

Abstract
Endometriosis is a benign gynecological condition that causes considerable morbidity due to associated infertility, debilitating pelvic pain and inflammatory dysfunctions. Diet is a highly modifiable risk factor for many chronic diseases, but its contribution to endometriosis has not been extensively investigated, due partly to the paradoxical inverse association between obesity and disease incidence. Nevertheless, chronic exposure to dietary high-fat intake has been linked to greater systemic inflammation and oxidative stress, both features of women with endometriosis. Here, we evaluated the effects of a high-fat diet (HFD) (45% fat kcal) on endometriosis progression using an immunocompetent mouse model where ectopic lesion incidence was induced in wild-type recipients by ip administration of endometrial fragments from transcription factor Krüppel-like factor 9-null donor mice. We show that HFD significantly increased ectopic lesion numbers in recipient mice with no significant weight gain and modifications in systemic ovarian steroid hormone and insulin levels, relative to control diet-fed (17% fat kcal) mice. HFD promotion of lesion establishment was associated with reductions in stromal estrogen receptor 1 isoform and progesterone receptor expression, increased F4/80-positive macrophage infiltration, higher stromal but not glandular epithelial proliferation, and enhanced expression of proinflammatory and prooxidative stress pathway genes. Lesion-bearing HFD-fed mice also displayed higher peritoneal fluid TNFα and elevated local and systemic redox status than control diet-fed counterparts. Our results suggest that HFD intake exacerbates endometriosis outcome in the absence of ovarian dysfunction and insulin resistance in mice and warrants further consideration with respect to clinical management of endometriosis progression and recurrence in nonobese patients.
Endometriosis is a benign gynecological disease characterized by the presence of endometrial fragments outside of the uterus, predominantly in the abdominal cavity and ovary (1). The disease affects approximately 10% of reproductive-aged women, 50% of whom show decreased fertility. Despite decades of research, the disease remains difficult to diagnose in its initial stages and can remain undetected (an average gap of 7.5 y) until the onset of overt symptoms which include debilitating pelvic pain and dysmenorrhea. The recent report that 11% of women in the general population have undiagnosed endometriosis and/or delayed diagnosis from onset of symptoms (2), precluding timely clinical care, has significant implications at many levels (eg, physical, emotional, financial, and societal), resulting in substantially reduced quality of life (3). The pathophysiology of endometriosis is complex and arises from a combination of genetic, hormonal, inflammatory, and immune dysfunctions (4–6).
Obesity is known to have detrimental effects on the endometrium. An increased body mass index (BMI) is the most significant risk factor for the development of endometrial cancer (7). Moreover, obesity has been shown to diminish pregnancy success due to poor embryo attachment and results in inferior in vitro fertilization outcomes as well as increased risks of pregnancy complications (8–10). Paradoxically, epidemiological data demonstrate an inverse association between adult BMI and the incidence of endometriosis (11–13). The mechanism(s) linking decreased BMI in women with endometriosis remains unknown. It has been proposed that the disparate relationship may stem from secondary conditions in obese women such as polycystic ovary syndrome, which results in anovulation and fewer/irregular menstrual cycles (14) and thus, decreased menstrual tissue reflux, the latter being the most accepted etiology for endometriosis (15). However, women with polycystic ovary syndrome are not protected from endometriosis (16). Moreover, there are sporadic reports on a positive association between high adiposity and endometriosis (17) and between prepubertal obesity and endometriosis recurrence risk (18).
Diet constitutes an important risk factor for many chronic diseases. Although endometriosis is considered a chronic and progressive condition, very few studies have focused on understanding the mechanistic relationship between specific dietary intake and endometriosis risk and recurrence. Nevertheless, a number of epidemiological studies have implicated specific diets with endometriosis in the general population (19). For instance, consumption of fish oil, green vegetables, fruits, and dairy products has been negatively associated with endometriosis risk (19–21). Conversely, intake of red meat and trans/saturated fats was positively linked to an increased risk of developing the disease (19, 22). Dietary influences on endometriosis are not surprising given that many physiological processes that contribute to disease progression such as estrogen levels, inflammation, and enhanced prostaglandin metabolism are affected by dietary components (23, 24). Specific fatty acids are known to increase systemic levels of IL-6 and other inflammatory markers that are found at higher concentrations among women with endometriosis (25). By contrast, the higher consumption of antioxidant-rich diets reported for women without endometriosis may protect against disease development by reducing systemic oxidative stress (26, 27). However, the link between obesogenic diets and endometriosis risk remains uncertain, driven in part by the inherent challenges associated with dietary recall, divergent patient manifestations, and varying severity of endometriosis in population-based studies.
We have recently shown that null mutation of endometrial Krüppel-like factor 9 (KLF9), a member of the specificity Protein/KLF family of transcription factors, promotes endometriotic lesion establishment in a mouse model (28). In this paradigm, wild-type (WT) mice with a fully intact immune system and which are ip administered with endometria from donor Klf9 null mice (29) display 100% lesion incidence 4 weeks after endometrial fragment injection. KLF9 is a mediator of both progesterone (P4) receptor (PGR) and estrogen receptor (ESR) signaling in uterine endometrial cells, primarily in stroma and glands (30–32). Klf9 null female mice are subfertile, due in part to P4 resistance (33, 34), a condition associated with endometriosis (1, 3). Importantly, women with endometriosis showed decreased endometrial KLF9 expression, relative to women without the disease (35). In human endometrial stromal cells, coloss of KLF9 and PGR by small interfering RNA targeting in vitro resulted in aberrant expression of components of inflammatory, WNT and IGF-associated pathways, all known features of endometriotic implants (35). Our mouse model, thus, provides a highly suitable system to investigate dietary effects on lesion progression and development.
In the current study, we used endometrial Klf9 null-recipient WT mice fed either a control diet (CD) or a high-fat diet (HFD), beginning at weaning (postnatal day [PND]21) and continuing through lesion harvest at PND84, to evaluate the influence of HFD consumption on endometriosis development. We analyzed ectopic lesions from mice of the 2 diet groups for proliferation, apoptosis, steroid receptor (PGR, ESR) levels, and macrophage marker F4/80 expression. We employed gene array expression profiling to identify gene cohorts in lesions that are altered by a HFD intake. Further, we evaluated select measures of redox status in sera, peritoneal fluids and lesions; inflammation (TNFα and soluble TNF receptor 1 [sTNFR1]) in peritoneal fluids; and ovarian steroid hormone synthetic competency (estradiol [E2], P4) in sera, as a function of dietary exposure. Our collective results provide support for an ovarian-independent negative impact of HFD exposure on lesion establishment occurring via the induction of prooxidative/proinflammatory status in systemic and local environments of lesions.
