

Abstract
Key points
Purkinje cells of the cerebellum receive ∼180,000 parallel fibre synapses, which have often been viewed as a homogeneous synaptic population and studied using single action potentials.
Many parallel fibre synapses might be silent, however, and granule cells in vivo fire in bursts. Here, we used trains of stimuli to study parallel fibre inputs to Purkinje cells in rat cerebellar slices.
Analysis of train EPSCs revealed two synaptic components, phase 1 and 2. Phase 1 is initially large and saturates rapidly, whereas phase 2 is initially small and facilitates throughout the train. The two components have a heterogeneous distribution at dendritic sites and different pharmacological profiles.
The differential sensitivity of phase 1 and phase 2 to inhibition by pentobarbital and NBQX mirrors the differential sensitivity of AMPA receptors associated with the transmembrane AMPA receptor regulatory protein, γ‐2, gating in the low‐ and high‐open probability modes, respectively.
Abstract
Cerebellar granule cells fire in bursts, and their parallel fibre axons (PFs) form ∼180,000 excitatory synapses onto the dendritic tree of a Purkinje cell. As many as 85% of these synapses have been proposed to be silent, but most are labelled for AMPA receptors. Here, we studied PF to Purkinje cell synapses using trains of 100 Hz stimulation in rat cerebellar slices. The PF train EPSC consisted of two components that were present in variable proportions at different dendritic sites: one, with large initial EPSC amplitude, saturated after three stimuli and dominated the early phase of the train EPSC; and the other, with small initial amplitude, increased steadily throughout the train of 10 stimuli and dominated the late phase of the train EPSC. The two phases also displayed different pharmacological profiles. Phase 2 was less sensitive to inhibition by NBQX but more sensitive to block by pentobarbital than phase 1. Comparison of synaptic results with fast glutamate applications to recombinant receptors suggests that the high‐open‐probability gating mode of AMPA receptors containing the auxiliary subunit transmembrane AMPA receptor regulatory protein γ‐2 makes a substantial contribution to phase 2. We argue that the two synaptic components arise from AMPA receptors with different functional signatures and synaptic distributions. Comparisons of voltage‐ and current‐clamp responses obtained from the same Purkinje cells indicate that phase 1 of the EPSC arises from synapses ideally suited to transmit short bursts of action potentials, whereas phase 2 is likely to arise from low‐release‐probability or ‘silent’ synapses that are recruited during longer bursts.
Key points
Purkinje cells of the cerebellum receive ∼180,000 parallel fibre synapses, which have often been viewed as a homogeneous synaptic population and studied using single action potentials.
Many parallel fibre synapses might be silent, however, and granule cells in vivo fire in bursts. Here, we used trains of stimuli to study parallel fibre inputs to Purkinje cells in rat cerebellar slices.
Analysis of train EPSCs revealed two synaptic components, phase 1 and 2. Phase 1 is initially large and saturates rapidly, whereas phase 2 is initially small and facilitates throughout the train. The two components have a heterogeneous distribution at dendritic sites and different pharmacological profiles.
The differential sensitivity of phase 1 and phase 2 to inhibition by pentobarbital and NBQX mirrors the differential sensitivity of AMPA receptors associated with the transmembrane AMPA receptor regulatory protein, γ‐2, gating in the low‐ and high‐open probability modes, respectively.
Abbreviations
- AMPAR
AMPA receptor
- d‐AP5
d‐(−)‐2‐amino‐5‐phosphonopentanoic acid
- CNQX
6‐cyano‐7‐nitroquinoxaline‐2,3‐dione
- CPCCOEt
7‐(hydroxyimino)cyclopropa[b]chromen‐1a‐carboxylate ethyl ester
- GYKI 53655
1‐(4‐aminophenyl)‐3‐methylcarbamyl‐4‐methyl‐7,8‐methylenedioxy‐3,4‐dihydro‐5H‐2,3‐benzodiazepine
- Iss
steady‐state current
- mGluR1
metabotropic glutamate receptor 1
- NBQX
2,3‐dioxo‐6‐nitro‐1,2,3,4‐tetrahydrobenzo(f)quinoxaline‐7‐sulfonamide
- PF
parallel fibre
- Popen
open probability
- SR 95531
2‐(3‐carboxypropyl)‐3‐amino‐6‐(4‐methoxyphenyl)pyridazinium bromide
- TARP
transmembrane AMPA receptor regulatory protein
Introduction
Purkinje cells are the sole output neurons of the cerebellar cortex. They receive ∼180,000 excitatory synapses from parallel fibres (PFs; one or two synapses per fibre; Harvey & Napper, 1988; Pitchitpornchai et al. 1994), and <1% of that number from a single powerful climbing fibre. Parallel fibre synapses are thought to be a major site of synaptic plasticity that underlies motor learning, and they have typically been studied and viewed as a population with homogeneous properties; however, plasticity and information storage must result in synaptic heterogeneity. Individual PF synapses identified in paired recordings typically exhibit high release probability, but such synapses are rare, leading to the conclusion that most of the synapses identified anatomically by electron microscopy are in fact silent (85%; Ekerot & Jörntell, 2001; Isope & Barbour, 2002), which has been suggested to offer optimal potential for information storage (Brunel et al. 2004). In contrast, a population study in which beams of PFs were stimulated ascribed a low release probability to PF synapses (Dittman et al. 2000; Foster et al. 2005). Although there is thus evidence that PF synapses form a heterogeneous population that is subject to modification, silent or virtually silent synapses are inherently difficult to study because they contribute little to the average synaptic response elicited by single stimuli. In addition, granule cells fire bursts of action potentials in vivo (Chadderton et al. 2004; Jörntell & Ekerot, 2006), whereas most of the analysis of PF synapses has been done with one or two stimuli, conditions that do not cover the physiological range.
l‐Glutamate is the transmitter at PF synapses, and rat Purkinje cells express a variety of glutamate receptors, comprising AMPA receptor (AMPAR) subunits [GluA1, 2 and 3 (Lambolez et al. 1992; Baude et al. 1994) and GluD2 (Petralia et al. 1998; Yamasaki et al. 2011)], as well as ionotropic subunits of NMDA receptors (NR1; Petralia et al. 1994) and kainate receptors and metabotropic glutamate receptor 1 (mGluR1). In addition, Purkinje cells are known to express the auxiliary AMPAR subunits, transmembrane AMPA receptor regulatory proteins (TARPs) γ‐2 and γ‐7 (Yamazaki et al. 2010). In principle, this array of subunits can generate a variety of receptors; however, no segregation has been reported between particular sites [except for GluD2, which is present at PF synapses only after 2 weeks postnatal (Zhao et al. 1998)]. Although immunogold labelling of AMPARs indicates a large variability of receptor number and density at PF synapses (Masugi‐Tokita et al. 2007), most identified synapses are labelled, suggesting that electrically silent synapses might generate signals that are too small to be resolved. Synaptic currents that correspond to different populations of receptors have not been reported in Purkinje cells.
Here, we have characterized the responses of PF–Purkinje cell connections to trains of high‐frequency stimulation in acute cerebellar slices. Our results show that PF train EPSCs are made up of two components, which exhibit different facilitation patterns and sensitivity to pharmacological agents. The relative contribution of these components varies substantially between different dendritic locations and sets of synapses. We show that TARP γ‐2 modulation of AMPAR gating contributes to the complexity of the responses. In particular, the low‐ and high‐open‐probability (P open) gating modes of TARP‐associated receptors have different sensitivities to AMPAR antagonists, which mirror the different pharmacology of phase 1 and phase 2 of the synaptic response. The high‐P open gating mode contributes to the second component of the train EPSC, which is initially small but increases steadily throughout the stimulus train. The two synaptic components seem ideally suited to distinguish between short and long presynaptic bursts of action potentials.
Methods
Ethical approval
Sprague–Dawley rats were provided by Janvier (St Berthevin, France) and subsequently housed in agreement with the European Directive 2010/63/UE regarding the protection of animals used for experimental and other scientific purposes.
Experimental procedures were approved by the French Ministry of Research and the ethical committee for animal experimentation of Paris Descartes.
Slice preparation
Experiments were performed in vitro on either transverse slices 300 μm thick or sagittal slices 220 μm thick cut from the cerebellum of 17‐ to 23‐day‐old Sprague–Dawley male or female rats. Briefly, rats were killed by decapitation under general anaesthesia following inhalation of the volatile anesthetic isoflurane in accordance with the Directive 2010/63/UE, and the cerebellum was quickly removed and cooled in ice‐cold solution. After removal of the brainstem, the tissue was glued to the stage of a vibrotome (Leica VT1200S, Germany). Slices were kept in a vessel bubbled with 95% O2–5% CO2 at 34°C for 1 h and then allowed to cool down to room temperature. Slice preparation and recordings were made in a bicarbonate‐buffered solution containing (mm): 115 NaCl, 2.5 KCl, 1.3 NaH2PO4, 26 NaHCO3 and 25 glucose. For preparation of the slices, the solution contained 4 mm MgSO4 and 0.5 mm CaCl2; for recovery and recording, 1 mm MgSO4 and 2 mm CaCl2, respectively.
Patch‐clamp recording from outside‐out patches
tsA201 cells were maintained in DMEM with 10% fetal bovine serum. Cells plated on poly‐L‐lysine‐coated glass coverslips were transiently transfected with 0.5–1.0 μg of total cDNA per coverslip using X‐tremeGENE 9 DNA transfection reagent (Roche) according to the manufacturer's instructions. Individual GluA and TARP cDNAs were co‐transfected at ratios of 1:1 to 1:3. For heteromer expression, GluA1, GluA2 and γ‐2 were co‐transfected at ratios of 1:1:1 to 1:1:3. In some experiments, GluA1–γ‐2 and GluA2–γ‐2 tandem receptors were co‐transfected to ensure complete incorporation of γ‐2 in receptor assemblies. The enhanced green fluorescent protein (eGFP; 0.2 μg) was included in the transfection mix to identify transfected cells. The GluA1 and GluA2 cDNAs encode the flip splice variants and a glutamine at the Q/R editing site in GluA2, unless stated otherwise. Outside‐out patch recordings were performed 24–72 h after transfection. For the experiments where the effect of pentobarbital was compared for receptors containing Q vs. R versions of GluA2, we expressed the receptors in Xenopus oocytes to increase the size of the currents and excised outside‐out patches after removal of the vitelline membrane (5–7 days after injecting the oocytes with cRNA for GluA1, GluA2 and γ‐2). The oocytes and cRNAs were kind gifts from Susumu Tomita (Yale University).
All recordings were made at room temperature with an EPC‐9 amplifier (HEKA) and PatchMaster acquisition software, essentially as described by Robert & Howe (2003). The recordings were made at a holding potential of −100 mV. The external solution contained (mm): 150 NaCl, 3 KCl, 2 CaCl2, 1 MgCl2, 5 glucose and 10 Hepes (pH adjusted to 7.4 with NaOH). Patch pipettes (open tip resistance 4–10 MΩ) were filled with a solution containing (mm): 135 CsF, 33 CsOH, 2 MgCl2, 1 CaCl2, 11 EGTA and 10 Hepes (pH adjusted to 7.4 with CsOH). The external solution with and without glutamate (10 mm) was applied to the outside‐out patch using theta glass pipettes mounted on a piezoelectric bimorph (Robert & Howe, 2003). The rise times of responses to fast application of 10 mm glutamate were 250–400 μs. The bath was superfused constantly with normal external solution at a rate of 1–2 ml min−1 (one to two bath volumes per minute). Antagonists were added to the external solution and were present both with and without glutamate unless noted otherwise. Ensemble currents were low‐pass filtered at 3 kHz, sampled at 20–40 kHz, and analysed as described by Robert & Howe (2003).
Patch‐clamp recording of synaptic currents
Whole‐cell patch‐clamp recordings were made from Purkinje neurons, identified by their size and location at the edge of the molecular and granule cell layers with an EPC9 or EPC10 amplifier (HEKA, Germany) and PatchMaster acquisition software. The internal solution contained (mm): 140 potassium gluconate, 10 KCl, 10 Hepes, 0.1 EGTA, 4.6 MgCl2, 4 ATPNa2 and 0.4 GTPNa, pH adjusted to 7.3 with KOH and osmolality to 295 mosmol kg−1. When filled with internal solution, recording pipettes had a resistance between 3 and 4.0 MΩ. Membrane currents were recorded at a pipette potential of −60 mV (not corrected for junction potential of approximately −12 mV pipette–bath). For recording train EPSCs at depolarized membrane potential, the internal solution contained in (mm): 125 caesium gluconate, 5 QX‐314Cl, 10 EGTA, 10 Hepes, 4 MgATP, 0.3 NaGTP, 1 CaCl2 and 0.1 spermine, pH adjusted to 7.3 with CsOH. Series resistance was 80% compensated. During experiments, the preparation was visualized on an upright microscope (Olympus BFX51; 60×0.9 NA water‐dipping objective) and the bath was continuously perfused at a rate of 1–2 ml min−1 (one to two bath volumes per minute) with solution equilibrated with 95% O2–5% CO2 to maintain pH.
Parallel fibres were stimulated with a patch pipette identical to those used for recordings and positioned at the surface of the molecular layer. The stimulation pipette was usually positioned in the central third of the molecular layer, except for data presented in Fig. 3, where the whole molecular layer was investigated in sagittal slices. Pulses of 100 μs duration and 40 V from an isolated stimulator were used in transverse slices, and 15 V in sagittal slices. Identical stimulus parameters and pipettes were used between experiments to achieve comparable numbers and density of active PFs. Trains of 10 stimuli at a frequency of 100 Hz were repeated once per minute.
Figure 3. Facilitation is a local property of dendritic sites .
