

Abstract
The objective of this study is to present a mathematical model which can describe the spatiotemporal progression of cerebral ischaemia and predict magnetic resonance observables including the apparent diffusion coefficient (ADC) of water and transverse relaxation time T2. This is motivated by the sensitivity of the ADC to the location of cerebral ischaemia and T2 to its time-course, and that it has thus far proven challenging to relate observations of changes in these MR parameters to stroke timing, which is of considerable importance in making treatment choices in clinics. Our mathematical model, called the cytotoxic oedema/dissociation (CED) model, is based on the transit of water from the extra- to the intra-cellular environment (cytotoxic oedema) and concomitant degradation of supramacromolecular and macromolecular structures (such as microtubules and the cytoskeleton). It explains experimental observations of ADC and T2, as well as identifying the rate of spread of effects of ischaemia through a tissue as a dominant system parameter. The model brings the direct extraction of the timing of ischaemic stroke from quantitative MRI closer to reality, as well as providing insight on ischaemia pathology by imaging in general. We anticipate that this may improve patient access to thrombolytic treatment as a future application.
Keywords: MRI, T2 relaxation, apparent diffusion coefficient, acute ischaemia
Introduction
MRI has the potential to make highly detailed and objective assessments of the brain in patients suffering ischaemic stroke.1,2 However, there yet exists no widely accepted method for the inversion of MRI observations to parameters describing ischaemia in a clinical sense, including the time of onset. It is therefore desirable to have a framework for modelling ischaemia that can be fitted to MRI data, as well as predict the spatio-temporal progress of ischaemia, in the time window for which patients are eligible for current drug protocols.1 For acute stroke, the time of onset is one of the key factors in decision-making for potentially beneficial and safe administration of thrombolytic therapy.3,4 However, as many as 30% of acute stroke patients lack the data allowing exact onset time to be determined at the time of admission to emergency medicine,5 thus prompting need for an objective means for timing of stroke. It is well established that absolute relaxation times of brain water change in a time dependent manner in focal brain ischaemia.6–9
Apparent diffusion coefficient (ADC) mapping (the voxel-wise fitting of ADC values from a set of diffusion-weighted images) has for some time been used as the gold standard for locating lesions caused by ischaemia. The ADC for water is highly sensitive to the location of ischaemia, reducing rapidly after onset, 10–13 but carries little information on the time of onset, stabilising rather quickly after onset. In hyperacute ischaemia, a shortened T2 is detected for up to 60 min after vascular insult,14 followed by an increase for the first 5–6 h of insult. Recent preclinical15,16 and clinical17 studies have demonstrated that the time course of T2 relaxation times in diffusion-positive stroke lesions can be described heuristically by linear15 or quadratic17 functions. Indeed, the potential of the T2 MRI signal to aid in clinical decision-making for thrombolytic therapy has been clinically trialled using T2-FLAIR/ADC mismatch as a proxy of stroke duration.18 While this approach has acceptable specificity for stroke timing, it suffers from rather low sensitivity for patient stratification.19 The reason for the low sensitivity is currently researched; however, our rodent study suggested that absolute relaxation times, but not weighted signal intensities, are able to provide accurate timing for ischaemia.16
A major difficulty in using MRI to assess ischaemia is relating its many observables to parameters of direct clinical utility. The complex relationships between MR acquisition parameters and ischaemia “system” parameters are not understood, yet at the very least the dominant system parameters should be identified before realistic attempts to make objective assessments of ischaemia from quantitative MRI data.9 There currently exists no combined mathematical model capable of explaining spatio-temporal changes in both ADC and T2 of the type observable in MRI experiments, though there has been useful progress. In particular, a model based upon oxygen metabolism has been able to explain reductions in T220 but was not spatio-temporal, whilst a recent approach used diffeomorphic mapping to explain spatiotemporal changes in diffusion/perfusion mismatch lesion morphology but not ADC or T2 values.21 An approach to determine the distinct penumbra and lesion, based on integrating various physiological processes, has also been proposed, which, although not constructed for the prediction of MRI parameters, is quite comprehensive and is able to model cytotoxic oedema.22
Part of the difficulty is that the biophysical mechanisms for the changes in ADC and T2 remain ambiguous from experimental findings.10,23,24 Early work identified a redistribution of water between intra- and extracellular spaces as a determinant of ADC in ischaemia.25–29 Theoretical and experimental models for ADC have considered changes in cell membrane permeability and intra/extracellular water distribution,24,30,31 restricted diffusion,32–34 and extracellular tortuosity.30,32,35 An emerging picture is that a redistribution of water from the extra- to intracellular space, although insufficient to account for the ADC changes in the acute phase of ischaemia alone, may do so, if concomitant changes in either the intracellular or extracellular environment are accounted for.29 However, to reiterate, no such model has yet carried its assumptions across to the simultaneous prediction of T2 from the same principles.
