

Abstract
Key points
Synaptic excitation and inhibition must be properly balanced in individual neurons and neuronal networks to allow proper brain function.
Disrupting this balance may lead to autism spectral disorders and epilepsy.
We show the basic helix–loop–helix transcription factor NeuroD2 promotes inhibitory synaptic drive but also decreases cell‐intrinsic neuronal excitability of cortical pyramidal neurons both in vitro and in vivo.
We identify two genes potentially downstream of NeuroD2‐mediated transcription that regulate these parameters: gastrin‐releasing peptide and the small conductance, calcium‐activated potassium channel, SK2.
Our results reveal an important function for NeuroD2 in balancing synaptic neurotransmission and intrinsic excitability.
Our results offer insight into how synaptic innervation and intrinsic excitability are coordinated during cortical development.
Abstract
Synaptic excitation and inhibition must be properly balanced in individual neurons and neuronal networks for proper brain function. Disruption of this balance during development may lead to autism spectral disorders and epilepsy. Synaptic excitation is counterbalanced by synaptic inhibition but also by attenuation of cell‐intrinsic neuronal excitability. To maintain proper excitation levels during development, neurons must sense activity over time and regulate the expression of genes that control these parameters. While this is a critical process, little is known about the transcription factors involved in coordinating gene expression to control excitatory/inhibitory synaptic balance. We show here that the basic helix–loop–helix transcription factor NeuroD2 promotes inhibitory synaptic drive but also decreases cell‐intrinsic neuronal excitability of cortical pyramidal neurons both in vitro and in vivo as shown by ex vivo analysis of a NeuroD2 knockout mouse. Using microarray analysis and comparing wild‐type and NeuroD2 knockout cortical networks, we identified two potential gene targets of NeuroD2 that contribute to these processes: gastrin‐releasing peptide (GRP) and the small conductance, calcium‐activated potassium channel, SK2. We found that the GRP receptor antagonist RC‐3059 and the SK2 specific blocker apamin partially reversed the effects of increased NeuroD2 expression on inhibitory synaptic drive and action potential repolarization, respectively. Our results reveal an important function for NeuroD2 in balancing synaptic neurotransmission and intrinsic excitability and offer insight into how these processes are coordinated during cortical development.
Key points
Synaptic excitation and inhibition must be properly balanced in individual neurons and neuronal networks to allow proper brain function.
Disrupting this balance may lead to autism spectral disorders and epilepsy.
We show the basic helix–loop–helix transcription factor NeuroD2 promotes inhibitory synaptic drive but also decreases cell‐intrinsic neuronal excitability of cortical pyramidal neurons both in vitro and in vivo.
We identify two genes potentially downstream of NeuroD2‐mediated transcription that regulate these parameters: gastrin‐releasing peptide and the small conductance, calcium‐activated potassium channel, SK2.
Our results reveal an important function for NeuroD2 in balancing synaptic neurotransmission and intrinsic excitability.
Our results offer insight into how synaptic innervation and intrinsic excitability are coordinated during cortical development.
Abbreviations
- AP
action potential
- AHP
afterhyperpolarization
- E/I
excitatory/inhibitory
- GRP
gastrin‐releasing peptide
- GRPR
gastrin‐releasing peptide receptor
- IEI
inter‐event interval
- mEPSC
miniature excitatory postsynaptic current
- mIPSC
miniature inhibitory postsynaptic current
- NeuroD2
neurogenic differentiation factor 2
- sEPSC
spontaneous excitatory postsynaptic current
- sIPSC
spontaneous inhibitory postsynaptic current
- TF
transcription factor
Introduction
Alterations in cortical circuit development can lead to neurodevelopmental disorders including autism spectral disorders and epilepsy (Hoischen et al. 2014; Lee et al. 2015). Proper neuronal circuit formation and function require coordination between excitatory and inhibitory synaptic innervation as well as the cell‐intrinsic excitability of individual neurons (Turrigiano, 2011). This means that individual neurons must sense and integrate synaptic activity over time and correspondingly control expression of genes regulating synaptic innervation and intrinsic excitability. While the transcription factors (TFs) that control the initial fating of neuronal cell types are well defined (Kohwi & Doe, 2013; Imayoshi & Kageyama, 2014), the TFs and downstream genes that coordinate neuronal excitability and synaptic connectivity during circuit formation, after neuronal cell fating, are less well understood.
Neurogenic differentiation factor 2 (NeuroD2) is a highly conserved TF of the basic helix–loop–helix protein family and is one of the first TFs expressed in post‐mitotic neurons (McCormick et al. 1996). NeuroD2 transactivation can be activated by calcium influx making it a good candidate for linking neuronal activity to the transcription of genes that regulate synaptic innervation and intrinsic excitability in developing neurons. It is well established that NeuroD2 promotes neuronal survival and excitatory synapse maturation (Olson et al. 2001; Ince‐Dunn et al. 2006; Wilke et al. 2012). However, its role in regulating inhibitory synapse development and intrinsic neuronal excitability has not been examined. Furthermore, the direct targets of this important late‐stage TF are largely unknown in post‐mitotic neurons.
We examined the role of NeuroD2 using gene knockdown and knockout (KO), and overexpression while measuring synaptic innervation and intrinsic excitability of pyramidal neurons in developing cortical networks both in vitro and ex vivo. In parallel, we performed microarray analysis on wild‐type (WT) and NeuroD2 KO cortical cultures to identify potential candidate target genes responsible for any observed changes in cellular electrophysiology. Using this parallel approach we found that NeuroD2 acts in a cell intrinsic manner to regulate neuronal excitability and promote synaptic innervation in developing cortical neurons. By comparing microarray results we identified two transcripts that are significantly decreased in expression in NeuroD2 KO tissue that can mechanistically link NeuroD2 levels to inhibitory synapse number and cellular excitability: gastrin‐releasing peptide (GRP) and the small conductance, calcium‐activated potassium channel, SK2.
GRP acts on the gastrin‐releasing peptide receptor (GRPR) to promote inhibitory neurotransmission (Cao et al. 2010). We found GRP mRNA was strongly decreased in NeuroD2 null neurons and the GRPR antagonist RC‐3059 partially blocked the increase in inhibitory synaptic inputs onto NeuroD2 overexpressing cells. The small conductance calcium‐activated potassium channel, SK2, which is also developmentally regulated (Gymnopoulos et al. 2014), regulates intrinsic excitability of neurons by mediating action potential (AP) after‐hyperpolarization (Garcia‐Junco‐Clemente et al. 2013). SK2 mRNA was also significantly decreased in the absence of NeuroD2 and NeuroD2 overexpression altered AP parameters in a manner consistent with increased SK2 expression. Moreover, these changes were partially blocked by the SK2 selective antagonist apamin. In brief, our data reveal that the activity‐dependent transcription factor NeuroD2 functions as a critical mediator of both neuronal excitability and synaptic innervation during cortical circuit development. Furthermore, NeuroD2 mediates these changes, at least in part, by contributing to the expression of SK2 and GRP.
Methods
Animal care and use
All experimental procedures were performed in accordance with the policies and recommendations of the International Association for the Study of Pain, and have been approved by the Institutional Animal Care and Use Committee of Tulane University, which is a fully AAALAC accredited institute. All efforts were made to minimize the number of animals used for study.
Neuronal cultures
Dissociated cortical neurons were harvested from timed‐pregnant embryonic day (E)18 rats or E16 mice. Briefly, for rat cortical cultures, a time‐pregnant animal was deeply anaesthetized with isoflurane and killed by cervical dislocation. Embryos were dissected in ice‐cold Hanks’ balanced salt solution buffer. Cortical caps were dissected (hippocampi removed) and then digested with trypsin for 20 min at 37°C before trituration. Multiple embryos were combined for plating into individual cultured wells, and thus individual cultured coverslips represented multiple animals of unspecified sex. Dissociated neurons were cultured on tissue culture dishes or glass coverslips coated with poly‐d‐lysine and laminin (BD Biosciences, San Jose, CA, USA). Rat neuron culture medium consisted of basal medium Eagle with 1× Glutamax, 1000 U ml−1 penicillin G and streptomycin sulfate, 5% FBS, 1 × N2 supplement and 25 μm β‐mercaptoethanol (Invitrogen, Carlsbad, CA, USA). Cultures were maintained at 37°C in 5% ambient CO2. Media were changed every other day. Mouse cultures were prepared using the same dissection and plating protocols, but with the following growth medium: Neurobasal Medium with 1 × Glutamax, 2% fetal bovine serum, 1 × B27 supplement, and 25 μm β‐mercaptoethanol (Invitrogen). Detailed protocols can be found in Hall et al. (2007).
