

Abstract
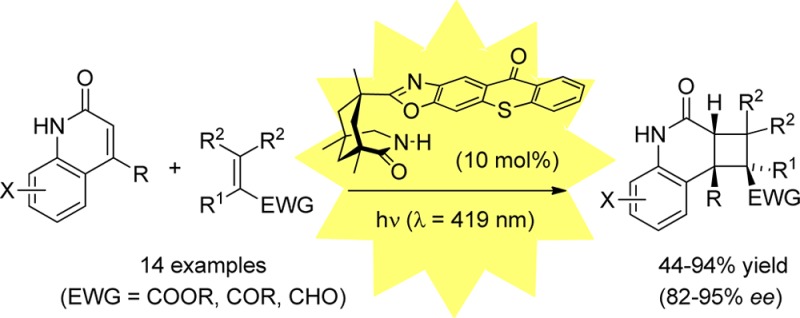
In the presence of a chiral thioxanthone catalyst (10 mol %) the title compounds underwent a clean intermolecular [2 + 2] photocycloaddition with electron-deficient olefins at λ = 419 nm. The reactions not only proceeded with excellent regio- and diastereoselectivity but also delivered the respective cyclobutane products with significant enantiomeric excess (up to 95% ee). Key to the success of the reactions is a two-point hydrogen bonding between quinolone and catalyst enabling efficient energy transfer and high enantioface differentiation. Preliminary work indicated that solar irradiation can be used for this process and that the substrate scope can be further expanded to isoquinolones.
The [2 + 2] photocycloaddition reaction1 has emerged over the past decades as a powerful tool in organic synthesis2 and offers a straightforward access to cyclobutanes generating up to four stereogenic centers in a single transformation. Intermolecular [2 + 2] photocycloaddition reactions offer wide flexibility regarding the choice of substrates but frequently suffer from relatively poor regio- and stereocontrol.1 In contrast to thermal cycloaddition reactions, the intermediate of a [2 + 2] photocycloaddition is short-lived and requires efficient trapping by its reaction partner.3 In recent years, interest in enantioselective photochemical reactions has significantly increased, and it is therefore not surprising that attempts have been made to perform the reaction enantioselectively by the use of a chiral auxiliary or a chiral template.4Catalytic enantioselective photochemical reactions5 are still scarce, however, and Scheme 1 provides the two only examples, in which notable enantioselectivities were achieved in an intermolecular [2 + 2] photocycloaddition reaction6,7 under catalytic conditions. The first example, which led to the formation of cyclobutanes 1, was reported in 2014 by the Yoon group.8 Its characteristic feature is the use of a ruthenium photoredox catalyst under visible light conditions, which generates in the presence of a chiral Lewis acid a radical anion that in turn can add enantioselectively to another enone. The relative configuration of the products could be controlled by the chiral ligand of the Lewis acid. In the same year it was found by our group that triplet energy transfer is a viable concept for asymmetric induction by a chiral triplet sensitizer in the intermolecular addition of pyridones to certain alkynes.9 The catalyst was a chiral xanthone, which required irradiation by UV-A light (λ = 366 nm).
Scheme 1. Previous Work on Enantioselective Intermolecular [2 + 2] Photocycloaddition Reactions.

In the present study, we have attempted to apply a chiral sensitizer to an intermolecular [2 + 2] photocycloaddition under visible light irradiation. We found the title compounds to be suitable substrates to react with a variety of olefins upon sensitization with a chiral thioxanthone at λ = 419 nm. Enantioselectivities exceeded in several cases 90% ee, and it was possible to substantiate the hypothesis that the reaction rate of the respective olefin with the photoexcited substrate is decisive for a successful chirality transfer. Our preliminary results are summarized in this communication.