Materials and Methods
Animals
All animal experiments were conducted in accordance with the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals, following protocols approved by the University of Arkansas for Medical Sciences Institutional Animal Care and Use Committee. At PND21, female C57BL/6J Klf9 WT pups were weaned and randomly assigned to one of 2 American Institute of Nutrition-93G-based pelleted diets with casein as the protein source: 1) CD (n = 7) containing 17% total kcal from lard fat (Harlan) and 2) HFD (n = 9) containing 45% of total kcal from lard fat (Harlan); the latter recapitulates the percent kcal from fat in a typical “Western diet” (Supplemental Table 1) (36). After 5 weeks on their assigned diets (at PND56), mice were injected ip with minced endometrium isolated from Klf9 null mice (28) and continued on their assigned diets. Tissue, sera, and peritoneal fluids were collected from recipient mice 4 weeks later (Figure 1A).
Figure 1. Experimental design and characterization of endometrial-like lesions derived from mice fed CD or HFD.

A, Schematic representation of the generation of endometrial ectopic lesions in mice and their analyses. Klf9 WT mice were used as recipients of endometrial fragments from Klf9 null mice (donors), as previously described (28), with the indicated numbers of mice used for each diet group. B, Representative bright-field images of ectopic lesions isolated from WT recipients assigned to either CD or HFD groups, 4 weeks after ip administration of Klf9 null endometrium. Scale bar, 1 mm. C, H&E-stained sections of representative CD or HFD lesions at ×400 magnification. Scale bar, 20 μm. D, Representative sections of CD or HFD ectopic lesions showing Ki-67-positive stromal cells. E, Representative sections of CD or HFD ectopic lesions showing TUNEL-positive stromal cells. Red arrows indicate Ki-67-positive (D) and TUNEL staining (E) stromal cells. Four lesions per dietary group, each representing an individual mouse, were analyzed. Scale bar, 20 μm. For D and E, the percentages of staining cells (expressed as mean ± SEM) were quantified by counting the number of immunopositive nuclei over the total number of stromal cells per field and are presented as bar graphs; *, P < .05 by Student's t test.
Imaging and collection of ectopic lesions
Lesions were visualized, counted, and photographed using a Carl Zeiss SteREO Discovery V8 steromicroscope (Carl Zeiss, Inc) equipped with a Canon EOS 1000D camera (Canon, Inc). Lesion volume was calculated as previously described (28) based on dimensions (length, width) measured using Axiovision software (Carl Zeiss). All lesions were snap-frozen in liquid nitrogen or placed in 10% neutral-buffered formalin for further processing (below).
RNA isolation, gene expression profiling with microarrays, and quantitative RT-PCR (QPCR)
Gene expression analyses used Mouse Genome 430 2.0 high-density oligonucleotide arrays (Affymetrix, Inc), with 54 120 probe sets to interrogate 38 500 well-characterized mouse transcripts. Total RNA was extracted from endometriotic lesions (n = 7 and 9, respectively, for CD and HFD, each representing an individual mouse) using the QIAGEN AllPrep kit (QIAGEN) following the manufacturer's protocol. RNA concentrations were determined using the Nanodrop ND-1000 spectrophotometer (Nanodrop). cRNA preparation, hybridization, washes, and detection followed the manufacturer's recommendations. RNAs were prepared using GeneChip 3′ IVT Express kit (Affymetrix). RNAs equally pooled from 3 or 4 mice per diet group constituted one biological replicate and were hybridized to one array; 2 independent replicates were evaluated per treatment group. Expression data were analyzed using GeneSpring GX version 11 software (Agilent Technologies). The .CEL files containing probe level intensities were processed using the GeneChip robust multiarray analysis with quantile normalization. The normalized data were subjected to pairwise comparisons. Gene sets were also subjected to gene set enrichment analyses (GSEA) as previously described (37). Biological function and ontology analyses used Affymetrix NetAffx and GeneSpring programs, and genes with more than or equal to 1.2-fold change (FC) and P ≤ .05 were analyzed for functional gene annotations using ingenuity pathway analysis (Ingenuity Systems). Data discussed here were deposited in Gene Expression Omnibus (GSE80603). For validation of microarray results and other RNA expression analyses, cDNA was synthesized from 1 μg of total RNA using the iScript cDNA Synthesis kit (Bio-Rad Laboratories), and subjected to QPCR using iTaq Universal SYBR Green Supermix (Bio-Rad) and the Bio-Rad CFX96 Real Time System module and c1000 Touch thermal cycler. Intron-flanking primers (available upon request) were designed to eliminate genomic DNA amplification using the Primer Express software (Applied Biosystems) and were obtained from Integrated DNA Technologies, Inc. A standard curve was generated by serially diluting pooled cDNAs (obtained in equal aliquots from all samples) beginning with the most concentrated cDNA pool designated as 10 000 arbitrary units. Target mRNA abundance in endometriotic lesions was normalized to a factor derived from the geometric mean of expression values for B-actin and Gapdh calculated using the GeNorm program (38).
Lesion morphometry and immunohistochemistry (IHC)
Endometriotic lesions were fixed in 10% neutral-buffered formalin as previously described (28). Sections (5 μm) were mounted on poly(lysine)-coated slides (Fisher Scientific) and used to evaluate lesion morphology and for IHC. For morphometric analyses, the sections were deparaffinized in xylene, rehydrated in a series of alcohols, and stained with hematoxylin and eosin (34). For detection and quantification of immunostaining cells, sections were treated with Citra Plus (Biogenex) to unmask antigen and with 3% hydrogen peroxide (30 min, room temperature) to block endogenous peroxidase activity. Sections were incubated in a blocking solution (VectaStain ABC kits; Vector Laboratories) for 30 minutes to reduce nonspecific staining and then with primary antibodies for 24 hours at 4°C in a humidity chamber. Antibodies were obtained from the following sources and used at the indicated dilutions: 1) rabbit antimouse ESR1 (1:250, sc-542; Santa Cruz Biotechnology, Inc); 2) rabbit antihuman PGR (1:300, sc-7208; Santa Cruz Biotechnology, Inc); and 3) rabbit antimouse Ki-67 (1:75, ab16667; Abcam). Incubation with biotinylated, antirabbit secondary antibody (1:200 dilution, VectaStain ABC kits; Vector Laboratories) was carried out for 30 minutes at room temperature. Sections were stained with 3,3′-diaminobenzidine (Chromogen; Dako), counterstained with hematoxylin, dehydrated, cleared, and coverslipped for examination under a microscope. TUNEL staining was performed using the ApopTag Peroxidase In Situ Apoptosis Detection kit following the manufacturer's instructions (Millipore Corp). Control sections for all IHCs were processed with omission of the primary antibody. A total of 3–4 slides, with each slide representing an endometriotic lesion from an individual donor mouse per diet group, were evaluated. Approximately 1000 stromal cells were counted on average from at least 4–5 randomly chosen fields (×400 magnification) per slide using an Axiovert 200M microscope with an Axiocam HRc camera and Axiovision software (Carl Zeiss). Approximately 400 glandular epithelial cells, representing a total of 9–10 glands from 3 independent lesions, were similarly analyzed. Results are expressed as the percent of nuclear-immunopositive stromal or epithelial cells (ratio of nuclear positively staining cells to total numbers of total cells counted × 100).
