

Abstract
The nicotinamide nucleotide adenylyltransferase 1 (NMNAT1) enzyme is essential for regenerating the nuclear pool of NAD+ in all nucleated cells in the body, and mounting evidence also suggests that it has a separate role in neuroprotection. Recently, mutations in the NMNAT1 gene were associated with Leber congenital amaurosis, a severe retinal degenerative disease that causes blindness during infancy. Availability of a reliable mammalian model of NMNAT1-Leber congenital amaurosis would assist in determining the mechanisms through which disruptions in NMNAT1 lead to retinal cell degeneration and would provide a resource for testing treatment options. To this end, we identified two separate N-ethyl-N-nitrosourea–generated mouse lines that harbor either a p.V9M or a p.D243G mutation. Both mouse models recapitulate key aspects of the human disease and confirm the pathogenicity of mutant NMNAT1. Homozygous Nmnat1 mutant mice develop a rapidly progressing chorioretinal disease that begins with photoreceptor degeneration and includes attenuation of the retinal vasculature, optic atrophy, and retinal pigment epithelium loss. Retinal function deteriorates in both mouse lines, and, in the more rapidly progressing homozygous Nmnat1V9M mutant mice, the electroretinogram becomes undetectable and the pupillary light response weakens. These mouse models offer an opportunity for investigating the cellular mechanisms underlying disease pathogenesis, evaluating potential therapies for NMNAT1-Leber congenital amaurosis, and conducting in situ studies on NMNAT1 function and NAD+ metabolism.
Leber congenital amaurosis (LCA), a genetically heterogeneous retinal degenerative disease associated with mutations in at least 18 genes, represents the most common cause (10% to 18%) of incurable blindness in children1, 2 and accounts for ≥5% of all inherited retinal degenerations.3 Across all forms of LCA, vision is impaired at birth or begins deteriorating during early infancy because of retinal degeneration.4 Patients often develop nystagmus, and, as the disease advances, amaurotic pupils may be observed2 because of the absence of functioning photoreceptors and intrinsically photosensitive ganglion cells.5, 6 Both rod and cone photoreceptors degenerate rapidly, and many patients have no detectable electroretinogram (ERG) response after the first year of life.4 Although LCA can be a feature of syndromic diseases, each form of this retinal degeneration is caused by mutations in a single gene, and most cases are recessively inherited2 (RetNet, https://sph.uth.edu/retnet/disease.htm, accessed March 7, 2016).
Genes that harbor mutations responsible for LCA have critical roles in different cell types throughout the retina.2, 7 A substantial portion (approximately 5%)8 of LCA cases are caused by mutations in the NMNAT1 gene,1, 8, 9, 10, 11, 12, 13, 14, 15 which encodes the nicotinamide nucleotide adenylyltransferase 1 (NMNAT1) enzyme that is essential for regenerating the nuclear pool of NAD+. Nuclear NAD+, synthesized from ATP and either nicotinic acid mononucleotide or nicotinamide mononucleotide, is important for processes related to DNA repair, gene expression, cell senescence, and cell signaling.16, 17, 18 In the Golgi complex and mitochondria, two other NMNAT isoforms that similarly serve to regenerate NAD+ are encoded by NMNAT2 and NMNAT3, respectively.17, 19 However, neither NMNAT2 nor NMNAT3 compensates for the loss of NMNAT1 function, considering that homozygous knockout of Nmnat1 in mice results in embryonic lethality.20 Moreover, multiple lines of evidence indicate that NMNAT1 may have neuroprotective chaperone and housekeeping functions that are separate from its role in the NAD+ synthesis pathway.21, 22, 23, 24, 25, 26 As an example, the Wallerian degeneration slow (WldS) fusion protein that delays axon degeneration after nerve injury in WldS mice includes full-length Nmnat1 fused to a portion of the Ube4b multi-ubiquitination factor.22, 24 Given that NMNAT1 is expressed ubiquitously,9, 19 it is unclear why abnormal NMNAT1 function causes a retina-specific disease.
The precise mechanism(s) underlying retinal cell degeneration in NMNAT1-LCA also has not been defined, and no treatment is available for patients. Thus far, in the absence of an animal model, mutant NMNAT1 has been studied in human embryonic kidney cells (HEK293T), human cervical cancer cells (HeLa), mouse dorsal root ganglia, NMNAT1-LCA patient fibroblasts, and NMNAT1-LCA patient red blood cells (nonnucleated),8, 10, 27, 28 with inconsistent results. Further, in vitro systems do not allow for the assessment of whether a treatment preserves vision and cannot address practical challenges associated with delivering therapeutic agents to the NMNAT1-LCA retina. For instance, determining which specific cell type(s) should be the therapeutic target may be critical for a successful intervention, given that the effects of NMNAT1 overexpression in otherwise healthy retinal cells are unknown.
Here, we report the discovery and characterization of two mutant Nmnat1 mouse lines derived from chemical mutagenesis programs.29, 30 Wild-type mice were injected with a potent mutagen, N-ethyl-N-nitrosourea (ENU), which introduces point mutations in DNA that are transmissible through the germline. Mutations in Nmnat1 were identified, and multiple outcrosses with wild-type mice served to eliminate most ENU-induced mutations at other chromosomal locations.31 One line harbors the p.V9M mutation, which was observed to cause LCA in patients from unrelated families and has been investigated in two separate studies.8, 27 The other line harbors a novel mutation, p.D243G, which has yet to be reported in the human population (ExAC Browser, http://exac.broadinstitute.org, accessed March 7, 2016). The retinal degenerative phenotype observed in both lines recapitulates key aspects of NMNAT1-LCA. These mouse models show promise for elucidating pathologic mechanisms associated with NMNAT1-LCA, developing therapies for patients with this disease, and understanding the roles of NMNAT1 in neuroprotection and NAD+ metabolism.
Materials and Methods
Identification of Nmnat1V9M Mice
Mice expressing the p.V9M variant of NMNAT1 were derived from a gene-driven screen of the Harwell ENU DNA archive to identify Nmnat1 missense alleles. The archive consists of pooled DNA samples from >10,000 ENU-mutagenized G1 male mice, which is paralleled by frozen sperm samples.29, 32 Oligonucleotide primers (Table 1) were designed to allow amplification of all protein-coding sequences within Nmnat1 and used for PCR amplification of the pooled DNA samples. Amplified products were assessed by high-resolution melting curve analysis with the use of the LightScanner platform (Idaho Technology, Salt Lake City, UT). Because of the reduced melting temperature of mismatched nucleotides, ENU-induced mutations are evident as left-shifted melt curves, which are then verified by Sanger sequencing. This approach identified the Nmnat1 c.25G>A allele (Nmnat1imh, hereafter referred to as Nmnat1V9M), which was subsequently re-derived by in vitro fertilization using cryopreserved sperm (mixed BALB/c and C3H/HeH)33 and wild-type C57BL/6J (B6) oocytes. The subsequent progeny were then bred into the wild-type B6 background. Before data collection, this mouse strain was outcrossed to B6 mice five times to eliminate approximately 97% of the ENU-induced mutations. On the basis of the mutation rate of approximately 0.67 mutations/Mbp for ENU mutagenesis and the size of the mouse genome (approximately 2.5 × 103 Mbp),31 it would be anticipated that in a mouse where no mutation is being selected for during outcrossing, only approximately 50 induced mutations would remain across the entire genome, of which just one would be expected to be in a coding region.34 The chance of one additional coding mutation that is physically linked to the known mutation on mouse chromosome 4 persisting after five outcrosses is 22%.34 To further mitigate the possibility of influence from the presence of an unidentified mutation, all experiments were performed on age-matched homozygous mutant Nmnat1V9M, heterozygous Nmnat1V9M, and wild-type mice generated by intercrossing heterozygous Nmnat1V9M mice.
Table 1.
Set of Oligonucleotide Primer Pairs that Provide Complete Coverage of the Nmnat1 Protein-Coding Sequences for PCR Amplification and Sequencing
| Exon | Forward | Reverse |
|---|---|---|
| 2 | 5′-GGTTGCATGTAGGTCAACAC-3′ | 5′-GTCTTTAATTAGCTGGGTCGC-3′ |
| 3 | 5′-TAAAGTCTGATTTGTTATGCCTAATATCG-3′ | 5′-ACAAGAACATGTGGACTGC-3′ |
| 4 | 5′-TCTGACATTAAGGAGTGTGCT-3′ | 5′-TTTGGAGTCCTGGTAGACATC-3′ |
| 5 | 5′-CCTGACCTTGTGCTTGATTC-3′ | 5′-TGGTGGACGAGATGTCATT-3′ |
| 5 | 5′-CAGAGCAACATCCACCT-3′ | 5′-GAGTCTCCAGACGAGCC-3′ |
These primers were used in the procedures that led to the identification of the p.V9M-Nmnat1 allele.