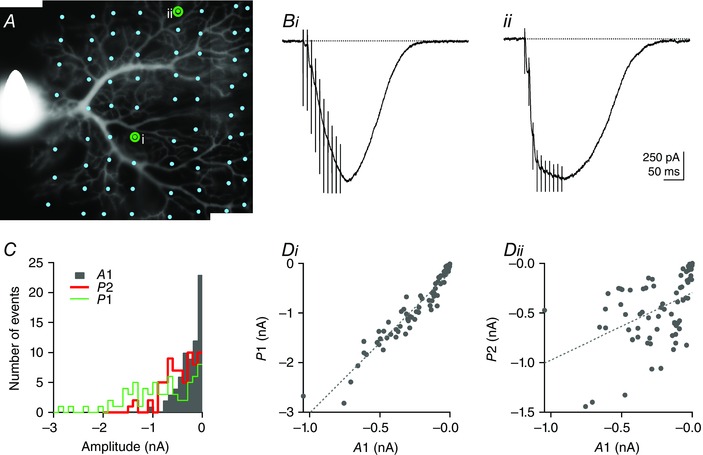
A. morphology of a Purkinje cell loaded with 20 μm Alexa‐488. The PF train EPSC was systematically probed at different locations of the dendritic tree by moving the stimulation pipette (dots denote the position of the tip of the pipette; 71 locations tested in this example) in sagittal slices. i and ii mark the positions of the stimulation pipette corresponding to the PF train EPSCs shown in B. The train EPSCs display similar maximal amplitudes but very different facilitation during the train. Bi, a PF train EPSC that builds up. Bii, a saturating example. C, histogram of A1, P1 and P2 showing the wide distribution of these parameters and the heterogeneity of the amplitude of the two phases. Di, plot of P1 vs. A1 showing a strong correlation between these parameters (Pearson's coefficient = 0.959, P ≪ 0.001), as observed for values from different cells (Fig. 1). Dii, plot of P2 vs. A1. P2 and A1 are significantly correlated (Pearson's coefficient = 0.456, P < 0.001), although less strictly and mostly because of a strong correlation for train EPSCs where A1 was very small. SR 95531 (3 μm) and d‐APV (50 μm).
Imaging
To obtain the morphology of the Purkinje cell dendritic tree, the internal solution was supplemented with 20 μm Alexa‐488 (Life Technologies, USA) and images were obtained with 0.1 s exposures by an Andor Ixon EMCCD camera (Andor Technology, Belfast, UK) and OptoLED light source (CAIRN Research, Faversham, UK) with 470/40 nm excitation and 530/40 nm emission. When recording intracellular calcium signals, the pipette solution was additionally supplemented with 500 μm fluo‐5‐F (pentapotassium salt; Life Technologies, USA). After focusing, a dendritic subregion of 150 × 200 pixels (40 μm × 53 μm) was imaged at 37.3 fps at EM gain 150. Analysis was by ΔF/F after subtraction of background fluorescence (Igor Pro; Wavemetrics Portland, USA; routines kindly provided by Brandon Stell).
Chemicals
2,3‐Dioxo‐6‐nitro‐1,2,3,4‐tetrahydrobenzo(f)quinoxaline‐7‐sulfonamide (NBQX), 6‐cyano‐7‐nitroquinoxaline‐2,3‐dione (CNQX), 2‐(3‐carboxypropyl)‐3‐amino‐6‐(4‐methoxyphenyl)pyridazinium bromide (SR 95531), cyclothiazide (CTZ) and d‐(−)‐2‐amino‐5‐phosphonopentanoic acid (d‐AP5) were purchased from Tocris Bioscience (Bristol, UK). Alternatively, SR 95531 and d‐AP5 and 7‐(hydroxyimino)cyclopropa[b]chromen‐1a‐carboxylate ethyl ester (CPCCOEt) were bought from Ascent Scientific (Cambridge, UK). Stocks were prepared in water, except for CPCCOEt and cyclothiazide, which were prepared in DMSO, and were diluted in saline immediately before use. Pentobarbital was purchased from Centravet (Maisons‐Alfort, France). All other chemicals were purchased from Sigma.
Data analysis
For the NBQX experiments with recombinant receptors in outside‐out patches, two types of protocols were used. In one, 100 ms applications of 10 mm glutamate were repeated at 0.4 s intervals, which allowed recovery from desensitization between applications. In most experiments, NBQX was included in both control and glutamate‐containing solutions. In a few experiments, NBQX was excluded from the glutamate‐containing solution, and longer (400–500 ms) applications of glutamate were made to estimate the off‐kinetics of NBQX block. In either case, 30 records were collected in steady‐state conditions and used to construct mean ensemble currents in PatchMaster (records that contained artefacts or jumps in the holding current were excluded). The mean records were exported to Igor Pro software (Wavemetrics), and peak and steady‐state currents during the glutamate application were measured manually (peak currents were defined as the difference between the peak inward current and the steady‐state current late in the application). In the second type of NBQX protocol, 2 ms applications of 10 mm glutamate were repeated at short intervals (typically, 10 applications at 100 Hz) to mimic the high‐frequency trains of PF stimulation in cerebellar slices. The trains were repeated once per second. In these studies, NBQX was included in both control and glutamate‐containing solutions. After digital averaging in PatchMaster (20–30 repetitions), the mean waveforms were exported to Igor Pro. Steady‐state currents were estimated from the current amplitudes at the end of the 2 ms glutamate applications during the last three to five applications in the train. The peak current was taken as the difference between this value and the maximal inward current evoked by the first 2 ms pulse of 10 mm glutamate.
We also tested the effect of 110 μm pentobarbital on recombinant receptors with protocols similar to those used for NBQX. We performed bi‐exponential fits to the decays of the currents evoked by 2 or 100 ms applications of 10 mm glutamate in the absence and presence of pentobarbital. Zero time was set at the end of the 2 ms applications or the peak of the inward current evoked by the 100 ms applications (deactivation and desensitization, respectively). This analysis allowed us to obtain percentage inhibition values for the steady‐state current (I ss) and the amplitudes of the fast and slow components of desensitization (a f and a s), as well as the relative amplitude of the slow component of deactivation (a 2). Mean percentage inhibition values for NBQX and pentobarbital block of the different parameters were calculated and compared with unpaired Student's t test and one‐ or two‐way ANOVA with the online software package VassarStats (http://vassarstats.net).
For the analysis of synaptic currents, raw current traces were exported to Igor Pro (Wavemetrics), and peak currents were measured as the average over a time window overlapping the peak and spanning a few sampling points. The amplitudes of the first, the third and the 10th response (A1, A3 and A10, respectively) within a train, were measured for each individual trace. The amplitude of phase 2 (P2 = A10 – A3) and ratios R1 = A3/A1 and R2 = P2/A1 were computed from these measurements. Individual values were then averaged for four or five consecutive trains except for Figs 3 and 10, where a single‐train EPSC was recorded at each location. Statistical significance was tested with non‐parametric methods where possible. These do not require assumptions about the nature of the distribution of the variables (as parametric tests do); the Wilcoxon signed rank test (non‐parametric, for paired samples) and the Wilcoxon Mann–Whitney test (non‐parametric, for unpaired samples). However, parametric tests can be more robust in the case of small samples and when differences are large and consistent. For this reason, Student's t test was used to test the statistical significance of differences for the data presented in Fig. 6. Tests were conducted using Igor Pro (Wavemetrics). All values given are means ± SEM.
Figure 10. Action potential generation in response to PF train stimulation .
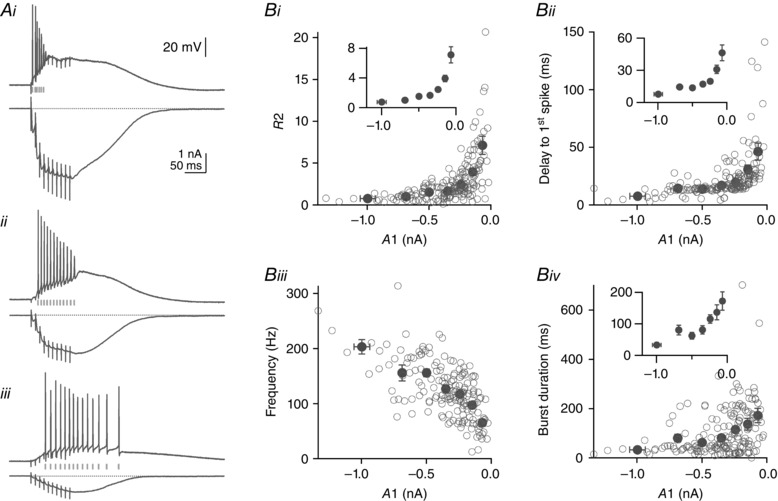
Parallel fibre train EPSCs and the corresponding current‐clamp responses of Purkinje cells were stimulated across the dendritic field of 10 Purkinje cells in sagittal slices. A, three examples of PF train EPSCs and the corresponding responses in current clamp from the same cell. Examples are of the three types described in Fig. 1: saturating (Ai); hybrid (Aii); and building up progressively (Aiii). Calibrations in each panel are identical. B, R2 = P2/A1 (Bi), the delay to the first spike (Bii), the average spike frequency during the elicited burst of action potentials (Biii), and burst duration (Biv) are plotted against the amplitude of the first response of the train EPSC, A1. Open circles are individual values; black circles are average values for bins of A1 amplitude (0 to −100, −100 to −200, −200 to −300, −300 to −400, −400 to −600, −600 to −800 and below −800 pA). Insets show average data only at a higher magnification. Small A1 amplitudes are associated with larger R2 values, longer delay to the first spike, lower spiking frequency and longer burst duration. On average, for A1 values of −68.4 ± 5.5 pA, R2 was 7.1 ± 1.1, delay to first spike was 46.3 ± 7.4 ms, spike frequency was 65.4 ± 6.6 Hz and burst duration was 172.2 ± 29.0 ms. For A1 values of −686.4 ± 13.6 pA, R2 was 1.0 ± 0.1, delay to first spike was 14.4 ± 1.4 ms, spike frequency was 155.5 ± 14.5 Hz and burst duration was 80.4 ± 15.7 ms. The results show that saturating train EPSCs can recruit the postsynaptic cell efficiently and precisely with short presynaptic bursts of activity, whereas building up train EPSCs require longer presynaptic trains. All cells were recorded in sagittal slices.
Figure 6. CNQX activates currents that are sensitive to pentobarbital .
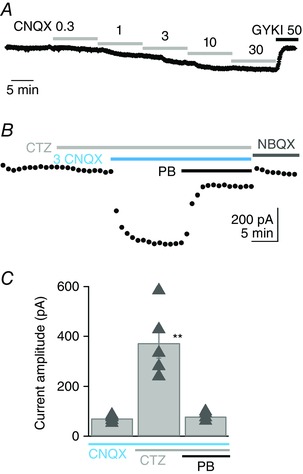
A, example of the membrane current activated in a Purkinje cell by addition of increasing concentrations of CNQX to the external solution. The addition of 3 μm CNQX activated an inward current of −76.4 pA. Currents were completely blocked by 50 μm GYKI 53655, the selective AMPAR non‐competitive antagonist. B, in another cell, 3 μm CNQX was applied in the presence of 100 μm cyclothiazide (CTZ) and activated a current of −430.3 pA. Application of 110 μm pentobarbital (PB) inhibited the current by 78.6%. The baseline was measured at the end of the experiment after application of 10 μm NBQX. C, on average, 3 μm CNQX activated a current of −72 ± 5.8 pA (n = 5) in control conditions, −372 ± 61.7 pA (n = 5) in the presence of 100 μm CTZ and only −82.4 ± 8.4 pA in the presence of both CTZ and 110 μm pentobarbital (n = 4). The CNQX current amplitudes in CTZ alone were significantly greater than control values and the data in pentobarbital, **P < 0.01, unpaired Student's t tests. Pentobarbital data were not significantly different from control values (P = 0.35). Bars indicate SEM. SR 95531 (3 μm) and d‐APV (50 μm). All cells were recorded in sagittal slices.
Results
Analysis of PF train EPSCs: two types of synaptic responses
We analysed parallel fibre to Purkinje cell synapses in sagittal or transverse cerebellar slices from postnatal day 17–23 rats. Parallel fibres were stimulated using a focal glass electrode, stimulating beams of parallel fibres while recording train EPSCs in Purkinje cells in the presence of GABAA and NMDA receptor antagonists. Granule cells (whose axons are PFs) fire in short trains, and to mimic this pattern, trains of 10 stimuli at 100 Hz were applied and repeated once per minute. Although we used reproducible stimulation intensity and pulse width for a given type of slice and distance, the amplitude of the first EPSC varied substantially between recordings. When Purkinje cells are loaded with a fluorescent indicator, it is obvious that the dendritic tree of the cell is very dense (see Fig. 3). Although EPSC amplitude is generally assumed to reflect the number of synapses activated, variations of the EPSC amplitude might reflect not only variation in the number of stimulated fibres and synapses but also variation in the response of individual synapses. While using trains to stimulate PFs, we noticed that sites that gave large responses to a single pulse were more likely to recruit rapidly saturating responses, whereas small initial responses facilitated steadily during the train to similarly large amplitudes. In all work presented here, the behaviour of PF train EPSCs was characterized whether responses to a single pulse were large or small.