Macrophage infiltration of ectopic lesions was evaluated using rabbit antihuman F4/80 polyclonal antibody (1:100 dilution, PA5-32399; Thermo Scientific) with incubation for 24 hours at 4°C in a humidity chamber, followed by incubation with secondary rabbit antirat IgG (Table 3). Staining with 3,3′-diaminobenzidine and hematoxylin followed the procedures described above. Immunopositive-areas (showing clusters of immunostained macrophages) were counted from 5 randomly chosen fields at ×200 magnification per slide; a total of 3 (CD) and 5 (HFD) slides, with each slide representing an endometriotic lesion from an individual mouse, were evaluated. Results are expressed as the ratio of the numbers of immunopositive areas over the total number of areas counted (15 for CD; 25 for HFD) × 100.
Table 3.
List of Antibodies Used for IHC
| Protein of Interest | Blocking | Primary Antibody (Concentration/Incubation) | Secondary Antibody (Concentration/Incubation) |
|---|---|---|---|
| ESR1 | 5% normal goat serum | 1:200; 24 h | 1:200 biotinylated antirabbit IgG; 30 min |
| F4/80 | 5% normal goat serum | 1:100; 24 h | 1:200 biotinylated antirabbit IgG; 30 min |
| Ki-67 | 5% normal goat serum | 1:100; 24 h | 1:200 biotinylated antirabbit IgG; 30 min |
| PGR | 5% normal goat serum | 1:200; 24 h | 1:200 biotinylated antirabbit IgG; 30 min |
Peritoneal fluid collection and ELISA
Peritoneal fluids were collected by injecting 1ml cold PBS-containing protease inhibitors (Halt Protease Inhibitor Cocktail; Thermo Scientific) into the peritoneum of recipient mice before lesion isolation, using a 27-gauge needle (Fisher Scientific). Fluid was retrieved using the same syringe after gentle massaging of the peritoneum, then centrifuged at 1500 rpm for 10 minutes at 4°C to remove peritoneal cells. Supernatant was stored at −80°C until analysis. Peritoneal fluid was quantified for insulin, TNFα and sTNFR1 levels (n = 7–9 mice/diet group) using mouse insulin (Millipore, Inc), mouse TNFα and mouse sTNFR1 (Quantikine ELISA; R&D Systems) ELISAs, respectively, following the manufacturer's instructions.
Serum RIA and ELISA
Approximately 500 μL of whole blood was collected by closed cardiac puncture from recipient mice at killing. Serum was separated by centrifugation of whole blood at 4600g for 1 hour and stored at −20°C before analysis. Serum E2, P4, and insulin levels were measured using the Ultrasensitive Estradiol kit (Beckman Coulter), Progesterone EIA kit (Cayman Chemical), and mouse insulin ELISA (Millipore, Inc), respectively, following the manufacturer's protocols.
Oxidative stress biomarker analysis
Sera and peritoneal fluids of recipient animals (all with ectopic lesions) were analyzed for levels of oxidative (reduced and oxidized glutathione; reduced and oxidized cysteine) and nitrosative (3′-nitrotyrosine) stress biomarkers and for measures of methylation capacity (aminothiols methionine [Met] and homocysteine [Hcy]) by HPLC with electrochemical detection (HPLC-ED), following previously published procedures (39). Briefly, 100 μL of 10% metaphosphoric acid were added to 50 μL of plasma or peritoneal fluid to precipitate protein; the solution was mixed well and incubated on ice for 30 minutes. After centrifugation for 15 minutes at 18 000g at 4°C, supernatants were passed through a 0.2-μm nylon membrane filter, and 20 μL were injected into the HPLC system. The analyses were performed using HPLC-ED model 5200A column (5 μm, 4.6 × 150 mm; MCM, Inc). Metabolite levels were quantified using the HPLC-ED software.
Genomic DNA 8-OH-guanosine levels
Tissue DNA was isolated using the QIAmp DNA Mini kit (QIAGEN) following the manufacturer's protocol. Approximately 1 μg of DNA was then treated with Ribonuclease A (Sigma) to a final concentration of 0.02 mg/mL and incubated at 37°C for 15 minutes. The purified DNA was then digested into component nucleotides by sequential treatments with nuclease P1, snake venom phosphodiesterase, and alkaline phosphatase as previously described (40). The digested nucleotides were stored at −20°C until analysis by LC-MS/MS. Base separation was performed with a Dionex HPLC system coupled to an electrospray ionization tandem mass spectrometer (Thermo-Finnigan LCQ) using a Phenomenex Gemini column (C18, 150 × 2.0 mm, 3-μm particle size), following established methodologies (40).
Statistical analysis
Statistical analysis was performed using SigmaStat software (version 3.5; Systat Software). Data were expressed as mean ± SEM. For comparison of lesion numbers and volumes between diet groups, data were analyzed using the Mann-Whitney rank sum test. For all others, data were compared by Student's t test. Differences with values of P < .05 and .05 < P < .10 were considered significant and with tendency for significance, respectively.
Results
HFD exposure increases number of endometriotic lesions
The schematic of the analyses conducted to evaluate the effects of dietary HF on endometriosis development in our mouse model is shown in Figure 1A. WT mice at weaning were randomly assigned to either CD or HFD and were continued on their diets for 5 weeks before induction of endometriotic-like lesions at PND56, at which time mice had undergone 4–5 estrous cycles. The temporal strategy for diet assignment and endometriosis-induction simulates the early exposure to HFD of the general population (ie, during early childhood) and the occurrence of endometriosis in reproductive-aged women. Mice at weaning (diet assignment) had comparable body weights (CD = 9.82 ± 0.33 g; HFD = 8.39 ± 0.61 g; P > .05), and provision of a HFD did not significantly increase body weights over those fed CD (P = .11) when measured at study termination (Table 1). Serum E2 and P4 (concordant with estrous cycle lengths of normal duration) (data not shown) and insulin levels did not differ between mice fed CD and HFD (Table 1). Representative bright-field macroscopic images and Hematoxylin & Eosin-stained sections of ectopic lesions are shown (Figure 1, B and C). Ectopic lesions display the presence of endometrial glands and stromal cells, confirming their endometrial origin. Consistent with our previous findings (28), lack of endometrial Klf9 expression in donor tissue resulted in 100% lesion incidence in WT recipients, and this occurred irrespective of diet (Table 1). Consumption of a HFD did not alter lesion volumes in mice, relative to CD-fed mice (P = .597). However, there was a significant increase (by an average of 1) (P = .038) in lesion numbers in mice fed a HFD (Table 1).
Table 1.