The objectives of this study were three-fold: (i) to develop a spatiotemporal model for ischaemia which simultaneously predicts T2 and ADC changes; (ii) to compare its predictions with measurements made in a rat model of ischaemia, and (iii) to establish an automated procedure to delineate volumes with abnormal T2 values inside the ischaemic lesion for an unambiguous assessment from combined diffusion and absolute T2 data. To the latter end, we have used quantitative diffusion and T2 MRI data from rat stroke model. Clinical applicability of the approach is discussed.
Materials and methods
Animal model for stroke and MRI data acquisition
Male Wistar rats (n = 5, weight 200–300 g) were exposed to permanent middle cerebral artery occlusion (MCAo) according to Longa et al.36 The occluding thread was left in place for the duration of the MRI scanning and the animal was sacrificed thereafter by decapitation in deep isoflurane anaesthesia. All animal procedures were approved by the Committee for the Welfare of Laboratory Animals of the University of Eastern Finland and the Provincial Government of Kuopio, Finland, and conducted in accordance with the guidelines set by the European Community Council Directives 86/609/EEC.
Scans were conducted using a horizontal 9.4 T Agilent MRI scanner (Agilent, Palo Alto, CA, USA) equipped with an actively decoupled linear volume transmitter and quadrature receiver coil pair (RAPID Biomedical GmbH, Rimpar, Germany). Each “measurement” comprised a three-plane (orthogonal) diffusion-weighted sequence (b-values = 0, 400, 1040 mm2 s−1; TE = 36 ms, TR = 2000 ms) and a multi-echo spin-echo sequence (12 echoes 10 ms inter-echo spacing, starting at 10 ms, TR = 2000 ms). Twelve slices were imaged with the following parameters: 0.5 mm gap, slice thickness = 1 mm, FOV = 2.56 cm × 2.56 cm was covered by 128 × 256 points. Such measurements, each taking ∼20 min, were made at times commencing 60, 120, 180, and 240 min after MCAo.
Image processing for rat data
To increase the confidence of parameter estimates, all data prior to fitting were filtered with a squared Hamming window function in the phase and frequency encoded dimensions in k-space. T2 fitting was performed by a mono-exponential fit in a logarithmic space, in which the system was solved on a voxel-wise basis using linear least squares by the Moore-Penrose pseudo-inverse. A bi-exponential function was also fitted, but f-tests at each voxel revealed virtually no voxels within the image for which the additional parameters could be justified (see Supplementary material). An image of p-values for these f-tests is provided in the SI.
Trace of diffusion tensor (ADC) maps were calculated from diffusion-weighted data by the analytical solution to the diffusion equation.
In order to define an ADC lesion, it was found to be easier to use reciprocal ADC values, such that the lesion is bright with high values akin to T2 maps later on during MCAo. Such an ADC map is proportional to the translational correlation time (and its cube the rotational correlation time). Lesion detection proceeds by selecting voxels which have values significantly higher than average in the reciprocal-ADC map, as well as fulfilling any additional filters in use (such as excluding unrealistic T2 values likely to be CSF, for instance). By “higher than average”, we found that 1 standard deviation above the median, when using reciprocal ADC values, was very effective. However, with such large datasets, equally stable results were obtainable using the mean or mode to define the “average”.