Initial work was performed with parent 2(1H)-quinolone10 as the substrate and methyl or ethyl acrylate as the reaction partner (Table 1). Previous experiments on the intramolecular [2 + 2] photocycloaddition reaction of quinolones had shown that the reactions are best performed at a concentration of c = 2.5 mM in trifluorotoluene solution and with 10 mol % of the catalyst.7a,7b,7e,7f As in most intermolecular [2 + 2] photocycloaddition reactions, the photochemically inactive olefin was used in excess to avoid [2 + 2] photodimerization. The triplet energy of 2(1H)-quinolone is tabulated as 276 kJ mol–1 and seemed sufficiently close to the triplet energy of thioxanthones11 to expect an energy transfer. This expectation was gratifyingly confirmed, as a smooth reaction was observed already with 10 equiv of methyl acrylate at λ = 419 nm.12 The reaction was complete after 10 h, and product 4a(13) was obtained in 74% ee. Remarkably, the regio- and simple diastereoselectivity of the reaction was high and a single isomer was obtained. The product is the formal head-to-tail (HT) product14 with the methoxycarbonyl substituent in the exo-position. An increase in the number of acrylate equivalents (entries 1–3) led to an increase in rate and an increase in enantioselectivity. The reaction with 50 equiv of methyl acrylate was complete after 6 h and delivered the product with 81% ee. A further increase in the olefin concentration was not attempted for practical purposes (removal of excess olefin, olefin polymerization, solubility). A decrease in temperature to −40 °C turned out to be equally impractical, as the rate decreased significantly (entry 4). The same observation was made for ethyl acrylate (entries 5, 6), which delivered at −40 °C a slightly improved enantioselectivity for product 4b at the expense of the space-time yield. There was no background reaction in the absence of a sensitizer (entry 7), which indicated that the thioxanthone is involved in the catalytic cycle (vide infra).
Table 1. Optimization of the Catalytic Enantioselective [2 + 2] Photocycloaddition of 2(1H)-Quinolone with Acrylates.

| entry | R | equiv (alkene) | t (h)a | temp (°C) | product | yield (%)b | ee (%)c |
|---|---|---|---|---|---|---|---|
| 1 | Me | 10 | 10 | –25 | 4a | 79 | 74 |
| 2 | Me | 20 | 7 | –25 | 4a | 74 | 80 |
| 3 | Me | 50 | 6 | –25 | 4a | 76 | 81 |
| 4 | Me | 50 | 22 | –40 | 4a | 85 | 83 |
| 5 | Et | 50 | 6 | –25 | 4b | 82 | 78 |
| 6 | Et | 50 | 45 | –40 | 4b | 79 | 80 |
| 7d | Et | 50 | 24 | –25 | 4b | — | — |
Irradiation time at λ = 419 nm in trifluorotoluene solution at −25 °C or in a 2/1 mixture (v/v) of hexafluoro-meta-xylene and trifluorotoluene at −40 °C (c = 2.5 mM).
Yield of isolated product.
The enantiomeric excess was calculated from the ratio of enantiomers (4/ent-4) as determined by chiral HPLC analysis.
No thioxanthone (3) was added to the reaction mixture.
Further studies were concerned with a preliminary evaluation of the substrate scope (Table 2). Expectedly, the ester substituent at the acrylate did not change yield or enantioselectivity and the benzyl acrylate [2 + 2] photocycloaddition product 4c was obtained in 79% yield and 80% ee. A positive influence on the enantioselectivity was noted upon employing an α-substituted acrylate (product 4d) or a stronger electron-withdrawing group at the olefinic double bond. Indeed, methyl vinyl ketone and ethyl vinyl ketone turned out to be excellent olefin components, providing the quinolone products 4e and 4f with enantioselecitivities exceeding 90% ee. Methyl substitution at the β-position of the α,β-unsaturated ketone had little impact on the enantioselectivity (product 4g), but the reaction remained incomplete even after an irradiation time of 8 h. 4-Pentylquinolone reacted smoothly with methyl vinyl ketone to furnish product 4h (94%, 90% ee). Methyl substitution was further probed at positions C5 to C8 of the quinolone core (products 4i, 4k, 4l, 4m).
Table 2. Influence of Substituents on the Catalytic Enantioselective [2 + 2] Photocycloaddition Reactions between 2(1H)-Quinolones and Olefinsa.