Figure 1 A shows representative examples of PF train EPSCs elicited by a train of 10 stimuli at 100 Hz. Three kinds of response were noted. Some responses tended to saturate after three stimuli (Fig. 1 Ai), whereas others showed facilitation over the train duration, progressively increasing in amplitude (Fig. 1 Aii). Yet others appeared to be hybrids, with a progressive build‐up superimposed on an initial saturating response (Fig. 1 Aiii). Train EPSCs appeared to be made of the sum of EPSCs with two kinds of facilitation properties during trains. Some responses reached a maximum by the third stimulus and thereafter maintained a plateau, whereas others built up from a very small initial amplitude and kept facilitating. The proportion of the two types of responses (saturating and steadily facilitating) varied for different cells; some EPSCs were almost purely saturating or facilitating, as in the examples of Fig. 1 Ai and ii, but most were a mixture, as in example Fig. 1 Aiii. The fast‐saturating type was best isolated during the first three stimuli, whereas the second type was best measured once the first reached a plateau.
Figure 1. Analysis of the parallel fibre (PF) train EPSC shows that it can be divided into two phases .
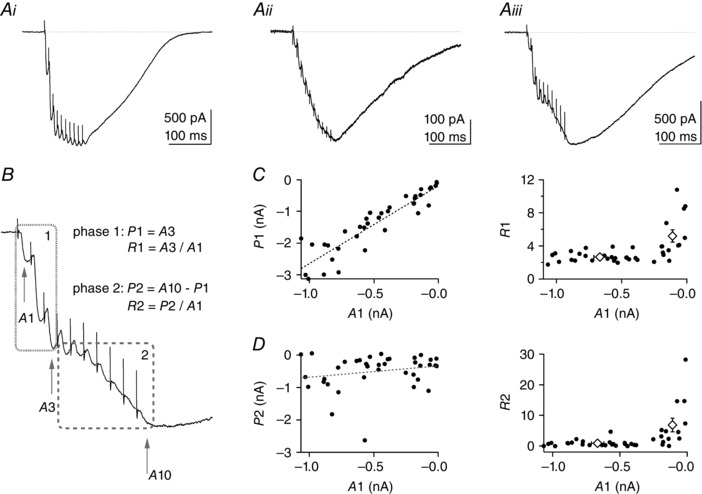
Parallel fibre train EPSCs were elicited by 10 stimuli at 100 Hz repeated once per minute. Stimulation artefacts have been partly removed for clarity. A, three examples of PF train EPSCs with different facilitation behaviour: i, saturating; ii, increasing progressively throughout the train; and iii, hybrid of the first two. B, example Aiii is used to illustrate the analysis of the PF train EPSC in two separate phases: phase 1 characterized by amplitudes A1 and A3 of the first and third responses, respectively; and phase 2 characterized by the amplitudes A3 and A10 of the 10th response. R1 = A3/A1 is the amplitude of phase 1 normalized to the initial response, A1. It is a measure of facilitation after three stimuli. P2 = A10 − A3 is the amplitude of phase 2. R2 = P2/A1 is the amplitude of phase 2 normalized to the initial response, A1. We come back to the significance of R2 in the last panel. C, left panel, plot of P1 vs. A1 for individual cells (n = 38) shows correlation between these parameters (Pearson's coefficient = 0.917, P < 0.001). The standard deviation of the baseline current noise was 3.05 pA (n = 38) and the smallest A1 current measured was −11.9 pA. Even the smallest A1 values were well distinguished from baseline. C, right panel, plot of R1 vs. A1. Open diamonds correspond to mean R1 values for PF train EPSCs with large and small A1 values (inward currents >200 and <200 pA, respectively). R1 is typically larger for small values of A1, where the train EPSC is dominated by progressively increasing responses (as in A ii). D, left panel, plot of amplitudes P2 vs. A1. There is no significant correlation of the amplitudes of the two phases (Pearson's coefficient = 0.215, P > 0.1). D, right panel, plot of R2 vs. A1. Given that A1 and P1 are strongly correlated, R2 = P2/A1 is a relative measure of the amplitude of phase 2 with respect to that of phase 1 and reports on the proportion of the two phases of the train EPSC. Open diamonds correspond to mean R2 values for PF train EPSCs analysed in the large and small A1 amplitude categories defined above. SR 95531 (3 μm) and d‐APV (50 μm) were present in all recordings; 32 of 38 cells were from transverse slices, six of 38 from sagittal slices.
Figure 1 B illustrates the analysis developed to characterize the PF train EPSC and measure the two components that were observed in isolation during train stimulation in some cells. The amplitudes A1, A3 and A10 of the first, third and 10th responses within the train were measured as absolute amplitudes, and we defined two phases. The initial phase of the train EPSC (phase 1, up to the third stimulation) is described by A1, A3 and the ratio of amplitudes A3/A1, which we call R1. We equated A3 to the amplitude of phase 1 (P1), and R1 is a measure of the facilitation during this phase. The second phase (phase 2, from the third to the 10th EPSC) is characterized by the absolute amplitude P2, taken as the difference between the peak amplitude of the response to the 10th and third stimuli (P2 = A10 – A3), a measure of the additional current recruited late in the train. Finally, we define R2, the ratio of P2 over A1, which is a measure of the amplitude of phase 2 relative to that of the response to a single stimulus. Given that A1 is proportional to phase 1 (see below for correlation between A1 and P1) and is the least contaminated by overlap with phase 2, R2 is also an indicator of the relative size of the two phases (steadily facilitating vs. saturating).
Figure 1 C shows a plot of P1 vs. the initial amplitude A1 in the left panel and a plot of R1 (A3/A1) vs. A1 on the right for 38 cells. P1 is highly correlated with the amplitude of A1 and, as a result, R1 is fairly constant for amplitudes of A1 larger than 200 pA. This reflects the fact that while phase 1 of the train EPSCs saturates (as illustrated in Fig. 1 Ai), facilitation is reproducible during the first three stimuli. In contrast, for small amplitudes (A1 below 200 pA, as in example Fig. 1 Aii), there is no saturation of the train EPSC, and R1 increases substantially, along with the relative size of the phase 2 component. The left panel of Fig. 1 D shows a plot of P2, the amplitude of the current recruited during phase 2 (A10 − A3), plotted vs. A1. In contrast to P1, P2 was not correlated significantly with A1 and varied independently from cell to cell. As a result, for cells that gave small A1 values, the PF train EPSC was dominated by the steadily facilitating phase 2 component, which resulted in larger values of R2. An example of such a cell is shown in Fig. 1 Aii. In such cases, where phase 2 made a large contribution to the train EPSC, R1 values were also large (Fig. 1 C, right), presumably because facilitation was greater for phase 2 (compared with phase 1). Most of the experiments presented in Fig. 1 were performed on transverse cerebellar slices (32 out of 38). However, the three patterns of train EPSC described were also observed in sagittal slices, and data from transverse and sagittal slices were pooled.
Our analysis shows that the PF train EPSCs elicited in response to high‐frequency stimulation are composed of variable proportions of two synaptic components with specific facilitation properties. One component usually dominates phase 1 of the response and quickly saturates, whereas a second component, typically very small initially, progressively increases throughout the train and dominates phase 2. The two synaptic components might correspond to the summed response of two different populations of synapses, which is suggested by the difference in facilitation during the train.
mGluR1s and PF train EPSCs
Parallel fibre train stimulation activates the AMPAR‐mediated EPSC described above, and also an mGluR1‐mediated slow EPSC through a Gq‐mediated pathway (Batchelor & Garthwaite, 1994; Tempia et al. 1998; Canepari & Ogden, 2003). The mGluR1 slow EPSC could therefore contribute to the currents described above, particularly during phase 2, although it activates on a slower time scale than AMPARs, rising slowly after the 100 ms train and peaking in ∼500 ms (Batchelor & Garthwaite, 1994; Canepari et al. 2004) and it is reduced in amplitude when AMPAR‐mediated transmission is intact (Auger & Ogden, 2010). However, because mGluR1 is also coupled via Gq to the phospholipase C cascade, if calcium is released from stores this might additionally influence the PF train EPSC.
To test directly the possible contribution of mGluR1 activation, we analysed the effect of CPCCOEt, an mGluR1 antagonist, on the PF train EPSC while imaging Ca2+ in parallel with the fluorescent indicator fluo‐5F. Figure 2 A shows an example of a train EPSC in control conditions and in 50 μm CPCCOEt. Although there is a slight reduction of the train EPSC, traces normalized to A10 show that the relative proportions of phase 1 and 2 are not affected by the antagonist. Figure 2 B shows the calcium signals associated with the train EPSC in control conditions and in the presence of CPCCOEt, as well as the mGluR1‐mediated signal isolated by subtraction. The mGluR1‐mediated calcium signal peaks ∼540 ms after the beginning of the train and is fully inhibited by CPCCOEt. Data are summarized in Fig. 2 C. In the presence of 50 or 100 μm CPCCOEt, A1 was 89.6 ± 3.0% (n = 14, P = 0.004), P1 was 94.7 ± 1.5% (n = 14, P = 0.005), P2 was 109.5 ± 4.9% (n = 14, P = 0.075) and R2 was 123.2 ± 7.4% (n = 14, P = 0.008) of control values. If the mGluR1 slow EPSC contributed significantly to phase 2, inhibition of the mGluR1 slow EPSC with CPCCOEt would be expected to decrease P2 and R2. In fact, we observed a small increase of both P2 and R2. The mGluR1 current is therefore distinct from the PF train EPSC, although it can overlap with its decay. Calcium signals associated with mGluR1 activation were slow compared with the train EPSCs from the same neurons and were fully inhibited by the mGluR1 antagonist CPCCOEt. The mGluR and AMPAR systems coexist and interact at PF synapses, but the EPSCs they generate can be clearly distinguished from each other.
Figure 2. Role of metabotropic glutamate receptor 1 (mGluR1) and voltage‐gated conductances .
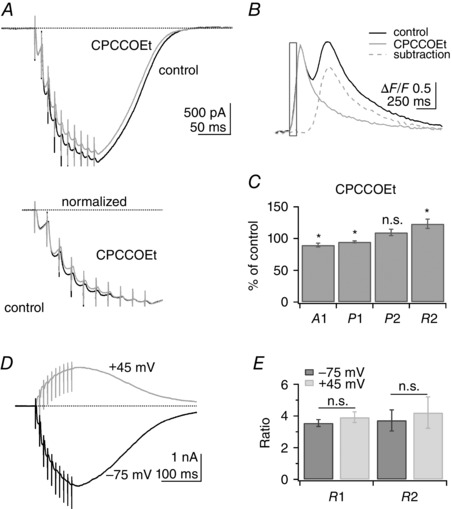
A, top panel, example of a PF train EPSC recorded in control conditions (black trace) and in the presence of the mGluR1 antagonist CPCCOEt (50 μm, grey trace). A, bottom panel, traces normalized to A10. mGluR1 inhibition has little effect on the train EPSC. B, ΔF/F signals associated with the train EPSC in control conditions (black trace) and CPCCOEt (grey trace). Subtraction of the signal in CPCCOEt from control conditions shows the inhibition of the mGluR1‐mediated calcium signal (dotted grey trace). C, A1, P1, P2 and R2 in CPCCOEt as a percentage of control values for 14 cells. Wilcoxon signed rank test, *P < 0.01; n.s., not significant (n = 14, P = 0.075). Although CPCCOEt slightly modifies the train EPSC, there is no inhibition of phase 2, indicating that this component of the train EPSC is not generated by activation of mGluR1 receptors. D, example of a PF train EPSC recorded at −60 mV (−75 mV after correction for liquid junction potential; black trace) and after 5 min at +60 mV (+45 mV after correction; grey trace). Steady‐state depolarization is expected to inactivate voltage‐gated conductances. E, average values of R1 and R2 at −75 and +45 mV were not significantly different, indicating that voltage‐gated conductances do not affect the relative proportion of phase 1 and 2 (n = 10; Wilcoxon signed rank test for R1, P = 0.084 and R2, P = 0.49). Bars indicate SEM. SR 95531 (3 μm) and d‐APV (50 μm).
Voltage‐gated conductances and PF train EPSCs
Given that the progressively rising train EPSC identified during phase 2 has not been described previously, we considered whether it arose spuriously from poor voltage control and consequent activation of dendritic voltage‐gated conductances. Ly et al. (2016) recently showed activation of T‐type calcium channels by PF EPSCs; however, block of the T‐type calcium channels did not modify the kinetics of the EPSCs, because of the rapid activation–deactivation kinetics of the T‐type channels.
To test this possibility further, we recorded train EPSCs at depolarized potentials where voltage‐gated conductances are largely inactivated. The PF train EPSC was recorded with CsGluc internal solution while the membrane potential was held at −60 or +60 mV (−75 and +45 mV after compensation for a junction potential of −15 mV) for a sufficient amount of time to inactivate voltage‐gated conductances (4–5 min). Figure 2 D shows an example of a PF train EPSC recorded at −75 and +45 mV. The train EPSC was not significantly altered by depolarization and, in particular, phase 2 currents were still observed at depolarized potentials. The ratios R1 and R2 report the relative amplitudes of phase 1 and phase 2 currents with respect to A1. R1 and R2 were analysed at −75 vs. +45 mV to determine whether membrane potential affects the proportion of the two phases. This analysis is summarized in Fig. 2 E. On average in 10 cells, R1 was 3.55 ± 0.23 at −75 mV and 3.92 ± 0.34 at +45 mV (P = 0.14, n = 10) and R2 was 3.72 ± 0.67 at −75 mV and 4.21 ± 0.99 at +45 mV (P = 0.37, n = 10). If phase 2 currents relied on activation of voltage‐gated conductances, R2 would be expected to decrease at depolarized potential. We therefore conclude that the PF train EPSCs we have identified are not influenced by activation of voltage‐gated dendritic conductances, a conclusion consistent with our pharmacological analysis of the train EPSC presented below.