The “lesion” is then stored as a binary mask. This will typically contain several distinct clusters of non-zero voxels, and even single voxels due to noise in the data. It is therefore filtered to remove clusters of voxels smaller than a prescribed cluster size, in order that clusters too small to realistically be ischaemic lesions are removed. The smallest acceptable lesion is a user-supplied parameter.
To complement average T2 in the ADC lesion, we introduce the parameter f2, given by
| (1) |
where NHigh is the number of high T2 voxels in the lesion, NLow is the number of low T2 voxels in the lesion, and NLesion is the total number of voxels in the ADC lesion. A T2 observation is counted as “high” if its value is more than one half-width at half-maximum higher than the modal T2, and “low” if less than one half-width at half-maximum below the modal T2 (this definition being numerically stable). Numbers of voxels can of course be replaced with volumes and the result is unchanged. The parameter f2 offers advantage over average T2 for lesion assessment being relatively insensitive to the B0 field variation. In addition, the subtraction of the NLow term allows for decreases in relaxation times due to ischaemia or other pathology. Its values are between −1 and 1.
All image pre-processing, computation of parametric maps, as well as lesion detection were conducted by software written in-house using the Matlab programming environment (Mathworks, Natick, MA, USA) unless otherwise specified.
Model for MRI parameters in ischaemic brain parenchyma
Our spatio-temporal model may be qualitatively summarised as follows: upon ischaemia with collapse in energy state, water moves from the extra- to the intracellular environment. However, as mentioned already, this is insufficient to explain either ADC or T2 changes. We therefore invoke, in the intracellular environment, a two-state model for water in which it exists in exchange between a “free” state and one in which it is bound to macromolecules and supra-macromolecular structures (such as microtubules and other cytoskeletal structures). The ADC of the latter state is assumed to increase with time after insult due to dissociation and/or proteolysis.29 The transit of water from the extra- to intracellular space may be interpreted as cytotoxic edema, whilst the concomitant disassembly of supra-macromolecular structures, collectively a dissociative process, gives rise to the name “cytotoxic oedema/dissociation” (CED) model. This is summarised in Figure 1, along with the terms that are used in its mathematical formulation. We do not consider vasogenic oedema, involving disruption of the blood-brain-barrier, in our model.
Figure 1.

The “Cytotoxic oEdema Dissociation” (CED) model for ischemia. Water is represented by red dots, the entire system by the black line, partitioned into the intra- and extra-cellular spaces. Macromolecules and supra-macromolecular structures are represented by blue objects. The various symbols of the model are indicated; KF(t) is the partition coefficient for water across the cell membrane with time dependence encoded by δ. KI is the (time-independent) equilibrium constant between bound and free intracellular water. DEF and DIF are the time-independent diffusion coefficients for the free extra- and free intracellular states, respectively, whilst DIB(t) is the time-dependent diffusion coefficient for the bound intracellular state with time dependence encoded by ɛ.
More rigorously, we consider three “pools” of water; the free extracellular (“EF”) water, the free intracellular (“IF”) water and the bound intracellular (“IB”) water. The EF and IF states exchange across in plasma membrane with time-dependent equilibrium constants KF, whilst the IF and IB states are in exchange with equilibrium constant KI. To obtain an MR parameter, we model magnetic moments using the Bloch–McConnell–Torrey equation for transverse magnetisation, given by
| (2) |
Here, Ω is a diagonal matrix of chemical shifts, R2 a diagonal matrix of transverse relaxation rate constants, D a diagonal matrix of (isotropic) diffusion coefficients, K the kinetic matrix, i the imaginary unit, γ the gyromagnetic ratio for the relevant nucleus (in this case 1H), G the magnetic field gradient, X a Cartesian coordinate vector, and the complex-valued transverse magnetisation at time t and position X. The field gradient and diffusion term is relevant only for ADC modelling. If there is no field gradient applied, then we can ignore the effect of translational diffusion. For numerical simulations, the time-evolution can be evaluated by means of
| (3) |
where L represents the time-evolution operator appropriate for the simulation, , and in the second line U and V are the eigenvector and eigenvalue matrices of the time evolution operator, respectively, such that U diagonalises . The time-zero magnetisation is given by the equilibrium water fraction vector . The T2 or ADC, as appropriate, is then the least-squares fit to the observable signal
| (4) |
In the case of ADC, typically we used only two b-values in the simulations, rather than the entire time-domain signal, in which case the ADC has the simple and well known form
| (5) |
where the b-value has the usual definition
| (6) |
The exchanging system is governed by the set of differential equations
| (7) |
where K is the kinetic matrix describing the system and is the vector-valued fractional concentration, ordered fIB, fIF, fEF. They satisfy the identity
| (8) |
The EF and IF states exchange across the plasma membrane, whilst the IF and IB states exchange intracellularly. Defining kmn as the reaction rate constant between states m and n, we can then write the rate matrix K as
| (9) |
For example, is the first-order rate constant for the exchange from the IB state to the IF state. We define the following equilibrium constants
| (10) |
| (11) |
We model cytotoxic oedema by making the parameter KF, the equilibrium constant for partition of water between the intra- and extracellular spaces, a function of time. We use the heuristic expression
| (12) |
where δ is the time constant for water uptake by cells, and and refer to the values of KF in the non-ischaemic state and at the end of the acute phase, respectively.