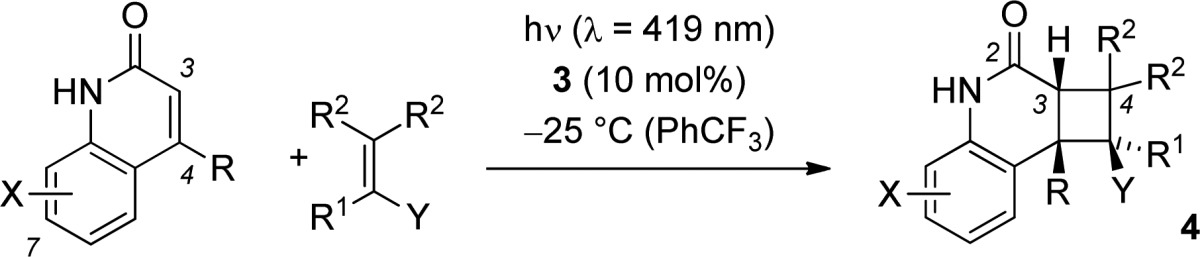
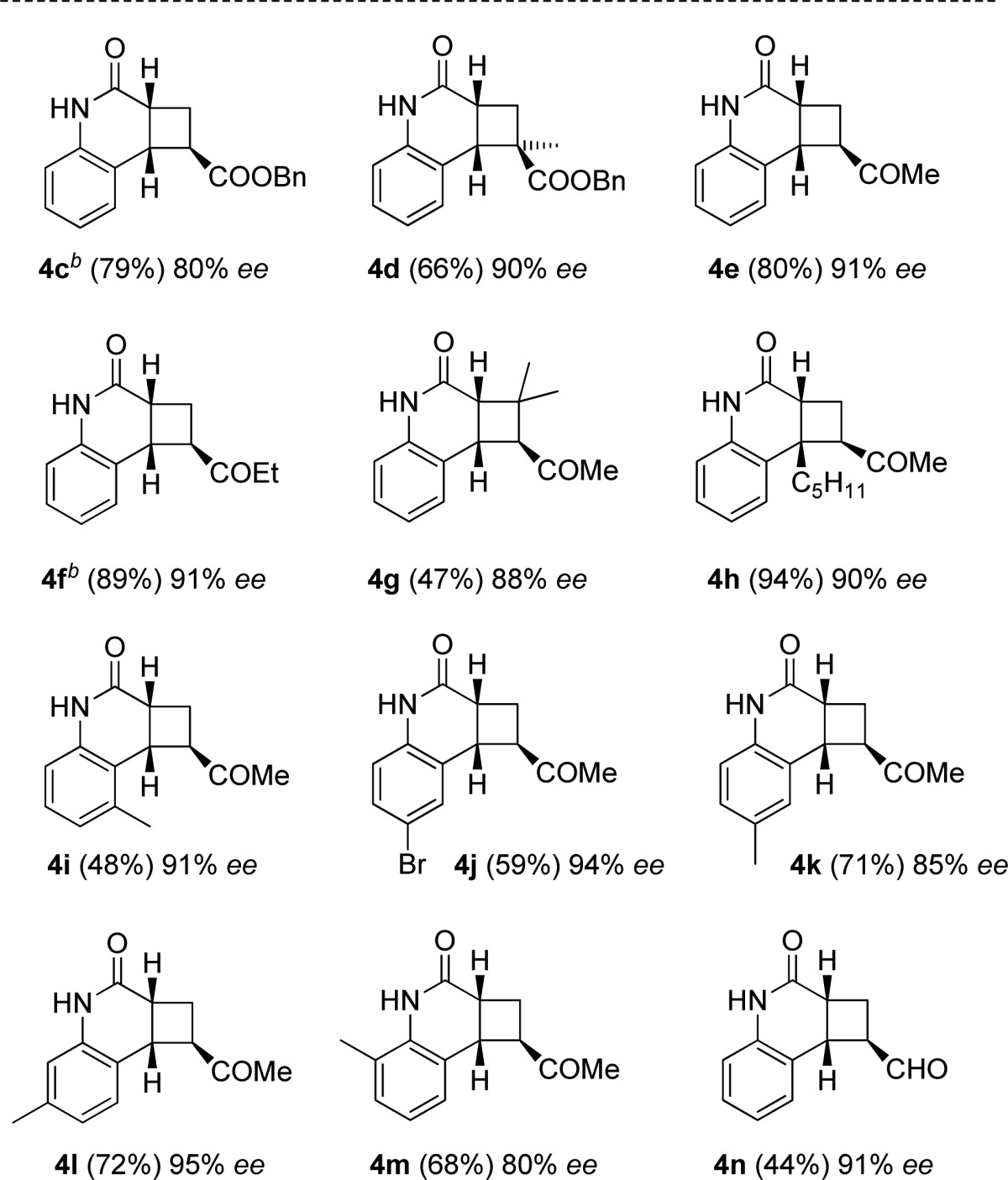
Irradiation times varied between 6 and 18 h. The reactions were performed on a scale of 25 μmol. For detailed information, see the Supporting Information.