Variability arises at dendritic sites
In the analysis presented in Fig. 1, a single dendritic site was tested for each cell. We next asked whether the observed variations in the relative size of phase 1 and 2 reflect cell‐to‐cell variability or local variations between sets of synapses on the dendritic tree of a Purkinje cell. Figure 3 shows the analysis of PF train EPSCs from a Purkinje cell loaded with the fluorophore Alexa‐488 to visualize the morphology of the dendritic tree. These experiments were performed in sagittal slices exclusively, to visualize the dendritic tree of the cell and position the stimulation pipette with respect to the tree. To probe local variations, the position of the stimulation pipette was systematically changed (n = 5 cells).
Figure 3 A shows the morphology of the Purkinje cell, and dots indicate the successive positions of the stimulation pipette (71 locations tested in this example). Stimulation intensity and duration were identical for all positions (15 V and 100 μs). The responses obtained from the stimulation sites marked i and ii in Fig. 3 A are shown in Fig. 3 B. Both responses reach a similar peak amplitude at the end of the train. Figure 3 Bi is an example of a small initial EPSC that progressively facilitates during the train, whereas Fig. 3 Bii is an example of a response that saturates after the third stimulation. Figure 3 C is a histogram illustrating the distributions of the amplitudes: A1, P1 and P2. The amplitudes of each component show remarkable variability (coefficients of variation: CV A1 = 91.4%, CV P1 = 74.8% and CV P2 = 72.0%) when examined over the entire cell. On average for five cells, the coefficient of variation of A1 was 99.8 ± 2.8%, the coefficient of variation of P1 was 81.3 ± 2.8% and the coefficient of variation of P2 was 76.3 ± 6.4%. Figure 3 D shows plots of the amplitude of phase 1 (P1 = A3) and phase 2 (P2 = A10 − A3) vs. A1. As observed for different cells (Fig. 1), P1 is highly correlated with A1 (Pearson's coefficient = 0.959, P ≪ 0.001), demonstrating the consistent facilitation of the phase 1 component; A1 and P2 are also significantly correlated (Pearson's coefficient = 0.456, P < 0.001), although less tightly. The correlation between A1 and P2 appears to reflect primarily the high P2 values for train EPSCs where A1 was small. This result might be expected if the small A1 train EPSCs were made up almost exclusively of building‐up currents with reproducible facilitation. In contrast, for currents composed of both saturating and building‐up components, A1 is dominated by the large saturating component whereas P2 is dominated by the building‐up component, and the correlation between A1 and P2 is lost, as seen for large A1 train EPSCs. Variations in facilitation of the phase 2 component between cells could explain why no such correlation was observed for the data in Fig. 1 D, where data from 38 cells were pooled. These experiments show that the heterogeneity of the PF EPSCs arises primarily from local variability of synaptic responses between dendritic sites of individual Purkinje cells and not from cell‐to‐cell variability. To our knowledge, such mapping of synaptic connections onto a single Purkinje cell has not been done before. It reveals a high degree of local tuning of synapses.
Facilitation and depression of EPSCs during repetitive stimuli are usually taken as indicators of the presynaptic release probability of the underlying synapses. Here, the EPSC facilitation during a train could be interpreted as indicating the presence of two different kinds of synapses, one with high release probability (saturating responses, highlighted in phase 1) and one with lower release probability (responses progressively increasing, highlighted in phase 2). Although it could be argued that extracellular stimulation efficiency could generate some of this heterogeneity (Merrill et al. 1978), we show below that the two types of responses also have different sensitivities to pharmacological compounds, suggesting that they report synapses with different receptor signatures, raising the possibility that differences in receptor behaviour may contribute to the different facilitation pattern of the two components.
Pentobarbital preferentially inhibits phase 2 of the PF train EPSC
In addition to its well‐known effects on GABAA receptors, anaesthetic concentrations of pentobarbital inhibit both native and recombinant AMPARs. The inhibition is selective for edited GluA2‐containing receptors that have an arginine rather than a glutamine at the tip of the pore helix in GluA2 subunits, receptors that also have greatly reduced permeability to Ca2+ (Taverna et al. 1994; Yamakura et al. 1995). The results of previous studies indicated that Purkinje cells express edited AMPARs (Tempia et al. 1996; Häusser & Roth, 1997). We therefore tested the effect of pentobarbital on the PF train EPSC.
Figure 4 A (top panel) shows average PF train EPSCs in control conditions and in 110 μm pentobarbital for one experiment. Pentobarbital does not inhibit the train EPSC uniformly and modifies the proportions of phase 1 and 2. This is highlighted when the train EPSCs are normalized to A10 (Fig. 4 A, bottom panel); the PF train EPSC shown exhibits a hybrid pattern in control solution, but saturates after the third stimulus in pentobarbital, reflecting the selective inhibition of the phase 2 current by pentobarbital. Figure 4 B shows plots of the amplitude of A1, P1 and P2 during the course of the experiment. There is almost complete block of P2, compared with only a modest reduction in A1 and P1 during the application of 110 μm pentobarbital, which results in a sharp decline in R2. Average data are presented in Fig. 4 C. Pentobarbital inhibited A1 by 15.7 ± 5.1% (n = 22) and P1 by 29.9 ± 3.4% (n = 22). The P2 values were reduced more and were even occasionally negative in pentobarbital (A10 decreasing below A3). Negative values would be expected if the saturating component of the train EPSC depresses to some extent during phase 2, although such depression would only be apparent when the amplitude of P2 became small. To minimize this complication arising from the overlap of the two components, the inhibition of P2 and R2 was calculated for train EPSCs with P2 values larger than 150 pA. In such cells, pentobarbital inhibited P2 by 77.3 ± 5.0% (n = 14) and R2 by 73.4 ± 6.3% (n = 14). The absolute amplitude of P2 was reduced from a mean of −553 to −129 pA (76.6% inhibition, n = 22), and R2 was decreased from 3.81 ± 1.67 to 1.02 ± 0.76 (n = 22), highlighting a stronger inhibition of phase 2 relative to phase 1 in the presence of the antagonist.
Figure 4. Pentobarbital inhibits phase 2 of the PF train EPSC .
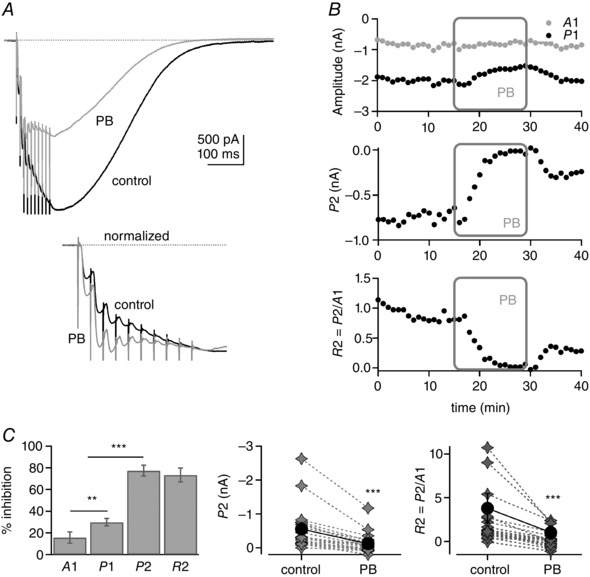
A, top panel, example of a PF train EPSC in control conditions (black trace) and after bath application of 110 μm pentobarbital (PB; grey). Inhibition is strongest at the end of the train. A, bottom panel, traces normalized to A10. In pentobarbital (grey trace), phase 2 is inhibited more potently than phase 1. This converts the train EPSC from a hybrid to a saturating pattern. B, plots of A1, P1, P2 and R2 during the course of the experiment. The pentobarbital application decreased P2 most efficiently but had little effect on A1. C, left panel, percentage inhibition by pentobarbital of A1 and P1 for 22 cells, and P2 and R2 for 14 cells. Some P2 values were negative in pentobarbital (A10 smaller than A3), presumably reflecting overlap in the two components (see Results). The inhibitions of P2 and R2 were calculated for train EPSCs with P2 values larger than 150 pA (14 cells). C, right panels, individual and average values of P2 and R2 for all 22 cells (grey diamonds, individual cells; black circles, mean values ± SEM). All changes were statistically significant. For the illustration, **P = 0.002 and ***P ≪ 0.001. Bars indicate SEM. SR 95531 (3 μm) and d‐APV (50 μm). Sixteen of 22 cells were from transverse slices and six from sagittal slices.
The results show that pentobarbital inhibits phase 2 of the PF train EPSC more potently than it inhibits phase 1. This observation supports the idea that the two components of the train EPSC described above are mediated by different populations of receptors. Pentobarbital preferentially inhibits AMPARs that contain the edited (R) version of GluA2 (Taverna et al. 1994; Yamakura et al. 1995), and phase 1 responses are relatively insensitive to pentobarbital. One interpretation of our results is that calcium‐permeable AMPARs contribute substantially to phase 1 responses; however, previous studies in Purkinje cells have concluded that they lack such receptors (Tempia et al. 1996; Häusser & Roth, 1997). We have not investigated the calcium permeability of the receptors underlying phase 1 currents, and their reduced sensitivity to pentobarbital might be influenced by other receptor properties or the reported use dependence of pentobarbital inhibition (Marzalec & Narahashi, 1993).
NBQX preferentially inhibits phase 1 of the train EPSC
Parallel fibre synapses onto Purkinje cells are equipped with AMPARs and, although Purkinje cells express GluN1 (Petralia et al. 1994) and GluK subunits (Wisden & Seeburg, 1993), functional NMDA receptors (in mice) and kainate receptors have been detected only at CF synapses (Huang et al. 2004; Piochon et al. 2007; Renzi et al. 2007). NBQX is a high‐affinity competitive antagonist of AMPA/KA receptors (Sheardown et al. 1990; Smith & Howe, 2000), commonly used to block AMPAR‐mediated synaptic currents and, owing to a slow unbinding rate, it does not unbind significantly on the time scale of a synaptic glutamate transient (Diamond & Jahr, 1997; Robert & Howe, 2003).
Figure 5 shows the effect of 200 nm NBQX on a PF train EPSC. Figure 5 A shows average traces in control conditions and in the presence of NBQX (top) and traces normalized to the 10th response of the train (A10; below). The normalized traces highlight the altered facilitation of the train EPSC, which builds up more progressively in NBQX than in control solution. The analysis presented in Fig. 5 B shows plots of the amplitudes A1, P1 and P2 over time from one recording, as well as the ratio R2. Although A1 and P1 are strongly inhibited and decrease from −449 to −105 pA and from −1158 to −370 pA, respectively, P2 is mostly unaffected. Following a transient increase of the inward current at the beginning of the NBQX application, P2 decreased only slightly from −370 to −360 pA. R2 increases during the NBQX application, reflecting the selective decrease of phase 1. On average, 200 nm NBQX inhibited A1 by 76.3 ± 2.1%, P1 by 69.0 ± 1.7% and P2 by 14.3 ± 6.8% (n = 9; Fig. 5 C). Figure 5 D shows the change of R2, a relative measure of phase 2 to phase 1. On average in 200 nm NBQX, R2 was 4.2 ± 0.8 times control values.
Figure 5. Differential inhibition of phases 1 and 2 by NBQX .
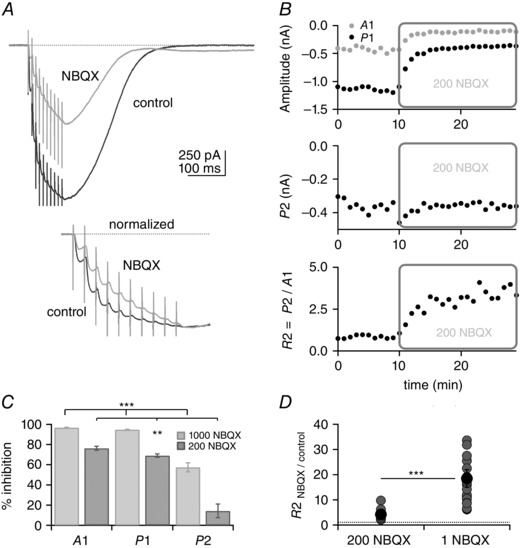
A, top panel, example of PF train EPSC inhibition by 200 nm NBQX. At this concentration, the train EPSC is partly blocked and the time course of EPSC build‐up is modified. A, bottom panel, traces normalized to A10. In NBQX (grey trace), the saturating component is reduced. B, plots of A1, P1, P2 and R2 vs. time. Application of 200 nm NBQX reduces A1 and P1 but has no effect on P2. Plot of R2 shows that phase 2 becomes bigger relative to phase 1 in NBQX. C, percentage inhibition of A1, P1 and P2 by 200 nm and 1 μm NBQX. D, individual (grey circles) and average values (black circles) of R2 normalized to control in 200 nm and 1 μm NBQX. Bars indicate SEM. SR 95531 (3 μm) and d‐APV (50 μm). The percentages of inhibition of A1, P1 and P2 by NBQX were all statistically different from each other at the two concentrations of NBQX tested, as was R2 (Wilcoxon signed rank test, **P = 0.004 for 200 nm NBQX data and ***P ≪ 0.001 for 1 μm NBQX). For the data with 1 μm NBQX, 17 out of 25 cells were from transverse slices and eight from sagittal slices. For the data with 200 nm NBQX, all cells were from sagittal slices.
Inhibition of the train EPSC by NBQX was also analysed at 1 μm. Given that inhibition by NBQX was strong at this concentration, experiments were performed on train EPSCs with large initial amplitude (A1), which were exclusively of the saturating or hybrid type. At 1 μm (Fig. 5 C), NBQX inhibited A1 by 96.7 ± 0.4% (n = 22) and P1 by 94.9 ± 0.4% (n = 22). In these conditions, the average A1 amplitude in NBQX was −33.5 ± 6.9 pA, still well above resolution. P2 was inhibited by 57.4 ± 4.4% (n = 22), producing a marked change in the ratio of the two phases reflected by the change of R2. R2 was 18.5 ± 3.5 times control values (Fig. 5 D). The results show that at concentrations of 0.2–1.0 μm, NBQX is a more effective inhibitor of phase 1 than of phase 2, and the selective block of phase 1 results in steady facilitation of the train EPSC during the course of the entire train. The results are compatible with the suggestion that the two components of the PF train EPSC are mediated by different populations of receptors, and the receptors that underlie phase 2 have a lower affinity for NBQX than those underlying phase 1.