Changes in the intracellular ADC for the bound state, DIB, are modelled by a similar heuristic function
| (13) |
where ɛ is the time constant for the changes in intracellular diffusion coefficient due to reduced averaged macromolecular size or other processes, again with its values in the non-ischaemic state and at the end of the acute phase required. The value of is then calculated from using the expression for dipolar relaxation
| (14) |
where d is the intramolecular dipolar coupling constant for the water protons and the term is related to through the Stokes–Einstein relations and can be written as
| (15) |
Here, kB is Botlzmann constant, T is the absolute temperature, and η is the solvent viscosity.
Finally, as a model of the spread of ischaemia through tissue, we assume the lesion to be spherical, spreading with kinetics described by
| (16) |
where L(t) is the radius of the lesion at time t, α is the final radius of the lesion, and β is the time constant for the spread of ischaemia. The total volume of a spherical lesion can then be expressed as
| (17) |
The simulations were designed and conducted using software written in-house in the Matlab programming environment. Further details can be found in the Supplementary material (equations (S1)-(S27)).
Results
MRI changes in rat MCAo
The ADC lesion, with T2 segmentation, along with the other images acquired at several time points for a typical rat brain which has undergone MCAo is shown in Figure 2. The ADC lesion detected by the current method is a realistic and well-defined volume. The ADC lesion expands with time, as does the fraction of elevated T2 voxels (shown in red in Figure 2(a), (f), (k), and (p)). It is clear that a considerable amount of tissue within the lesion retains a T2 relaxation signature characteristic of healthy tissue. Growth of high-T2 area within the ADC lesion has a time dependence associated with it, which may reflect a transition to irreversible ischaemic damage.6,8,9 Inspection of the T2-weighted images shows that the T2 segmentation is not merely a separation of tissue types (such as gray and white matter), though this may come into play to some extent in other species.
Figure 2.
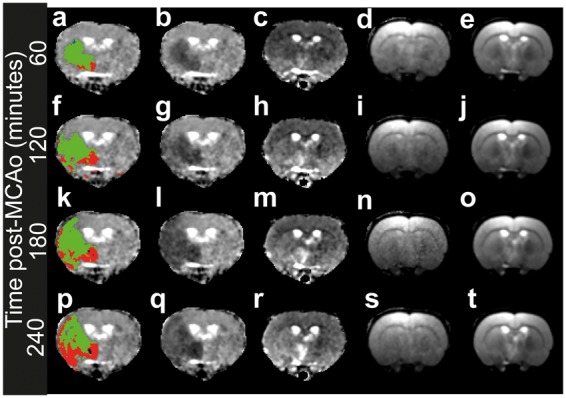
Example of lesion detection and T2 segmentation over time in rat brain. The MRI data in rows were recorded at different time points, whilst columns are different image types. Row 1 (a–e), 60 min post-MCAO, row 2 (f–j), 120 min post-MCAO, row 3 (k–o) 180 min post-MCAO, row 4 (p–t) 240 min post-MCAO. The first column shows the ADC map with the lesion shown. Green denotes regions with “normal” T2, red with elevated T2, blue (apparent only in a) reduced T2 (see text). Column 2 shows the ADC map alone, column 3 shows the T2 map, column 4 the I(0) map from T2 fitting, and column 5 the T2-weighted images.