The reaction was performed on a scale of 0.15 mmol.
Enantioselectivities remained high and reached a peak for substitution at position C7 with product 4l being formed in 95% ee. A decrease in selectivity was noted for product 4m derived from 8-methylquinolone. Notably, the [2 + 2] photocycloaddition was compatible with a bromine substituent at the quinolone core (product 4j). Even the sensitive olefin acrolein could be brought to react in the enantioselective [2 + 2] photocycloaddition. Product 4n was isolated in 44% yield and with 91% ee.
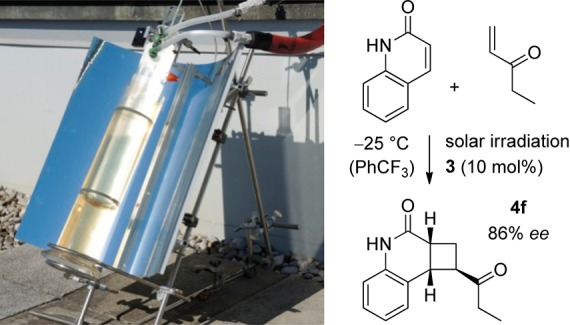
Given the fact that visible light enables the enantioselective [2 + 2] photocycloaddition it seemed desirable to attempt the reaction also with solar irradiation. A parabolic reflector was employed to focus the sun beam on the reaction vessel (Scheme 2). An Fe2(SO4)3 filter solution15 was applied to avoid undesired direct excitation by short wavelength photons. Due to the low sunlight intensity in winter, the reaction was slower than with the artificial light sources. After 3 h, the yield for 4f was 81% at 21% conversion but the enantioselectivity remained high (86% ee).
Scheme 2. Apparatus for Solar Irradiation and Result of an Intermolecular [2 + 2] Photocycloaddition.
The absolute configuration of the photocycloaddition products was derived from their specific rotation. It has been earlier reported that the helical arrangement of the conjugated π-system in dihydro-2-quinolones is responsible for the chiroptical properties of this compound class.7b,16The specific rotation of 3,6-dihydrocyclobuta[c]-2-quinolones 4b, 4c, 4d, 4f, and 4j was determined, and they were found to be levorotatory suggesting an (R)-configuration at carbon atom C3 (Table 2). This absolute configuration indicates an attack at the prostereogenic quinolone carbon atom C3 from the Re face. Indeed, based on previous results,7b,7e it is assumed that quinolone is excited into its reactive triplet state by triplet energy transfer in complex 5 (Scheme 3). In line with the mode of action of related templates,17 the approach to the photoexcited quinolone in 5* is only feasible for the olefin to occur from the Re (bottom) face relative to carbon atom C3 leading to the putative 1,4-diradical 6, from which product formation occurs by C–C bond formation at C4.
Scheme 3. Mechanistic Picture of the Enantioselective [2 + 2] Photocycloaddition and Origin for the Formation of Racemic Product rac-4.