Parameters of Lesions and Recipients Exposed to CD or HFD
| Diet |
||
|---|---|---|
| CD | HFD | |
| Lesion parameters | ||
| Number (per mouse) | 1.4 ± 0.3 | 2.3 ± 0.2a |
| Volume (mm3) (per lesion) | 16.9 ± 5.6 | 14.2 ± 3.9 |
| Incidence | 100% (7/7) | 100% (9/9) |
| Body weights (g) | ||
| PND21 | 9.8 ± 0.3 | 8.4 ± 0.6 |
| PND84 (sac) | 24.0 ± 0.9 | 24.9 ± 1.2 |
| Serum indices | ||
| Estrogen (pg/mL) | 26.8 ± 0.9 | 28.8 ± 2.1 |
| P4 (ng/mL) | 11.0 ± 1.7 | 9.7 ± 1.5 |
| Insulin (ng/dL) | 1.78 ± 0.3 | 2.20 ± 0.4 |
| Peritoneal fluid indices | ||
| TNFα (pg/mL) | 33.1 ± 3.6 | 45.5 ± 3.2a |
| sTNFR1 (pg/mL) | 177.9 ± 23.6 | 184.3 ± 23.6 |
| Insulin (ng/dL) | 0.93 ± 0.01 | 0.86 ± 0.03 |
P ≤ .05.
HFD lesions display higher proliferation and lower apoptotic status
To determine if the promotion of lesion establishment (ie, increase in lesion numbers) with HFD exposure involves alterations in proliferative and apoptotic parameters, lesions from mice of both diet groups were evaluated. HFD lesions displayed a higher percentage of stromal Ki-67 immunoreactivity (Figure 1D) and lower percentage of stromal TUNEL-positive cells (Figure 1E), compared with CD lesions. Glandular epithelial cells from the same lesions, however, did not display the same changes noted for stromal cells. In particular, lesion epithelial cells for mice of the 2 diet groups showed comparable percentages of Ki-67 immunoreactivity (CD = 26.1 ± 1.1 vs HFD = 22.4 ± 3.3; P = .565) and of TUNEL positivity (CD = 8.2 ± 3.5 vs HFD = 7.5 ± 3.5; P = .894).
HFD lesions show increased macrophage recruitment
Macrophages are important inflammatory cells that can compromise normal function in the reproductive tract and cause pathology (41, 42). Given that endometriosis is associated with inflammation (1, 3), the infiltration of endometrial implants by host macrophages was compared by evaluating the numbers of F4/80-positive areas in HFD and CD lesions (Figure 2A). A total of 15–25 independent stromal areas (5 random field per lesion and 3–5 independent mouse lesions per diet group) were evaluated. HFD lesions had greater percentage of F4/80+ areas, indicative of presence of mature macrophages, than CD lesions (Figure 2B). In the latter, the presence of F4/80+ cells was sporadic and largely undetected.
Figure 2. Macrophage F4/80+ cells in CD and HFD ectopic lesions in mice.
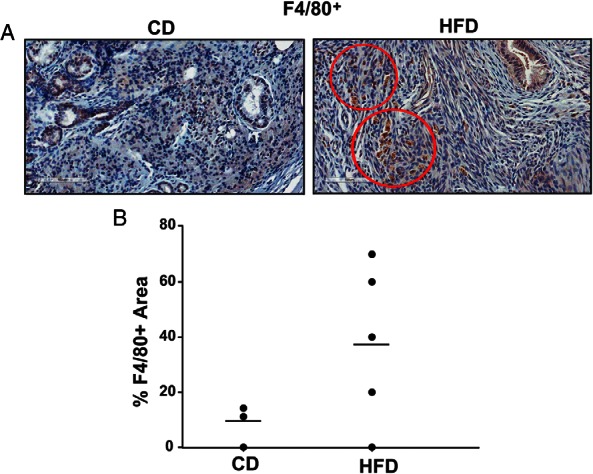
A, Representative sections of ectopic lesions harvested from CD- and HFD-fed mice, showing positive immunostaining for F4/80 in HFD lesion-stromal cells. Scale bar, 100 μm. B, Graph represents percentages of F4/80+ areas in stromal cells of CD or HFD ectopic lesions, determined by counting the number of immunopositive areas over the total number of stromal areas evaluated ×100. Each point represents a lesion from an individual mouse.
HFD and CD lesions differ in steroid hormone receptor expression
Aberrant expression of both PGR and ESR isoforms has been reported in lesions of women with endometriosis and in implants generated in mouse models of the disease (1, 3, 28, 35). Compared with CD lesions, those derived from HFD-fed recipients had lower levels of total Pgr, Pgr-B, and Esr1 transcripts, with no differences noted for Esr2 (Figure 3A). IHC analysis demonstrated a significant decrease in stromal nuclear-localized immunopositive PGR and ESR1 within HFD relative to CD lesions, thus mimicking the changes noted in corresponding mRNA abundance (Figure 3, B and C). Lesions were further analyzed by QPCR for expression of 2 known PGR down-regulated targets, namely Wnt4 and stanniocalcin 1 (Stc1) (28, 35). The lower PGR levels in HFD relative to CD lesions were not accompanied by higher Wnt4 and Stc1 transcript levels (Supplemental Figure 1).
Figure 3. ESR and PGR are differentially expressed in CD and HFD ectopic lesions in a mouse model.

A, Transcript levels of Pgr and Esr isoforms were determined in CD or HFD lesions by QPCR. Data (mean ± SEM) are expressed as FC from CD lesion group and were obtained from n = 7 lesions, with each lesion isolated from an individual mouse per diet group. B and C, Representative sections of CD and HFD ectopic lesions showing PGR-positive (B) and ESR1-positive (C) stromal cells (indicated by red arrowheads). Four lesions per dietary group, each representing an individual mouse, were analyzed. Scale bar, 20 μm. For B and C, the percentages of immunostaining cells (expressed as mean ± SEM) were quantified by counting the number of immunopositive stromal nuclei over the total number of stromal cells per field and are presented as bar graphs; *, P < .05 by Student's t test.
HFD altered gene expression profiles of endometriotic lesions
To investigate gene networks perturbed directly or indirectly by HFD and which may be partly responsible for the induction of ectopic lesion numbers with HFD intake, we performed microarray analysis on lesions derived from CD- or HFD-fed mice. Unsupervised hierarchical clustering of gene expression profiles indicated that CD samples grouped separately from those of the HFD group (Figure 4A). A total of 359 transcripts were significantly regulated (P < .05) by at least 1.2-fold with dietary intake of increased fat. Of these, 169 were up-regulated and 190 were down-regulated. Ingenuity pathway analysis indicated that altered gene expression included those of genes associated with angiogenesis, oxidative stress and immune response, inflammation, cytokine/chemokine activity, and lipid transport (Table 2). We confirmed gene expression changes for a subset of genes belonging to immune/inflammatory and oxidative stress pathways, by QPCR. HFD resulted in up-regulated Cxcl4, Il17a, and Csf2 and in down-regulated Il17b and Cd136 transcript levels, relative to CD (Figure 4B). Expression of a subset of genes related to the synthesis of the antioxidant glutathione (GSH) was either lower (glutamate-cysteine ligase catalytic subunit [Gclc], glutathione peroxidase 1 [Gpx1]) or tended to be lower (glutathione reductase [Gsr] and glutathione synthetase [Gss]) in HFD than in CD lesions (Figure 3C). By contrast, transcript abundance for carbonic anhydrase III (Ca3) with proproliferative and antiapoptotic functions under oxidative stress conditions (43) and for Gpx3, whose induced expression is associated with increased production of reactive oxygen species (44), were up-regulated in HFD than in CD lesions (Figure 4C).