Identification of Nmnat1D243G Mice
The Nmnat1tvrm113 mutant, hereafter referred to as Nmnat1D243G, was identified in a B6 G3 ENU mutagenesis screen from the Translational Vision Research Models program30 at The Jackson Laboratory (Bar Harbor, ME) by indirect ophthalmoscopy. The fundus examination revealed an abnormal retina with pigmentary changes and a grainy appearance with light spots, and this phenotype segregated in a monogenic, recessive manner. The genomic location of the mutation was determined by linkage analysis. Genomic DNA from progeny of an F2 intercross of the mutant strain with DBA/2J (DBA) was extracted as described.35 DNA from 10 affected and 10 unaffected F2 offspring was pooled and assayed genome-wide with 48 simple sequence length polymorphic markers known to differ between B6 and DBA. Once a map position was identified on chromosome 4, it was refined in a high-resolution intercross that involved 765 F2 mice of the intercross described.
To identify the causative mutation, a whole mouse exome library was constructed, using fragmented genomic DNA (1 μg) to a peak size of 300 bp by sonicating on low power for 30 seconds with power on, 30 seconds with power off for a total of 10 minutes using a Diagenode Bioruptor UCD-200TM-EX (Denville, NJ). The precapture paired end library was constructed with the TruSeq DNA Sample Preparation Kit (part number FC-121-100; Illumina, San Diego, CA) with no size selection step and 18 cycles of PCR. The precapture library was hybridized to the Roche NimbleGen Mouse Exome (Reference no. 9999042611) capture probe set (Roche NimbleGen, Madison, WI) according to the manufacturer's instructions. The sequencing library was subjected to real-time quantitative PCR, pooled with two similar libraries, and sequenced on a single lane of an Illumina HiSeq 2000 (Illumina) using a 2 × 100 bases (paired end) sequencing protocol. High-throughput sequence data were sorted with a local JAX Galaxy interface pipeline.36, 37, 38 Sequence reads were quality assessed using FastQC version 0.5 (Babraham Bioinformatics, Babraham, UK; http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and aligned to the mouse reference genome (mm10) from the University of California Santa Cruz, released December 2011, using BWA version 1.2.3.39 PCR duplicates were removed with SAMtools rmdup version 1.0.0.40 Single nucleotide polymorphisms and insertions/deletions were called with SAMtools mpileup version 1.0.0,40 and genomic and functional annotations were assigned to the variants using SnpEff version 0.9.41 Sequence analysis revealed no additional coding variants within the critical chromosomal region. To confirm the identified variant in both the segregating mapping and the maintenance colonies, PCR amplification and analysis were performed. Phenotypic characterization was performed on age-matched B6-Nmnat1 mutant (N5) and B6 control mice.
Mouse Husbandry
Mice, bred and housed in the Schepens Eye Research Institute (Nmnat1V9M mice) and Research Animal Facility at The Jackson Laboratory (Nmnat1D243G mice), were provided ad libitum 4% or 6% fat rodent diet, respectively, and water in a vivarium with a 12-h light/12-h dark cycle. This study conformed to the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research. All procedures were approved by the Animal Care and Use Committees of the Schepens Eye Research Institute and of The Jackson Laboratory.
Genotyping
For the Nmnat1V9M line, genomic DNA was isolated from tissue collected from each mouse with the use of the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). PCR was then performed with a forward primer 5′-ACGTATTTGCCCACCTGTCT-3′ (intron 1) and a reverse primer 5′-TGGGGTTAAAAGAGCCACAG-3′ (exon 2) to amplify a 194-bp region of Nmnat1 that contained codon nine. The 20-μL PCR reactions had final concentrations of 200 μmol/L for each primer, 200 nmol/L for each of the dNTPs (dATP, dGTP, dTTP, and dCTP), 2 mmol/L MgCl2, and 1 unit of Hot FirePol DNA polymerase (Solis BioDyne, Tartu, Estonia). The thermocycling protocol was 95°C for 14 minutes; 30 cycles of 95°C for 45 seconds, 53°C for 45 seconds, 72°C for 30 seconds; 72°C for 7 minutes. The PCR product was subjected to Sanger sequencing, and the electropherograms were analyzed at nucleotide c.25 to identify each mouse as wild-type, heterozygous, or homozygous Nmnat1V9M (Figure 1A).
Figure 1.

Identification of the Nmnat1V9M and Nmnat1D243G mutations by Sanger sequencing of genomic DNA. A: Electropherograms of w/w, wt/V9M, and V9M/V9M mice, where codon 9 is bracketed and base c.25 is highlighted in yellow. B: Electropherograms of w/w, wt/D243G, and D243G/D243G mice, where codon 243 is bracketed and base c.728 is highlighted in yellow. D243G/D243G, homozygous Nmnat1D243G; V9M/V9M, homozygous Nmnat1V9M; wt/D243G, heterozygous Nmnat1D243G; wt/V9M, heterozygous Nmnat1V9M; w/w, wild-type littermate.
For Nmnat1D243G mice, the Nmnat1 genomic region that contained the mutation was amplified with the forward primer 5′-GAACGAGTGGATCACCAATGA-3′ (exon 5), reverse primer 5′-GCAAGTCCTTCCCTGAGTTT-3′ (3′ untranslated region), and the NEB taq polymerase kit (New England BioLabs, Ipswich, MA). The 11-μL PCR reactions had final concentrations of 180 nmol/L for each primer, 225 nmol/L dNTPs, and 0.55 units of Taq DNA polymerase. The thermocycling protocol was 97°C for 3 minutes; 40 cycles of 95°C for 15 seconds, 55°C for 15 seconds, 72°C for 30 seconds; 72°C for 3 minutes. The 278-bp PCR product was subjected to Sanger sequencing, and concordance of the retinal phenotype with the A>G transition at c.727 was assessed (Figure 1B).
Both mutant Nmnat1 lines were found to be negative for the Crb1rd8 mutation.42
Electroretinography
After having been dark-adapted overnight, each mouse was anesthetized by intraperitoneal injection of ketamine and xylazine diluted in sterile saline, and the eyes were dilated by topical application of 1% tropicamide (Nmnat1V9M mice) or 1% atropine sulfate (Nmnat1D243G mice) approximately 10 minutes before data collection. The mice were placed on a heated stage to maintain body temperature, and a gold ring electrode (Diagnosys, Lowell, MA) was gently placed on the center of each cornea with a small application of ophthalmic lubricant. Full-field ERGs were recorded simultaneously from both eyes in response to 4-ms broadband stimuli with the use of the ColorDome Ganzfeld system (Diagnosys). For Nmnat1V9M mice, ERGs were collected from 7 to 10 mice per genotype at 1, 2, 3, 4, 7.5, and 10 months of age, and for Nmnat1D243G mice, ERGs were collected across 12 months from three to seven mice per genotype per time point. The stimulus parameters used to test each mouse line (described in the next paragraph) were selected independently at the respective testing facility; however, both sets of parameters generate responses from the target class(es) of photoreceptors. As in all experiments described here, both male and female mice were tested.
In dark-adapted Nmnat1V9M mice, rod-driven ERGs were recorded in response to a 0.01 cd.s/m2 light stimulus (10 flashes at 0.2 Hz), and mixed rod/cone ERGs were recorded in response to a 10 cd.s/m2 light stimulus (three flashes at 0.03 Hz). For dark-adapted Nmnat1D243G mice, rod-driven ERGs were recorded in response to a 0.006 cd.s/m2 light stimulus (5 flashes at 0.1 Hz), and mixed rod/cone ERGs were recorded in response to a 0.25 cd.s/m2 stimulus (five flashes at 0.1 Hz). Next, mice were light-adapted by exposure to a steady, rod-suppressing background light that was set to 5 cd/m2 43, 44 for the Nmnat1V9M mice and 110 cd/m2 for the Nmnat1D243G mice. This background light then remained on during the acquisition of cone-driven ERGs that were recorded in response to a 20 cd.s/m2 light stimulus (20 flashes at 0.5 Hz) for the Nmnat1V9M mice and a 10 cd.s/m2 light stimulus (10 flashes at 1 Hz) for the Nmnat1D243G mice.
For all conditions, the magnitude of the ERG b-wave was measured as the absolute voltage change from the trough of the a-wave (or from the voltage measured at the expected implicit time of that trough, should the a-wave be undetectable) to the b-wave peak.45 The a-wave was measured as the absolute voltage change from prestimulus baseline to the initial post-stimulus trough.46 Analysis of the a-wave is reported for brighter stimulus intensities (ie, 0.25 cd.s/m2 and 10 cd.s/m2) in the dark-adapted condition for which this component of the ERG is robust in normal mice, as opposed to light-adapted ERGs47 and dim (ie, 0.006 cd/m2 and 0.01 cd/m2) dark-adapted ERGs.48 For the Nmnat1V9M line, a one-way analysis of variance was used for a multiple comparison across genotypes, and post hoc testing was performed with the two-tailed, unpaired t-test for which only P values <0.0167 were considered significant after Bonferroni correction. The two-tailed, unpaired t-test was used to determine significance for the Nmnat1D243G line.
In Vivo Retinal Imaging
Anesthesia and dilation procedures were as described in the section above for electroretinography. Images were collected at 0.75, 1, 2, 3, 4, 8, 10, and 15 months of age in the Nmnat1V9M mice and at ages up to 12 months in the Nmnat1D243G mice. Fundus images were acquired with the Micron retinal imaging microscope system (Phoenix Biosystems, Pleasanton, CA) and StreamPix software version 5.8.1.4 (NorPix, Montreal, Canada). The Micron model III was used for assessing the Nmnat1V9M mice, and model IV was used for assessing the Nmnat1D243G mice. GenTeal lubricating ointment was applied to the cornea to minimize light refraction between the eye and microscope objective.