We have previously speculated that dissociation of the triplet quinolone (T1) from complex 5* leads to a deterioration of the enantioselectivity as the former species will not encounter any asymmetric induction thus forming racemic products rac-4.7b In the present reactions, we noted that more electron-rich olefins such as vinyl acetate produced the respective products with lower selectivity. Product 4o(10c) was the formal HT product of the vinyl acetate [2 + 2] photocycloaddition to quinolone and was formed as an exo/endo mixture. Under the conditions given in Table 2, the exo-product (42% yield) was obtained with 58% ee, and the endo-product (36% yield) with 43% ee. In order to support the hypothesis that a lower reaction rate might be responsible for the decrease in enantioselectivity as compared for example to product 4f (91% ee), a competition experiment was performed (Scheme 4). The reaction was conducted at room temperature in acetonitrile solution, which facilitated the isolation and quantification of suitable samples in short time intervals. Triplet sensitization was achieved with the tert-butyl analogue 7 (10 mol %) of the chiral catalyst 3.
Scheme 4. Competing Intermolecular [2 + 2] Photocycloaddition of Vinyl Acetate vs Ethyl Vinyl Ketone and 2(1H)-Quinolone.
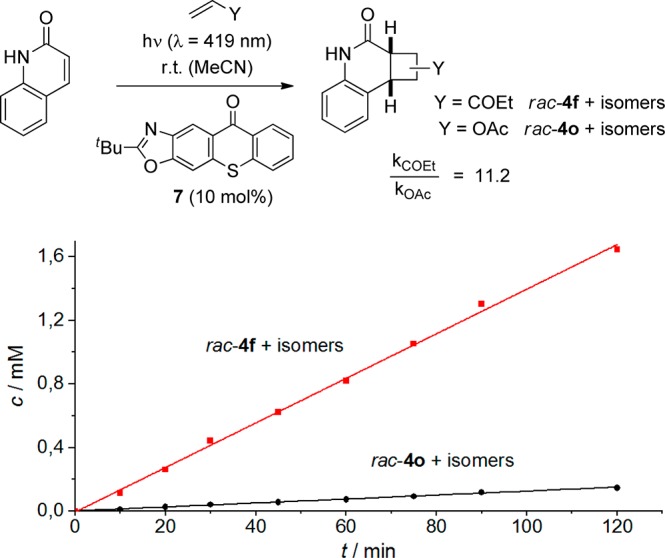
Parent 2(1H)-quinolone (1 equiv) was reacted in the presence of 25 equiv of vinyl acetate and ethyl vinyl ketone. Product analysis was performed by GLC employing dodecane as the internal standard. It is evident from the rate profile (Scheme 4) that formation of product rac-4f was significantly faster than formation of product rac-4o. Quantification of the data delivered a relative reaction rate kCOEt/kOAc of 11.2 in favor of the former reaction, which is in line with the higher enantioselectivity obtained with ethyl vinyl ketone as the olefinic reaction partner. If steric hindrance interferes with the hydrogen bonding event, the enantioselectivity of the [2 + 2] photocycloaddition can also decrease. The comparably low enantioselectivity for product 4m (Table 2) is likely due to the methyl group at carbon atom C8 of the quinolone core.
The kinetic data show that insufficient enantioface differentiation in complex 5* is not responsible for a lack of enantioselectivity with certain olefins but their low reaction rates. With this in mind, other substrates which contain a lactam binding motif and which show a high intermolecular reaction rate should be equally suitable for the enantioselectively catalyzed intermolecular [2 + 2] photocycloaddition reaction. A preliminary experiment with parent 1(2H)-isoquinolone18 and methyl vinyl ketone revealed that a high enantioselectivity can be achieved also with isoquinolone substrates (Scheme 5). Product 8(18d) was obtained as a single isomer in 91% ee. The conversion remained incomplete after 9 h, and varying amounts of starting material were recovered. In the best case, the conversion was 86% and the yield of product was 74% (86% based on conversion).
Scheme 5. Enantioselective Intermolecular [2 + 2] Photocycloaddition of 1(2H)-Isoquinolone and Methyl Vinyl Ketone.