Given that NBQX is a competitive antagonist, an alternative explanation for the different facilitation of the PF train EPSC in NBQX is that dissociation of the antagonist and subsequent competition for binding with synaptic glutamate give rise to a progressive recruitment of receptors, as NBQX block re‐equilibrates in the presence of glutamate that lingers in the synaptic cleft in high‐release conditions. However, the rate at which NBQX unbinds is slow (2–3 s−1; MacLean et al. 2014), seemingly too slow to account for rapid facilitation of phase 2 during a 100 ms train. Presynaptic kainate receptors have been described at the PF to Purkinje cell synapse (Delaney & Jahr, 2002), where they facilitate release with increasing stimulation frequency. However, block of kainate receptors by NBQX would be expected to reduce facilitation of the PF train EPSC, decreasing R1 and R2, whereas we observed an increase in these measures of facilitation.
We have discussed earlier the possibility that phase 2 currents are generated by poor voltage clamp and activation of voltage‐gated dendritic conductances. The decrease of the train EPSC amplitude in the presence of NBQX would be expected to reduce voltage‐clamp errors and therefore decrease activation of voltage‐gated conductances, decreasing the proportion of phase 2 currents. However, NBQX increased R2 (the relative proportion of phase 2 current), and there is no reason to expect that the activation of voltage‐gated conductances would be differentially sensitive to block of the train EPSC by pentobarbital and NBQX.
In total, our observations are compatible with the conclusion that PF train EPSCs are composed of two components with different sensitivities to pentobarbital and NBQX. Phase 1 is selectively blocked by NBQX, whereas phase 2 is selectively blocked by pentobarbital.
TARPed AMPARs: CNQX‐evoked currents and pentobarbital sensitivity
One possible source of receptor heterogeneity is the relative inclusion of auxiliary subunits, such as TARPs (Milstein & Nicoll, 2008; Kato et al. 2010; Jackson & Nicoll, 2011; Yan & Tomita, 2012). CNQX has been shown to be a partial agonist at AMPARs associated with TARP γ‐2 and other type I TARPs (Menuz et al. 2007). Purkinje cells express both TARP γ‐2 and γ‐7 (Yamazaki et al. 2010), and CNQX activates an AMPAR‐mediated current in these cells (Menuz et al. 2007). However, GluA1, GluA3 or GluA4 co‐expressed with TARP γ‐7 are not activated by CNQX (Bats et al. 2012). The available data therefore suggest that CNQX‐activated currents in Purkinje cells are carried by AMPARs associated with TARP γ‐2. To identify further the receptors that underlie the PF train EPSC, we compared their sensitivity to inhibition by pentobarbital with the corresponding sensitivity of the CNQX‐activated current.
Figure 6 shows that bath application of CNQX, in the presence of GABAA and NMDA receptor antagonists, activates an inward current with an EC50 of the order of 2 μm (n = 3). In control conditions, CNQX (3 μm) activated a mean inward current of −72.0 ± 5.8 pA (n = 5) that was blocked by 50 μm GYKI 53655, a selective non‐competitive AMPAR antagonist. When applied in the presence of 100 μm cyclothiazide (CTZ) to block AMPAR desensitisation, the mean CNQX‐activated current was −373.2 ± 61.4 pA (n = 5), confirming that CNQX is a partial agonist, which both activates and desensitizes AMPARs (Menuz et al. 2007; MacLean & Bowie, 2011). The application of pentobarbital (110 μm) inhibited the CNQX current by 72.4 ± 7% (n = 4), similar to the extent by which pentobarbital inhibited P2 of the train EPSC. In total, the results suggest that phase 2 of the train EPSC is mediated by AMPARs associated with the auxiliary TARP subunit, γ‐2.
Receptors that contain TARP γ‐2 generate a sustained current with reduced sensitivity to NBQX
Our results with CNQX indicate that phase 2 of the train EPSC is mediated by AMPARs that are associated with TARP γ‐2, suggesting the possibility that the different sensitivities of phase 1 and phase 2 to NBQX were related to TARP modulation of receptor properties, especially given that TARPs are known to alter AMPAR pharmacology and sensitivity to antagonists (Menuz et al. 2007; Cokić & Stein, 2008; Milstein & Nicoll, 2008: Kato et al. 2010). To test this possibility directly, tsA201 cells were transfected with GluA1 and GluA2, either with or without γ‐2, and AMPAR currents were evoked by 10 mm glutamate in outside‐out patches from the cells with a piezoelectric fast application system.
We first set out to mimic repetitive synaptic stimulation by applying trains of ten 2 ms pulses of glutamate at 10 ms intervals. Examples of the results with and without TARP γ‐2 are shown in Fig. 7. In the absence of γ‐2 co‐expression, the rapid rise and decline of the currents through GluA1/GluA2 heteromers resulted in nearly complete decay of the currents during the interapplication interval and, therefore, very small, sustained inward currents (Fig. 7 A). Co‐expression of γ‐2 slowed the deactivation decay at the end of each 2 ms application, primarily by promoting the appearance of a slow component of decay, making the decline in the current clearly bi‐exponential (see Fig. 7 B and Tomita et al. 2005; Zhang et al. 2006; Cho et al. 2007; Milstein et al. 2007). As a result, the responses decayed less completely before the next application, and this incomplete decay resulted in a substantial sustained inward current during the trains (what we will refer to as ‘pedestal’ current), which was increased relative to that seen when GluA1 and GluA2 were co‐expressed without TARP γ‐2. Decreasing the interval between applications to 4 ms, an interval at which most of the fast decay component but little of the TARP‐associated slow component had declined, resulted in less complete decay of the responses and larger pedestal currents during the train, although peak currents were more reduced at 167 Hz stimulation (Fig. 7 C). The results are consistent with the conclusion that the larger pedestal currents seen with TARP γ‐2 result from TARP‐mediated slowing of deactivation (Howe, 2015). Although shortening the interpulse interval increased the pedestal currents, the application system was less stable, and we therefore used 100 Hz trains in all subsequent studies.
Figure 7. Co‐expression of TARP γ‐2 enhances a pedestal current that is resistant to NBQX .
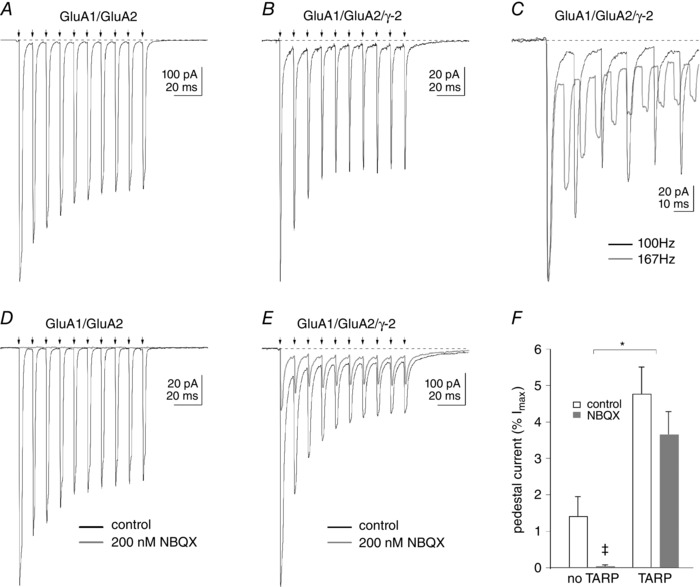
A and B, inward currents evoked by ten 2 ms applications of 10 mm glutamate (arrows) repeated every 10 ms to outside‐patches from tsA201 cells expressing GluA1 and GluA2 either without (A) or with co‐expression of the type I TARP, γ‐2 (B). If deactivation is not complete between pulses, there is still a residual current that persists at the start of the next application. We refer to this current as the pedestal current. In the absence of γ‐2, the currents decay almost completely before the next application, and there is only a small inward pedestal current during the train (A). With γ‐2 co‐expression (B), the decay of the current is slower and bi‐exponential, and the relative size of the pedestal current is enhanced. C, the size of the pedestal current is increased when the applications are repeated at 6 ms intervals, where the decay of the responses is less complete between applications, owing to the presence of the TARP‐associated slow component. D and E, trains of responses in two other patches expressing GluA1 and GluA2 without or with γ‐2. In the absence of γ‐2 co‐expression (D), both the peak and pedestal currents are abolished by the continuous application of 200 nm NBQX. When γ‐2 was co‐expressed (E), the peak current (maximal inward current evoked by the first application minus the pedestal current) is inhibited by ∼75%, whereas the enhanced pedestal current is inhibited by ∼40%. F, mean percentages of the pedestal current (expressed as a percentage of the maximal current evoked by the first control application in the same patch) in the absence and presence of 200 nm NBQX in patches from cells that did not and did express TARP γ‐2. The results are from seven and 10 patches. Bars indicate SEM. *Both control and NBQX values without TARP γ‐2 were significantly different from the corresponding values with TARP γ‐2 (two‐way ANOVA, P < 0.0001). ‡The means with and without NBQX were not significantly different; however, the percentage inhibition of the pedestal current without γ‐2 (98.4 ± 1.0%, n = 7) was significantly different from zero (P < 0.0001, Student's two‐tailed t test), whereas the corresponding inhibition with γ‐2 (22.8 ± 11.3%, n = 10) was not (P = 0.075).
We next tested the effect of 200 nm NBQX on the receptor responses to repeated stimulation without and with TARP γ‐2 co‐expression. As is shown in Fig. 7 D, 200 nm NBQX completely blocked glutamate‐evoked currents, both peak and sustained, when GluA1 and GluA2 were co‐expressed without γ‐2. In contrast, when γ‐2 was present, 200 nm NBQX reduced the initial peak response to glutamate (maximal inward current minus the pedestal current) by ∼75%, and reduced the slow pedestal current that developed during the train by only ∼40% (Fig. 7 E). Figure 7 F shows the mean pedestal current amplitude (estimated from the last three to five applications during the train; see Methods) in the absence and presence of 200 nm NBQX for GluA1/GluA2 heteromeric receptors without and with co‐expression of γ‐2. As can be seen, inclusion of γ‐2 in receptor assemblies enhanced the relative amplitude of the sustained pedestal currents [expressed as a percentage of the maximal current (I max) evoked by the first control application] and also reduced their sensitivity to NBQX. In the absence of γ‐2 co‐expression, 200 nm NBQX reduced the peak and pedestal currents through GluA1/GluA2 receptors to similar extents (97.6 ± 1.5 and 98.5 ± 1.0%, respectively; n = 7). When the same pore‐forming subunits were co‐expressed with γ‐2, 200 nm NBQX reduced the peak current evoked by the first glutamate application by 86.0 ± 2.4%, whereas the pedestal currents were reduced on average by 22.7 ± 11.3%. The inhibition of the pedestal current with γ‐2 was not significantly different from zero, but was significantly different from NBQX inhibition of the initial peak current (n = 10, P < 0.0003).
The TARP‐associated high‐P open gating is relatively insensitive to NBQX
We have shown recently that γ‐2 and other type I TARPs modulate AMPAR gating at the single‐receptor level by promoting a distinct gating mode that is characterized by longer activations and increased frequency of openings to the two largest open levels (high‐P open mode; Zhang et al. 2014). In patches containing only one active GluA/γ‐2 tandem receptor (in which γ‐2 is fused directly to the C‐terminus of the pore‐forming subunit), the receptor spends 30–40% of its time in the high‐P open gating mode, but during the remaining time it gates with kinetics that are indistinguishable from receptors composed of only GluA subunits (low‐P open mode). We further showed that the low‐P open gating mode underlies the fast component seen in the bi‐exponential deactivation and desensitization decays observed with TARP co‐expression (Cho et al. 2007; Milstein et al. 2007), whereas the TARP‐associated slow component and enhanced steady‐state currents seen during sustained applications arise almost exclusively from the high‐P open mode (Zhang et al. 2014; Howe, 2015). The pedestal currents seen here during trains of repetitive applications appeared to arise primarily from the incomplete decay of the slow component seen with TARPs (Fig. 7 B and C), suggesting that the pedestal currents arise primarily from receptors gating in the high‐P open mode. To test whether the resistance of the pedestal currents to NBQX arose because the high‐P open gating mode is relatively insensitive to NBQX inhibition, we compared the extent to which NBQX blocked the peak and steady‐state currents evoked by 100 ms applications of glutamate (having shown before that the peak and steady‐state currents in such experiments primarily reflect the low‐ and high‐P open gating modes, respectively).