Cross-sectional retinal images were acquired with the InVivoVue OCT system (Bioptogen, Morrisville, NC). Both eyes from 7 to 10 mice from each genotype were tested at each time point for the Nmnat1V9M line, with the exception of 15 months when five mice were tested per genotype, whereas one eye from four or five mutant or three to eight B6 mice was tested at each age for the Nmnat1D243G line. For both mouse lines, a rectangular OCT volume centered on the optic nerve head was captured, and a B-scan located approximately 200 μm ventral to the ONH was selected for measurement. Retinal thickness was measured as the distance between the nerve fiber layer and the hyporeflective boundary between the retinal pigment epithelium (RPE) and choroid. Significance testing of the data was performed as described in the section above for electroretinography.
Ex Vivo Retinal Imaging
Nmnat1V9M Mice
Mice were euthanized with a lethal dose of Euthasol (Virbac, Fort Worth, TX), and tissues were fixed by intracardiac perfusion with the use of a Masterflex peristaltic pump (Cole-Parmer, Vernon Hills, IL). Each mouse was perfused first with 0.13 mol/L phosphate-buffered saline (PBS) pH 7.2 to 7.4 that contained 2 U heparin/mL until the perfusate became clear, then with approximately 30 mL of 4% paraformaldehyde in PBS, followed by approximately 30 mL of one-half Karnovsky's fixative (2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 mol/L sodium cacodylate buffer, pH 7.4). All solutions were warmed to approximately 37°C at the time of perfusion, and the rate of injection was approximately 6.7 mL/min. Immediately after perfusion, each eye was marked for orientation with permanent ink and enucleated. After 1 hour of post-fixation in one-half Karnovsky's fixative, the anterior segment was removed, and the eye cup was further post-fixed overnight and then washed three times with sodium cacodylate buffer. Next, the eyecup was incubated with 2% osmium tetroxide, dehydrated with graded ethyl alcohol solutions, and blocked in epoxy resin.
Eyes were sectioned and prepared for both light microscopy and transmission electron microscopy (TEM). For light microscopy, semithin (1 μm) transverse sections were cut at the level of the optic nerve head so that each slice contained the temporal and nasal retina. The sections were stained with 1% toluidine blue in 1% sodium tetraborate aqueous solution and imaged with an Eclipse Ti microscope (Nikon, Tokyo, Japan). Two mice (one eye each) per genotype at the same time points described in In Vivo Retinal Imaging were analyzed, with the exception that three mice per genotype were evaluated at 15 months. Images were acquired from the nasal retina with the use of a 20× open air objective with a DS-Fi1 camera (Nikon ) set to 1.5× optical zoom. For specimens from the 1-, 4-, and 15-month time points, ultrathin sections (60 to 90 nm) were cut from the epoxy blocks using an EM UC7 ultra-microtome (Leica Microsystems, Buffalo Grove, IL), stained with uranyl acetate and Sato's lead citrate stain, and then imaged by TEM using a Tecnai G2 Spirit transmission electron microscope (FEI, Hillsboro, OR) and AMT XR41 digital CCD camera (Advanced Microscopy Techniques, Woburn, MA). For some light microscopy and TEM images, an additional brightness/contrast adjustment was made with Photoshop (Adobe Systems, Mountain View, CA).
Tissue preparation and immunofluorescence analyses on sagittal vibratome sections was performed as described previously,49 with the exceptions that the mice were euthanized by carbon dioxide asphyxiation, eye orientation was marked with a mild corneal burn with the use of a small vessel cauterizer (Fine Scientific Tools, Foster City, CA), and Fluoromount G was used as the slide mounting medium. Images were acquired from the ventral retina using a Leica SP5 confocal microscope (Leica Microsystems), and 10-μm Z-stacks (0.5-μm spacing between image slices) were assembled in ImageJ version 1.50g (NIH, Bethesda, MD). Final images were generated by brightest point projection and sharpened with the Unsharp Mask function (1.0 pixel radius, 0.3 mask weight).
Nmnat1D243G Mice
Tissue preparation for light microscopy was performed as previously described50 in three or four mice per genotype at ages 1, 3, 6, 9, and 12 months. Briefly, after carbon dioxide asphyxiation, eyes were enucleated and placed in cold acetic acid/methanol solution overnight at 4°C. The eyes were then embedded in paraffin, sectioned at 5 μm, stained with hematoxylin and eosin, and examined under light microscopy. Slides were scanned with a NanoZoomer 2.0HT (Hamamatsu, Japan) at 40× magnification with the use of a 20× objective and 2× digital zoom. Post-acquisition processing was completed with Fiji51 with images taken within 500 μm on either side of the optic nerve head for each sample and sharpened with the Unsharp Mask function (1.0 pixel radius, 0.7 mask weight). Three to five independent samples were assessed for each time point.
For immunofluorescence, eyes were enucleated and fixed overnight in cold methanol/acetic acid/phosphate-buffered solution (3:1:4 v/v/v). Tissues were then dehydrated, paraffin-embedded, and sliced into 5-μm sections before being deparaffinated and blocked with normal donkey serum (Jackson ImmunoResearch, West Grove, PA). The primary antibodies used were goat anti–S-opsin (sc-14363; Santa Cruz Biotechnology, Santa Cruz, CA) and rabbit anti–M/L-opsin (AB5405; Millipore, Billerica, MA), both diluted 1:200. After overnight incubation in primary antibody at 4°C, the sections were washed in PBS and incubated with secondary antibodies for 1 hour at room temperature. The secondary antibody for the S-opsin antibody was Alexa Fluor 488-labeled donkey anti-goat IgG (Life Technologies, Carlsbad, CA), and the secondary antibody for the L/M-opsin antibody was cyanine 3-labeled donkey anti-rabbit IgG (Jackson ImmunoResearch), both diluted 1:200. Specimens were stained with DAPI nuclear stain, washed several times in PBS, and mounted in Vectashield anti-fade medium (Vector Laboratories, San Mateo, CA). Fluorescent staining was visualized with a DMLB upright microscope (Leica Microsystems) with the use of the QImaging camera system (QImaging, Surrey, BC, Canada).
TEM analysis was performed on 1-month-old eyes that were fixed and dissected as previously described.52 The tissue was then post-fixed in osmium tetroxide, dehydrated, embedded in plastic, and sectioned. Ultrathin sections were stained with uranyl acetate and lead citrate and then imaged with a JEM-1230 (JEOL, Tokyo, Japan) transmission electron microscope.
Pupillary Light Response
Video clips of pupil constriction in response to the onset of steady light exposure were recorded with the Micron retinal imaging microscope system (model III for Nmnat1V9M mice; model IV for Nmnat1D243G) and built-in light source. The light stimulus was set to approximately 50% of the maximum brightness (approximately 60,000 lux) without filters. In Fiji, single frames from the Nmnat1D243G recordings were cropped, sharpened with Unsharp Mask (radius 1, mask weight 0.8), and contrast enhanced with Enhance Local Contrast with the use of default settings, except for a maximum slope of 2.00.
Results
The Nmnat1V9M and Nmnat1D243G mice were generated by ENU mutagenesis, as described in Materials and Methods. Neither homozygous Nmnat1V9M nor Nmnat1D243G mice presented an extraocular phenotype. Size, weight, activity level, motor coordination, breeding success, and life expectancy were normal.
Loss of Retinal Function in Nmnat1 Mutant Mice
Measurements of retinal function with the use of electroretinography showed that both Nmnat1V9M and Nmnat1D243G mutant mice experienced rapid loss of rod and cone function. Rod-driven, mixed rod/cone, and cone-driven ERGs were collected at 1, 2, 3, 4, 7.5, and 10 months from wild-type, heterozygous, and homozygous Nmnat1V9M mice littermates (Figure 2). In all three stimulus conditions, the b-wave measurements in wild-type and heterozygous Nmnat1V9M littermates were statistically equivalent (two-tailed, unpaired t-test) at every time point, and both sets of mice show an age-dependent decrease that was consistent with previous studies.49, 53, 54 Although the responses in homozygous Nmnat1V9M mice were decreased by approximately 35% at 1 month in both dark-adapted conditions (rod-driven and mixed rod/cone) and by 40% in the light-adapted condition, these difference were not statistically significant (one-way analysis of variance, P = 0.06 for rod-driven ERG; P = 0.14 for mixed rod/cone ERG, P = 0.03 for cone-driven ERG). By 2 months, the decrements in the rod-driven, mixed rod/cone, and cone-driven ERGs in homozygous Nmnat1V9M mice were >70% (P = 1.8 × 10−7, 3.8 × 10−6, and 3.1 × 10−8, respectively), and by 4 months, ERGs were undetectable across all conditions. Homozygous Nmnat1D243G mice also showed greatly reduced retinal function in comparison with age-matched wild-type B6 mice in rod-driven (−72%, unpaired t-test: P = 1.6 × 10−3), mixed rod/cone (−87%, unpaired t-test: P = 3.3 × 10−7), and cone-driven (−71%, unpaired t-test: P = 1.7 × 10−4) conditions by 12 months; this occurred along a slower time scale than for homozygous Nmnat1V9M mice (Supplemental Figure S1).
Figure 2.