In summary, chiral thioxanthone 3 was shown to be a competent catalyst to mediate the enantioselective intermolecular [2 + 2] photocycloaddition of various quinolones and electron-deficient alkenes. Association between the catalyst and the substrate by hydrogen bonding is critical for the success of the reaction, and dissociation of the photoexcited substrate from the catalyst is likely responsible for a loss in enantioselectivity as observed for vinyl acetate as the reaction partner. The kinetic parameters of these processes are currently being studied in more detail and will be reported together with possible applications of the enantioselective [2 + 2] photocycloaddition in due course.
Acknowledgments
Financial support by the European Research Council under the European Union’s Horizon 2020 research and innovation programme (Grant Agreement No. 665951 – ELICOS) and the Deutsche Forschungsgemeinschaft (Reinhart Koselleck program) is gratefully acknowledged. A.T. thanks the research training group (Graduiertenkolleg) 1626 “Chemical Photocatalysis” for a scholarship. R.A. acknowledges the Alexander von Humboldt Foundation for a research fellowship. Dr. S. Breitenlechner is acknowledged for his help with the rate measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b03221.
Experimental procedures, analytical data for all new compounds, proof of constitution and configuration, competition experiment, NMR spectra (PDF)
Author Contributions
† A.T. and R.A. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Reviews:; a Bauslaugh P. G. Synthesis 1970, 287–300. 10.1055/s-1970-21606. [DOI] [Google Scholar]; b Baldwin S. W. Org. Photochem. 1981, 5, 123–225. [Google Scholar]; c Crimmins M. T. Chem. Rev. 1988, 88, 1453–1473. 10.1021/cr00090a002. [DOI] [Google Scholar]; d Becker D.; Haddad N. Org. Photochem. 1989, 10, 1–162. [Google Scholar]; e Crimmins M. T.; Reinhold T. L. Org. React. 1993, 44, 297–588. 10.1002/0471264180.or044.02. [DOI] [Google Scholar]; f Mattay J.; Conrads R.; Hoffmann R.. [2 + 2] Photocycloadditions of α,β-Unsaturated Carbonyl Compounds. In Methoden der Organischen Chemie (Houben-Weyl), 4th ed.; Vol. E 21c; Helmchen G., Hoffmann R. W., Mulzer J., Schaumann E., Eds.; Thieme: Stuttgart, 1995; pp 3087–3132. [Google Scholar]; g Pete J.-P. Adv. Photochem. 1996, 21, 135–216. 10.1002/9780470133521.ch2. [DOI] [Google Scholar]; h Fleming S. A.; Bradford C. L.; Gao J. J.. Regioselective and Stereoselective [2 + 2] Photocycloadditions. In Organic Photochemistry, Molecular and Supramolecular Photochemistry, Vol. 1; Ramamurthy V., Schanze K. S., Eds.; Dekker: New York, 1997; pp 187–244. [Google Scholar]; i Bach T. Synthesis 1998, 683–703. 10.1055/s-1998-2054. [DOI] [Google Scholar]; j Margaretha P.Photocycloaddition of Cycloalk-2-enones to Alkenes. In Synthetic Organic Photochemistry, Molecular and Supramolecular Photochemistry, Vol. 12; Griesbeck A. G., Mattay J., Eds.; Dekker: New York, 2005; pp 211–237. [Google Scholar]; k Hehn J. P.; Müller C.; Bach T.. Formation of a Four-Membered Ring: From a Carbonyl-Conjugated Alkene. In Handbook of Synthetic Photochemistry; Albini A., Fagnoni M., Eds.; Wiley-VCH: Weinheim, 2010; pp 171–215. [Google Scholar]; l Poplata S.; Tröster A.; Zou Y.