Plasmids and transfection
Mouse NeuroD2 cNDA was cloned into the pCS2+ plasmid under control of a CMV promoter (Wilke et al. 2012). To generate NeuroD2 shRNA the sequence corresponding to nucleotides 911 to 931 of rat NeuroD2 (GCTCTGTCTCAACGGCAACTT) was cloned into the pLKO.1 expression vector (Wilke et al. 2012). Mouse and rat NeuroD2 are 100% conserved in this target region. Cultured neurons were transfected at 8 days in vitro (DIV) using Lipofectamine 2000 (Invitrogen). An enhanced green fluorescent protein (eGFP) expressing plasmid (pBos) was used as a transfection marker in all experiments, including control (GFP alone). Positive transfected neurons were confirmed to be excitatory pyramidal neurons by the lack of glutamic acid decarboxylase 65 (GAD65) staining (data not shown).
RNA isolation, microarray analysis and qPCR
RNA was isolated using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). For microarray analysis, seven samples for each WT and KO mouse cortical cultures were prepared and hybridized to SurePrint G3 mouse GE 8 × 60 K arrays (Agilent, Santa Clara, CA, USA). Array data were processed and analysed similarly to published protocols (Han & Jones, 2014). GeneSpring GX software version 12 (Agilent) was used to analyse microarray data. A probe was selected if it met the following criteria: fold change more than 1.2 and significance P < 0.05 with Student's unpaired t test. False discover rates (FDR) were calculated using Benjamini–Hochberg multiple testing correction in Genespring software (presented in Table 1). Two FDR values are reported for each gene, FDR Select and FDR Total. To get values for FDR Select, ∼600 genes, which had fold change > 1.2 and significance P < 0.05, were assessed using Benjamini–Hochberg multiple testing correction. For FDR Total, total probes (∼60 K on chip) were assessed using Benjamini–Hochberg multiple testing correction, without cut‐off for fold change (= 1.0) nor significance (P = 1.0). Gene expression changes were then tested for biological significance using qPCR analysis (Table 1).
Table 1.
Selected genes that are downregulated (>1.2‐fold) in NeuroD2 KO vs. WT cortical tissue
| Gene symbol | Fold change | P | FDR (Select) | FDR (Total) | Description |
|---|---|---|---|---|---|
| Neurod2†‡ | −8.81 | 9.41 × 10−11 | 1.78 × 10−8 | 5.25 × 10−6 | Neurogenic differentiation factor 2 |
| Grp†‡ | −2.48 | 0.008090833 | 0.02335726 | 0.7937901 | Gastrin releasing peptide |
| Kif6 | −1.77 | 0.039458968 | 0.04267144 | 0.85842603 | Kinesin family member 6 |
| Kcns3 | −1.69 | 0.025873655 | 0.035351716 | 0.79384476 | Voltage‐gated channel, delayed‐rectifier, subfamily S, member 3 |
| Plxndl†‡ | −1.48 | 0.005626001 | 0.016380074 | 0.7004802 | Plexin Dl |
| Kcnd3 | −1.42 | 0.002585986 | 0.011132563 | 0.5020108 | Voltage‐gated channel, Shal‐related family, member 3 |
| Epha6 | −1.42 | 0.002643762 | 0.011132563 | 0.6172144 | Eph receptor A6 |
| Nefm† | −1.42 | 0.007119948 | 0.02335726 | 0.7937901 | Neurofilament, medium polypeptide |
| Gabra2† | −1.41 | 0.032407083 | 0.03593437 | 0.79715264 | GABA‐A receptor, subunit α2 |
| Kcnt2 | −1.39 | 0.001616994 | 0.009913773 | 0.15403655 | Potassium channel, subfamily T, member 2 |
| Foxo6 | −1.39 | 0.043240737 | 0.045719594 | 0.8979501 | Forkhead box O6 |
| Kcnip4 | −1.37 | 0.002442279 | 0.010243218 | 0.41595492 | Kv channel interacting protein 4 |
| Kcnjll | −1.37 | 0.034641597 | 0.038337044 | 0.8065073 | Potassium inwardly rectifying channel, subfamily J, member 11 |
| Stxbp5l | −1.37 | 0.048574124 | 0.04882949 | 0.95340574 | Syntaxin binding protein 5‐like |
| Gabbr2 | −1.35 | 0.006926423 | 0.019712156 | 0.7937901 | GABA‐B receptor, 2 |
| Dixdcl†‡ | −1.35 | 0.01726422 | 0.031362873 | 0.7937901 | DIX domain containing 1 |
| Foxol | −1.34 | 0.002442279 | 0.010243218 | 0.49281004 | Forkhead box O1 |
| Camk2a† | −1.33 | 0.042989478 | 0.045495648 | 0.8933901 | Calcium/calmodulin‐dependent protein kinase II α |
| Ppplrlb | −1.32 | 0.00460536 | 0.012099204 | 0.63865095 | Protein phosphatase 1, regulatory (inhibitor) subunit 1B |
| Ephb6 | −1.32 | 0.005699302 | 0.016380074 | 0.7109885 | Eph receptor B6 |
| Kcnk4 | −1.31 | 0.005386524 | 0.016380074 | 0.6707512 | Potassium channel, subfamily K, member 4 |
| Atp2bl | −1.31 | 0.007285039 | 0.02335726 | 0.7937901 | ATPase, Ca2+ transporting, plasma membrane 1 |
| Bok | −1.31 | 0.021214584 | 0.034998067 | 0.7937901 | BCL2‐related ovarian killer protein |
| Sla | −1.3 | 0.04818842 | 0.04882949 | 0.92200255 | Src‐like adaptor |
| Slitrk3 | −1.29 | 0.03366219 | 0.036189463 | 0.8025127 | SLIT and NTRK‐like family, member 3 |
| Kcncl | −1.29 | 0.041096598 | 0.045084268 | 0.88167274 | Potassium voltage‐gated channel, Shaw‐related subfamily, member 1 |
| Cplx2†‡ | −1.28 | 0.008529506 | 0.02335726 | 0.7937901 | Complexin 2 |
| Ier5 | −1.28 | 0.012563508 | 0.030142762 | 0.7937901 | Immediate early response 5 |
| Arrdc2 | −1.28 | 0.012955907 | 0.030538168 | 0.7937901 | Arrestin domain containing 2 |
| Itprl | −1.28 | 0.013064444 | 0.0312032 | 0.7937901 | Inositol 1,4,5‐trisphosphate receptor 1 |
| Nrxnl | −1.28 | 0.024665145 | 0.035351716 | 0.7937901 | Neurexin I |
| Grin2c | −1.28 | 0.03399089 | 0.036776014 | 0.8042854 | Glutamate receptor, ionotropic, NMDA2C |
| Mylip | −1.27 | 0.021410765 | 0.035166554 | 0.7937901 | Myosin regulatory light chain interacting protein |
| Ptpn21 | −1.26 | 0.037434865 | 0.042422105 | 0.85433745 | Protein tyrosine phosphatase, non‐receptor type 21 |
| Camk4† | ‐1.25 | 0.002294663 | 0.010165026 | 0.32551256 | Calcium/calmodulin‐dependent protein kinase IV |
| Kcnh3 | −1.25 | 0.004905365 | 0.012099204 | 0.6438913 | Potassium voltage‐gated channel, subfamily H (eag‐related), member 3 |
| Cacnala | −1.24 | 0.010805262 | 0.02335726 | 0.7937901 | Calcium channel, voltage‐dependent, P/Q type, α1A subunit |
| Klf5 | −1.24 | 0.03539486 | 0.038420103 | 0.8133888 | Kruppel‐like factor 5 |
| Kcnj9 | −1.24 | 0.03978704 | 0.044782665 | 0.8609039 | Potassium inwardly rectifying channel, subfamily J, member 9 |
| Slc2a6 | −1.23 | 0.020900369 | 0.034003094 | 0.7937901 | Solute carrier family 2 (facilitated glucose transporter), member 6 |
| Efna5 | −1.23 | 0.03539486 | 0.03897731 | 0.82933736 | Ephrin A5 |
| Chka | −1.23 | 0.036945086 | 0.040957764 | 0.8425187 | Choline kinase α |
| Ppp2r5b | −1.22 | 0.01254365 | 0.02633919 | 0.7937901 | Protein phosphatase 2, regulatory subunit B |
| Epha4 | −1.22 | 0.017394949 | 0.032909703 | 0.7937901 | Eph receptor A4 |
| Phactr2 | −1.22 | 0.027841333 | 0.035731114 | 0.79680043 | Phosphatase and actin regulator 2 |
| Kcnc3 | −1.22 | 0.046944622 | 0.047048863 | 0.91154444 | Potassium voltage‐gated channel, Shaw‐related subfamily, member 3 |
| Scnlb | −1.22 | 0.049731918 | 0.049957633 | 0.99979216 | Sodium channel, voltage‐gated, type I, β |
| Pik3r6 | −1.21 | 0.012251093 | 0.023756737 | 0.7937901 | Phosphoinositide‐3‐kinase, regulatory subunit 6 |
| Npdcl | −1.21 | 0.03686852 | 0.03927016 | 0.8302656 | Neural proliferation, differentiation and control gene 1 |
| Stxbp5 | −1.21 | 0.048574124 | 0.049407627 | 0.99979216 | Syntaxin binding protein 5 |
| Κcnn2†‡ | −1.2 | 9.04 × 10−4 | 0.005928889 | 0.068098344 | Potassium intermediate/small conductance calcium‐activated channel |
| Bai2 | −1.2 | 0.006960979 | 0.021744039 | 0.7937901 | Brain‐specific angiogenesis inhibitor 2 |
| Ppmlf | −1.2 | 0.036424253 | 0.03927016 | 0.8300124 | Protein phosphatase 1F |
| Pddcl | −1.2 | 0.04594794 | 0.047048863 | 0.9028201 | Parkinson disease 7 domain containing 1 |
| Lrfn5 | −1.2 | 0.04753913 | 0.0478091 | 0.91255474 | Leucine rich repeat and fibronectin type III domain containing 5 |
| Gcat | −1.2 | 0.04825415 | 0.04882949 | 0.938082 | Glycine C‐acetyltransferase |
Over 50 genes were selected, based on their possible involvement in central nervous development from a list of over 600 probes whose expression was decreased in microarray between NeuroD2 WT and KO with a significant fold change larger than 1.2. Seven pairs of WT and KO culture tissues were included in the microarray analysis. †Genes whose expression we further assessed by qPCR; ‡genes whose change in expression was verified by qPCR (6/10).