Retinal function declines with age in V9M/V9M mice. ERG data from wt/wt, wt/V9M , and V9M/V9M mice across 10 months. A–C: B-wave measurements from rod (A), mixed rod/cone (B), and cone (C) ERGs that were generated with full-field, broadband stimuli (4 ms) at 0.01 cd.s/m2 (dark-adapted), 10 cd.s/m2 (dark-adapted), and 20 cd.s/m2 (light-adapted), respectively. D: A-wave measurements from the mixed rod/cone condition. Data are expressed as means ± SEM. ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001. ERG, electroretinogram; V9M/V9M, homozygous Nmnat1V9M; wt/V9M, heterozygous Nmnat1V9M; wt/wt, wild-type littermate.
To directly confirm the loss of photoreceptor function, the a-wave was measured in the dark-adapted ERGs generated in response to the 10-cd.s/m2 stimulus for the Nmnat1V9M mice and the 0.25-cd.s/m2 stimulus for the Nmnat1D243G mice. Although the a-wave was not significantly different across genotypes in the Nmnat1V9M line at 1 month (one-way analysis of variance, P = 0.06), it was decreased in homozygous Nmnat1V9M mice by 91% (P = 1.0 × 10−10) in comparison with the wild-type and heterozygous littermates at 2 months (Figure 2). By 4 months, the a-wave was absent in homozygous Nmnat1V9M mice, as was the b-wave. The a-waves of the Nmnat1D243G homozygous mutant and B6 wild-type control mice were equivalent (unpaired t-test, P = 0.86) at 1 month (Supplemental Figure S1). However, the a-wave of the homozygous mutant mice declined rapidly between 3 and 4 months, and by 12 months, this component of the ERG was decreased by 83% (P = 7.4 × 10−5) in comparison with that of the control mice.
Retinal Degeneration in Nmnat1 Mutant Mice
In vivo retinal imaging of heterozygous and homozygous Nmnat1V9M mice and wild-type littermates at 0.75, 1, 2, 3, 4, 8, 10, and 15 months of age showed a progressive and severe chorioretinal degeneration. The representative pairs of fundus and OCT images shown for each time point in Figure 3A were collected from the same retina; images from heterozygous Nmnat1V9M mice are not presented because they were indistinguishable from those of the wild-type littermates. At 3 weeks, the fundus and OCT images from homozygous Nmnat1V9M mice appeared normal. However, despite an unremarkable fundus image at 1 month, the retina was noticeably thinner, particularly at the level of the photoreceptors. By 2 months, retinal vasculature was attenuated, and by 3 months, there was evidence of optic atrophy. At 4 months, OCT showed an advanced degeneration of the photoreceptor layer, and the polygonal lattice of the RPE could be seen through the remaining neural retina in the fundus image. By 8 months, RPE degeneration was suggested by depigmented patches that spread to cover much of the retina in the 10- and 15-month-old mice. Similar changes in retinal morphologic structure occurred in homozygous Nmnat1D243G mice (Figure 3B) but at a slower rate.
Figure 3.

In vivo retinal imaging shows rapid and severe chorioretinal degeneration. A: Representative pairs of fundus and OCT images from the Nmnat1V9M mouse line across 15 months at ages indicated. Optic atrophy is evident at 3 months (black arrow). Boxed areas are shown at higher magnification below. Bottom row: At 4 months, the polygonal lattice of the RPE is visible, and at 8 months, regions of RPE degeneration are observed (asterisk). Larger regions of RPE degeneration are indicated with black arrowheads in the images at 10 and 15 months. B: Representative pairs of fundus and OCT images from the Nmnat1D243G and control mice across 12 months. Boxed areas are shown at higher magnification below. C: Retinal thickness at different ages, measured as the distance from the nerve fiber layer to the RPE (orange arrow and gray arrow, respectively, in images in A and B). The left panel shows measurements for wt/wt, wt/V9M , and V9M/V9M mice. The right panel shows measurements for B6 and D243G/D243G mice. The initial loss of retinal thickness is due to outer retinal degeneration; region between the blue and gray arrows in images in A and B. Data are expressed as means ± SEM. ∗∗P < 0.01, ∗∗∗P < 0.001. Scale bars: approximately 100μm (magnified images); approximately 150μm (OCT images, x and y dimensions); approximately 250μm (fundus images). D243G/D243G, homozygous Nmnat1D243G; RPE, retinal pigment epithelium; V9M/V9M, homozygous Nmnat1V9M; wt/V9M, heterozygous Nmnat1V9M; wt/wt, wild-type.
The progressive retinal thinning in the homozygous Nmnat1V9M and Nmnat1D243G mice was quantified by measuring the distance from the nerve fiber layer to the RPE. Although the retinal thickness remained stable in wild-type littermates and heterozygous Nmnat1V9M mice, loss of retinal structure in homozygous Nmnat1V9M mice was especially rapid during the first 4 months of life (Figure 3C). Normal retinal thickness was observed in 3-week-old homozygous Nmnat1V9M mice (one-way analysis of variance, P = 0.78), but 1 week later, the retinas were approximately 18% thinner than those of the littermate controls (P = 5.1 × 10−12). Retinal thickness in homozygous Nmnat1V9M mice was then reduced by approximately 35% (P = 1.1 × 10−29) at 2 months, approximately 45% (P = 1.7 × 10−38) at 4 months, and approximately 57% (P = 1.6 × 10−26) at 15 months. In contrast, the retinas of homozygous Nmnat1D243G mice were not significantly thinner than wild-type at 1 month (unpaired, two-tailed t-test, P = 0.07), and were 47% (P = 1.5 × 10−6) thinner than normal at 12 months (Figure 3C).
The in vivo retinal imaging results were corroborated by retinal histology. Light microscopy of toluidine blue–stained, semithin sections showed progressive retinal degeneration in homozygous Nmnat1V9M mice across 15 months, whereas the morphologic structure of both wild-type littermates and heterozygous Nmnat1V9M retinas remained unchanged (Figure 4A). Although homozygous Nmnat1V9M retinas appeared normal at 3 weeks, the photoreceptor outer segment layer and outer nuclear layer showed thinning at 1 month and nearly complete degeneration by 4 months; only one row of photoreceptor nuclei was visible in the representative image. The inner nuclear layer, which contains the bipolar cell nuclei, maintained a normal appearance for the first several months but then became increasingly disorganized and thinner by 15 months. Similarly, the RPE was morphologically normal for at least the first 4 months of life, but by month 8 there were patches that had degenerated entirely (Figure 4A). These areas in which the RPE was absent were consistent with the pigmentation pattern observed in the fundi of older mice (Figure 3). A similar, albeit more gradual, outer retinal degeneration was observed in homozygous Nmnat1D243G mice (Figure 4B). Although neither ex vivo nor in vivo imaging data established whether development was completely normal in the mutant Nmnat1 retina, ERG data (Loss of Retinal Function in Nmnat1 Mutant Mice) and immunolabeling of the cone opsins (Supplemental Figure S2) confirmed that these retinas developed and had function.
Figure 4.

Light microscopy shows retinal degeneration. A:Nmnat1V9M. Representative bright-field images of semithin (1 μm) sections obtained from mice at the indicated ages stained with toluidine blue, which were acquired from the nasal retina at the plane of the optic nerve head. B:Nmnat1D243G. Representative bright-field images of 5-μm paraffin sections stained with hematoxylin and eosin. Rapid photoreceptor loss is observed in both mutant mouse lines. RPE degeneration is also evident in the V9M/V9M mice by 8 months of age (arrow). Scale bars: 50 μm (A); 25 μm (B). Original magnification: ×30 (A); ×40 (B). chrd, choroid; D243G/D243G, homozygous Nmnat1D243G; GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; ONL, outer nuclear layer; OPL, outer plexiform layer; PIS, photoreceptor inner segments; POS, photoreceptor outer segments; RPE, retinal pigment epithelium; V9M/V9M, homozygous Nmnat1V9M; wt/wt, wild-type.
TEM imaging further validated the results from light microscopy. A decrease in photoreceptor outer segment length was evident at 1 month of age in the homozygous Nmnat1V9M mice (Figure 5A), and regions of RPE cell degeneration were evident at 15 months (Figure 5B). In places where the RPE remained, the apical processes that would ordinarily interdigitate with the photoreceptor outer segments were coiled, and the basal infoldings had been largely replaced by basal deposits. In 1-month-old homozygous Nmnat1D243G mice, the photoreceptor outer segments and RPE had a normal appearance (Supplemental Figure S3).
Figure 5.

Transmission electron microscopy shows morphologic changes in V9M/V9M photoreceptors and RPE. A: Representative images from 1-month-old mice show photoreceptor outer segments of normal length in the wt/wt retina compared with shortened outer segments in the V9M/V9M retina. B: Representative images acquired at the level of the RPE at 4 and 15 months in wt/wt and V9M/V9M retina, as indicated. Coiled apical processes are marked with arrowheads. Scale bars: 10 μm (A); 3 μm (B). Original magnification: ×1400 (A); ×6800 (B). chrd, choroid; POS, photoreceptor outer segment; RPE, retinal pigment epithelium; V9M/V9M, homozygous Nmnat1V9M; wt/wt, wild-type.