-Q.; Bach T.. Chem. Rev. 2016, 116, in press, doi: 10.1021/acs.chemrev.5b00723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviews:; a Iriondo-Alberdi J.; Greaney M. F. Eur. J. Org. Chem. 2007, 4801–4815. 10.1002/ejoc.200700239. [DOI] [Google Scholar]; b Hoffmann N. Chem. Rev. 2008, 108, 1052–1103. 10.1021/cr0680336. [DOI] [PubMed] [Google Scholar]; c Bach T.; Hehn J. P. Angew. Chem., Int. Ed. 2011, 50, 1000–1045. 10.1002/anie.201002845. [DOI] [PubMed] [Google Scholar]
- Schuster D. I.Mechanistic Issues in [2 + 2]-Photocycloadditions of Cyclic Enones to Alkenes. In CRC Handbook of Photochemistry and Photobiology, 2nd ed.; Horspool W. M., Lenci F., Eds.; CRC Press: Boca Raton, 2004; pp 72/1–72/24. [Google Scholar]
- a Inoue Y., Ed. Chiral Photochemistry, Molecular and Supramolecular Photochemistry, Vol. 11; Dekker: New York, 2004. [Google Scholar]; b Ramamurthy V., Inoue Y., Eds. Supramolecular Photochemistry; Wiley: Hoboken, 2011. [Google Scholar]
- Reviews:; a Yang C.; Inoue Y. Chem. Soc. Rev. 2014, 43, 4123–4143. 10.1039/C3CS60339C. [DOI] [PubMed] [Google Scholar]; b Brimioulle R.; Lenhart D.; Maturi M. M.; Bach T. Angew. Chem., Int. Ed. 2015, 54, 3872–3890. 10.1002/anie.201411409. [DOI] [PubMed] [Google Scholar]
- For a recent review on catalytic enantioselective [2 + 2] cycloaddition reactions, see:Xu Y.; Conner M. L.; Brown M. K. Angew. Chem., Int. Ed. 2015, 54, 11918–11928. 10.1002/anie.201502815. [DOI] [PubMed] [Google Scholar]
- For catalytic enantioselective (≥90% ee) intramolecular [2 + 2] photocycloaddition reactions, see:; a Müller C.; Bauer A.; Bach T. Angew. Chem., Int. Ed. 2009, 48, 6640–6642. 10.1002/anie.200901603. [DOI] [PubMed] [Google Scholar]; b Müller C.; Bauer A.; Maturi M. M.; Cuquerella M. C.; Miranda M. A.; Bach T. J. Am. Chem. Soc. 2011, 133, 16689–16697. 10.1021/ja207480q. [DOI] [PubMed] [Google Scholar]; c Brimioulle R.; Guo H.; Bach T. Chem. - Eur. J. 2012, 18, 7552–7560. 10.1002/chem.201104032. [DOI] [PubMed] [Google Scholar]; d Brimioulle R.; Bach T. Science 2013, 342, 840–843. 10.1126/science.1244809. [DOI] [PubMed] [Google Scholar]; e Maturi M. M.; Wenninger M.; Alonso R.; Bauer A.; Pöthig A.; Riedle E.; Bach T. Chem. - Eur. J. 2013, 19, 7461–7472. 10.1002/chem.201300203. [DOI] [PubMed] [Google Scholar]; f Alonso R.; Bach T. Angew. Chem., Int. Ed. 2014, 53, 4368–4371. 10.1002/anie.201310997. [DOI] [PubMed] [Google Scholar]; g Vallavoju N.; Selvakumar S.; Jockusch S.; Sibi M. P.; Sivaguru J. Angew. Chem., Int. Ed. 2014, 53, 5604–5608. 10.1002/anie.201310940. [DOI] [PubMed] [Google Scholar]; h Vallavoju N.; Selvakumar S.; Jockusch S.; Prabhakaran M. T.; Sibi M. P.; Sivaguru J. Adv. Synth. Catal. 2014, 356, 2763–2768. 10.1002/adsc.201400677. [DOI] [Google Scholar]; i Brimioulle R.; Bach T. Angew. Chem., Int. Ed. 2014, 53, 12921–12924. 10.1002/anie.201407832. [DOI] [PubMed] [Google Scholar]; j Brimioulle R.; Bauer A.; Bach T. J. Am. Chem. Soc. 2015, 137, 5170–5176. 10.1021/jacs.5b01740. [DOI] [PubMed] [Google Scholar]