Figure 4. HFD intake altered inflammatory and oxidative stress signaling pathways in endometriotic lesions in a mouse model.

A, The top 50 up- or down-regulated genes by HFD in ectopic lesions were determined from microarray analysis. Confirmation of gene expression changes for genes associated with inflammation (B) or oxidative stress (C) from CD or HFD lesions was performed by QPCR. Data (mean ± SEM) are expressed as FC from CD lesions and were obtained from n = 7 (CD) and 8 (HFD) lesions; *, P < .05 by Student's t test. Bars with numerical P values show tendency for significance.
Table 2.
Summary of GSEA With FDR ≤ 0.25
| Gene Set | FDR |
|---|---|
| Enriched in HFD | |
| Extracellular matrix | 0.073 |
| Metalloendopeptidase activity | 0.081 |
| Collagen | 0.138 |
| Vasculature development | 0.189 |
| Oxygen and reactive oxygen species metabolic process | 0.218 |
| Angiogenesis | 0.235 |
| Diminished in HFD | |
| Generation of a signal involved in cell-cell signaling | 0.001 |
| Hormone activity | 0.042 |
| Cytokine activity | 0.048 |
| T-cell activation | 0.123 |
| Immune response | 0.129 |
| Inflammatory response | 0.131 |
| Cell-cell signaling | 0.132 |
| Regulation of apoptosis | 0.172 |
| Oxidoreductase activity | 0.177 |
For FDR, see Ref. 37.
Redox status differs in endometriotic CD- and HFD-fed mice
Given the gene expression changes in oxidative stress signaling pathways in ectopic lesions as a function of diet (Figure 4, B and C), we evaluated the redox status in sera and peritoneal fluids of mice with lesions. Sera from the HFD group showed higher cystine to cysteine ratio (due to lower cysteine levels), lower Met to Hcy ratio (due to lower Met and higher Hcy levels, respectively), and higher 3′-nitrotyrosine levels than sera from the CD group (Figure 5A). No difference in serum levels of GSH and GSSG were found between diet groups. Peritoneal fluids from HFD group showed lower GSH to GSSG ratio, higher cystine to cysteine ratio and higher 3′-nitrotyrosine levels than those of the CD group, with no differences noted for the Met to Hcy ratio between diet groups (Figure 5B). The increased redox status (decreased GSH to GSSG ratio; increased cystine to cysteine ratio) in peritoneal fluids of HFD mice was accompanied by higher levels of TNFα with no corresponding changes in sTNFR1 levels, when compared with CD-fed counterparts (Table 1).
Figure 5. HFD induces systemic and local alterations in antioxidant/oxidant balance in recipient mice with lesions.

Levels of oxidants and antioxidants and their ratios in sera (A) and peritoneal fluids (B) of lesion-bearing mice fed CD or HFD are shown. Data are expressed as mean ± SEM and were obtained from n = 7 (CD) or 9 (HFD) mice. C, Levels of 8-OH-guanosine in DNA isolated from CD or HFD lesions. Data are expressed as mean ± SEM from n = 4 mice per diet group; *, P < .05 by Student's t test.
We also determined the levels of 8-OH-guanosine, the oxidized form of guanosine, as a biomarker of DNA damage in lesions as a function of diet. Lesions from mice fed HFD showed higher content of 8-OH-guanosine in DNA than those of CD-fed group (Figure 5C).
Discussion
In this study, we used a recently described immunocompetent mouse model of endometriosis (28) to address the contribution of high dietary fat intake from postweaning through early adulthood, on endometriosis development. In this model, minced endometrial tissues from Klf9 null mutant mice injected into the peritoneum of WT recipient mice resulted in 100% ectopic lesion formation. Because the only difference between recipients is the fat content of their diets after weaning, this dietary paradigm provides a relevant model to experimentally address the consequence of lifetime HFD intake, beginning at childhood and within the period of reproductive competence, to the pathogenesis of endometriosis. We found that recipient mice fed a HFD had a significant albeit modest increase in lesion numbers compared with CD littermates. Given that recipients were only fed HFD for a total of 9 weeks, with no significant differences in body weights at sac relative to those fed CD, and did not manifest systemic dysfunctions in ovarian steroid hormone and insulin production, our findings suggest that in women with normal reproductive cycles and insulin sensitivity, high dietary fat intake may constitute a risk factor for endometriosis progression.
Our findings established that HFD exposure increased inflammatory markers within the peritoneum (eg, TNFα) and ectopic lesions (eg, F4/80+ macrophages) in mice, findings that are consistent with the human disease. Elevated TNFα levels within the peritoneal fluid are commonly observed in endometriosis patients and are thought to contribute to angiogenesis and proliferation by stimulating cytokine and growth factor signals from resident macrophages and neutrophils (45, 46). In agreement with the latter, we found an increase in the numbers of areas with infiltrating macrophages in HFD relative to CD lesions. Further, we found by gene array analyses and confirmed by QPCR, that HFD lesions manifested greater gene expression of the cytokines Cxcl4, Cxcl12, and Il17a. Chemokine (C-X-C motif) ligand 4 (CXCL4), known for its role in wound healing and tumor formation through its regulation of angiogenesis and immune cell function (47), has been localized to infiltrating macrophages of endometriotic lesions (48). CXCL12, which is expressed by natural killer cells of deep infiltrating lesions, is considered to impair immune response and clearance of implants (49). Moreover, IL-17a, secreted in response to TNFα from peritoneal fluids, facilitates the proliferation of endometriotic stromal cells (50). IL-17a also induces chronic inflammation in endometrioma cells by triggering the production of proinflammatory cytokines and angiogenic factors (51) and stimulating neutrophil recruitment through growth-regulated oncogene-α (52). A recent study has implicated a central role for macrophage-directed inflammation in the progression of endometriosis in women (53). However, because the expression levels of other macrophage-related genes such as Il17b and Cd136 were decreased in ectopic lesions with HFD, further studies will be necessary to clarify whether, and if so how, HFD may regulate macrophage polarization to support lesion establishment and growth. Additionally, although we were unable to visually detect significant differences in angiogenesis and indeed, found no differences in the expression levels of 2 proangiogenic genes namely c-fos induced growth factor (Figf) and vascular endothelial growth factor receptor 1 (Flt1) (Supplemental Figure 1) in CD and HFD lesions, the proangiogenic effects of HFD on lesion progression constitute another important direction for future studies.