Amaurosis in Nmnat1 Mutant Mice
Although the ERG revealed a complete loss of outer retinal function for homozygous Nmnat1V9M mice, these measurements did not specifically assess inner retinal cell function. Because photoreceptors and intrinsically photosensitive ganglion cells ordinarily contribute to the pupillary light response, the intrinsically photosensitive ganglion cells could be evaluated functionally by observing this reflex after photoreceptor degeneration. In advanced cases, homozygous Nmnat1V9M mice were found to have attenuated pupillary constriction in response to light, whereas wild-type littermates and heterozygous Nmnat1V9M mice maintained normal pupillary light responses (Figure 6). This aspect of the phenotype was observed as early as 11.5 months of age. No homozygous NmnatD243G mice were identified as having lost the pupillary light response during the first 15 months of life (Supplemental Figure S4), which is consistent with a slower progression of retinal degeneration, relative to the homozygous NmnatV9M mice.
Figure 6.
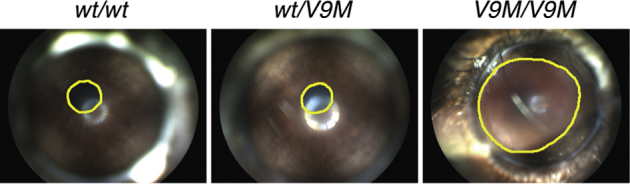
V9M/V9M mice develop amaurosis. Frames from video recordings of the eyes of 11.5-month-old wt/wt, wt/V9M , and V9M/V9M littermates on exposure to a steady light stimulus. Pupils are outlined in yellow. V9M/V9M, homozygous Nmnat1V9M; wt/V9M, heterozygous Nmnat1V9M; wt/wt, wild-type.
Discussion
The retinal degenerative phenotype in both homozygous Nmnat1V9M and Nmnat1D243G mice follows a monogenic recessive inheritance pattern without exception, showing a clear causal connection between the mutant Nmnat1 alleles and disease. This early-onset chorioretinal degeneration fully recapitulates key aspects of human NMNAT1-LCA in that there is a severe loss of retinal function associated with widespread thinning of the retina which is first observed at the level of the photoreceptor cells, followed by attenuation of retinal vasculature, optic atrophy, and a nonuniform degeneration of the RPE. This RPE phenotype may directly correspond to the atrophic macular lesions with RPE loss that is observed in patients.1, 8, 9, 10 To our knowledge, the homozygous Nmnat1V9M line is the only LCA mouse model that has been shown to develop pupillary amaurosis, a feature of all genetic forms of LCA2 that arises from the loss of photoreceptor and intrinsically photosensitive ganglion cell function.5, 6, 55, 56, 57 As in human patients, the disease in mice appears to be confined to the retina. Affected animals are otherwise healthy, are of normal size and weight, show normal levels of activity and motor coordination, breed normally, and live equally as long as their littermates; this implies that NMNAT1 serves an especially important role in the retina.
Because these mouse models were generated and discovered in ENU-mutagenesis screens, confirming that the phenotype is caused entirely by each mutation under investigation is difficult. Another unknown genetic variant that persists even after multiple outcrosses with wild-type mice could affect the results.31, 34 This study accounts for that concern in two complementary ways. First, the homozygous Nmnat1V9M mice were always compared with littermate controls, which would be expected to harbor the same additional variants. Second, having been generated independently greatly reduces the likelihood that the Nmnat1V9M and Nmnat1D243G lines would share the same secondary ENU-induced mutations. The chance of both mouse lines harboring an additional, physically linked mutation that causes or contributes to the phenotype is low.
In both homozygous Nmnat1V9M and Nmnat1D243G mutant mice, degeneration occurs after retinal development. Data collected from these mice suggest that photoreceptors are most vulnerable to disruptions in NMNAT1 because they are the first cell type to show signs of disease, with shortening of outer segments evident at 1 month in the homozygous Nmnat1V9M mice. In both mouse lines, the loss of photoreceptor cell nuclei is precipitous and appears to precede the changes observed in other retinal cell types. Photoreceptors may be hypersensitive to decreased levels of nuclear NAD+ because of their extremely high metabolic activity.58, 59, 60 Recent work has shown that a mutation in NMNAT1 which changes the charge of a critical amino acid residue likely destabilizes the secondary protein structure when stressed by heat shock,27 and environmental challenges in cells with high metabolic activity could potentially trigger a similar effect. The metabolic demands of light exposure after eye opening61 that require processes supported by NMNAT1 may overwhelm mutant photoreceptors, which is consistent with these cells degenerating after development. This idea is supported by data from Drosophila, showing that photoreceptor degeneration caused by a loss of the nmnat protein can be significantly reduced by blocking phototransduction.21 Although to a lesser extent than photoreceptors, homozygous Nmnat1V9M and Nmnat1D243G mice show that the RPE cells also depend on NMNAT1 as they undergo degeneration, as do inner retinal cells (eg, bipolar cells) in the homozygous Nmnat1V9M mice. The development of pupillary amaurosis in homozygous Nmnat1V9M mice may indicate loss of intrinsically photosensitive ganglion cells in the advanced stage of the disease.5, 6, 55, 56, 57
Two hypotheses, which are not mutually exclusive, have been proposed to explain why mutations in NMNAT1 variants cause retinal disease. First, abnormal enzymatic activity in mutant NMNAT1 alters nuclear NAD+ levels, and this loss of nuclear NAD+ homeostasis interferes with important cellular processes. For example, DNA repair, gene transcription, and programed cell death are modulated by PARP1-mediated poly(ADP-ribosyl)ation, which is the main consumer of this NAD+ pool.62, 63, 64, 65, 66 Similarly, nuclear sirtuins (eg, SIRT1, SIRT6, and SIRT7), which are important to cell survival because of their histone and transcription factor deacetylase activity, also rely on nuclear NAD+.67, 68 Failure of sirtuin function may be secondary to depletion of nuclear NAD+ by activated PARP1.69 Evidence suggests that the degree to which NMNAT1 is catalytically dysfunctional depends on the mutation harbored,8, 10, 27 and such variability across mutants may explain individual differences in the rate of disease progression within the patient population. Second, NMNAT1 is hypothesized to act as a stress-response protein, and mutations could disrupt associated neuroprotective activity. This alternative role for NMNAT1 was observed in Drosophila where nmnat acts as a chaperone to facilitate neuronal protection and neuronal maintenance when faced with axon injury or induced neurodegeneration in a manner independent of its NAD+ synthase function.1, 21, 23 Similarly, the WldS fusion protein, containing full-length Nmnat1 and a fragment of multiubiquitination factor Ube4b, was found to delay neurodegeneration after nerve injury26 in mice,24 rats,25 and flies.21
The homozygous Nmnat1V9M and Nmnat1D243G mice are promising resources for understanding pathogenesis of retinal degeneration in NMNAT1-associated LCA. These mouse models may help to distinguish whether and how altered NMNAT1 enzymatic and/or stress-response activity cause disease. The difference in the natural history of disease between these mouse strains, which are on the same genetic background (B6), should allow for correlative studies that answer questions about allelic variation and the disease progression. In addition, these mice may also be useful for defining the normal roles of NMNAT1 in the brain and other organs throughout the body.
The development of animal models that faithfully recapitulate NMNAT1-associated LCA will facilitate the development of therapies for patients with this severe form of retinal degeneration.1, 8, 9, 10 Gene augmentation therapy that uses adeno-associated virus vectors is especially attractive for treating inherited retinal degenerations on the basis of positive results from early clinical trials of adeno-associated virus–mediated gene therapy for RPE65-associated LCA, choroideremia, X-linked androleukodystrophy, and hemophilia,70, 71, 72, 73 as well as the success of gene therapy in other animal models of inherited retinal degenerations.74, 75, 76, 77 Studies are now in progress to test this approach using homozygous Nmnat1V9M mice. Given that amaurosis is observed in homozygous Nmnat1V9M mice, recovery of the pupillary light response could provide an additional noninvasive assessment of therapeutic efficacy78; this method was used in gene augmentation for RPE65-LCA in dogs79 and humans.72, 80 Moreover, identifying the disease mechanisms underlying retinal degeneration in the homozygous Nmnat1V9M and Nmnat1D243G mice may lead to more therapeutic options for patients with NMNAT1-LCA.
Acknowledgments
We thank Carlos Aguilar, Joanne Dorning, Jeanie Hansen, and Philip Seifert for their technical assistance with the study and Scientific Services at The Jackson Laboratory, the Frozen Embryo and Sperm Archive (FESA) team at MRC Harwell, and the Mary Lyon Center at MRC Harwell.
Footnotes
Supported by NIH/National Eye Institute grants EY012910 (E.A.P.), 5T32 EY-007145-16 (S.H.G.), EY016501 (P.M.N.), P30 EY003790 (Schepens Ophthalmology Core), P30 EY014104 (Massachusetts Eye and Ear Infirmary Ophthalmology Core); the Foundation for Retinal Research/Gavin R. Stevens Foundation (E.A.P.); Fight for Sight (PD15001; S.H.G.); and NIH/National Cancer Institute grant CA34196 (The Jackson Laboratory).
Disclosures: None declared.
Current address of M.S., Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wroclaw, Poland.
Supplemental material for this article can be found at http://dx.doi.org/10.1016/j.ajpath.2016.03.013.
Supplemental Data
Supplemental Figure S1.