For qPCR, 1 μg RNA was harvested from WT and KO mouse cultures and reverse transcribed with iScript cDNA Synthesis kit (Bio‐Rad, Hercules, CA, USA). cDNA was subsequently used for real‐time quantitative PCR performed in an iCycler Optical Module (Bio‐Rad) using SsoFast EvaGreen Supermix (Bio‐Rad). Technical replicates as well as no template and no RT negative controls were included and each individual experiment was performed in triplicate samples. Expression of the gene of interest was normalized to ActinB. The sequences of the primers used were: ActinB, F: 5′‐CCACACCCGCCACCAGTTCG‐3′, R: 5′‐CTAGGGCGGCCCACGATGGA‐3′; NeuroD2 F: 5′‐CAAGAAGCGCGGGCCGAAGA‐3′, R: 5′‐TTGGCCTTCTGTCGCCGCAG‐3′; GRP F: 5′‐GTCACTGGGCTGTGGGACACTTA‐3′, R: 5′‐CTTCCCAGCGGACGTACCCC‐3′; SK2 F: 5′‐GTCGCTGTATTCTTTAGCTCTG‐3′, R: 5′‐ACGCTCATAAGTCATGGC‐3′.
Immunohistochemistry
Primary antibodies used were against GAD65 (mouse, 1:1000, Chemicon, MilliporeSigma, St. Louis, MO, USA) and the GABAARγ2 subunit (rabbit, 1:500, Chemicon). Secondary antibodies were Alexa Fluor 594 and 633‐conjugated (1:1000, Invitrogen). Cultured neurons were fixed with 4% paraformaldehyde and 2% sucrose in phosphate buffered saline (PBS) for 15 min at room temperature. Cultures were then blocked in 1× blocking buffer (1× PBS, 0.1% Triton X‐100, 5% normal donkey serum), incubated with primary antibody in blocking buffer overnight at 4°C and incubated with the secondary antibody in blocking buffer for 1 h at room temperature. Images (1024 × 1024 pixels) were acquired on a Nikon A1 confocal microscope with a 60× objective. For each set of antibodies in individual experiments, images were acquired with identical settings for laser power, detector gain, amplifier offset and pinhole diameter. Inhibitory synapse number was quantified following published protocols (Lin et al. 2008).
Acute brain slice preparation and electrophysiology
Pups (P13–16) from NeuroD2 WT and KO littermates were anaesthetized with isoflurane and rapidly decapitated. Brains were removed and immediately placed into ice‐cold artificial cerebrospinal fluid (ACSF) containing (mm): 124 NaCl, 4 KCl, 26 NaHCO3, 1.26 NaH2PO4, 6 MgCl2 and 1 CaCl2 and bubbled with 95% O2–5% CO2. Coronal slices 350 μm thick were obtained using a Leica VT1200 vibrating blade microtome. Slices were transferred to an incubation chamber containing bicarbonate‐buffered ACSF containing 3 mm MgCl2 and 2 mm CaCl2 and bubbled with 95% O2–5% CO2. All recordings were performed at room temperature. TTX 0.5 μm was included to record miniature currents. Whole‐cell pipette solution for mIPSC voltage‐clamp recordings contained (mm): 120 CsCl, 30 Hepes, 2 MgCl2, 1 CaCl2, 11 EGTA, and 4 ATP (Haam et al. 2014). For mEPSC recordings, the internal solution contained (mm): 10 CsCl, 105 CsMeSO3, 0.5 ATP, 0.3 GTP, 10 Hepes, 5 glucose, 2 MgCl2 and 1 EGTA (Hall et al. 2007). To record mEPSCs and mIPSCs from the same neurons, internal solution contained (mm): 20 KCl, 100 CsMeSO3, 10 Hepes, 4 ATP, 0.3 GTP, 10 sodium phosphocreatine and 3 QX‐314 (Maffei and Turrigiano, 2008). Reversal potentials (−49 mV for GABAergic currents and +10 mV for glutamatergic currents) were predicted and then empirically confirmed by isolating mIPSCs or mEPSCs pharmacologically (mIPSC: 20 μm 6,7‐dinitroquinoxaline‐2,3‐dione (DNQX) and 50 μm (2R)‐amino‐5‐phosphonovaleric acid (APV); mEPSC: 50 μm picrotoxin) followed by stepping the holding voltage in 5 mV increments around the predicted reversal potentials. For current‐clamp recordings, the internal solution contained (mm): 125 potassium gluconate, 8 NaCl, 5 d‐glucose, 5 Hepes, 4 ATP, 0.3 GTP, 2 MgCl2 and 1 EGTA (Ince‐Dunn et al. 2006). The liquid junction potential for each internal solution was calculated in Clampfit (Molecular Devices, Sunnyvale, CA, USA): mIPSC internal solution: 2.7 mV; mEPSC internal solution: 8.7 mV; internal solution for same cell recordings: 20 mV; and internal solution for current‐clamp recordings: 10 mV. In presented data the junction potential was only corrected for in same cell recordings. For slice recordings, layer II/III pyramidal neurons in barrel cortex were identified via IR DIC visualization. Pipette resistances ranged from 4 to 7 MΩ. Access resistance ranged from 8 to 20 MΩ and was monitored for consistency; for analysed cells change was never > 20%. Data were acquired using LabChart software (ADInstruments, Sydney, Australia) or Igor software (Wavemetrics, Lake Oswego, OR, USA) (digitization at 10 kHz and filtering at 2 kHz). Recordings were discarded if the initial leak current was above 300 pA or changed by more than 20% over the course of recording.
Statistics
Data are presented as means ± SD. For microarray and qPCR experiments, expression levels were normalized to WT and tested using Student's t‐test. The rest of the data (except for in Fig. 6 D, G and K) were nested for analysis using a Linear Mixed Model in IBM SPSS, Statistics 22 (IBM, Armonk, NY, USA). Data in Fig. 6 D, G and K were tested with two‐way ANOVA using UNIANOVA of General Linear Model in SPSS. Data from culture experiments were nested by individual experiment and individual culture (N) and overall cell number (n). Data from acute slices were nested by animal and cell number (n). Multiple comparisons were corrected by post hoc LSD test. *P < 0.05, **P < 0.01 and ***P < 0.001.
Figure 6. Increased intrinsic excitability and higher spontaneous AP rates in layer II/III pyramidal neurons of NeuroD2 null neurons .