Another important finding of our study is the impairment of redox status caused by HFD in mice with ectopic implants. The loss of redox balance and methylation capacity, demonstrated by changes in serum and/or peritoneal levels of cysteine, cystine, Met, Hcy, and by the increased levels of the nitrosative stress biomarker 3′-nitrotyrosine with HFD, is concordant with the GSEA showing that HFD promotes reactive oxygen species-mediated metabolic processes (false discovery rate [FDR] = 0.189) in ectopic lesions (Table 2). Women with endometriosis display lower levels of GSH as well as increased levels of oxidative stress biomarkers in peritoneal fluids compared with healthy women (26), suggesting that these disturbances are contributory to disease development and progression. Our data did not show a similar decrease in peritoneal GSH levels, although GSH to GSSG ratio was significantly reduced with HFD. HFD also elicited perturbations in key enzymes essential for the synthesis of the antioxidant glutathione. In particular, HFD lesions had significantly reduced transcript levels for the Gclc, which is the rate-limiting enzyme for the first step in glutathione synthesis. Transcript levels for other enzymes in this pathway including Gsr and Gss, tended to be similarly diminished in HFD lesions, consistent with impairment of glutathione synthesis. Another antioxidant enzyme whose transcript was decreased in HFD lesions, Gpx1, is responsible for detoxification of hydrogen peroxide molecules; its expression was previously reported to be inversely associated with apoptosis and decreased in many cancer types (54). Increased oxidative stress is known to induce proliferation and to reduce apoptosis and thus, may confer a selective survival advantage to endometrotic lesions (55). This is consistent with our findings of increased Ki-67 (proliferation) and decreased TUNEL (apoptosis) staining within stromal areas of HFD lesions. The increased expression of Gpx3 with HFD may be a consequence of GPX3's role to counterbalance HFD's promotion of reactive oxygen species. Moreover, the significantly elevated levels of 8-OH-guanosine, indicative of increased DNA damage in HFD lesions, may reflect the elevated localized oxidative stress imposed by HFD, are consistent with the recent report that higher DNA damage measured as an increase in 8-OH-guanosine levels can be predictive of endometriosis progression (55), and suggest that HFD lesions may have defects in base-excision repair mechanisms, which have been attributed to transcriptional repression of the DNA damage response (56). Our results indicate that the increased oxidative stress and inflammatory pathways induced by HFD exposure may in tandem, create a potent feed-forward loop to maintain a prosurvival environment locally (peritoneum, lesion) and, systemically, to promote lesion establishment.
In this study, we show that HFD resulted in the loss of specific PGR and ESR isoform expression, namely Pgrb and Esr1 in ectopic lesions, whereas Esr2 expression was not perturbed by dietary type. In a previous study (28), we found that in lesions generated in WT recipients from endometria of donor WT and Klf9 null mice, Esr2 expression was increased concomitant with decreased Esr1 expression. These results are consistent with the specific contributions of ESR2 in disease establishment, mechanistically addressed in a recent study by O'Malley and coworkers (57) and raise the interesting possibility that an increase in lesion numbers by HFD may be dependent on the loss of stromal ESR1 induction of PGR-B expression. Interestingly, the reduction of total PGR in HFD lesions was not accompanied by increased expression of Wnt4 and Stc1, 2 genes which are negatively regulated by P4 (35). Because Wnt4 and Stc1 transcripts were previously shown to display higher levels in lesions concomitant with decreased levels of Pgra transcript (28), these results suggest that the loss of Pgrb isoform is closely aligned with HFD effects on lesion progression. Our findings that HFD lesions with lower PGR expression show reduced apoptosis and higher proliferation status are also consistent with P4 inhibition of proliferation and induction of apoptosis in cancer cells through negative modulation of reactive oxygen species (58). Inflammation is also well recognized to contribute to P4 resistance by down-regulating the expression of PGR (59). Furthermore, activation of proinflammatory transcription factors could compete for a limited pool of nuclear receptor coregulators and/or disrupt the interaction of PGR with critical transcriptional partners to diminish PGR transactivity (60). On the other hand, the link between loss of ESR1 and increase in lesion number in this study is less clear. Although it has been reported that ESR1 is subject to redox-dependent modifications of cysteine residues in the protein's DNA binding domain leading to loss of DNA-binding function (61), this effect is more reflected at the level of transcriptional activity, rather than of protein or transcripts. Estrogens have been shown to mediate oxidative DNA damage in breast cancer cells (62), and it is possible that induction by HFD of local estrogen synthesis by increasing Cyp19A1 (aromatase) expression in lesions may underlie the higher oxidative DNA damage in HFD lesions. Clearly, more studies are required to further dissect the prooxidant effects of HFD unrelated to systemic hormone levels.
In summary, we found that a higher dietary fat intake increased lesion numbers in an immunocompetent mouse model of endometriosis. HFD consumption, in the absence of significant weight gains, enhanced gene networks associated with inflammatory and oxidative stress pathways within systemic and local environments to promote lesion establishment. Our studies support a link between diet and endometriosis that is independent of overt obesity and insulin resistance, reinforce the potential value of maintaining a healthy diet early in life to minimize adult endometriosis risk, and raise the need for further mechanistic investigations in tandem with population-based studies to identify specific types of dietary fat with negative impact on the disease.
Acknowledgments
We thank Olekssandra Pavliv and Ariel Noble for technical assistance and members of Dr R. C. M. Simmen's laboratory for helpful discussions during the course of this work.
This work was supported by National Institutes of Health Grants RO1HD21961, R01CA136493, and UL1TR000039 and by the Arkansas Biosciences Institute Intramural Grant Program.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- BMI
- body mass index
- CD
- control diet
- E2
- estradiol
- ESR
- estrogen receptor
- FC
- fold change
- FDR
- false discovery rate
- Gpx1
- glutathione peroxidase 1
- GSEA
- gene set enrichment analysis
- GSH
- glutathione
- Hcy
- homocysteine
- HFD
- high-fat diet
- HPLC-ED
- HPLC with electrochemical detection
- IHC
- immunohistochemistry
- KLF9
- Krüppel-like factor 9
- Met
- methionine
- P4
- progesterone
- PGR
- P4 receptor
- PND
- postnatal day
- QPCR
- quantitative RT-PCR
- sTNFR1
- soluble TNF receptor 1
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick end labeling
- WT
- wild type.