Retinal function declines with age in homozygous Nmnat1D243G mice. ERG data from B6 (black open circles, solid lines) and homozygous Nmnat1D243G (gray open circles, dotted lines) mice across 12 months. B-wave measurements from rod (A), mixed rod/cone (B), and cone (C) ERGs recorded from using a 0.006 cd.s/m2 (dark-adapted), 0.25 cd.s/m2 stimulus (dark-adapted), and 10 cd.s/m2 stimulus (light-adapted), respectively. D: A-wave measurements from ERGs collected in the mixed rod/cone condition. Data are expressed as means ± SEM. ∗P < 0.05, ∗∗P < 0.01. ERG, electroretinogram.
Supplemental Figure S2.

Immunofluorescence analysis of cones. Images acquired from the ventral retinas of a 3-week-old mice incubated with antibodies against L/M-opsin (red) and S-opsin (green). Double labeling (yellow) occurs in cones that co-express both classes of opsin. A: Wild-type control retina and homozygous Nmnat1V9M retina. B: Wild-type control retina and homozygous Nmnat1D243G. Although the homozygous Nmnat1V9M retina shown has evidence of early degeneration, the opsin labeling in both mouse lines indicates that functional cones develop before degeneration. Scale bars = 50 μm.
Supplemental Figure S3.
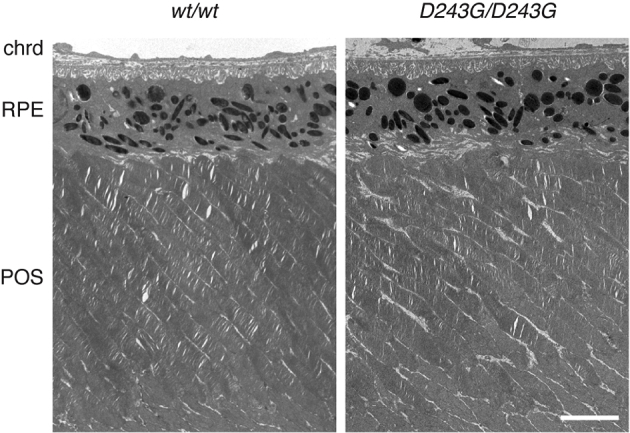
Transmission electron microscopic images show that photoreceptor outer segment and RPE structure in wild-type mice and homozygous Nmnat1D243G mice are equivalent at 1 month. Scale bar = 4 μm. Original magnification, ×5080. chrd, choroid; POS, photoreceptor outer segment; RPE, retinal pigment epithelium.
Supplemental Figure S4.
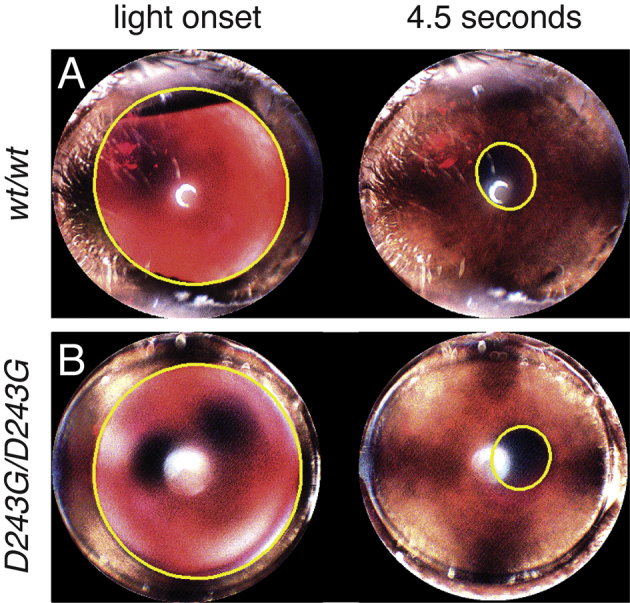
Amaurosis is not observed in homozygous Nmnat1D243G mice. Frames from video recordings of the eye of a 14-month-old wild-type mouse (A) and age-matched homozygous Nmnat1D243G mouse (B) at light onset and at 4.5 seconds of exposure to a steady light stimulus. Pupils are outlined in yellow.
References
- 1.Perrault I., Hanein S., Zanlonghi X., Serre V., Nicouleau M., Defoort-Delhemmes S., Delphin N., Fares-Taie L., Gerber S., Xerri O., Edelson C., Goldenberg A., Duncombe A., Le Meur G., Hamel C., Silva E., Nitschke P., Calvas P., Munnich A., Roche O., Dollfus H., Kaplan J., Rozet J.M. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet. 2012;44:975–977. doi: 10.1038/ng.2357. [DOI] [PubMed] [Google Scholar]
- 2.den Hollander A.I., Roepman R., Koenekoop R.K., Cremers F.P. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res. 2008;27:391–419. doi: 10.1016/j.preteyeres.2008.05.003. [DOI] [PubMed] [Google Scholar]
- 3.Koenekoop R.K. An overview of Leber congenital amaurosis: a model to understand human retinal development. Surv Ophthalmol. 2004;49:379–398. doi: 10.1016/j.survophthal.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 4.den Hollander A.I., Black A., Bennett J., Cremers F.P. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest. 2010;120:3042–3053. doi: 10.1172/JCI42258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Markwell E.L., Feigl B., Zele A.J. Intrinsically photosensitive melanopsin retinal ganglion cell contributions to the pupillary light reflex and circadian rhythm. Clin Exp Optom. 2010;93:137–149. doi: 10.1111/j.1444-0938.2010.00479.x. [DOI] [PubMed] [Google Scholar]
- 6.Gooley J.J., Ho Mien I., St Hilaire M.A., Yeo S.C., Chua E.C., van Reen E., Hanley C.J., Hull J.T., Czeisler C.A., Lockley S.W. Melanopsin and rod-cone photoreceptors play different roles in mediating pupillary light responses during exposure to continuous light in humans. J Neurosci. 2012;32:14242–14253. doi: 10.1523/JNEUROSCI.1321-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weleber R.G., Francis P.J., Trzupek K.M., Beattie C. University of Washington; Seattle, WA: 2013. Leber Congenital Amaurosis (GeneReviews) [Google Scholar]
- 8.Falk M.J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D.S., Zeitz C., Borman A.D., Staniszewska M., Shukla R., Palavalli L., Mohand-Said S., Waseem N.H., Jalali S., Perin J.C., Place E., Ostrovsky J., Xiao R., Bhattacharya S.S., Consugar M., Webster A.R., Sahel J.A., Moore A.T., Berson E.L., Liu Q., Gai X., Pierce E.A. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiang P.W., Wang J., Chen Y., Fu Q., Zhong J., Chen Y., Yi X., Wu R., Gan H., Shi Y., Chen Y., Barnett C., Wheaton D., Day M., Sutherland J., Heon E., Weleber R.G., Gabriel L.A., Cong P., Chuang K., Ye S., Sallum J.M., Qi M. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet. 2012;44:972–974. doi: 10.1038/ng.2370. [DOI] [PubMed] [Google Scholar]
- 10.Koenekoop R.K., Wang H., Majewski J., Wang X., Lopez I., Ren H., Chen Y., Li Y., Fishman G.A., Genead M., Schwartzentruber J., Solanki N., Traboulsi E.I., Cheng J., Logan C.V., McKibbin M., Hayward B.E., Parry D.A., Johnson C.A., Nageeb M., Finding of Rare Disease Genes (FORGE) Canada Consortium. Poulter J.A., Mohamed M.D., Jafri H., Rashid Y., Taylor G.R., Keser V., Mardon G., Xu H., Inglehearn C.F., Fu Q., Toomes C., Chen R. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44:1035–1039. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng Y., Huang H., Wang Y., Liu Z., Li N., Chen Y., Li X., Li M., Zhou X., Mu D., Zhong J., Wu J., Su Y., Yi X., Zhu J. A novel missense NMNAT1 mutation identified in a consanguineous family with Leber congenital amaurosis by targeted next generation sequencing. Gene. 2015;569:104–108. doi: 10.1016/j.gene.2015.05.038. [DOI] [PubMed] [Google Scholar]
- 12.Jin X., Qu L.H., Meng X.H., Xu H.W., Yin Z.Q. Detecting genetic variations in hereditary retinal dystrophies with next-generation sequencing technology. Mol Vis. 2014;20:553–560. [PMC free article] [PubMed] [Google Scholar]
- 13.Corton M., Nishiguchi K.M., Avila-Fernandez A., Nikopoulos K., Riveiro-Alvarez R., Tatu S.D., Ayuso C., Rivolta C. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS One. 2013;8:e65574. doi: 10.1371/journal.pone.0065574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Siemiatkowska A.M., van den Born L.I., van Genderen M.M., Bertelsen M., Zobor D., Rohrschneider K., van Huet R.A., Nurohmah S., Klevering B.J., Kohl S., Faradz S.M., Rosenberg T., den Hollander A.I., Collin R.W., Cremers F.P. Novel compound heterozygous NMNAT1 variants associated with Leber congenital amaurosis. Mol Vis. 2014;20:753–759. [PMC free article] [PubMed] [Google Scholar]
- 15.Coppieters F., Van Schil K., Bauwens M., Verdin H., De Jaegher A., Syx D., Sante T., Lefever S., Abdelmoula N.B., Depasse F., Casteels I., de Ravel T., Meire F., Leroy B.P., De Baere E. Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med. 2014;16:671–680. doi: 10.1038/gim.2014.24. [DOI] [PubMed] [Google Scholar]
- 16.Belenky P., Bogan K.L., Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 17.Lau C., Niere M., Ziegler M. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front Biosci (Landmark Ed) 2009;14:410–431. doi: 10.2741/3252. [DOI] [PubMed] [Google Scholar]
- 18.Chiarugi A., Dolle C., Felici R., Ziegler M. The NAD metabolome–a key determinant of cancer cell biology. Nat Rev Cancer. 2012;12:741–752. doi: 10.1038/nrc3340. [DOI] [PubMed] [Google Scholar]
- 19.Berger F., Lau C., Dahlmann M., Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J Biol Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 20.Conforti L., Janeckova L., Wagner D., Mazzola F., Cialabrini L., Di Stefano M., Orsomando G., Magni G., Bendotti C., Smyth N., Coleman M. Reducing expression of NAD+ synthesizing enzyme NMNAT1 does not affect the rate of Wallerian degeneration. FEBS J. 2011;278:2666–2679. doi: 10.1111/j.1742-4658.2011.08193.x. [DOI] [PubMed] [Google Scholar]