The T2 kinetics of ischaemia are summarised for several different animals (Figure 3). It is striking that lesions expand into healthy-appearing regions at very different rates, and that this is apparently manifested in the T2 absolute percentage difference (APD) between voxels in the ADC lesion and those outside, and likewise f2 (additional figures are provided in the Supplementary material). The lesion volume, despite the aspherical nature of the lesions, could be described by equation (17), enabling extraction of effective ischaemic spread time constants and radii (which are later compared to theoretical predictions of the CED model).
Figure 3.
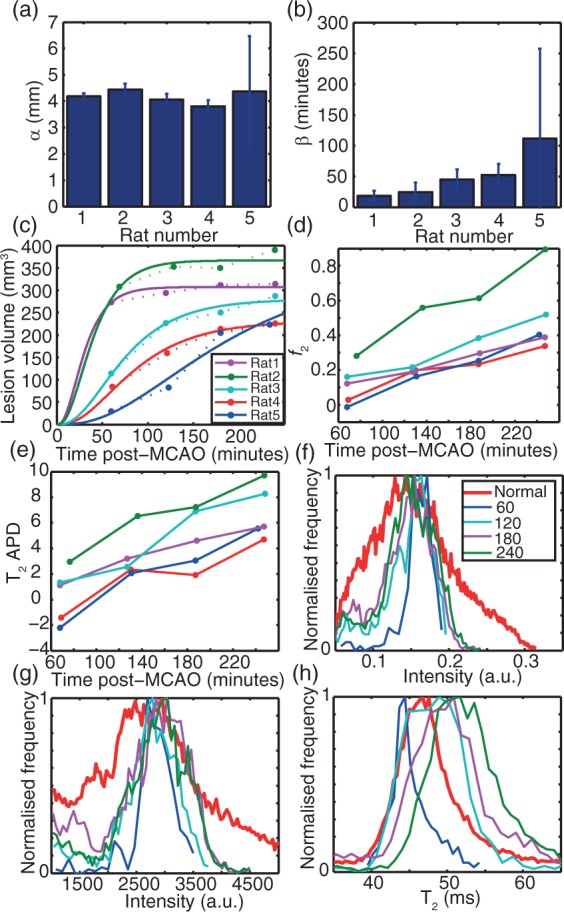
Time dependence of MRI parameters relating to ischaemia in rat MCAo. (a–e) findings for all animals; (f–h) detailed findings for a typical animal (Rat 3, f–h all use the legend as in f). (a,b) Fitted effective lesion radii and time constants, respectively, using equation (16), showing 95% confidence intervals on fitted parameters; (c) lesion volume by automated detection including the fit using the parameters in (a and b) and equation (16). Solid lines indicate the fit, dotted lines connect points for visual guidance; (d) f2 for all animals plotted separately; (e) absolute percentage difference (APD) for T2 between lesion and non-lesion for all animals. Panels (c) to (e) share the legend shown in (c). (f) In a typical animal, the distribution of I(0) derived from T2 fitting as a function of time; (g) Echo-summed T2-weighted signal intensities obtained by summing all 12 echoes with TEs ranging from 10 ms to 120 ms; (h) absolute T2 distribution. In (f) to (h), the “normal” reference histogram (red) is derived from all non-lesion voxels for the 60-min post-MCAO scan.
The f2 parameter is very stable across all animals (Figure 3d). Comparison to APD between average lesion and non-lesion T2 (Figure 3e) shows that f2 kinetics were better separated between animals. We therefore surmise that f2 is a potentially sensitive marker of the time dependence of ischaemia, more numerically stable than T2 APD.
The change in T2 distribution shows a progression towards higher values as time elapses, with approximately normal distributions but with increasing widths. T2 distribution therefore becomes wider as time progresses, a manifestation of the differential kinetics of ischaemia at different spatial coordinates (Figure 3h). Such clear time dependence is not apparent in the I(0) (Figure 3f) or T2-weighted intensities (Figure 3g).