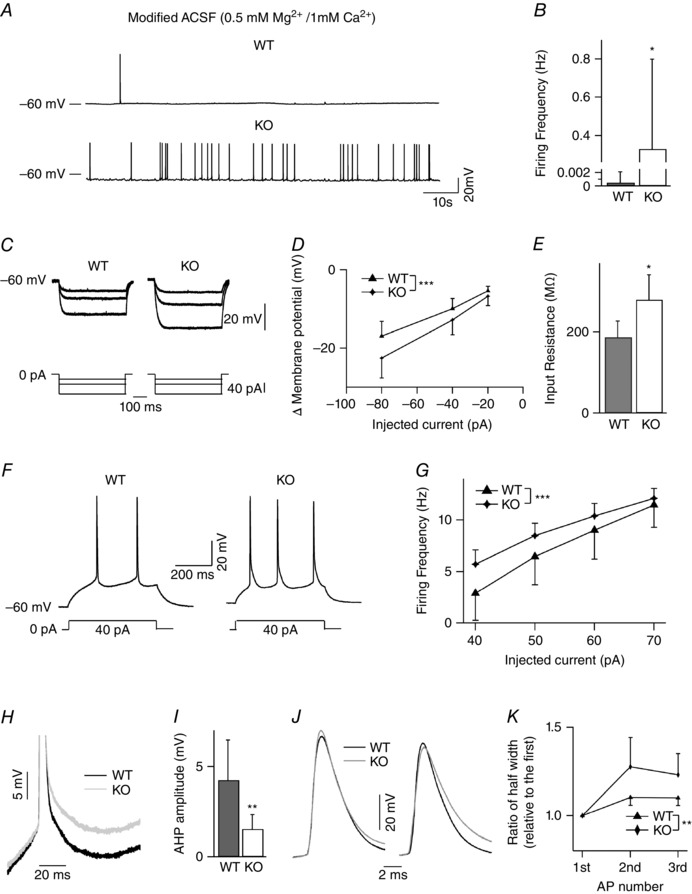
A, representative spontaneous AP firing for WT and NeuroD2 null cells. B, average spontaneous firing rate was significantly higher in NeuroD2 KO neurons than WT neurons (n = 10 for WT and n = 12 for KO; *P < 0.05). C, superimposed voltage traces depicting the response to 500 ms hyperpolarizing step currents in one WT and KO neurons. D, current–voltage (I–V) curves of the steady‐state voltage in relation to injected currents (n = 6 for both WT and KO; ***P < 0.001). E, input resistance of mutant neurons was significantly higher than WT neurons (*P < 0.05). F, example current‐clamp recordings in response to 40 pA depolarizing current steps for WT and KO layer II/III cortical neurons. G, average firing frequency as a function of the current injected showing an increase in NeuroD2 KO neurons (n = 6 for both WT and KO; ***P < 0.001). H, representative traces of AHP after the first AP (in 8 Hz AP trains) in NeuroD2 WT and KO neurons. I, average data showed decreased AHP after the first AP in NeuroD2 KO neurons compared to WT neurons (**P < 0.01). J, representative traces of the first and third APs (in 8 Hz AP trains) from NeuroD2 WT and KO trains shown at expanded time base. K, the ratio of the half‐widths of the second and the third APs relative to the first AP is significantly greater in the KOs (**P < 0.01).
Results
NeuroD2 promotes inhibitory synapses onto cortical pyramidal neurons in vitro
Excitatory synaptic innervation must be countered by inhibitory synapses to maintain proper excitatory/inhibitory (E/I) balance on developing neurons (Eichler & Meier, 2008). Because NeuroD2 can be transactivated by calcium influx, which is an integrated signal of excitatory activity in neurons, we hypothesized that NeuroD2 might help balance excitation by promoting synaptic inhibition. To test if NeuroD2 is involved postsynaptically in maintaining inhibitory synaptic tone, we modified its expression in cultured rat cortical neurons by transfecting with NeuroD2‐cDNA (overexpression) or plasmid‐based RNAi constructs targeting NeuroD2 (knockdown) (Wilke et al. 2012) at 8 days in vitro (DIV) and then recording miniature IPSCs (mIPSCs) between 18 and 20 DIV. In all experiments, cells were co‐transfected with GFP for identification and compared with GFP‐alone expressing control neurons. Overexpression of NeuroD2 significantly increased mIPSC frequency while NeuroD2 knockdown decreased mIPSC frequency (Fig. 1 A–C, n = 30 cells from N = 8 multiple embryo‐derived individual cultures for GFP, n = 36 cells from N = 11 for NeuroD2‐cDNA and n = 26 cells, N = 9 for NeuroD2‐RNAi; P = 0.021 between GFP and NeuroD2‐cDNA, P = 0.004 between GFP and NeuroD2‐RNAi, all in 4 individual experiments). Meanwhile there was a minor change in mIPSC amplitude (Fig. 1 A–C, P = 0.012 between GFP and NeuroD2‐cDNA, P = 0.06 between GFP and NeuroD2‐RNAi). Together these data suggest that NeuroD2 positively regulates inhibitory synapse number onto developing cortical pyramidal neurons.
Figure 1. NeuroD2 regulates inhibitory synaptic development through GRP‐GRPR signalling in vitro .
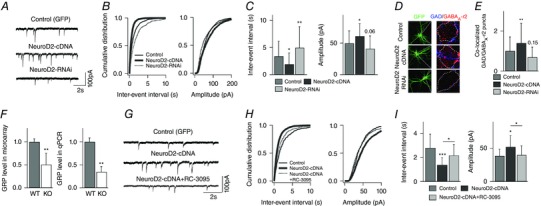
A, representative mIPSCs recorded from cultured cortical neurons transfected with GFP (control), NeuroD2‐cDNA or NeuroD2‐RNAi plasmid. Scale bar, 100 pA, 2 s. B, cumulative distributions of mIPSC inter‐event intervals and amplitudes from neurons under the same conditions as in A. C, summary of mIPSC inter‐event intervals and amplitudes from neurons under the same conditions as in A (n = 30 for GFP, n = 36 for NeuroD2‐cDNA and n = 26 for NeuroD2‐RNAi. *P < 0.05; **P < 0.01). D, representative images of immunofluorescent labeling of GAD65 and GABAA‐γ2 in control, NeuroD2‐cDNA or NeuroD2‐RNAi neurons. E, summary of inhibitory synaptic puncta analysis in control, NeuroD2‐cDNA or NeuroD2‐RNAi neurons (n = 36 for GFP, n = 46 for NeuroD2‐cDNA and n = 32 for NeuroD2‐RNAi, **P < 0.01). F, GRP mRNA levels measured by microarray (n = 7 for both WT and KO) and by qPCR (n = 3 experiments each in triplicate format for both WT and KO) in 14 DIV cultured cortical neurons (**P < 0.01). G, representative mIPSCs recorded from cultured cortical neurons transfected with GFP and NeuroD2‐cDNA in a separate set of experiments. The GRPR antagonist RC‐3095 was added into the growth medium after transfection (scale bar, 100 pA, 2 s). H, cumulative distributions of mIPSC inter‐event intervals and amplitudes recorded from neurons transfected with GFP, NeuroD2‐cDNA or NeuroD2‐cDNA+RC‐3095. I, summary of mIPSC inter‐event intervals and amplitudes recorded from neurons transfected with GFP, NeuroD2‐cDNA or NeuroD2‐cDNA+RC‐3095 (n = 8 for GFP, n = 14 for NeuroD2‐cDNA and n = 11 for NeuroD2‐cDNA+RC‐3095; *P < 0.05, ***P < 0.001).
To support our inference that a change in mIPSC frequency is due to a change in inhibitory synapse number, we used antibodies against GAD65 and GABA receptor to anatomically define inhibitory synapses by measuring colocalization of these markers on GFP transfected pyramidal neurons (Lin et al. 2008; Hong et al. 2008). As in the electrophysiological recordings, cultured rat cortical neurons were transfected at 8 DIV but then fixed and immunostained between 18 and 20 DIV with antibodies that recognize the presynaptic inhibitory marker GAD65 and the postsynaptic inhibitory marker GABAA receptor γ2 subunit (GABAARγ2). The number of anatomically defined inhibitory synapses was estimated by measuring the number of co‐localized GAD65 and GABAARγ2 puncta overlapping with the somatic GFP signal. Consistent with the mIPSC data, overexpression of NeuroD2 increased the number of anatomically defined inhibitory synapses while knockdown of NeuroD2 showed only a trend to decrease (Fig. 1 D and E, n = 36 cells, N = 7 for GFP; n = 46 cells, N = 7 for NeuroD2‐cDNA; n = 32 cells, N = 8, for NeuroD2‐RNAi; P = 0.008 between GFP and NeuroD2‐cDNA, P = 0.15 between GFP and NeuroD2‐RNAi, all from 3 independent experiments).