- 21.Zhai R.G., Cao Y., Hiesinger P.R., Zhou Y., Mehta S.Q., Schulze K.L., Verstreken P., Bellen H.J. Drosophila NMNAT maintains neural integrity independent of its NAD synthesis activity. PLoS Biol. 2006;4:e416. doi: 10.1371/journal.pbio.0040416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhai R.G., Rizzi M., Garavaglia S. Nicotinamide/nicotinic acid mononucleotide adenylyltransferase, new insights into an ancient enzyme. Cell Mol Life Sci. 2009;66:2805–2818. doi: 10.1007/s00018-009-0047-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhai R.G., Zhang F., Hiesinger P.R., Cao Y., Haueter C.M., Bellen H.J. NAD synthase NMNAT acts as a chaperone to protect against neurodegeneration. Nature. 2008;452:887–891. doi: 10.1038/nature06721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mack T.G., Reiner M., Beirowski B., Mi W., Emanuelli M., Wagner D., Thomson D., Gillingwater T., Court F., Conforti L., Fernando F.S., Tarlton A., Andressen C., Addicks K., Magni G., Ribchester R.R., Perry V.H., Coleman M.P. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 25.Adalbert R., Gillingwater T.H., Haley J.E., Bridge K., Beirowski B., Berek L., Wagner D., Grumme D., Thomson D., Celik A., Addicks K., Ribchester R.R., Coleman M.P. A rat model of slow Wallerian degeneration (WldS) with improved preservation of neuromuscular synapses. Eur J Neurosci. 2005;21:271–277. doi: 10.1111/j.1460-9568.2004.03833.x. [DOI] [PubMed] [Google Scholar]
- 26.Gillingwater T.H., Ribchester R.R. Compartmental neurodegeneration and synaptic plasticity in the Wld(s) mutant mouse. J Physiol. 2001;534:627–639. doi: 10.1111/j.1469-7793.2001.00627.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sasaki Y., Margolin Z., Borgo B., Havranek J.J., Milbrandt J. Characterization of Leber's congenital amaurosis-associated NMNAT1 mutants. J Biol Chem. 2015;290:17228–17238. doi: 10.1074/jbc.M115.637850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Siemiatkowska A.M., Schuurs-Hoeijmakers J.H., Bosch D.G., Boonstra F.N., Riemslag F.C., Ruiter M., de Vries B.B., den Hollander A.I., Collin R.W., Cremers F.P. Nonpenetrance of the most frequent autosomal recessive leber congenital amaurosis mutation in NMNAT1. JAMA Ophthalmol. 2014;132:1002–1004. doi: 10.1001/jamaophthalmol.2014.983. [DOI] [PubMed] [Google Scholar]
- 29.Coghill E.L., Hugill A., Parkinson N., Davison C., Glenister P., Clements S., Hunter J., Cox R.D., Brown S.D. A gene-driven approach to the identification of ENU mutants in the mouse. Nat Genet. 2002;30:255–256. doi: 10.1038/ng847. [DOI] [PubMed] [Google Scholar]
- 30.Won J., Shi L.Y., Hicks W., Wang J., Naggert J.K., Nishina P.M. Translational vision research models program. Adv Exp Med Biol. 2012;723:391–397. doi: 10.1007/978-1-4614-0631-0_50. [DOI] [PubMed] [Google Scholar]
- 31.Wansleeben C., van Gurp L., Feitsma H., Kroon C., Rieter E., Verberne M., Guryev V., Cuppen E., Meijlink F. An ENU-mutagenesis screen in the mouse: identification of novel developmental gene functions. PLoS One. 2011;6:e19357. doi: 10.1371/journal.pone.0019357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Quwailid M.M., Hugill A., Dear N., Vizor L., Wells S., Horner E., Fuller S., Weedon J., McMath H., Woodman P., Edwards D., Campbell D., Rodger S., Carey J., Roberts A., Glenister P., Lalanne Z., Parkinson N., Coghill E.L., McKeone R., Cox S., Willan J., Greenfield A., Keays D., Brady S., Spurr N., Gray I., Hunter J., Brown S.D., Cox R.D. A gene-driven ENU-based approach to generating an allelic series in any gene. Mamm Genome. 2004;15:585–591. doi: 10.1007/s00335-004-2379-z. [DOI] [PubMed] [Google Scholar]
- 33.Thornton C.E., Brown S.D., Glenister P.H. Large numbers of mice established by in vitro fertilization with cryopreserved spermatozoa: implications and applications for genetic resource banks, mutagenesis screens, and mouse backcrosses. Mamm Genome. 1999;10:987–992. doi: 10.1007/s003359901145. [DOI] [PubMed] [Google Scholar]
- 34.Keays D.A., Clark T.G., Flint J. Estimating the number of coding mutations in genotypic- and phenotypic-driven N-ethyl-N-nitrosourea (ENU) screens. Mamm Genome. 2006;17:230–238. doi: 10.1007/s00335-005-0101-4. [DOI] [PubMed] [Google Scholar]
- 35.Buffone G.J., Darlington G.J. Isolation of DNA from biological specimens without extraction with phenol. Clin Chem. 1985;31:164–165. [PubMed] [Google Scholar]
- 36.Blankenberg D., Von Kuster G., Coraor N., Ananda G., Lazarus R., Mangan M., Nekrutenko A., Taylor J. Galaxy: a web-based genome analysis tool for experimentalists. Curr Protoc Mol Biol. 2010;Chapter 19 doi: 10.1002/0471142727.mb1910s89. Unit 19.10.1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Giardine B., Riemer C., Hardison R.C., Burhans R., Elnitski L., Shah P., Zhang Y., Blankenberg D., Albert I., Taylor J., Miller W., Kent W.J., Nekrutenko A. Galaxy: a platform for interactive large-scale genome analysis. Genome Res. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goecks J., Nekrutenko A., Taylor J., Galaxy Team Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cingolani P., Platts A., Wang le L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mattapallil M.J., Wawrousek E.F., Chan C.C., Zhao H., Roychoudhury J., Ferguson T.A., Caspi R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Krishna V.R., Alexander K.R., Peachey N.S. Temporal properties of the mouse cone electroretinogram. J Neurophysiol. 2002;87:42–48. doi: 10.1152/jn.00489.2001. [DOI] [PubMed] [Google Scholar]
- 44.Xu L., Ball S.L., Alexander K.R., Peachey N.S. Pharmacological analysis of the rat cone electroretinogram. Vis Neurosci. 2003;20:297–306. doi: 10.1017/s0952523803203084. [DOI] [PubMed] [Google Scholar]
- 45.Bush R.A., Sieving P.A. Inner retinal contributions to the primate photopic fast flicker electroretinogram. J Opt Soc Am A Opt Image Sci Vis. 1996;13:557–565. doi: 10.1364/josaa.13.000557. [DOI] [PubMed] [Google Scholar]
- 46.Kjellstrom S., Bush R.A., Zeng Y., Takada Y., Sieving P.A. Retinoschisin gene therapy and natural history in the Rs1h-KO mouse: long-term rescue from retinal degeneration. Invest Ophthalmol Vis Sci. 2007;48:3837–3845. doi: 10.1167/iovs.07-0203. [DOI] [PubMed] [Google Scholar]
- 47.Peachey N.S., Goto Y., al-Ubaidi M.R., Naash M.I. Properties of the mouse cone-mediated electroretinogram during light adaptation. Neurosci Lett. 1993;162:9–11. doi: 10.1016/0304-3940(93)90547-x. [DOI] [PubMed] [Google Scholar]
- 48.Saszik S.M., Robson J.G., Frishman L.J. The scotopic threshold response of the dark-adapted electroretinogram of the mouse. J Physiol. 2002;543:899–916. doi: 10.1113/jphysiol.2002.019703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Greenwald S.H., Kuchenbecker J.A., Roberson D.K., Neitz M., Neitz J. S-opsin knockout mice with the endogenous M-opsin gene replaced by an L-opsin variant. Vis Neurosci. 2014;31:25–37. doi: 10.1017/S0952523813000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee Y., Kameya S., Cox G.A., Hsu J., Hicks W., Maddatu T.P., Smith R.S., Naggert J.K., Peachey N.S., Nishina P.M. Ocular abnormalities in Large(myd) and Large(vls) mice, spontaneous models for muscle, eye, and brain diseases. Mol Cell Neurosci. 2005;30:160–172. doi: 10.1016/j.mcn.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 51.Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.Y., White D.J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Won J., Gifford E., Smith R.S., Yi H., Ferreira P.A., Hicks W.L., Li T., Naggert J.K., Nishina P.M. RPGRIP1 is essential for normal rod photoreceptor outer segment elaboration and morphogenesis. Hum Mol Genet. 2009;18:4329–4339. doi: 10.1093/hmg/ddp385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gresh J., Goletz P.W., Crouch R.K., Rohrer B. Structure-function analysis of rods and cones in juvenile, adult, and aged C57bl/6 and Balb/c mice. Vis Neurosci. 2003;20:211–220. doi: 10.1017/s0952523803202108. [DOI] [PubMed] [Google Scholar]
