293. [DOI] [PubMed] [Google Scholar]
- François A, Schüetter N, Laffray S, Sanguesa J, Pizzoccaro A, Dubel S, Mantilleri A, Nargeot J, Noël J, Wood John N, Moqrich A, Pongs O & Bourinet E (2015). The low‐threshold calcium channel Cav3.2 determines low‐threshold mechanoreceptor function. Cell Reports 10, 370–382. [DOI] [PubMed] [Google Scholar]
- Garcia‐Caballero A, Gadotti VM, Stemkowski P, Weiss N, Souza IA, Hodgkinson V, Bladen C, Chen L, Hamid J, Pizzoccaro A, Deage M, Francois A, Bourinet E & Zamponi GW (2014). The deubiquitinating enzyme USP5 modulates neuropathic and inflammatory pain by enhancing Cav3.2 channel activity. Neuron 83, 1144–1158. [DOI] [PubMed] [Google Scholar]
- Grauert A, Engel D & Ruiz AJ (2014). Endogenous zinc depresses GABAergic transmission via T‐type Ca2+ channels and broadens the time window for integration of glutamatergic inputs in dentate granule cells. J Physiol 592, 67–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen PB (2015). Functional importance of T‐type voltage‐gated calcium channels in the cardiovascular and renal system: news from the world of knockout mice. Am J Physiol Regul Integr Comp Physiol 308, R227–237. [DOI] [PubMed] [Google Scholar]
- Hilaire C, Lucas O, Valmier J & Scamps F (2012). Neurotrophin‐4 modulates the mechanotransducer Cav3.2 T‐type calcium current in mice down‐hair neurons. Biochem J 441, 463–471. [DOI] [PubMed] [Google Scholar]
- Hirzel K, Müller U, Latal AT, Hülsmann S, Grudzinska J, Seeliger MW, Betz H & Laube B (2006). Hyperekplexia phenotype of glycine receptor α1 subunit mutant mice identifies Zn2+ as an essential endogenous modulator of glycinergic neurotransmission. Neuron 52, 679–690. [DOI] [PubMed] [Google Scholar]
- Hu H, Bandell M, Petrus MJ, Zhu MX & Patapoutian A (2009). Zinc activates damage‐sensing TRPA1 ion channels. Nat Chem Biol 5, 183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Huang S, Peers C, Du X, Zhang H & Gamper N (2015). GABAB receptors inhibit low‐voltage activated and high‐voltage activated Ca2+ channels in sensory neurons via distinct mechanisms. Biochem Biophys Res Commun 465, 188–193. [DOI] [PubMed] [Google Scholar]
- Huc S, Monteil A, Bidaud I, Barbara G, Chemin J & Lory P (2009). Regulation of T‐type calcium channels: signalling pathways and functional implications. Biochim Biophys Acta 1793, 947–952. [DOI] [PubMed] [Google Scholar]
- Jeong SW, Park BG, Park JY, Lee JW & Lee JH (2003). Divalent metals differentially block cloned T‐type calcium channels. Neuroreport 14, 1537–1540. [DOI] [PubMed] [Google Scholar]
- Kang HW, Park JY, Jeong SW, Kim JA, Moon HJ, Perez‐Reyes E & Lee JH (2006). A molecular determinant of nickel inhibition in Cav3.2 T‐type calcium channels. J Biol Chem 281, 4823–4830. [DOI] [PubMed] [Google Scholar]
- Kang HW, Vitko I, Lee SS, Perez‐Reyes E & Lee JH (2010). Structural determinants of the high affinity extracellular zinc binding site on Cav3.2 T‐type calcium channels. J Biol Chem 285, 3271–3281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawabata A, Ishiki T, Nagasawa K, Yoshida S, Maeda Y, Takahashi T, Sekiguchi F, Wada T, Ichida S & Nishikawa H (2007). Hydrogen sulfide as a novel nociceptive messenger. Pain 132, 74–81. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Chen WY & Yang YC (2004). Block of tetrodotoxin‐resistant Na+ channel pore by multivalent cations: gating modification and Na+ flow dependence. J Gen Physiol 124, 27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Gomora JC, Cribbs LL & Perez‐Reyes E (1999). Nickel block of three cloned T‐type calcium channels: low concentrations selectively block α1H. Biophys J 77, 3034–3042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y‐F, Tsai M‐L, Chen C‐C & Yen C‐T (2011). Involvement of the Cav3.2 T‐type calcium channel in thalamic neuron discharge patterns. Mol Pain 7, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Walker JS & Tracey DJ (1999). Zinc alleviates thermal hyperalgesia due to partial nerve injury. Neuroreport 10, 1619–1623. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Castelli L, Taglietti V & Tanzi F (2003). Dual effect of Zn2+ on multiple types of voltage‐dependent Ca2+ currents in rat palaeocortical neurons. Neuroscience 117, 249–264. [DOI] [PubMed] [Google Scholar]