NeuroD2 promotes inhibitory synapses through regulation of GRP expression
In parallel experiments, we employed microarray analysis in an attempt to identify gene targets of NeuroD2 transcription that might be involved in synapse development. Interestingly, in our microarray screen performed at 14 DIV on cultured NeuroD2 WT and KO mouse cortical neurons, we found that gastrin‐releasing peptide (GRP) was one of the strongest affected mRNAs in terms of fold change, being significantly decreased in NeuroD2 KO neuronal cultures compared to WT control cultures (Table 1). This decrease in GRP mRNA was confirmed by qPCR analysis (Fig. 1 F, n = 7 samples for each WT and KO mouse cultures in microarray, P = 0.001; t test and n = 3 samples, each sample run in triplicate, for each WT and KO mouse cultures in qPCR, P = 0.002). We also observed a decrease in GRP mRNA in cortical tissue taken from P14 KO animals compared to WT littermates (WT = 1 ± 0.05 vs. KO = 0.66 ± 0.1; mean ± SD, P = 0.02). GRP has a positive effect on inhibitory neurotransmission through activation of its receptor GRPR (Shumyatsky et al. 2002; Cao et al. 2010). To test the involvement of GRP‐GRPR in mediating the effects of NeuroD2 overexpression in regulating inhibitory synaptic tone, we used the GRPR antagonist RC‐3095 (2 μm) (Shumyatsky et al. 2002). Chronic incubation (8–18 DIV) with RC‐3059 partially reversed the increase in mIPSC frequency in NeuroD2 overexpressing cultured rat cortical neurons, supporting the hypothesis that the GRP‐GRPR system is involved downstream of NeuroD2 to regulate inhibitory synapse development (Fig. 1 G–I, n = 8 cells for GFP, n = 14 cells for NeuroD2‐cDNA and n = 11 cells for NeuroD2‐cDNA+RC‐3095, N = 2–4 for each condition; P = 0.0008 between GFP and NeuroD2‐cDNA, P = 0.02 between NeuroD2‐cDNA and NeuroD2‐cDNA+RC‐3095). Interestingly, RC‐3059 alone did not decrease mIPSC frequency significantly in control neurons (data not shown), suggesting that basal activity of GRP‐GRPR signalling is low in cultured cortical neurons and that it needs to be activated or enhanced, such as under NeuroD2 overexpressing conditions, in order to see an effect.
In vivo deletion of NeuroD2 decreases frequency of inhibitory and excitatory quantal neurotransmission
To confirm that NeuroD2 also regulates inhibitory synapse development in vivo, we generated acute brain slices from a NeuroD2 KO mouse line (Olson et al. 2001) and recorded mIPSCs ex vivo from layer II/III cortical neurons in somatosensory barrel cortex at postnatal day (P)13–16. Consistent with our in vitro knockdown data, we observed a decrease in mIPSC frequency in KO neurons compared to WT neurons in acute brain slices generated from littermate control animals (Fig. 2, n = 24 cells for WT and n = 32 cells for KO from four pairs of littermate animals; P = 0.04). Prior studies demonstrated that excitatory synapse maturation is altered in NeuroD2 KO animals (Ince‐Dunn et al. 2006; Wilke et al. 2012). To determine how loss of NeuroD2 affects synaptic E/I balance in layer II/III neurons, we also recorded mEPSCs from the same brain area in KO and WT animals. We observed a decrease in mEPSC frequency in KO animals compared to WT controls (Fig. 3, n = 28 cells for WT and n = 32 cells for KO from four pairs of littermate animals, P = 0.0005). Because NeuroD2 KO animals are prone to seizures, suggestive of an increased E/I ratio, such a compensatory decrease in excitatory transmission in KO animals would not necessarily be predicted. Thus, we wanted to examine this question further.
Figure 2. Reduced frequency of inhibitory quantal transmission in layer II/III pyramidal neurons of NeuroD2 null mice .
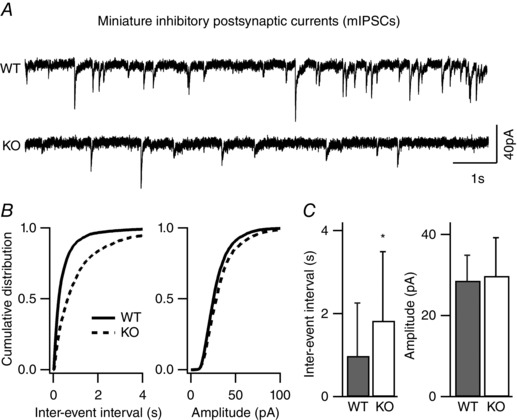
mIPSCs recorded from layer II/III pyramidal neurons in the presence of TTX, APV, and DNQX. A, sample traces from WT and mutant neurons voltage clamped at −70 mV (scale bar, 40 pA, 1 s). B, cumulative distributions of inter‐event interval (IEI) and amplitude from all mIPSCs events of 24 WT neurons and 32 mutant neurons showed a significant rightward shift in the mIPSC frequency for mutant neurons. C, average data of mIPSC inter‐event intervals and amplitudes showed a significant decrease in mIPSC frequency (increased IEI) in KO neurons (n = 24 for WT and n = 32 for KO; *P < 0.05).
Figure 3. Reduced frequency of excitatory quantal transmission in layer II/III pyramidal neurons of NeuroD2 null mice .
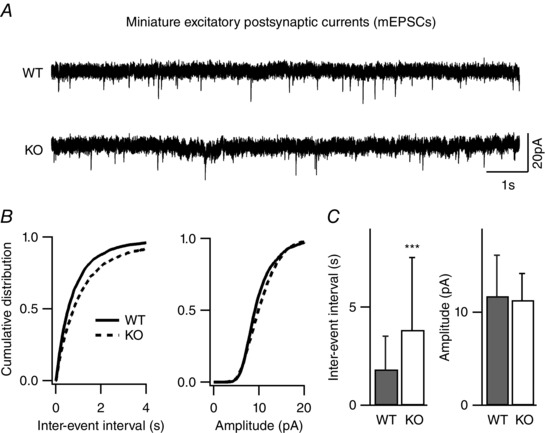
mEPSCs recorded from layer II/III pyramidal neurons in the presence of TTX, and picrotoxin. A, sample traces from WT and mutant neurons voltage clamped at −65 mV (scale bar, 20 pA, 1 s). B, cumulative distributions of inter‐event interval and amplitude from all mEPSCs events of 28 WT neurons and 32 mutant neurons showed a significant rightward shift in the mEPSC frequency for mutant neurons. C, average data of mEPSC inter‐event intervals and amplitudes showed a significant decrease in mEPSC frequency (increased IEI) in KO neurons (n = 28 for WT and n = 32 for KO; ***P < 0.001).
Since these mEPSC and mIPSC measurements are mean values across many cells and may not properly reflect the absolute E/I balance onto individual neurons, we also measured E/I synaptic balance at the level of individual cortical neurons. To do this we recorded mIPSCs and mEPSCs from single somatosensory layer II/III pyramidal neurons in brain slices by changing the holding potential in voltage‐clamp between the reversal potentials for inhibitory and excitatory synaptic currents (−49 mV and +10 mV, determined empirically, see Methods) to isolate excitatory and inhibitory currents, respectively. Interestingly, we observed a similar effect at the single cell level, which was a decrease in both mIPSC and mEPSC frequency (Fig. 4 A–E, n = 11 cells for WT and n = 13 cells for KO recorded from two pairs of littermate animals; P = 0.02 for mIPSC frequency comparison and P = 0.04 for mEPSC comparison). To determine whether there is an overall change in E/I balance in NeuroD2 KO neurons compared to WT neurons, we also calculated an E/I ratio based upon both frequency and amplitude values and saw no difference between NeuroD2 WT and KO neurons (Fig. 4 F, P = 0.76 for E/I ratio in frequency, P = 0.07 for E/I ratio in amplitude).
Figure 4. Frequency of both inhibitory and excitatory quantal events is reduced in layer II/III pyramidal neurons of NeuroD2 null mice .
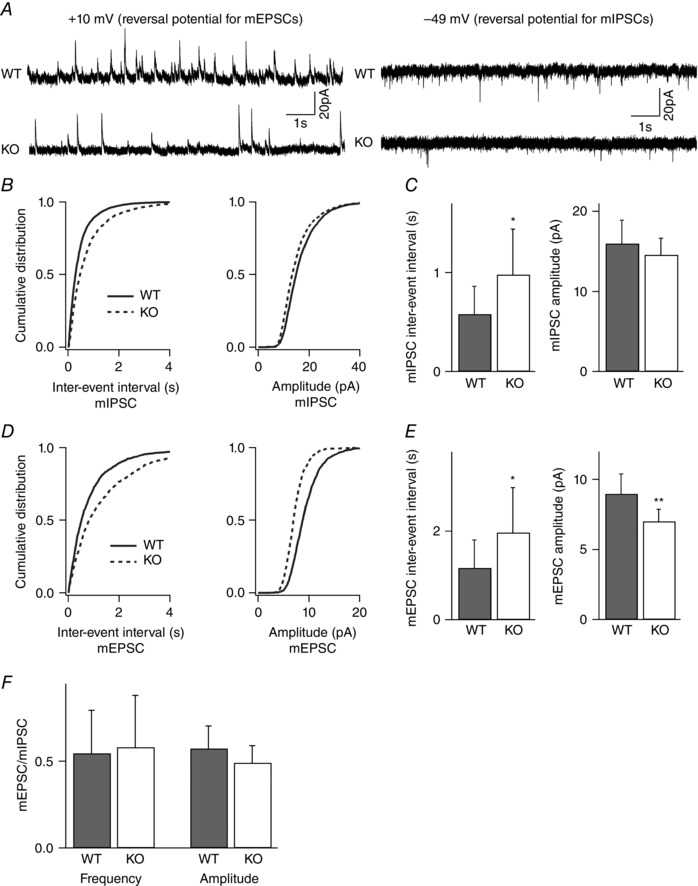
mIPSCs and mEPSCs recorded from the same layer II/III pyramidal neurons in the presence of TTX. A, sample traces from WT and mutant neurons voltage clamped at +10 mV for mIPSCs and −49 mV for mEPSCs (scale bar, 20 pA, 1 s). B, cumulative distributions of inter‐event interval and amplitude from all mIPSCs events of 11 WT neurons and 13 mutant neurons showed a significant rightward shift in the mIPSC frequency for mutant neurons. C, average data of mIPSC inter‐event intervals and amplitudes showed a significant decrease in mIPSC frequency (increased IEI) in KO neurons (n = 11 for WT and n = 13 for KO; *P < 0.05). D, cumulative distributions of inter‐event interval and amplitude from all mEPSCs events of 11 WT neurons and 13 mutant neurons showed a significant rightward shift in the mEPSC frequency and a leftward shift in the mEPSC amplitude for mutant neurons. E, average data of mEPSC inter‐event intervals and amplitudes showed a significant decrease in mEPSC frequency (increased IEI) and amplitude in KO neurons (n = 11 for WT and n = 13 for KO; *P < 0.05, **P < 0.01). F, the ratio of mEPSC to mIPSC IEI and amplitude was unchanged in NeuroD2 mutant neurons compared to WT.