- 54.Williams G.A., Jacobs G.H. Cone-based vision in the aging mouse. Vision Res. 2007;47:2037–2046. doi: 10.1016/j.visres.2007.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lucas R.J., Douglas R.H., Foster R.G. Characterization of an ocular photopigment capable of driving pupillary constriction in mice. Nat Neurosci. 2001;4:621–626. doi: 10.1038/88443. [DOI] [PubMed] [Google Scholar]
- 56.Lucas R.J., Hattar S., Takao M., Berson D.M., Foster R.G., Yau K.W. Diminished pupillary light reflex at high irradiances in melanopsin-knockout mice. Science. 2003;299:245–247. doi: 10.1126/science.1077293. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Y., Tu D.C., Denner D., Shane T., Fitzgerald C.M., Van Gelder R.N. Melanopsin-dependent persistence and photopotentiation of murine pupillary light responses. Invest Ophthalmol Vis Sci. 2007;48:1268–1275. doi: 10.1167/iovs.06-0925. [DOI] [PubMed] [Google Scholar]
- 58.Ames A., 3rd Energy requirements of CNS cells as related to their function and to their vulnerability to ischemia: a commentary based on studies on retina. Can J Physiol Pharmacol. 1992;70 Suppl:S158–S164. doi: 10.1139/y92-257. [DOI] [PubMed] [Google Scholar]
- 59.Ames A., 3rd, Li Y.Y., Heher E.C., Kimble C.R. Energy metabolism of rabbit retina as related to function: high cost of Na+ transport. J Neurosci. 1992;12:840–853. doi: 10.1523/JNEUROSCI.12-03-00840.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niven J.E., Laughlin S.B. Energy limitation as a selective pressure on the evolution of sensory systems. J Exp Biol. 2008;211:1792–1804. doi: 10.1242/jeb.017574. [DOI] [PubMed] [Google Scholar]
- 61.Wong-Riley M.T. Energy metabolism of the visual system. Eye Brain. 2010;2:99–116. doi: 10.2147/EB.S9078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Berger F., Lau C., Ziegler M. Regulation of poly(ADP-ribose) polymerase 1 activity by the phosphorylation state of the nuclear NAD biosynthetic enzyme NMN adenylyl transferase 1. Proc Natl Acad Sci U S A. 2007;104:3765–3770. doi: 10.1073/pnas.0609211104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bouchard V.J., Rouleau M., Poirier G.G. PARP-1, a determinant of cell survival in response to DNA damage. Exp Hematol. 2003;31:446–454. doi: 10.1016/s0301-472x(03)00083-3. [DOI] [PubMed] [Google Scholar]
- 64.Yu S.W., Andrabi S.A., Wang H., Kim N.S., Poirier G.G., Dawson T.M., Dawson V.L. Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc Natl Acad Sci U S A. 2006;103:18314–18319. doi: 10.1073/pnas.0606528103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu S.W., Wang H., Poitras M.F., Coombs C., Bowers W.J., Federoff H.J., Poirier G.G., Dawson T.M., Dawson V.L. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 66.Kim M.Y., Zhang T., Kraus W.L. Poly(ADP-ribosyl)ation by PARP-1: ‘PAR-laying’ NAD+ into a nuclear signal. Genes Dev. 2005;19:1951–1967. doi: 10.1101/gad.1331805. [DOI] [PubMed] [Google Scholar]
- 67.Houtkooper R.H., Pirinen E., Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chalkiadaki A., Guarente L. Sirtuins mediate mammalian metabolic responses to nutrient availability. Nat Rev Endocrinol. 2012;8:287–296. doi: 10.1038/nrendo.2011.225. [DOI] [PubMed] [Google Scholar]
- 69.Canto C., Auwerx J. Interference between PARPs and SIRT1: a novel approach to healthy ageing? Aging (Albany NY) 2011;3:543–547. doi: 10.18632/aging.100326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nathwani A.C., Tuddenham E.G., Rangarajan S., Rosales C., McIntosh J., Linch D.C., Chowdary P., Riddell A., Pie A.J., Harrington C., O'Beirne J., Smith K., Pasi J., Glader B., Rustagi P., Ng C.Y., Kay M.A., Zhou J., Spence Y., Morton C.L., Allay J., Coleman J., Sleep S., Cunningham J.M., Srivastava D., Basner-Tschakarjan E., Mingozzi F., High K.A., Gray J.T., Reiss U.M., Nienhuis A.W., Davidoff A.M. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;365:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maguire A.M., Simonelli F., Pierce E.A., Pugh E.N., Jr., Mingozzi F., Bennicelli J., Banfi S., Marshall K.A., Testa F., Surace E.M., Rossi S., Lyubarsky A., Arruda V.R., Konkle B., Stone E., Sun J., Jacobs J., Dell'Osso L., Hertle R., Ma J.X., Redmond T.M., Zhu X., Hauck B., Zelenaia O., Shindler K.S., Maguire M.G., Wright J.F., Volpe N.J., McDonnell J.W., Auricchio A., High K.A., Bennett J. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cideciyan A.V., Aleman T.S., Boye S.L., Schwartz S.B., Kaushal S., Roman A.J., Pang J.J., Sumaroka A., Windsor E.A., Wilson J.M., Flotte T.R., Fishman G.A., Heon E., Stone E.M., Byrne B.J., Jacobson S.G., Hauswirth W.W. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci U S A. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L., Mahlaoui N., Kiermer V., Mittelstaedt D., Bellesme C., Lahlou N., Lefrere F., Blanche S., Audit M., Payen E., Leboulch P., l'Homme B., Bougneres P., Von Kalle C., Fischer A., Cavazzana-Calvo M., Aubourg P. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 74.Min S.H., Molday L.L., Seeliger M.W., Dinculescu A., Timmers A.M., Janssen A., Tonagel F., Tanimoto N., Weber B.H., Molday R.S., Hauswirth W.W. Prolonged recovery of retinal structure/function after gene therapy in an Rs1h-deficient mouse model of x-linked juvenile retinoschisis. Mol Ther. 2005;12:644–651. doi: 10.1016/j.ymthe.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 75.Pang J.J., Boye S.L., Kumar A., Dinculescu A., Deng W., Li J., Li Q., Rani A., Foster T.C., Chang B., Hawes N.L., Boatright J.H., Hauswirth W.W. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008;49:4278–4283. doi: 10.1167/iovs.07-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Alexander J.J., Umino Y., Everhart D., Chang B., Min S.H., Li Q., Timmers A.M., Hawes N.L., Pang J.J., Barlow R.B., Hauswirth W.W. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007;13:685–687. doi: 10.1038/nm1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tam L.C., Kiang A.S., Kennan A., Kenna P.F., Chadderton N., Ader M., Palfi A., Aherne A., Ayuso C., Campbell M., Reynolds A., McKee A., Humphries M.M., Farrar G.J., Humphries P. Therapeutic benefit derived from RNAi-mediated ablation of IMPDH1 transcripts in a murine model of autosomal dominant retinitis pigmentosa (RP10) Hum Mol Genet. 2008;17:2084–2100. doi: 10.1093/hmg/ddn107. [DOI] [PubMed] [Google Scholar]
- 78.Park J.C., Moura A.L., Raza A.S., Rhee D.W., Kardon R.H., Hood D.C. Toward a clinical protocol for assessing rod, cone, and melanopsin contributions to the human pupil response. Invest Ophthalmol Vis Sci. 2011;52:6624–6635. doi: 10.1167/iovs.11-7586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Acland G.M., Aguirre G.D., Ray J., Zhang Q., Aleman T.S., Cideciyan A.V., Pearce-Kelling S.E., Anand V., Zeng Y., Maguire A.M., Jacobson S.G., Hauswirth W.W., Bennett J. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- 80.Aguirre G.K., Komaromy A.M., Cideciyan A.V., Brainard D.H., Aleman T.S., Roman A.J., Avants B.B., Gee J.C., Korczykowski M., Hauswirth W.W., Acland G.M., Aguirre G.D., Jacobson S.G. Canine and human visual cortex intact and responsive despite early retinal blindness from RPE65 mutation. PLoS Med. 2007;4:e230. doi: 10.1371/journal.pmed.0040230. [DOI] [PMC free article] [PubMed] [Google Scholar]