References
- 1. Giudice LC, Kao LC. Endometriosis. Lancet. 2004;364:1789–1799. [DOI] [PubMed] [Google Scholar]
- 2. Buck Louis GM, Hediger ML, Peterson CM, et al. Incidence of endometriosis by study population and diagnostic method: the ENDO study. Fertil Steril. 2011;96:360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Simoens S, Dunselman G, Dirksen C, et al. The burden of endometriosis: costs and quality of life of women with endometriosis and treated in referral centres. Hum Reprod. 2012;27:1292–1299. [DOI] [PubMed] [Google Scholar]
- 4. Burney RO, Giudice LC. Pathogenesis and pathophysiology of endometriosis. Fertil Steril. 2012;8:511–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nyholt DR, Low SK, Anderson CA, et al. Genome-wide association meta-analysis identifies new endometriosis risk loci. Nat Genet. 2012;44:1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sinaii N, Cleary SD, Ballweg ML, Nieman LK, Stratton P. High rates of autoimmune and endocrine disorders, fibromyalgia, chronic fatigue syndrome and atopic diseases among women with endometriosis: a survey analysis. Hum Reprod. 2002;17:2715–2724. [DOI] [PubMed] [Google Scholar]
- 7. Fader AN, Arriba LN, Frasure HE, von Gruenigen VE. Endometrial cancer and obesity: epidemiology, biomarkers, prevention and survivorship. Gynecol Oncol. 2009;114:121–127. [DOI] [PubMed] [Google Scholar]
- 8. McPherson NO, Bell VG, Zander-Fox DL, et al. When two obese parents are worse than one! Impacts on embryo and fetal development. Am J Physiol Endocrinol Metab. 2015;309:E568–E581. [DOI] [PubMed] [Google Scholar]
- 9. Cardozo ER, Karmon AE, Gold J, Petrozza JC, Styer AK. Reproductive outcomes in oocyte donation cycles are associated with donor BMI. Hum Reprod. 2016;31:385–392. [DOI] [PubMed] [Google Scholar]
- 10. Bodnar LM, Parks WT, Perkins K, et al. Maternal prepregnancy obesity and cause-specific stillbirth. Am J Clin Nutr. 2015;102:858–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Darrow SL, Vena JE, Batt RE, Zielezny MA, Michalek AM, Selman S. Menstrual cycle characteristics and the risk of endometriosis. Epidemiology 193;4:135–142. [DOI] [PubMed] [Google Scholar]
- 12. Missmer SA, Hankinson SE, Spiegelman D, et al. Reproductive history and endometriosis among premenopausal women. Obstet Gynecol. 2004;104:965–974. [DOI] [PubMed] [Google Scholar]
- 13. Shah DK, Correia KF, Vitonis AF, Missmer SA. Body size and endometriosis results from 20 years of follow-up within the Nurses' Health Study II prospective cohort. Hum Reprod. 2013;28:1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McAllister JM, Legro RS, Modi BP, Strauss JF., 3rd Functional genomics of PCOS: from GWAS to molecular mechanisms. Trends Endocrinol Metab. 2015;26:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sampson JA. Peritoneal endometriosis due to the menstrual dissemination of endometrial tissues into the peritoneal cavity. Am J Obstet Gynecol. 1927;14:422–469. [Google Scholar]
- 16. de Oliveira R, Adami F, Mafra FA, Bianco B, Vilarino FL, Barbosa CP. Causes of endometriosis and prevalent infertility in patients undergoing laparoscopy without achieving pregnancy. Minerva Ginecol. 2015;67:289–296. [PubMed] [Google Scholar]
- 17. Nava-González EJ, de la Garza-Casas YE, Salazar-Montalvo RG, Gallegos-Cabriales EC. [Relationship among anthropometric and gluco-metabolic parameters, bone mineral density and endometriosis]. Rev Med Inst Mex Seguro Soc. 2013;51:522–531. [PubMed] [Google Scholar]
- 18. Nagle CM, Bell TA, Purdie DM, et al. Relative weight at ages 10 and 16 years and risk of endometriosis: a case-control analysis. Hum Reprod. 2009;24:1501–1506. [DOI] [PubMed] [Google Scholar]
- 19. Parazzini F, Chiaffarino F, Surace M, et al. Selected food intake and risk of endometriosis. Hum Reprod. 2004;19:1755–1759. [DOI] [PubMed] [Google Scholar]
- 20. Covens AL, Christopher P, Casper RF. The effect of dietary supplementation with fish oil fatty acids on surgically induced endometriosis in the rabbit. Fertil Steril. 1988;49:698–703. [DOI] [PubMed] [Google Scholar]
- 21. Harris HR, Chavarro JE, Malspeis S, Willett WC, Missmer SA. Dairy-food, calcium, magnesium, and vitamin D intake and endometriosis: a prospective cohort study. Am J Epidemiol. 2013;177:420–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Missmer SA, Chavarro JE, Malspeis S, et al. A prospective study of dietary fat consumption and endometriosis risk. Hum Reprod. 2010;25:1528–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baer DJ, Judd JT, Clevidence BA, Tracy RP. Dietary fatty acids affect plasma markers of inflammation in healthy men fed controlled diets a randomized crossover study Am J Clin Nutr. 2004;79:969–973. [DOI] [PubMed] [Google Scholar]
- 24. Attaman JA, Stanic AK, Kim M, Lynch MP, Rueda BR, Styer AK. The anti-inflammatory impact of omega-3 polyunsaturated fatty acids during the establishment of endometriosis-like lesions. Am J Reprod Immunol. 2014;72:392–402. [DOI] [PubMed] [Google Scholar]
- 25. Bedaiwy MA, Falcone T, Sharma RK, et al. Prediction of endometriosis with serum and peritoneal fluid markers: a prospective controlled trial. Hum Reprod. 2002;17:426–431. [DOI] [PubMed] [Google Scholar]
- 26. Mier-Cabrera J, Aburto-Soto T, Burrola-Méndez S, et al. Women with endometriosis improved their peripheral antioxidant markers after the application of a high antioxidant diet. Reprod Biol Endocrinol. 2009;28:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Carvalho LF, Samadder AN, Agarwal A, Fernandes LF, Abrão MS. Oxidative stress biomarkers in patients with endometriosis: systematic review. Arch Gynecol Obstet. 2012;286:1033–1040. [DOI] [PubMed] [Google Scholar]
- 28. Heard ME, Simmons CD, Simmen FA, Simmen RC. Krüppel-like factor 9 deficiency in uterine endometrial cells promotes ectopic lesion establishment associated with activated notch and hedgehog signaling in a mouse model of endometriosis Endocrinology. 2014;155:1532–1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Morita M, Kobayashi A, Yamashita T, et al. Functional analysis of basic transcription element binding protein by gene targeting technology. Mol Cell Biol. 2003;23:2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang D, Zhang XL, Michel FJ, Blum JL, Simmen FA, Simmen RC. Direct interaction of the Krüppel-like family (Klf) member, Bteb1, and PR mediates progesterone-responsive gene expression in endometrial epithelial cells. Endocrinology. 2002;143:162–173. [DOI] [PubMed] [Google Scholar]
- 31. Zhang XL, Zhang D, Michel FJ, Blum JL, Simmen FA, Simmen RC. Selective interactions of Kruppel-like factor 9/basic transcription element-binding protein with progesterone receptor isoforms A and B determine transcriptional activity of progesterone-responsive genes in endometrial epithelial cells. J Biol Chem. 2003;278:21474–21482. [DOI] [PubMed] [Google Scholar]