In vivo deletion of NeuroD2 does not alter spontaneous neurotransmission measured ex vivo
The data above clearly suggest that inhibitory synapse number is decreased in the absence of NeuroD2 but that this is matched by a decrease in excitatory synapse number, potentially in a homeostatic manner to maintain E/I input ratio between KO and WT animals. This was surprising to us and seemingly incongruent with evidence from previous studies showing that NeuroD2 KO animals are hyperexcitable and prone to seizures. We next turned to measuring spontaneous neurotransmission, since this is a better indicator of network levels of excitability. We recorded sIPSCs and sEPSCs from single somatosensory layer II/III pyramidal neurons by alternating the holding potential in voltage‐clamp between the reversal potentials for inhibitory and excitatory synaptic currents, as before, but in the absence of TTX. Strikingly, we observed no change in either frequency or amplitude of sIPSCs and sEPSCs between NeuroD2 WT and KO neurons (Fig. 5 A–E, n = 14 cells for WT and n = 13 cells for KO from two pairs of littermate animals; P = 0.82 and P = 0.87 for sIPSC frequency and amplitude, respectively, P = 0.37 and 0.64 for sEPSC frequency and amplitude, respectively).
Figure 5. Spontaneous inhibitory and excitatory synaptic neurotransmission in layer II/III pyramidal neurons is unchanged in the absence of NeuroD2 .
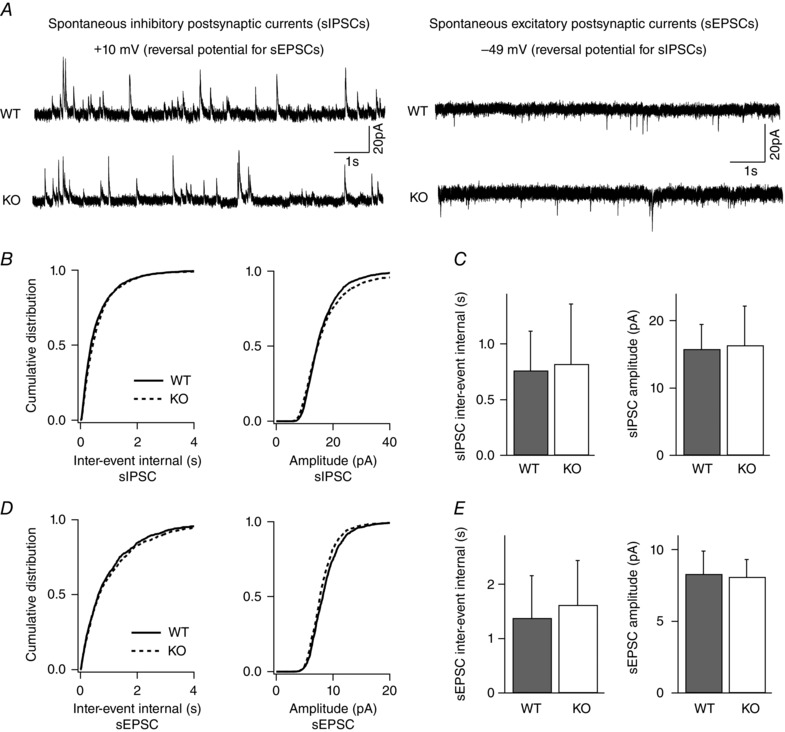
sIPSCs and sEPSCs recorded from single layer II/III pyramidal neurons. A, sample traces from a WT and KO neuron voltage clamped at +10 mV for sIPSCs and −49 mV for sEPSCs (scale bar, 20 pA, 1 s). B, cumulative distributions of inter‐event intervals and amplitudes of all sIPSCs events for 14 WT neurons and 13 KO neurons revealed no difference between WT and mutant neurons. C, average data of sIPSC inter‐event intervals and amplitudes in WT and mutant neurons (n = 14 cells for WT and n = 13 cells for KO; P = 0.82 and P = 0.87 for sIPSC frequency and amplitude, respectively). D, cumulative distributions of inter‐event intervals and amplitudes from all sEPSC events of 14 WT neurons and 13 mutant neurons showed no difference between WT and mutant neurons. E, average data of sEPSC inter‐event intervals and amplitudes in WT and mutant neurons (n = 14 cells for WT and n = 13 cells for KO; P = 0.37 and 0.64 for sEPSC frequency and amplitude, respectively).
NeuroD2 regulates the intrinsic excitability of developing pyramidal neurons
Despite a reduction in frequency of mIPSCs and mEPSCs in NeuroD2 KO neurons, the frequency of sIPSC and sEPSC remained unchanged. Excitability is also controlled by cell‐intrinsic parameters, which are not reflected in mEPSC and mIPSC recordings. Therefore, a lack of reduction in spontaneous event frequency could suggest an increase in AP frequency in the network, which is also suggested by a previous report where NeuroD2 KO animals displayed low seizure threshold (Olson et al. 2001). To determine whether or not the overall excitability of cortical neurons is altered after the loss of NeuroD2 function, we measured spontaneous action potential firing in somatosensory layer II/III pyramidal neurons in barrel cortex from NeuroD2 KO and WT littermates in acute slices. As layer II/III pyramidal neurons incubated in standard ACSF exhibit very low levels of spontaneous activity, we adopted a modified ACSF recipe which is believed to be more similar to in vivo rodent CSF than standard ACSF (Nani et al. 2005). This modified ACSF was applied to both WT and KO neurons to assess AP activity between these genotypes. Since these recordings are done under different conditions from the sEPSC/IPSC analysis it is not directly comparable but rather these experiments were meant to measure relative differences in AP firing. AP analysis revealed a significant increase in the mean firing rate of NeuroD2 KO compared with WT neurons (Fig. 6 A and B; n = 10 cells for WT and n = 12 cells for KO from two pairs of littermate animals; P = 0.04). To determine the underlying mechanisms associated with the changes in AP rates in layer II/III pyramidal neurons, we injected a series of 500 ms current steps and recorded the voltage response in current‐clamp recording in the presence of pharmacological blockers of excitatory and inhibitory synaptic transmission (APV, DNQX and picrotoxin) (Fig. 6 C). Voltage responses were plotted against injected current amplitude and revealed that NeuroD2 KO neurons responded more strongly to current steps than WT neurons (Fig. 6 D; n = 6 cells for each WT and KO from two pairs of littermate animals; P = 0.0003). Membrane input resistance, calculated from the slope of the subthreshold I–V curve, was significantly higher in NeuroD2 KO neurons than WT neurons (Fig. 6 E, P = 0.01). To compare intrinsic excitability between WT and KO neurons, a series of 500 ms depolarizing current steps were injected to elicit AP trains in the presence of synaptic blockers. Although there was no significant difference between WT and KO neurons in many parameters such as resting membrane potential measured after break‐in, AP threshold, AP amplitude and half‐width amplitude of the first evoked AP of a train, the number of APs evoked as a function of current injected was significantly increased in NeuroD2 KO neurons (Fig. 6 F and G; P = 0.0007). Notably, we also observed a significant decrease in the amplitude of the after‐hyperpolarization (AHP) following the first AP in NeuroD2 KO neurons compared to WT cells (Fig. 6 H and I; P = 0.009). Moreover, the ratio of the half‐widths of the second and the third APs relative to the first AP of a train was significantly greater in NeuroD2 KO neurons (Fig. 6 J and K; P = 0.003).