Figure 8 shows the effect of NBQX on GluA1/GluA2 heteromers with and without γ‐2. In accordance with our results with high‐frequency trains of stimuli, 200 nm NBQX almost completely blocked both peak and steady‐state currents in the absence of γ‐2 (Fig. 8 A). In marked contrast, the sustained current through GluA1/GluA2/γ‐2 heteromers was resistant to block by 200 nm NBQX, although the peak current was inhibited by ∼90% (Fig. 8 B; the amplitude of the peak current was measured as the difference between the maximal inward current and the steady‐state current). Indeed, when TARP γ‐2 was present, in some cases the steady‐state current was increased in the presence of NBQX, even though the peak current was markedly reduced (Fig. 8 C). To estimate the relative potency of NBQX to inhibit peak vs. sustained currents, different concentrations of NBQX were applied to heteromers with and without γ‐2. The concentration–response data for NBQX inhibition of the peak current are shown in Fig. 8 D. A Hill‐type fit to the results gave an IC50 value of 36 nm for NBQX inhibition of the peak current, which was completely blocked by 500 nm NBQX. The results are similar to previous data for GluA1 and GluA4 homomeric receptors (Robert and Howe, 2003), and the sensitivity of the peak current through GluA1/GluA2 heteromers was similar whether γ‐2 was co‐expressed or not (Fig. 8 D, grey and open symbols, respectively). In contrast, over a range of concentrations (50–500 nm) at which NBQX blocked 50–100% of the peak currents, on average the steady‐state currents increased in the presence of γ‐2 (Fig. 8 E). Interestingly, this effect was sometimes observed on the PF train EPSCs during the initial perfusion of the recording chamber with NBQX (see Fig. 5 B). In the presence of TARP γ‐2, inhibition of the steady‐state current was obtained only with NBQX concentrations of 1 μm and above, and the block remained incomplete even at 50 μm NBQX. Both NBQX‐mediated inhibition and enhancement of glutamate‐evoked currents were fully reversible.
Figure 8. The steady‐state current (Iss) during sustained applications of glutamate is resistant to inhibition by NBQX .
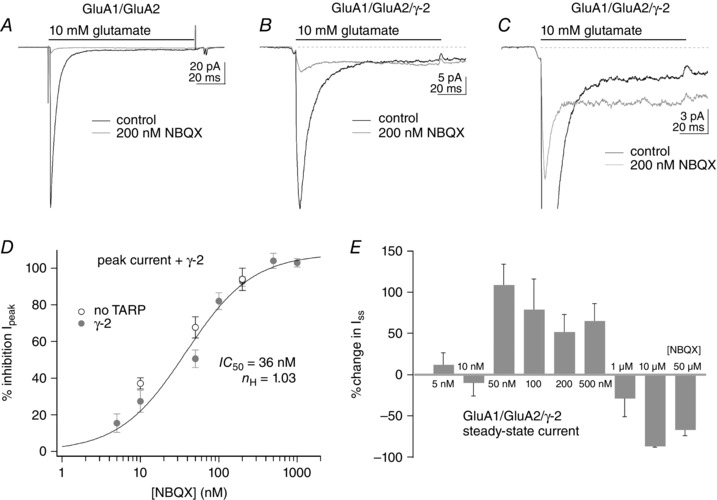
A–C, responses to 100 ms applications of 10 mm glutamate before (black traces) and during the continuous application of 200 nm NBQX (grey traces). In patches from cells expressing GluA1 and GluA2 alone (A), NBQX markedly reduced both peak and steady‐state currents. In patches from cells that also expressed γ‐2, 200 nm NBQX markedly reduced the peak responses, but the steady‐state currents were either little affected (B) or even larger than control currents (C). D, concentration–response data for NBQX inhibition of the peak current in patches from cells expressing GluA1 and GluA2 alone (open circles) or co‐expressing γ‐2 (filled grey circles). A Hill‐type fit to the data from patches co‐expressing γ‐2 gave IC50 and n H values of 36 nm and 1.03, respectively. The data for GluA1 and GluA2 without γ‐2 (open circles) gave percentage inhibition values similar to the corresponding values with γ‐2 at the three concentrations tested. Bars indicate SEM (three to eight values per point). E, mean percentage change in the I ss for GluA1/GluA2/γ‐2 heteromeric receptors over a 10,000‐fold range of NBQX concentrations. Note that, on average, NBQX concentrations that substantially reduce peak currents (D) increase the size of the I ss. The data for NBQX concentrations ≤1 μm are from the same patches as in D. The data for 10 and 50 μm NBQX are from two patches each (both concentrations completely inhibited the peak current). Bars indicate SEM.
One possible explanation for the incomplete block of the steady‐state current when TARP γ‐2 was present, even at concentrations up to 50 μm NBQX, is that the reduced affinity of NBQX for the high‐P open mode results in significant re‐equilibration of receptor occupancy during 100 ms applications of near‐saturating glutamate. To test this hypothesis, we compared the rate at which the steady‐state current recovered for GluA1/GluA2 heteromeric receptors without and with γ‐2 co‐expression. Outside‐out patches were equilibrated in an NBQX concentration sufficient to block virtually all the current at the beginning of the applications and were then jumped into 10 mm glutamate in the absence of NBQX for 400 or 500 ms. To obtain an estimate of the time it took for the currents to recover, we fitted the steady‐state currents with single exponential functions (the small size of the currents prohibited a detailed analysis of the time course and the impact of conductance substates; Smith & Howe, 2000). In the absence of γ‐2, the mean time constant from these fits was 395 ± 50 ms (n = 3). The corresponding rate constant (2.5 s−1) agrees well with previous reports for inhibition of peak AMPAR currents from triple‐jump experiments (MacLean et al. 2014). When the receptors contained γ‐2, the steady‐state current recovered substantially faster. Exponential fits to the results from six patches gave a mean time constant of 85 ± 20 ms, which differed significantly from the corresponding value in the absence of TARP γ‐2 (P < 0.001). The ∼5‐fold faster dissociation of NBQX results in significant relief of NBQX block of the steady‐state current during a 100 ms application of near‐saturating glutamate and is likely to account for the incomplete block at concentrations of 10 and 50 μm when the receptors contain the auxiliary subunit TARP γ‐2 (Fig. 8 E).
Pentobarbital selectively blocks the high‐P open mode seen with TARP γ‐2
To test whether TARP‐associated differences in receptor pharmacology contributed to the selective block by pentobarbital of phase 2 of the PF train EPSC, we determined the effect of 110 μm pentobarbital on glutamate‐evoked currents in outside‐out patches. Figure 9 A shows data obtained in a patch containing receptors composed of GluA1, GluA2‐R and γ‐2. This concentration of pentobarbital reduced the initial peak current by 34% and the steady‐state pedestal current (inset) by 53%. As found previously (Taverna et al. 1994; Yamakura et al. 1995), pentobarbital was significantly less effective at blocking both peak and pedestal currents when the receptors contained the unedited version of GluA2, which has a glutamine (Q) rather than an arginine (R) at the tip of the pore helix (Fig. 9 B). The selective inhibition by pentobarbital of the phase 2 component of the PF train EPSCs suggests that the AMPARs that underlie this component contain the edited (R) version of GluA2.
Figure 9. Pentobarbital inhibition is selective for the high‐open‐probability (Popen) gating mode .
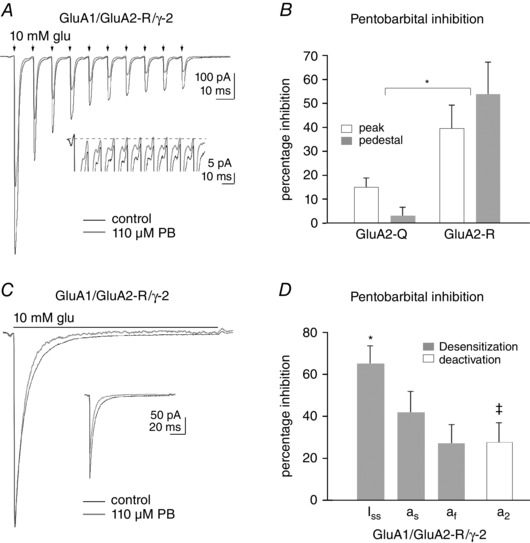
A, inward currents in an outside‐out patch from an oocyte co‐injected with cRNA for GluA1, GluA2‐R and γ‐2. The currents were evoked by ten 2 ms applications (arrows) of 10 mm glutamate applied every 10 ms in control solution (black trace) and in the continuous presence of 110 μm pentobarbital (PB; grey trace). Pentobarbital blocks the initial peak current by 34%, whereas the pedestal current is reduced by 53% (inset). B, mean percentage inhibition of the peak and pedestal currents produced by 110 μm pentobarbital for GluA1/GluA2/γ‐2 heteromeric receptors for versions of GluA2 that encoded either a glutamine (Q) or arginine (R) at the Q/R editing site at the tip of the pore helix (n = 5 and 4, respectively). Bars indicate SEM. *The percentage inhibition by pentobarbital, of both peak and pedestal currents, was greater for receptors containing GluA2‐R than for GluA2‐Q‐containing receptors (P < 0.0003, two‐way ANOVA, four or five patches per group). C, inward currents evoked by a 100 ms application of 10 mm glutamate in the absence (black trace) and presence of 110 μm pentobarbital (grey trace). The control and pentobarbital traces have been scaled so that they have the same peak amplitude. The inset shows the pair of responses on the same vertical scale. Note that the slow component of desensitization is reduced more than the peak response. D, mean values obtained from bi‐exponential fits to the decay of currents evoked by 100 ms (grey bars) or 2 ms (open bars) applications of 10 mm glutamate for pentobarbital (110 μm) inhibition of the steady‐state current (I ss), the fast and slow component of desensitization (a f and a s) and the slow component of deactivation (a 2). *The inhibition of the steady‐state current was significantly different from inhibition of the fast component (P < 0.05, one‐way ANOVA, n = 5). ‡The relative amplitude of the slow component of deactivation (a 2) was reduced by 110 μm pentobarbital (percentage change significantly different from zero, P = 0.022, Student's two‐tailed t test, n = 7).
The difference in the mean percentage inhibition for the peak and pedestal currents was modest and was not statistically significant when the two groups were treated as independent samples (Fig. 9 B). However, for the receptors containing GluA2‐R, the percentage inhibition of the pedestal current was in all cases greater than the corresponding inhibition of the peak current in the same patch, and the mean difference between the paired inhibition values obtained for four patches was significantly greater than zero (16.5 ± 6.5%, P < 0.05).
The dependence of pentobarbital inhibition on the presence of a charged residue within the transmembrane electric field (Taverna et al. 1994; Yamakura et al. 1995), as well as the use dependence of block (Marzalec & Narahashi, 1993), are features of open‐channel blockers. However, pentobarbital is largely uncharged at physiological pH, and the pH and voltage dependence of the block is inconsistent with an open‐channel blocking mechanism (Miljkovic & MacDonald, 1986; Jackson et al. 2003). Although the mechanism by which pentobarbital inhibits AMPAR currents remains somewhat unclear, Jackson and co‐workers (2003) found that pentobarbital selectively reduced steady‐state currents in hippocampal neurons (compared with initial peak currents activated by glutamate), suggesting the possibility that the high‐P open mode, which is largely responsible for the I ss for TARPed receptors (Zhang et al. 2014; Howe, 2015), is more sensitive to inhibition by pentobarbital.
To test this possibility more directly, we evaluated the effect of 110 μm pentobarbital on responses to 100 ms applications of 10 mm glutamate. The results from one patch are shown in Fig. 9 C, where the control currents and those in pentobarbital (inset) have been scaled to have the same peak amplitude. As can be seen, the portion of the trace corresponding to the slow component of desensitization and also the I ss are preferentially reduced by pentobarbital. Bi‐exponential fits to similar desensitization decays with and without 110 μm pentobarbital were obtained from five patches. The grey bars in Fig. 9 D show the mean percentage inhibition values for the I ss, as well as the amplitudes of the fast and slow exponential components of desensitization (a f and a s, respectively). The inhibition was greatest for the I ss and least for the fast component (P < 0.05). To test whether this difference arose from the known use dependence of pentobarbital block, we also tested seven patches with 2 ms applications, reasoning that any use dependence of the block would be minimal during such brief exposure to glutamate. Bi‐exponential fits to the deactivation decays were performed for the currents in each patch before and during 110 μm pentobarbital. The relative amplitude of the slow component (a 2) was significantly reduced by pentobarbital (Fig. 9 D; P = 0.022, n = 7). The comparison demonstrates that the slow component of decay, which arises from TARP‐mediated increases in the effective rate constant for channel opening when the receptor gates in the high‐P open mode (Cho et al, 2007; Howe, 2015), is selectively inhibited by pentobarbital. All the effects of pentobarbital were completely reversible.
Functional consequences: action potential generation by train EPSCs
We have shown that PF stimulation can recruit two types of train EPSCs, one with large initial amplitude that saturates after three stimuli (phase 1) and the other that is initially small but builds up progressively during a train of 10 stimuli (phase 2). Parallel fibre stimulation often recruits both types of train EPSCs, and the synaptic response is a mixture of phase 1 and phase 2 components. We next asked how the different patterns of the train EPSC affect the action potential discharge of the postsynaptic Purkinje cell. To evaluate further the functional impact of the currents we have identified, we measured responses to trains of PF stimuli in sequential voltage‐ and current‐clamp recordings. Current was injected to maintain the baseline membrane potential near −70 mV. Parallel fibre train EPSCs were stimulated at various locations of the dendritic tree in voltage clamp. For each location, the recording was then switched to current clamp to record the action potential discharge of the Purkinje cell in response to the train.
Figure 10 A presents three examples of train EPSCs and corresponding current‐clamp responses from the same cell. The examples show: a saturating train EPSC, which in current clamp triggers a 30.1 ms burst of action potentials at 233 Hz after a 3.3 ms delay (Fig. 10 Ai); a hybrid train EPSC, which triggers a longer burst of action potentials (84.0 ms) at lower frequency (143 Hz) and after a longer delay (16.2 ms; Fig. 10 Aii); and a train EPSC that builds up throughout the train and triggers a 173.2 ms burst of action potentials at 83.7 Hz after a delay of 30.8 ms (Fig. 10 Aiii). Virtually all positions tested in 10 cells triggered a burst of action potentials.