- Mathie A, Sutton GL, Clarke CE & Veale EL (2006). Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther 111, 567–583. [DOI] [PubMed] [Google Scholar]
- Matsunami M, Kirishi S, Okui T & Kawabata A (2011). Chelating luminal zinc mimics hydrogen sulfide‐evoked colonic pain in mice: possible involvement of T‐type calcium channels. Neuroscience 181, 257–264. [DOI] [PubMed] [Google Scholar]
- Maturana A, Lenglet S, Python M, Kuroda S & Rossier MF (2009). Role of the T‐type calcium channel CaV3.2 in the chronotropic action of corticosteroids in isolated rat ventricular myocytes. Endocrinology 150, 3726–3734. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Perez‐Reyes E & Todorovic SM (2005). The endogenous redox agent l‐cysteine induces T‐type Ca2+ channel‐dependent sensitization of a novel subpopulation of rat peripheral nociceptors. J Neurosci 25, 8766–8775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Joksovic PM, Su P, Kang HW, Van Deusen A, Baumgart JP, David LS, Snutch TP, Barrett PQ, Lee JH, Zorumski CF, Perez‐Reyes E & Todorovic SM (2007. a). Molecular mechanisms of subtype‐specific inhibition of neuronal T‐type calcium channels by ascorbate. J Neurosci 27, 12577–12583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez‐Reyes E, Lee JH, Shin HS & Todorovic SM (2007. b). Reducing agents sensitize C‐type nociceptors by relieving high‐affinity zinc inhibition of T‐type calcium channels. J Neurosci 27, 8250–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevin ST, Cromer BA, Haddrill JL, Morton CJ, Parker MW & Lynch JW (2003). Insights into the structural basis for zinc inhibition of the glycine receptor. J Biol Chem 278, 28985–28992. [DOI] [PubMed] [Google Scholar]
- Nozaki C, Vergnano AM, Filliol D, Ouagazzal AM, Le Goff A, Carvalho S, Reiss D, Gaveriaux‐Ruff C, Neyton J, Paoletti P & Kieffer BL (2011). Zinc alleviates pain through high‐affinity binding to the NMDA receptor NR2A subunit. Nat Neurosci 14, 1017–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P, Ascher P & Neyton J (1997). High‐affinity zinc inhibition of NMDA NR1‐NR2A receptors. J Neurosci 17, 5711–5725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Reyes E (2003). Molecular physiology of low‐voltage‐activated T‐type calcium channels. Physiol Rev 83, 117–161. [DOI] [PubMed] [Google Scholar]
- Prost AL, Bloc A, Hussy N, Derand R & Vivaudou M (2004). Zinc is both an intracellular and extracellular regulator of KATP channel function. J Physiol 559, 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynders A, Mantilleri A, Malapert P, Rialle S, Nidelet S, Laffray S, Beurrier C, Bourinet E & Moqrich A (2015). Transcriptional profiling of cutaneous MRGPRD free nerve endings and C‐LTMRs. Cell Rep 10, 1007–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose KE, Lunardi N, Boscolo A, Dong X, Erisir A, Jevtovic‐Todorovic V & Todorovic SM (2013). Immunohistological demonstration of CaV3.2 T‐type voltage‐gated calcium channel expression in soma of dorsal root ganglion neurons and peripheral axons of rat and mouse. Neuroscience 250, 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safieh‐Garabedian B, Poole S, Allchorne A, Kanaan S, N SaadÉ & Woolf CJ (1996). Zinc reduces the hyperalgesia and upregulation of NGF and IL‐1β produced by peripheral inflammation in the rat. Neuropharmacology 35, 599–603. [DOI] [PubMed] [Google Scholar]