- 32. Velarde MC, Zeng Z, McQuown JR, Simmen FA, Simmen RC. Kruppel-like factor 9 is a negative regulator of ligand-dependent estrogen receptor α signaling in Ishikawa endometrial adenocarcinoma cells. Mol Endocrinol. 2007;21:2988–3001. [DOI] [PubMed] [Google Scholar]
- 33. Simmen RC, Eason RR, McQuown JR, et al. Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Krüppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J Biol Chem 2004;279:29286–29294. [DOI] [PubMed] [Google Scholar]
- 34. Velarde MC, Geng Y, Eason RR, Simmen FA, Simmen RC. Null mutation of Krüppel-like factor9/basic transcription element binding protein-1 alters peri-implantation uterine development in mice. Biol Reprod. 2005;73:472–481. [DOI] [PubMed] [Google Scholar]
- 35. Pabona JM, Simmen FA, Nikiforov MA, et al. Krüppel-like factor 9 and progesterone receptor coregulation of decidualizing endometrial stromal cells: implications for the pathogenesis of endometriosis. J Clin Endocrinol Metab. 2012;97:E376–E392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wilson CR, Tran MK, Salazar KL, Young ME, Taegtmeyer H. Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem J. 2007;406:457–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002; 3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Melnyk S, Fuchs GJ, Schulz E, et al. Metabolic imbalance associated with methylation dysregulation and oxidative damage in children with autism. J Autism Dev Disord. 2012;42:367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Friso S, Choi SW, Dolnikowski GG, Selhub J. A method to assess genomic DNA methylation using high-performance liquid chromatography/electrospray ionization mass spectrometry. Anal Chem. 2002;74:4526–4531. [DOI] [PubMed] [Google Scholar]
- 41. Hadi T, Bardou M, Mace G, et al. Glutathione prevents preterm parturition and fetal death by targeting macrophage-induced reactive oxygen species production in the myometrium. FASEB J. 2015;29:2653–2666. [DOI] [PubMed] [Google Scholar]
- 42. Rakhila H, Girard K, Leboeuf M, Lemyre M, Akoum A. Macrophage migration inhibitory factor is involved in ectopic endometrial tissue growth and peritoneal-endometrial tissue interaction in vivo: a plausible link to endometriosis development. PLoS One. 2014;9:e110434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Räisänen SR, Lehenkari P, Tasanen M, Rahkila P, Härkönen PL, Väänänen HK. Carbonic anhydrase III protects cells from hydrogen peroxide-induced apoptosis. FASEB J. 1999;13:513–522. [DOI] [PubMed] [Google Scholar]
- 44. Wang H, Luo K, Tan LZ, et al. p53-induced gene 3 mediates cell death induced by glutathione peroxidase 3. J Biol Chem. 2012;287:16890–16902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chae SJ, Kim H, Jee BC, Suh CS, Kim SH, Kim JG. Tumor necrosis factor (TNF)-TNF receptor gene polymorphisms and their serum levels in Korean women with endometriosis. Am J Reprod Immunol. 2008;60:432–439. [DOI] [PubMed] [Google Scholar]
- 46. Lin YJ, Lai MD, Lei HY, Wing LY. Neutrophils and macrophages promote angiogenesis in the early stage of endometriosis in a mouse model. Endocrinology. 2006;147:1278–1286. [DOI] [PubMed] [Google Scholar]
- 47. Pilatova K, Greplova K, Demlova R, Bencsikova B, Klement GL, Zdrazilova-Dubska L. Role of platelet chemokines, PF-4 and CTAP-III, in cancer biology. J Hematol Oncol. 2013;6:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Furuya M, Tanaka R, Miyagi E, et al. Impaired CXCL4 expression in tumor-associated macrophages (TAMs) of ovarian cancers arising in endometriosis. Cancer Biol Ther. 2012;13:671–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bellelis P, Barbeiro DF, Rizzo LV, Baracat EC, Abrão MS, Podgaec S. Transcriptional changes in the expression of chemokines related to natural killer and T-regulatory cells in patients with deep infiltrative endometriosis Fertil Steril. 2013;99:1987–1993. [DOI] [PubMed] [Google Scholar]
- 50. Hirata T, Osuga Y, Yoshino O, et al. Development of an experimental model of endometriosis using mice that ubiquitously express green fluorescent protein. Hum Reprod. 2006;20:2092–2096. [DOI] [PubMed] [Google Scholar]
- 51. Ahn SH, Edwards AK, Singh SS, Young SL, Lessey BA, Tayade C. IL-17A contributes to the pathogenesis of endometriosis by triggering proinflammatory cytokines and angiogenic growth factors. J Immunol. 2015;195:2591–2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Takamura M, Osuga Y, Izumi G, et al. Interleukin-17A is present in neutrophils in endometrioma and stimulates the secretion of growth-regulated oncogene-α (Gro-α) from endometrioma stromal cells. Fertil Steril. 2012;98:1218–1224.e1–2. [DOI] [PubMed] [Google Scholar]
- 53. Beste MT, Pfäffle-Doyle N, Prentice EA, et al. Molecular network analysis of endometriosis reveals a role for c-Jun-regulated macrophage activation. Sci Transl Med. 2014;6:222ra16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Brigelius-Flohé R, Kipp AP. Physiological functions of GPx2 and its role in inflammation-triggered carcinogenesis. Ann NY Acad Sci. 2012;1259:19–25. [DOI] [PubMed] [Google Scholar]
- 55. Carvalho LF, Abrão MS, Biscotti C, Sharma R, Nutter B, Falcone T. Oxidative cell injury as a predictor of endometriosis progression. Reprod Sci. 2013;20:688–698. [DOI] [PubMed] [Google Scholar]
- 56. Kao J, Rosenstein BS, Peters S, Milano MT, Kron SJ. Cellular response to DNA damage. Ann NY Acad Sci. 2005;1066:243–258. [DOI] [PubMed] [Google Scholar]
- 57. Han SJ, Jung SY, Wu SP, et al. Estrogen receptor β modulates apoptosis complexes and the inflammasome to drive the pathogenesis of endometriosis. Cell. 2015;163:960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Nguyen H, Syed V. Progesterone inhibits growth and induces apoptosis in cancer cells through modulation of reactive oxygen species. Gynecol Endocrinol. 2011;27:830–836. [DOI] [PubMed] [Google Scholar]
- 59. Guzeloglu-Kayisli O, Kayisli UA, Semerci N, et al. Mechanisms of chorioamnionitis-associated preterm birth: interleukin-1β inhibits progesterone receptor expression in decidual cells. J Pathol. 2015;237:423–434. [DOI] [PubMed] [Google Scholar]
- 60. Zelenko Z, Aghajanova L, Irwin JC, Giudice LC. Nuclear receptor, coregulator signaling, and chromatin remodeling pathways suggest involvement of the epigenome in the steroid hormone response of endometrium and abnormalities in endometriosis. Reprod Sci. 2012;19:152–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Atsriku C, Benz CC, Scott GK, Gibson BW, Baldwin MA. Quantification of cysteine oxidation in human estrogen receptor by mass spectrometry. Anal Chem. 2007;79:3083–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mobley JA, Brueggemeier RW. Estrogen receptor-mediated regulation of oxidative stress and DNA damage in breast cancer. Carcinogenesis. 2004;25:3–9. [DOI] [PubMed] [Google Scholar]