Figure 10 B shows the analysis of results obtained from 10 cells. Parameters are plotted against the amplitude of the first EPSC of the train (A1), with average values for the 10 cells. Figure 10 Bi–iv shows the following plots: R2 (P2/A1), measured for the train EPSCs recorded in voltage clamp (Fig. 10 Bi); the delay to action potential firing (Fig. 10 Bii); the spike frequency during the burst (Fig. 10 Biii); and the duration of the action potential burst, measured for the corresponding current clamp responses (Fig. 10 Biv). Figure 10 Bi shows that R2, a measure of the relative proportion of phase 2, increases as the amplitude of A1 decreases, as seen earlier in the data shown in Fig. 1 D. The delay to the first spike in current clamp, shown in Fig. 10 Bii, also increases as A1 decreases, whereas the spike frequency, plotted in Fig. 10 Biii, decreases, and the duration of the spike burst, plotted in Fig. 10 Biv, increases.
The increased delay and decreased firing frequency for train EPSCs with small A1 values is expected, because such train EPSCs build up progressively from initially small amplitudes (Figs 1 and 3). As a result, the corresponding depolarizations are slower, taking more time to reach threshold for action potential generation. Surprisingly, however, action potential burst duration was longer for train EPSCs with small A1 values, presumably because the large depolarization associated with large‐A1 train EPSCs resulted in sodium channel inactivation and termination of action potential generation. Consistent with this explanation, reductions in spike amplitude preceded termination of the burst (Fig. 10 Ai and ii). Taken together, the data suggest that large and saturating inputs require only short presynaptic bursts of activity to recruit spiking of the postsynaptic Purkinje cell efficiently, whereas the initially small, progressively facilitating inputs generate delayed, lower frequency spike trains that are sustained for longer times.
Discussion
Two types of parallel fibre synapses with different AMPAR signatures
Our analysis of the PF train EPSC defines two phases, with phase 1 dominated by a peak response that saturates after the third stimulation and phase 2 dominated by a component that increases progressively throughout the train. The initial amplitude of the second component is usually small, but it can grow to large amplitudes and becomes more obvious as the first component levels off. Both presynaptic and postsynaptic factors might contribute to this behaviour. For example, phase 1 may arise from synapses with high release probability, which give initially large peak EPSCs that saturate rapidly. Conversely, phase 2 may arise from synapses with low release probability, leading to small initial amplitudes but slow and progressive recruitment. However, it is also possible that the properties of the different postsynaptic receptor populations identified here underlie the facilitation of phase 1 and 2 of the train EPSC.
Our measurements of the sensitivity of the train EPSC to the AMPAR antagonists pentobarbital and NBQX show that the properties of the postsynaptic receptors activated during the two phases are different. Phase 1 is less sensitive than phase 2 to block by pentobarbital and more sensitive to block by the competitive antagonist NBQX. Purkinje cells express the pore‐forming subunits GluA1, 2 and 3, the auxiliary subunits TARP γ‐2 and γ‐7, as well as GluD2 (Lambolez et al. 1992; Baude et al. 1994; Petralia et al. 1998; Yamazaki et al. 2010; Yamasaki et al. 2011). Although it seems likely that the composition of the AMPARs studied here in outside‐out patches (GluA1/GluA2/γ‐2) is representative of Purkinje cell AMPARs that are postsynaptic to PF inputs, in principle there are many potential molecular compositions and stoichiometries. The large currents produced by CNQX in whole‐cell recordings, and the similar sensitivity of the CNQX‐evoked currents and phase 2 of the synaptic responses to pentobarbital, suggest that phase 2 responses largely result from activation of receptors that contain TARP γ‐2. The similar resistance to block by NBQX of phase 2 of the synaptic response and the pedestal and steady‐state currents seen for γ‐2‐containing receptors in outside‐out patches also support the conclusion that the AMPARs underlying phase 2 contain γ‐2. The sensitivity of phase 2 to block by pentobarbital and the selective block by pentobarbital of TARPed receptors that contain GluR2‐R (vs. GluR2‐Q) support the conclusion that phase 2 of the train EPSC arises, primarily at least, from receptors that also contain the edited version of GluA2.
The identity of the receptors that underlie phase 1 of the train EPSC is less clear. The initial ESPC, A1, was relatively insensitive to 110 μm pentobarbital, raising the possibility that receptors that lack the edited version of GluA2 could contribute to phase 1, although previous studies have concluded that Purkinje cells lack such receptors (Häusser & Roth, 1997; Tempia et al. 1996). The data presented here do not allow us to reach a conclusion on that point. Our results with pentobarbital and NBQX in outside‐out patches show that differences in the sensitivity of phase 1 and phase 2 to block by antagonists may also reflect functional heterogeneity of individual receptors. The results strongly suggest that the low‐ and high‐P open gating modes described previously for TARP‐containing receptors (Zhang et al. 2014; Howe, 2015) have different sensitivities to NBQX, as well as to pentobarbital. In TARP γ‐2‐containing receptors, the pedestal current seen during trains of repeated brief applications of glutamate, the I ss during sustained glutamate applications and the slow component present in deactivation decays all reflect receptors that are gating in the high‐P open mode, which we show here are 5‐ to 10‐fold less sensitive to block by NBQX and more sensitive to block by pentobarbital than receptors gating in the low‐P open mode. In the presence of TARPs, the fast components in the bi‐exponential deactivation and desensitization decays arise from receptors gating in the low‐P open mode, which exhibit gating behaviour indistinguishable from that of AMPARs that lack TARPs. Even for GluA/TARP tandem receptors (which contain four TARP molecules per receptor), the low‐P open mode predominates in single‐channel records (60–70% of the total time) and accounts for most of the peak ensemble current (Zhang et al. 2014; Howe, 2015). In short, even when TARPs are included in receptor assemblies, at any given moment most receptors in a population will behave as though TARPs were not present. The greater sensitivity of the peak currents and phase 1 of the train EPSCs to NBQX inhibition, and their reduced sensitivity to block by pentobarbital, does not imply that the receptors giving rise to the peak current and phase 1 lack γ‐2; instead, they may simply be gating in the low‐P open mode. Together with our prior single‐channel data (Zhang et al. 2014), the results in outside‐out patches presented here clearly indicate that sensitivity to block by NBQX or pentobarbital differs for the low‐ and high‐P open modes, presumably because the two modes reflect different sets of receptor conformations.
When the peak currents are defined as the difference between the maximal inward current and the I ss, they are completely blocked by submicromolar concentrations of NBQX, similar to currents through receptors that do not contain TARPs (Fig. 8; Robert & Howe, 2003), whereas the pedestal and steady‐state currents are about an order of magnitude less sensitive. In contrast, the slow components of deactivation and desensitization, which arise from the high‐P open gating mode (Howe, 2015), exhibit increased sensitivity to block by pentobarbital (Fig. 9). Together with the different pharmacology of phase 1 and phase 2, the results suggest that the high‐P open gating mode of TARPed receptors makes a substantial contribution to phase 2 of the synaptic response to high‐frequency PF stimulation.
The kinetic heterogeneity inherent in the modal gating seen for TARP‐associated AMPARs contributes to the pedestal currents that develop during trains of brief glutamate applications, and the lower affinity for NBQX of AMPARs in the high‐P open mode is likely to contribute to the increase in the steady‐state currents we often observed during sustained 100 ms applications in the presence of NBQX (attributable to enhanced re‐equilibration of the block in competing glutamate); however, this does not explain why the steady‐state currents were larger at NBQX concentrations of 50–500 nm than in the absence of antagonist. Our results provide no explanation for this apparent recruitment by NBQX of receptors into the high‐P open gating mode. Recently, however, Carbone & Plested (2016) reported that AMPARs that contain TARPs exhibit evidence of a ‘superactivated’ state, which is recruited by repeated activation during a train of glutamate applications. The superactivated state appears to be what we have referred to as the high‐P open mode. Such activity‐dependent recruitment of superactivated or high‐P open gating might contribute to the build‐up of the phase 2 component during train EPSCs, and perhaps also to the enhanced steady‐state currents we saw in patches at NBQX concentrations in the 50–500 nm range.
Although our results suggest that modal gating contributes to the different pharmacology of phase 1 and phase 2 of the train EPSCs, it does not account for the large variability we observed for the relative amplitude of phase 1 and 2 for different stimulation sites in the same Purkinje cell. Synapse‐to‐synapse differences in TARP incorporation in the receptor population might contribute to this variability, or it may reflect other differences in molecular composition. Nonetheless, our results indicate that PF synapses are equipped with at least two populations of postsynaptic AMPARs that differ in their sensitivity to antagonists, kinetic behaviour and, perhaps, molecular composition. We did not try to isolate single synapse responses; therefore, we cannot show unambiguously whether these populations of receptors co‐localize or whether they are segregated at independent synaptic sites. However, the analysis shows that the two components of the PF train EPSC, saturating and progressively facilitating, are present in variable proportions at different dendritic sites on single Purkinje cells. Moreover, both components have been observed in near isolation. For these reasons, we suggest that the receptor populations underlying components 1 and 2, and highlighted by the differential sensitivity to antagonists, correspond to two different populations of synapses.
Could spillover generate phase 2 currents?
When we presented the results describing the different sensitivity of phase 1 and 2 to NBQX, we discussed the possibility that phase 2 might be generated by spillover of glutamate. We argued that the NBQX unbinding rate is too slow (2–3 s−1; MacLean et al. 2014) to account for rapid facilitation of component 2 during a 100 ms train. However, this NBQX dissociation rate appears to apply to receptors in the low‐P open gating mode and be substantially faster for TARPed receptors gating in the high‐P open mode (which exhibit reduced affinity for NBQX). Other results, however, support the conclusion that spillover is unlikely to contribute substantially to the phase 2 current. Takayasu et al. (2004) used trains of three PF stimuli (corresponding to phase 1 of our train EPSCs). In the presence of the glutamate transporter blocker TBOA (200 μm), spillover is pushed to the extreme and activates AMPARs for hundreds of milliseconds. Even in these conditions, a concentration of NBQX (250 nm) that has little effect on the phase 2 current inhibited the spillover phase of the EPSC to the same extent as the fast three stimuli EPSC (Takayasu et al. 2004). These results indicate that the spillover current does not have a reduced sensitivity to NBQX, and also suggests that there is no substantial unbinding and displacement of the antagonist by concentrations of glutamate reached during spillover.
Finally, we have argued that phase 2 currents are carried by AMPARs that contain the TARP γ‐2 subunit. If TARPed receptors were extrasynaptic, as seen for γ‐8 in the hippocampus (Inamura et al. 2006), they could detect spillover glutamate. The subcellular localization of γ‐2 has been investigated by Yamazaki et al. (2010) using immunogold labelling; γ‐2 is enriched at postsynaptic sites but is not detectable above background levels at extrasynaptic sites, making it unlikely that the phase 2 current arises from extrasynaptic receptors. In addition, the density of extrasynaptic AMPARs appears to be very low in Purkinje cells (Masugi‐Tokita et al. 2007).
Spillover has been shown to be prominent at a number of synapses in the cerebellum, including those made by PFs (Barbour et al. 1994; Carter & Regehr, 2000; Balakrishan et al. 2014) and, although we think it does not account for the phase 2 component, it may contribute to the slow decay of the train EPSCs at the end of PF train stimulation. We have not studied that aspect of the train EPSCs, and the basis of the slow decay remains unknown.
Synaptic correlates
Both in vivo sensory stimulation by Ekerot & Jörntell (2001) and paired recordings in slices by Isope & Barbour (2002) have suggested that most of the PF synapses are electrically silent. The average amplitude of identified connections between parallel fibre and Purkinje cells determined by Isope & Barbour (2002) was 8.4 ± 7.1 pA, but the connection rate was low, only 15% of that expected from morphological analysis. The saturating component of the EPSC would be the likely correlate of the synapse identified in paired recordings and described by Isope & Barbour (2002) as a high‐release‐probability synapse. The amplitude (A1) of the first EPSC in the trains we recorded varied greatly from cell to cell. However, the largest train EPSCs (A1 a∼1 nA; Fig. 1) give an estimate of 125 for the number of granule cell axons activated if their A1 amplitude is compared with the average EPSC amplitude measured by Isope & Barbour (2002) for stimulation of single granule cells (8.4 pA). Assuming that we stimulated a comparable number of fibres between experiments, for train EPSCs with A1 amplitudes of 200 pA, the amplitude for a single connection would be 1.6 pA (a very small current, corresponding to activation of one or two receptors) if the stimulation recruited 125 fibres, and PF train EPSCs dominated by the phase 2 current were typically smaller than 200 pA. Therefore, the individual connections that (as a population) give rise to the phase 2 current might be too small to detect. Given that the initial response of slowly facilitating synapses is very small, we suggest that they have usually remained unnoticed. Indeed, to our knowledge, slowly facilitating train EPSCs or hybrids of saturating and facilitating trains like those shown in Fig. 1 have not been reported. This suggests that our understanding of PF inputs to Purkinje cells is biased towards the small population of conspicuous synapses.
Physiological consequences
Facilitating phase 2 synapses are recruited efficiently only during a train of presynaptic activity. It has been shown that granule cells generate outputs that are very different depending on the sensory modalities conveyed by mossy fibres (reviewed by Arenz et al. 2009). Some sensory inputs are conveyed by brief on‐response bursts (Chadderton et al. 2004), whereas others generate sustained bursts of action potentials (Jörntell & Ekerot, 2006). Large initial saturating EPSCs would be ideally suited to transmit brief high‐frequency bursts and to respond with high temporal precision, as the data in Fig. 10 demonstrate. In contrast, the progressively facilitating EPSCs seen in phase 2 would be expected to filter brief or low‐frequency activity, only effectively transmitting sustained or repetitive bursts of activity, an expectation confirmed by our voltage‐ and current‐clamp recordings from the same Purkinje cells (Fig. 10). The two types of synapses appear well suited to transmit different types of presynaptic activity patterns. We have also shown that the distribution of the two types of responses on the dendritic tree of a single cell is highly heterogeneous. This may reflect the tuning of postsynaptic properties to the function of individual synapses or local dendritic areas.