NeuroD2 regulates AHP partially through SK2 gene expression
The above changes in AP parameters suggested to us an activity‐dependent decrement in repolarization in NeuroD2 null cells. In line with this, we noted in our microarray data that mRNA transcript expression of one member of the small conductance Ca2+‐activated K+ channel subfamily, SK2 (Kcnn2), was significantly decreased in NeuroD2 KO cultures. Furthermore, we could confirm this difference by qPCR analysis of mouse cortical cultures (Fig. 7 A; n = 7 samples from each WT and KO mouse cultures for microarray, P = 0.003, t test; and n = 5 samples from each WT and KO mouse cultures for qPCR, P = 0.04). We also observed a strong trend to decreased expression of SK2 in KO P14 animals compared to littermate WT cortex (WT = 1 ± 0.1 vs. KO = 0.73 ± 0.04; mean ± SD, P = 0.06). To test the hypothesis that NeuroD2 regulates the intrinsic excitability of cortical pyramidal neurons through control of SK2 levels, we introduced NeuroD2‐cDNA into cultured cortical neurons and then acutely treated cells with the SK2 specific antagonist apamin (300 nm). As predicted, overexpression of NeuroD2 significantly increased AHP amplitude after the first AP of evoked trains, and this was partially dependent on Kcnn2 activity as shown by its sensitivity to apamin (Fig. 7 B and C; n = 15 cells for GFP, n = 11 cells for NeuroD2‐cDNA, and n = 9 cells for NeuroD2‐cDNA + apamin from N = 2–3 for each condition; P = 0.0001 between GFP and NeuroD2‐cDNA, P = 0.047 between NeuroD2‐cDNA and NeuroD2‐cDNA + apamin). These data suggest that NeuroD2 regulates neuronal AHP, and therefore intrinsic excitability (at least in part) by regulating SK2 gene expression.
Figure 7. NeuroD2 regulates AHP through SK2 .
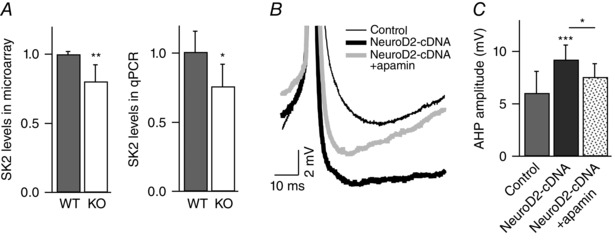
A, SK2 mRNA levels are decreased in NeuroD2 KO tissue as measured by microarray (n = 7 for both WT and KO) and qPCR (n = 5 experiments with each in triplicate for both WT and KO). Data are from 14 DIV cultured cortical neurons (*P < 0.05; **P < 0.01). B, representative traces of AHP after the first AP (in 8 Hz AP trains) in control, NeuroD2‐cDNA, and NeuroD2‐cDNA + apamin. C, summary of AHP measurements after the first AP in control, NeuroD2‐cDNA and NeuroD2‐cDNA + apamin (*P < 0.05, ***P < 0.001).
Discussion
Our experiments show that NeuroD2 regulates the developmental excitability of cortical pyramidal neurons by influencing both the amount of inhibitory synaptic innervation they receive and their cell‐intrinsic excitability. In NeuroD2 KO neurons, we observed a decrease in both excitatory and inhibitory synaptic tone, but also increased cell‐intrinsic excitability. Together, these data not only provide insight about the links between synaptic and cell‐intrinsic excitability but also clearly point to a critical role for NeuroD2 in coordinating these processes and maintaining proper levels of neuronal excitability in developing cortex.
In addition to describing a role for NeuroD2 in maintaining neuronal excitability, we identified potential gene targets that could be involved mechanistically. Using microarray analysis and qPCR confirmation, we identified GRP and SK2 as transcripts that are decreased in the absence of NeuroD2. Knowledge of the direct transcriptional targets of NeuroD2 in neurons has been lacking and to our knowledge thus far, this is the first evidence of an activity‐dependent transcription factor that regulates both synaptic and cell‐intrinsic properties of pyramidal neuron development.
Our data suggest that NeuroD2 promotes synaptic inhibition in a manner that requires GRPR activation. GRPR is the receptor for GRP, which is a 29 amino acid‐long mammalian homolog of the amphibian peptide bombesin and may serve as a cotransmitter with glutamate in pyramidal neurons in the rodent brain (Lee et al. 1999; Shumyatsky et al. 2002; Cao et al. 2010). GRP expresses across many brain areas including cortex, hippocampus and amygdala (Wada et al. 1990), and is required for proper social behaviour (Merali et al. 2014), adult neurogenesis (Walton et al. 2014) and fear conditioned learning (Bédard et al. 2007). GRP released from pyramidal neurons acts on GRPRs expressed on inhibitory neurons to modulate GABAergic vesicle release (Shumyatsky et al. 2002). We confirmed that NeuroD2 also expresses exclusively in pyramidal neurons in mouse cortex (data not shown). These data suggest that NeuroD2 supports GRP expression to promote inhibitory synapse number as measured by mIPSC frequency. While mIPSC frequency can reflect a change in synapse number it is also dependent upon presynaptic vesicle release probability. While we did not test inhibitory synapse release probability, our anatomical synapse staining data support the idea that the number of inhibitory synapses onto NeuroD2 null cells is decreased.
As mentioned above, NeuroD2 KO also resulted in a decrease in mEPSC frequency. However, based upon the absence of any obvious regulators of excitatory synapse development on our NeuroD2 KO microarray along with the observations that NeuroD2 overexpression in cultured cortical neurons decrease mEPSC frequency (data not shown) and synaptic scaling is intact in NeuroD2 KO neurons (data not shown), we infer that this may be a compensatory effect, due to a homeostatic plasticity mechanism in response to decreased inhibitory synaptic tone. Despite a decrease in both inhibitory and excitatory synaptic events, NeuroD2 KO mice have low seizure thresholds when induced by kainic acid (Olson et al. 2001). How might a perceived balanced decrease in synaptic E/I input lead to seizure susceptibility? Neurons also regulate their excitability through cell‐intrinsic mechanisms, which are developmentally regulated and susceptible to plasticity (Turrigiano, 2011). AP‐mediated Ca2+ influx opens potassium channels that control AP firing properties. While delayed‐rectifier potassium channels are responsible for the majority of repolarization during a single AP, calcium‐activated potassium channels determine the degree of AHP and become more dominant during sustained firing (Adelman et al. 2012). SK2 (also known as KCa2.2) is encoded by the KCNN2 gene and is a member of the potassium intermediate/small conductance calcium‐activated channel subfamily. It is exclusively activated by intracellular calcium ions and triggers potassium flux to regulate cell‐intrinsic excitability. The channel is important for learning and memory and is expressed in neurons across different brain areas including pyramidal neurons in all layers of the cortex (Sailer et al. 2004; Gymnopoulo et al. 2014). Activation of SK2 channels contributes to AHPs of medium duration (mAHPs) and directly affects neuronal intrinsic excitability. The decreased expression of SK2 in NeuroD2 KO neurons predicts a hyper‐excitation phenotype and could contribute to epileptic activity in these animals. In addition, we observed an increase in input impedance in NeuroD2 null neurons. It remains to be determined if this is related to decreased expression of a direct transcriptional target of NeuroD2.
NeuroD2 is a neuronal activity‐dependent transcription factor. In our unpublished data we have seen Npas4 expression is increased dramatically following KCl stimulation in WT cortical cultures and that this increase is significantly attenuated in NeuroD2 null cultures. Yet, under conditions of basal activity we observed no difference in Npas4 levels between NeuroD2 KO and WT cultures (data not shown). Moreover, in preliminary experiments we noted that if we knock down Npas4 with a siRNA plasmid in single cortical pyramidal neurons in vitro, this can block the ability of NeuroD2 overexpression to increase mIPSC event number (data not shown). Bidirectional manipulation of the activity‐dependent transcription factor Npas4 results in similar effects on mIPSC frequency in hippocampal neurons in vitro; increased inhibition in response to increased Npas4 levels and decreased inhibitory events following knockdown of Npas4 (Lin et al. 2008). It is also of interest to note that Npas4 has been shown to be required for synaptic changes downstream of changes in cell‐intrinsic excitability in adult‐born neurons (Sim et al. 2013). Together these data suggest a complex interaction between Npas4 and NeuroD2. Understanding the relationship and potential coordination of these two transcription factors in regulating synaptic development will be a very interesting avenue for future research.