Simulations of MRI parameter changes in ischaemia using the CED model
The predictions of the three-state CED model for brain tissue within ischaemic ADC lesion, where T2 changes take place within minutes of insult, are shown during the first 30 min after onset (Figure 4). In all the simulations we present, a lesion radius (α) of 3.5 mm is used, motivated by the experimental data (Figure 3a), with the ischaemic spread time constant (β) having values from 10 to 180 min, again in the range of our experimental findings (Figure 3b). Further details of values of simulation parameters can be found in the Discussion section. In the Supplementary material, video simulations are also provided. At an individual voxel, for argument’s sake the site of onset, the time course of the ADC is predicted to comprise a rapid drop initially stabilising at the low level. By contrast, the T2 first drops, then increases monotonically, eventually rising well above the baseline value. The timescale on which cells increase permeability to water, described by the parameter δ, was influential only in the first 30 min after onset of ischaemia (Figure 4). Thereafter, over at least an order of magnitude, the model was stable to the value of δ.
Figure 4.
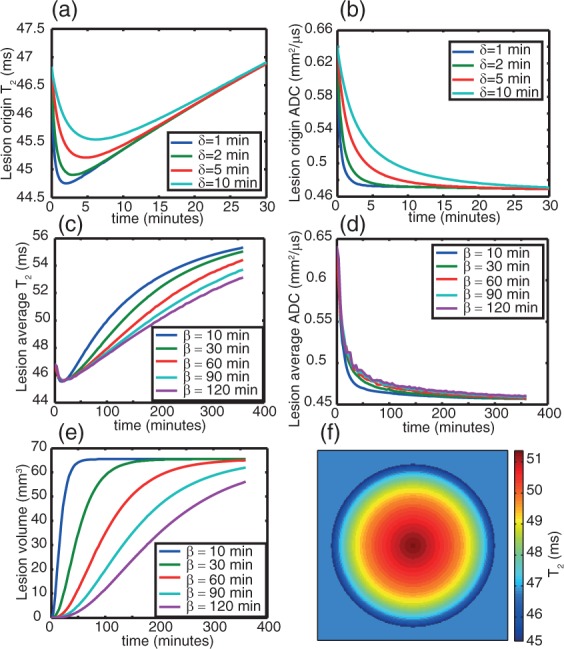
Simulation of the time-course of T2 and ADC using the CED model. Panels (a) and (b) show the early time dependence of T2 (a) and ADC (b) respectively, demonstrating different behaviour for different oedema timescales (given by δ). Panels (c) and (d) shows T2 (c) and ADC (d) kinetics over a longer timescale, for a fixed value of δ = 2 min but different values of β (the lesion growth coefficient), which is the main determinant of T2 and ADC kinetics after 30 min. Panel (e) shows the lesion volume kinetics for different β values; panel (f) shows the heterogeneity of T2 within a simulated circular lesion 2 h after onset at β = 60 min.
It is common practice to report the average ADC or T2 (or any other parameter) over a lesion, rather than values recorded at any particular voxel. For this reason, we have also simulated the time-course of the average (i.e. over an entire lesion) ADC, T2, I(0), sum-of-echoes intensities and I(TEoptimal), where I(0) is the signal intensity before any T2 decay and TEoptimal denotes the TE calculated to give optimal contrast between healthy and ischaemic tissue (this TE lies between that of the healthy and ischemic tissue and is ∼50 ms at 9.4T). These results are shown in Figure 4c-f, where the heterogeneity of T2 within the lesion is also simulated (Figure 4f). From Figure 4(f), we can see that an average parameter over a lesion is a complex term, dependent on the entire history of the lesion, discussed further in the Supplementary material (equations (S26) to (S27), Figures S1–S2).