Additional information
Competing interests
None declared.
Author contributions
S.P.S.D., J.R.H. and C.A.: conception and design; collection and assembly of data; data analysis and interpretation; and writing the manuscript. All authors approved the final version of the manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the EC Sixth Framework Programme grant (LSHM‐CT‐2007‐037765), the Agence Nationale de la Recherche grant (ANR‐2010‐BLAN‐143601 and the National Center for Scientific Research (France) to C.A. The work done by S.P.S.D. and J.R.H. was supported by funds from Yale University.
Acknowledgements
We thank David Ogden for continuous discussion and critical reading of the manuscript.
References
- Arenz A, Bracey EF & Margrie TW (2009). Sensory representations in cerebellar granule cells. Curr Opin Neurobiol 19, 445–451. [DOI] [PubMed] [Google Scholar]
- Auger C & Ogden DC (2010). AMPA receptor activation controls type I metabotropic glutamate receptor signalling via a tyrosine kinase at parallel fibre–Purkinje cell synapses. J Physiol 588, 3063–3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan S, Dobson KL, Jackson C & Bellamy TC (2014). Ectopic release of glutamate contributes to spillover at parallel fibre synapses in the cerebellum. J Physiol 592, 1493–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbour B, Keller BU, Llano I & Marty A (1994). Prolonged presence of glutamate during excitatory synaptic transmission to cerebellar Purkinje cells. Neuron 12, 1331–1343. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Madge DJ & Garthwaite J (1994). Synaptic activation of metabotropic glutamate receptors in the parallel fibre‐Purkinje cell pathway in rat cerebellar slices. Neuroscience 63, 911–915. [DOI] [PubMed] [Google Scholar]
- Bats C, Soto D, Studniarczyk D, Farrant M & Cull‐Candy SG (2012). Channel properties reveal differential expression of TARPed and TARPless AMPARs in stargazer neurons. Nat Neurosci 15, 853–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baude A, Molnár E, Latawiec D, McIlhinney RA & Somogyi P (1994). Synaptic and nonsynaptic localization of the GluR1 subunit of the AMPA‐type excitatory amino acid receptor in the rat cerebellum. J Neurosci 14, 2830–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunel N, Hakim V, Isope P, Nadal JP & Barbour B (2004). Optimal information storage and the distribution of synaptic weights: perceptron versus Purkinje cell. Neuron 43, 745–757. [DOI] [PubMed] [Google Scholar]
- Canepari M, Auger C & Ogden D (2004). Ca2+ ion permeability and single‐channel properties of the metabotropic slow EPSC of rat Purkinje neurons. J Neurosci 24, 3563–3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canepari M & Ogden D (2003). Evidence for protein tyrosine phosphatase, tyrosine kinase, and G‐protein regulation of the parallel fiber metabotropic slow EPSC of rat cerebellar Purkinje neurons. J Neurosci 23, 4066–4071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carbone AL & Plested AJ (2016). Superactivation of AMPA receptors by auxiliary proteins. Nat Commun 7, 10178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG & Regehr WG (2000). Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci 20, 4423–4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW & Häusser M (2004). Integration of quanta in cerebellar granule cells during sensory processing. Nature 428, 856–860. [DOI] [PubMed] [Google Scholar]
- Cho CH, St‐Gelais F, Zhang W, Tomita S & Howe JR (2007). Two families of TARP isoforms that have distinct effects on the kinetic properties of AMPA receptors and synaptic currents. Neuron 55, 890–904. [DOI] [PubMed] [Google Scholar]
- Cokić B & Stein V (2008). Stargazin modulates AMPA receptor antagonism. Neuropharmacology 54, 1062–1070. [DOI] [PubMed] [Google Scholar]
- Delaney AJ & Jahr CE (2002). Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron 36, 475–482. [DOI] [PubMed] [Google Scholar]
- Diamond JS & Jahr CE (1997). Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci 17, 4672–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman SJ, Kreitzer AC & Regehr WG (2000). Interplay between facilitation, depression, and residual calcium at three presynaptic terminals. J Neurosci 20, 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekerot CF & Jörntell H (2001). Parallel fibre receptive fields of Purkinje cells and interneurons are climbing fibre‐specific. Eur J Neurosci 13, 1303–1310. [DOI] [PubMed] [Google Scholar]
- Foster KA, Crowley JJ & Regehr WG (2005). The influence of multivesicular release and postsynaptic receptor saturation on transmission at granule cell to Purkinje cell synapses. J Neurosci 25, 11655–11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RJ & Napper RM (1988). Quantitative study of granule and Purkinje cells in the cerebellar cortex of the rat. J Comp Neurol 274, 151–157. [DOI] [PubMed] [Google Scholar]
- Häusser M & Roth A (1997). Dendritic and somatic glutamate receptor channels in rat cerebellar Purkinje cells. J Physiol 501, 77–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe JR (2015). Modulation of non‐NMDA receptor gating by auxiliary subunits. J Physiol 593, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YH, Dykes‐Hoberg M, Tanaka K, Rothstein JD & Bergles DE (2004). Climbing fiber activation of EAAT4 transporters and kainate receptors in cerebellar Purkinje cells. J Neurosci 24, 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamura M, Itakura M, Okamoto H, Hoka S, Mizoguchi A, Fukazawa Y, Shigemoto R, Yamamori S & Takahashi M (2006). Differential localization and regulation of stargazin‐like protein, γ‐8 and stargazin in the plasma membrane of hippocampal and cortical neurons. Neurosci Res 55, 45–53. [DOI] [PubMed] [Google Scholar]
- Isope P & Barbour B (2002). Properties of unitary granule cell→Purkinje cell synapses in adult rat cerebellar slices. J Neurosci 22, 9668–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC & Nicoll RA (2011). The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron 70, 178–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MF, Joo DT, Al‐Mahrouki AA, Orser BA & Macdonald JF (2003). Desensitization of α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA) receptors facilitates use‐dependent inhibition by pentobarbital. Mol Pharmacol 64, 395–406. [DOI] [PubMed] [Google Scholar]
- Jörntell H & Ekerot C‐F (2006). Properties of somatosensory synaptic integration in cerebellar granule cells in vivo . J Neurosci 26, 11786–11797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato AS, Gill MB, Yu H, Nisenbaum ES & Bredt DS (2010). TARPs differentially decorate AMPA receptors to specify neuropharmacology. Trends Neurosci 33, 241–248. [DOI] [PubMed] [Google Scholar]
- Lambolez B, Audinat E, Bochet P, Crépel F & Rossier J (1992). AMPA receptor subunits expressed by single Purkinje cells. Neuron 9, 247–258. [DOI] [PubMed] [Google Scholar]
- Ly R, Bouvier G, Szapiro G, Prosser HM, Randall AD, Kano M, Sakimura K, Isope P, Barbour B & Feltz A (2016). Contribution of postsynaptic T‐type calcium channels to parallel fibre‐Purkinje cell synaptic responses. J Physiol 594, 915–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean DM & Bowie D (2011). Transmembrane AMPA receptor regulatory protein regulation of competitive antagonism: a problem of interpretation. J Physiol 589, 5383–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean DM, Ramaswamy SS, Du M, Howe JR & Jayaraman V (2014). Stargazin promotes closure of the AMPA receptor ligand‐binding domain. J Gen Physiol 144, 503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marszalec W & Narahashi T (1993). Use‐dependent pentobarbital block of kainate and quisqualate currents. Brain Res 608, 7–15. [DOI] [PubMed] [Google Scholar]
- Masugi‐Tokita M, Tarusawa E, Watanabe M, Molnár E, Fujimoto K & Shigemoto R (2007). Number and density of AMPA receptors in individual synapses in the rat cerebellum as revealed by SDS‐digested freeze‐fracture replica labeling. J Neurosci 27, 2135–2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menuz K, Stroud RM, Nicoll RA & Hays FA (2007). TARP auxiliary subunits switch AMPA receptor antagonists into partial agonists. Science 318, 815–817. [DOI] [PubMed] [Google Scholar]
- Merrill EG, Wall PD & Yaksh TL (1978). Properties of two unmyelinated fibre tracts of the central nervous system: lateral Lissauer tract, and parallel fibres of the cerebellum. J Physiol 284, 127–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic Z & MacDonald JF (1986). Voltage‐dependent block of excitatory amino acid currents by pentobarbital. Brain Res 376, 396–399. [DOI] [PubMed] [Google Scholar]
- Milstein AD & Nicoll RA (2008). Regulation of AMPA receptor gating and pharmacology by TARP auxiliary subunits. Trends Pharmacol Sci 29, 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milstein AD, Zhou W, Karimzadegan S, Bredt DS & Nicoll RA (2007). TARP subtypes differentially and dose‐dependently control synaptic AMPA receptor gating. Neuron 55, 905–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Yokotani N & Wenthold RJ (1994). Light and electron microscope distribution of the NMDA receptor subunit NMDAR1 in the rat nervous system using a selective anti‐peptide antibody. J Neurosci 14, 667–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Zhao HM, Wang YX & Wenthold RJ (1998). Variations in the tangential distribution of postsynaptic glutamate receptors in Purkinje cell parallel and climbing fibre synapses during development. Neuropharmacol 37, 1321–1334. [DOI] [PubMed] [Google Scholar]
- Piochon C, Irinopoulou T, Brusciano D, Bailly Y, Mariani J & Levenes CJ (2007). NMDA receptor contribution to the climbing fiber response in the adult mouse Purkinje cell. J Neurosci 27, 10797–10809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitchitpornchai C, Rawson JA & Rees S (1994). Morphology of parallel fibres in the cerebellar cortex of the rat: an experimental light and electron microscopic study with biocytin. J Comp Neurol 54, 206–220. [DOI] [PubMed] [Google Scholar]
- Renzi M, Farrant M & Cull‐Candy SGJ (2007). Climbing‐fibre activation of NMDA receptors in Purkinje cells of adult mice. J Physiol 585, 91–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert A & Howe JR (2003). How AMPA receptor desensitization depends on receptor occupancy. J Neurosci 23, 847–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheardown MJ, Nielsen EO, Hansen AJ, Jacobsen P & Honore T (1990). 2,3‐Dihydroxy‐6‐nitro‐7‐sulfamoyl‐benzo(F)quinoxaline: a neuroprotectant for cerebral ischemia. Science 247, 571–574. [DOI] [PubMed] [Google Scholar]
- Smith TC & Howe JR (2000). Concentration‐dependent substate behavior of native AMPA receptors. Nat Neurosci 3, 992–997. [DOI] [PubMed] [Google Scholar]
- Takayasu Y, Iino M & Ozawa S (2004). Roles of glutamate transporters in shaping excitatory synaptic currents in cerebellar Purkinje cells. Eur J Neurosci 19, 1285–1295. [DOI] [PubMed] [Google Scholar]
- Taverna FA, Cameron BR, Hampson DL, Wang LY & MacDonald JF (1994). Sensitivity of AMPA receptors to pentobarbital. EurJ Pharmacol 267, R3–R5. [DOI] [PubMed] [Google Scholar]
- Tempia F, Kano M, Schneggenburger R, Schirra C, Garaschuk O, Plant T & Konnerth A (1996). Fractional calcium current through neuronal AMPA‐receptor channels with a low calcium permeability. J Neurosci 16, 456–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempia F, Miniaci MC, Anchisi D & Strata P (1998). Postsynaptic current mediated by metabotropic glutamate receptors in cerebellar Purkinje cells. J Neurophysiol 80, 520–528. [DOI] [PubMed] [Google Scholar]
- Tomita S, Adesnik H, Sekiguchi M, Zhang W, Wada K, Howe JR, Nicoll RA & Bredt DS (2005). Stargazin modulates AMPA receptor gating and trafficking by distinct domains. Nature 435, 1052–1058. [DOI] [PubMed] [Google Scholar]
- Wisden W & Seeburg PH (1993). A complex mosaic of high‐affinity kainate receptors in rat brain. J Neurosci 13, 3582–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Sakimura K, Mishina M & Shimoji K (1995). The sensitivity of AMPA‐selective glutamate receptor channels to pentobarbital is determined by a single amino acid residue of the α2 subunit. FEBS Lett 374, 412–414. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Miyazaki T, Azechi H, Abe M, Natsume R, Hagiwara T, Aiba A, Mishina M, Sakimura K & Watanabe M (2011). Glutamate receptor δ2 is essential for input pathway‐dependent regulation of synaptic AMPAR contents in cerebellar Purkinje cells. J Neurosci 31, 3362–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki M, Fukaya M, Hashimoto K, Yamasaki M, Tsujita M, Itakura M, Abe M, Natsume R, Takahashi M, Kano M, Sakimura K & Watanabe M (2010). TARPs γ‐2 and γ‐7 are essential for AMPA receptor expression in the cerebellum. Eur J Neurosci 31, 2204–2220. [DOI] [PubMed] [Google Scholar]
- Yan D & Tomita S (2012). Defined criteria for auxiliary subunits of glutamate receptors. J Physiol 590, 21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Devi SP, Tomita S & Howe JR (2014). Auxiliary proteins promote modal gating of AMPA‐ and kainate‐type glutamate receptors. Eur J Neurosci 39, 1138–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Robert A, Vogensen SB & Howe JR (2006). The relationship between agonist potency and AMPA receptor kinetics. Biophys J 91, 1336–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao HM, Wenthold RJ & Petralia RS (1998). Glutamate receptor targeting to synaptic populations on Purkinje cells is developmentally regulated. J Neurosci 18, 5517–5528. [DOI] [PMC free article] [PubMed] [Google Scholar]