- Sekiguchi F, Miyamoto Y, Kanaoka D, Ide H, Yoshida S, Ohkubo T & Kawabata A (2014). Endogenous and exogenous hydrogen sulfide facilitates T‐type calcium channel currents in Cav3.2‐expressing HEK293 cells. Biochem Biophys Res Commun 445, 225–229. [DOI] [PubMed] [Google Scholar]
- Shcheglovitov A, Vitko I, Lazarenko RM, Orestes P, Todorovic SM & Perez‐Reyes E (2012). Molecular and biophysical basis of glutamate and trace metal modulation of voltage‐gated Cav2.3 calcium channels. J Gen Physiol 139, 219–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MF & Hanck DA (1992). Mechanisms of extracellular divalent and trivalent cation block of the sodium current in canine cardiac Purkinje cells. J Physiol 454, 299–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin JB, Martinez‐Salgado C, Heppenstall PA & Lewin GR (2003). A T‐type calcium channel required for normal function of a mammalian mechanoreceptor. Nat Neurosci 6, 724–730. [DOI] [PubMed] [Google Scholar]
- Talley EM, Cribbs LL, Lee JH, Daud A, Perez‐Reyes E & Bayliss DA (1999). Differential distribution of three members of a gene family encoding low voltage‐activated (T‐type) calcium channels. J Neurosci 19, 1895–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorovic SM & Jevtovic‐Todorovic V (2007). Regulation of T‐type calcium channels in the peripheral pain pathway. Channels (Austin) 1, 238–245. [DOI] [PubMed] [Google Scholar]
- Todorovic SM & Jevtovic‐Todorovic V (2013). Neuropathic pain: role for presynaptic T‐type channels in nociceptive signaling. Pflugers Arch 465, 921–927. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Jevtovic‐Todorovic V, Meyenburg A, Mennerick S, Perez‐Reyes E, Romano C, Olney JW & Zorumski CF (2001). Redox modulation of T‐type calcium channels in rat peripheral nociceptors. Neuron 31, 75–85. [DOI] [PubMed] [Google Scholar]
- Todorovic SM, Meyenburg A & Jevtovic‐Todorovic V (2002). Mechanical and thermal antinociception in rats following systemic administration of mibefradil, a T‐type calcium channel blocker. Brain Res 951, 336–340. [DOI] [PubMed] [Google Scholar]
- Traboulsie A, Chemin J, Chevalier M, Quignard JF, Nargeot J & Lory P (2006). Subunit‐specific modulation of T‐type calcium channels by zinc. J Physiol 578, 159–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lonnerberg P, Lou D, Hjerling‐Leffler J, Haeggstrom J, Kharchenko O, Kharchenko PV, Linnarsson S & Ernfors P (2015). Unbiased classification of sensory neuron types by large‐scale single‐cell RNA sequencing. Nat Neurosci 18, 145–153. [DOI] [PubMed] [Google Scholar]
- Vergnano AM, Rebola N, Savtchenko LP, Pinheiro PS, Casado M, Kieffer BL, Rusakov DA, Mulle C & Paoletti P (2014). Zinc dynamics and action at excitatory synapses. Neuron 82, 1101–1114. [DOI] [PubMed] [Google Scholar]
- Wang R & Lewin GR (2011). The Cav3.2 T‐type calcium channel regulates temporal coding in mouse mechanoreceptors. J Physiol 589, 2229–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss N, Hameed S, Fernandez‐Fernandez JM, Fablet K, Karmazinova M, Poillot C, Proft J, Chen L, Bidaud I, Monteil A, Huc‐Brandt S, Lacinova L, Lory P, Zamponi GW & De Waard M (2012). A Cav3.2/syntaxin‐1A signaling complex controls T‐type channel activity and low‐threshold exocytosis. J Biol Chem 287, 2810–2818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White G, Lovinger DM & Weight FF (1989). Transient low‐threshold Ca2+ current triggers burst firing through an afterdepolarizing potential in an adult mammalian neuron. Proc Natl Acad Sci USA 86, 6802–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Lory P & Perez‐Reyes E (2010). Role of voltage‐gated calcium channels in epilepsy. Pflugers Arch 460, 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kehl SJ & Fedida D (2001). Modulation of Kv1.5 potassium channel gating by extracellular zinc. Biophys J 81, 125–136. [DOI] [PMC free article] [PubMed] [Google Scholar]