In summary, our data point to NeuroD2 as a critical regulator of cortical pyramidal neuron excitability, consistent with previously reported increased susceptibility to seizure in the NeuroD2 KO animals. We also found these animals exhibit altered social behavioure (data not shown). The data raise very compelling questions including how other aspects of intrinsic excitability and synaptic innervation are linked. It will be interesting to determine in future experiments what type of inhibition is specifically regulated by NeuroD2 transcription and discover exactly what the mechanism of stabilization is downstream of GRPR activation. Furthermore, are these processes recapitulated in adult‐born neurons as it is believed that similar developmental processes must be evoked during circuit integration in the developed brain (Scobie et al. 2009) Recent work, published during preparation and review of this article, has used ChIP‐seq to identify gene expression changes during embryonic development (E14.5) in NeuroD2 null embryos (Bayam et al. 2015). Interestingly, taken together with our results, the data suggest that NeuroD2 may affect expression of different genes depending upon developmental age of neurons. Future studies will be required to determine the answers to these questions and piece together the multiple ways in which cortical pyramidal neurons maintain E/I balance and cell excitability including via regulation of NeuroD2 levels during postnatal synapse formation. A complete understanding of these mechanisms will be critical for understanding autism spectral disorders and epilepsy and thus determining therapeutic interventions.
Additional information
Competing interests
The authors declare no conflict of interest.
Author contributions
F.C.: conception and design; collection and assembly of data; data analysis and interpretation; manuscript writing. J.M.: collection and assembly of data. Y.Z.: collection and assembly of data. K.A.: collection and assembly of data. D.Y.: collection and assembly of data. L.S.: provision of study materials. P.D.: provision of study materials; data analysis and interpretation. F.J.: provision of study materials. B.H.: conception and design; financial support; collection and assembly of data; data analysis and interpretation; manuscript writing. Experiments were performed at Tulane University. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was funded by NIMH R01 (MH099378‐01), NSF CAREER Award (0952455) and Brain and Behavior Research Foundation YIA (18996) all to B.J.H.
References
- Adelman JP, Maylie J & Sah P (2012). Channels: Form and function. Annu Rev Physiol 74, 245–269. [DOI] [PubMed] [Google Scholar]
- Bayam E, Sahin GS, Guzelsoy G, Guner G, Kabakcioglu A & Ince‐Dunn G (2015). Genome‐wide target analysis of NEUROD2 provides new insights into regulation of cortical projection neuron migration and differentiation. BMC Genomics 16, 681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bédard T, Mountney C, Kent P, Anisman H & Merali Z (2007). Role of gastrin‐releasing peptide and neuromedin B in anxiety and fear‐related behaviour. Behav Brain Res 179, 133–140. [DOI] [PubMed] [Google Scholar]
- Cao X, Mercaldo V, Li P, Wu L‐J & Zhuo M (2010). Facilitation of the inhibitory transmission by gastrin‐releasing peptide in the anterior cingulate cortex. Mol Pain 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler SA & Meier JC (2008). E‐I balance and human diseases – from molecules to networking. Front Mol Neurosci 1, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Junco‐Clemente P, Chow DK, Tring E, Lazaro MT, Trachtenberg JT & Golshani P (2013). Overexpression of calcium‐activated potassium channels underlies cortical dysfunction in a model of PTEN‐associated autism. Proc Natl Acad Sci USA 110, 18297–18302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymnopoulos M, Cingolani LA, Pedarzani P & Stocker M (2014). Developmental mapping of small‐conductance calcium‐activated potassium channel expression in the rat nervous system. J Comp Neurol 522, 1072–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haam J, Halmos KC, Di S & Tasker JG (2014). Nutritional state‐dependent ghrelin activation of vasopressin neurons via retrograde trans‐neuronal‐glial stimulation of excitatory GABA circuits. J Neurosci 34, 6201–6213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall BJ, Ripley B & Ghosh A (2007). NR2B signaling regulates the development of synaptic AMPA receptor current. J Neurosci 27, 13446–13456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W & Jones FE (2014). HER4 selectively coregulates estrogen stimulated genes associated with breast tumor cell proliferation. Biochem Biophys Res Commun 443, 458–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoischen A, Krumm N & Eichler EE (2014). Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat Neurosci 17, 764–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE & Greenberg ME (2008). A biological function for the neuronal activity‐dependent component of Bdnf transcription in the development of cortical inhibition. Neuron 60, 610–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I & Kageyama R (2014). bHLH factors in self‐renewal, multipotency, and fate choice of neural progenitor cells. Neuron 82, 9–23. [DOI] [PubMed] [Google Scholar]
- Ince‐Dunn G, Hall BJ, Hu S‐C, Ripley B, Huganir RL, Olson JM, Tapscott SJ & Ghosh A (2006). Regulation of thalamocortical patterning and synaptic maturation by NeuroD2. Neuron 49, 683–695. [DOI] [PubMed] [Google Scholar]
- Kohwi M & Doe CQ (2013). Temporal fate specification and neural progenitor competence during development. Nat Rev Neurosci 14, 823–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BH, Smith T & Paciorkowski AR (2015). Autism spectrum disorder and epilepsy: Disorders with a shared biology. Epilepsy Behav 47, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Dixon AK, Gonzalez I, Stevens EB, McNulty S, Oles R, Richardson PJ, Pinnock RD & Singh L (1999). Bombesin‐like peptides depolarize rat hippocampal interneurones through interaction with subtype 2 bombesin receptors. J Physiol 518, 791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Bloodgood BL, Hauser JL, Lapan AD, Koon AC, Kim T‐K, Hu LS, Malik AN & Greenberg ME (2008). Activity‐dependent regulation of inhibitory synapse development by Npas4. Nature 455, 1198–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A & Turrigiano GG (2008). Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci 28, 4377–4384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick MB, Tamimi RM, Snider L, Asakura A, Bergstrom D & Tapscott SJ (1996). NeuroD2 and neuroD3: distinct expression patterns and transcriptional activation potentials within the neuroD gene family. Mol Cell Biol 16, 5792–5800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merali Z, Presti‐Torres J, MacKay JC, Johnstone J, Du L, St‐Jean A, Levesque D, Kent P, Schwartsmann G, Roesler R, Schroder N & Anisman H (2014). Long‐term behavioural effects of neonatal blockade of gastrin‐releasing peptide receptors in rats: Similarities to autism spectrum disorders. Behav Brain Res 263, 60–69. [DOI] [PubMed] [Google Scholar]
- Nani VS, Chang Q, Maffei A, Turrigiano GG, Jaenisch R & Nelson SB (2005). Reduced cortical activity due to a shift in the balance between excitation and inhibition in a mouse model of Rett syndrome. Proc Natl Acad Sci USA 102, 12560–12565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson JM, Asakura A, Snider L, Hawkes R, Strand A, Stoeck J, Hallahan A, Pritchard J & Tapscott SJ (2001). NeuroD2 is necessary for development and survival of central nervous system neurons. Dev Biol 234, 174–187. [DOI] [PubMed] [Google Scholar]
- Sailer CA, Kaufmann WA, Marksteiner J & Knaus HG (2004). Comparative immunohistochemical distribution of three small‐conductance Ca2+‐activated potassium channel subunits, SK1, SK2, and SK3 in mouse brain. Mol Cell Neurosci 26, 458–469. [DOI] [PubMed] [Google Scholar]
- Scobie KN, Hall BJ, Wilke SA, Klemenhagen KC, Fujii‐Kuriyama Y, Ghosh A, Hen R & Sahay A (2009). Krüppel‐like factor 9 is necessary for late‐phase neuronal maturation in the developing dentate gyrus and during adult hippocampal neurogenesis. J Neurosci 29, 9875–9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shumyatsky GP, Tsvetkov E, Malleret G, Vronskaya S, Hatton M, Hampton L, Battey JF, Dulac C, Kandel ER & Bolshakov VY (2002). Identification of a signaling network in lateral nucleus of amygdala important for inhibiting memory specifically related to learned fear. Cell 111, 905–918. [DOI] [PubMed] [Google Scholar]
- Sim S, Antolin S, Lin CW, Lin Y & Lois C (2013). Increased cell‐intrinsic excitability induces synaptic changes in new neurons in the adulte dentate gyrus that require Npas4. J Neurosci 33, 7928–7940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G (2011). Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci 34, 89–103. [DOI] [PubMed] [Google Scholar]
- Wada E, Way J, Lebacq‐Verheyden AM & Battey JF (1990). Neuromedin B and gastrin‐releasing peptide mRNAs are differentially distributed in the rat nervous system. J Neurosci 10, 2917–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton NM, de Koning A, Xie X, Shin R, Chen Q, Miyake S, Tajinda K, Gross AK, Kogan JH, Heusner CL, Tamura K & Matsumoto M (2014). Gastrin‐releasing peptide contributes to the regulation of adult hippocampal neurogenesis and neuronal development. Stem Cells 32, 2454–2466. [DOI] [PubMed] [Google Scholar]
- Wilke SA, Hall BJ, Antonios JK, DeNardo LA, Otto S, Yuan B, Chen F, Robbins EM, Tiglio K, Williams ME, Qiu Z, Biederer T & Ghosh A (2012). NeuroD2 regulates the development of hippocampal mossy fibre synapses. Neural Dev 7, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]