- a Du J.; Skubi K. L.; Schultz D. M.; Yoon T. P. Science 2014, 344, 392–396. 10.1126/science.1251511. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Neier R. Science 2014, 344, 368–369. 10.1126/science.1252965. [DOI] [PubMed] [Google Scholar]
- Maturi M. M.; Bach T. Angew. Chem., Int. Ed. 2014, 53, 7661–7664. 10.1002/anie.201403885. [DOI] [PubMed] [Google Scholar]
- For early work on racemic intermolecular [2 + 2] photocycloaddition reactions of 2(1H)-quinolones, see:; a Evanega G. R.; Fabiny D. L. Tetrahedron Lett. 1968, 9, 2241–2246. 10.1016/S0040-4039(00)89729-4. [DOI] [Google Scholar]; b Loev B.; Goodman M. M.; Snader K. M. Tetrahedron Lett. 1968, 9, 5401–5404. 10.1016/S0040-4039(00)89789-0. [DOI] [Google Scholar]; c Evanega G. R.; Fabiny D. L. J. Org. Chem. 1970, 35, 1757–1761. 10.1021/jo00831a009. [DOI] [Google Scholar]; d Buchardt O.; Christensen J. J.; Harrit N. Acta Chem. Scand. 1976, 30, 189–192. 10.3891/acta.chem.scand.30b-0189. [DOI] [Google Scholar]; e Kaneko C.; Naito T.; Somei M. J. Chem. Soc., Chem. Commun. 1979, 804–805. 10.1039/c39790000804. [DOI] [Google Scholar]; f Kaneko C.; Naito T. Chem. Pharm. Bull. 1979, 27, 2254–2256. 10.1248/cpb.27.2254. [DOI] [Google Scholar]
- The triplet energy (ET) of parent thioxanthone is tabulated as ET = 265 kJ mol–1:Murov S. L.; Carmichael I.; Hug G. L.. Handbook of Photochemistry, 2nd ed.; Dekker: New York, 1993; p 80. [Google Scholar]
- For an emission spectrum of the irradiation lamps, see Supporting Information.
- All compounds 4a–4n have not yet been reported and were therefore prepared in racemic form for comparison (see Supporting Information).
- Parent 2(1H)-quinolone (carbosytril) has been previously reported to react with acrylonitrile exclusively to the HT product:; a Reference (10c); b Lewis F. D.; Reddy G. D.; Elbert J. E.; Tillberg B. E.; Meltzer J. A.; Kojima M. J. Org. Chem. 1991, 56, 5311–5318. 10.1021/jo00018a020. [DOI] [Google Scholar]
- Pellicori S. F. Appl. Opt. 1964, 3, 361–366. 10.1364/AO.3.000361. [DOI] [Google Scholar]
- Bauer A.; Westkämper F.; Grimme S.; Bach T. Nature 2005, 436, 1139–1140. 10.1038/nature03955. [DOI] [PubMed] [Google Scholar]
- a Bach T.; Bergmann H.; Grosch B.; Harms K. J. Am. Chem. Soc. 2002, 124, 7982–7990. 10.1021/ja0122288. [DOI] [PubMed] [Google Scholar]; b Bakowski A.; Dressel M.; Bauer A.; Bach T. Org. Biomol. Chem. 2011, 9, 3516–3529. 10.1039/c0ob01272f. [DOI] [PubMed] [Google Scholar]
- For some key references on intermolecular isoquinolone [2 + 2] photocycloaddition reactions, see:; a Evanega G. R.; Fabiny D. L. Tetrahedron Lett. 1971, 12, 1749–1752. 10.1016/S0040-4039(01)87451-7. [DOI] [Google Scholar]; b Chiba T.; Takada Y.; Kaneko C.; Kiuchi F.; Tsuda Y. Chem. Pharm. Bull. 1990, 38, 3317–3325. 10.1248/cpb.38.3317. [DOI] [Google Scholar]; c Al-Jalal N.; Covell C.; Gilbert A. J. Chem. Res., Synop. 1998, 678. 10.1039/a805642k. [DOI] [Google Scholar]; d Coote S. C.; Bach T. J. Am. Chem. Soc. 2013, 135, 14948–14951. 10.1021/ja408167r. [DOI] [PubMed] [Google Scholar]; e Coote S. C.; Pöthig A.; Bach T. Chem. - Eur. J. 2015, 21, 6906–6912. 10.1002/chem.201500173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.