The “average” ADC kinetics are, unsurprisingly, similar to those at a single coordinate and in agreement with the experimental findings here and by others.8,9 The predictions for the T2 kinetics both at a single coordinate and across an entire lesion are also consistent with experiment; initially, the T2 drops, then increases again monotonically after the minimum has been passed. The shape of this function, under the assumption of a spherical, cylindrical or circular model for the lesion morphology, depended most strongly upon the parameter β, describing the rate of lesion growth. The prediction is in good agreement with the experimental finding that the T2 increases can be heuristically described with a linear or quadratic function, though rigorously it is neither. Interestingly, the T2 kinetics at different β values, when sampled at times longer than 60 min, are approximately parallel lines, very similar to the experimental findings (Figure 3 d and e), such that the rate of lesion growth, that is, the rate at which effects of ischaemia extends throughout a tissue, may be able to explain the different time courses of T2 in the different test animals. In the case of ADC, however, this parameter made almost no difference. These simulations are shown in Figure 4.
Combining the CED model with the lesion detection method
We next used a set of T2-weighted and diffusion-weighted scans measured for a control rat to simulate an ischaemic stroke with the CED model, including a realistic noise level measured experimentally (the SD of signal intensity in an ROI placed in a single tissue). Using these detailed simulations, we sought to detect the simulated lesion with the approach described in the Methods section as a means of both validating the detection approach and examining the validity of the model under the most realistic circumstances. The simulated lesion was spherical with known size and position, so that the accuracy of its “experimental” determination could be assessed.
Figures 5 and 6 show the results from these simulations, which should be compared to Figures 2 and 3. The lesion masks in Figure 5 are very close to the simulated lesions, with only a few voxels spuriously missed or erroneously included, a finding expected in the presence of noise. This is an encouraging sign that the approach to automated detection of lesions is robust. Figure 6 demonstrates simultaneously the robustness of the detection method and accuracy of the model; the changes in f2 and APD as a function of lesion growth are very similar to that seen experimentally. The distributions of T2 with time are also similar to the experimental observations; one hour after onset of ischaemia, the T2 distribution of the lesion is rather narrow, before becoming broader but longer on average, then contracting in width again. The α and β input parameters are reproduced well by the automated lesion detection when equation (17) is fitted to the resulting lesion volume data, as seen in Figure 6(b) to (d).
Figure 5.
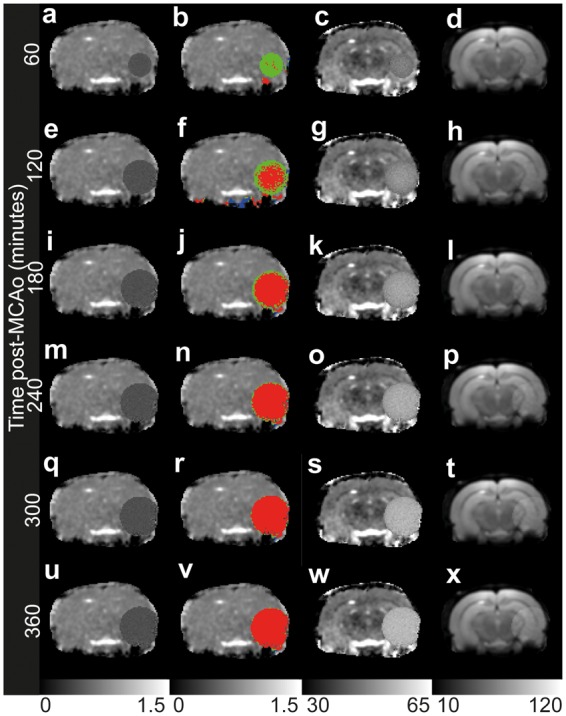
Lesions simulated in diffusion-weighted and T2-weighted data from a control rat and detected using the automated procedure given in the Methods section at different times post-onset. Panels (a, e, i, m, q, u): ADC maps with scale bar in mm2/µs. Panels (b, f, j, n, r, v): ADC maps with automatically detected lesions, coloured by proximity of T2 to healthy values (for details see Figure 1 legend) with scale bar in mm2/µs. Panels (c, g, l, o, s, w): T2 maps with scale bar in ms. Panels (d, h, k, p, t, x): T2-weighted maps at a TE of 50 ms, calculated as the optimum TE, with scale bar in arbitrary units.
Figure 6.
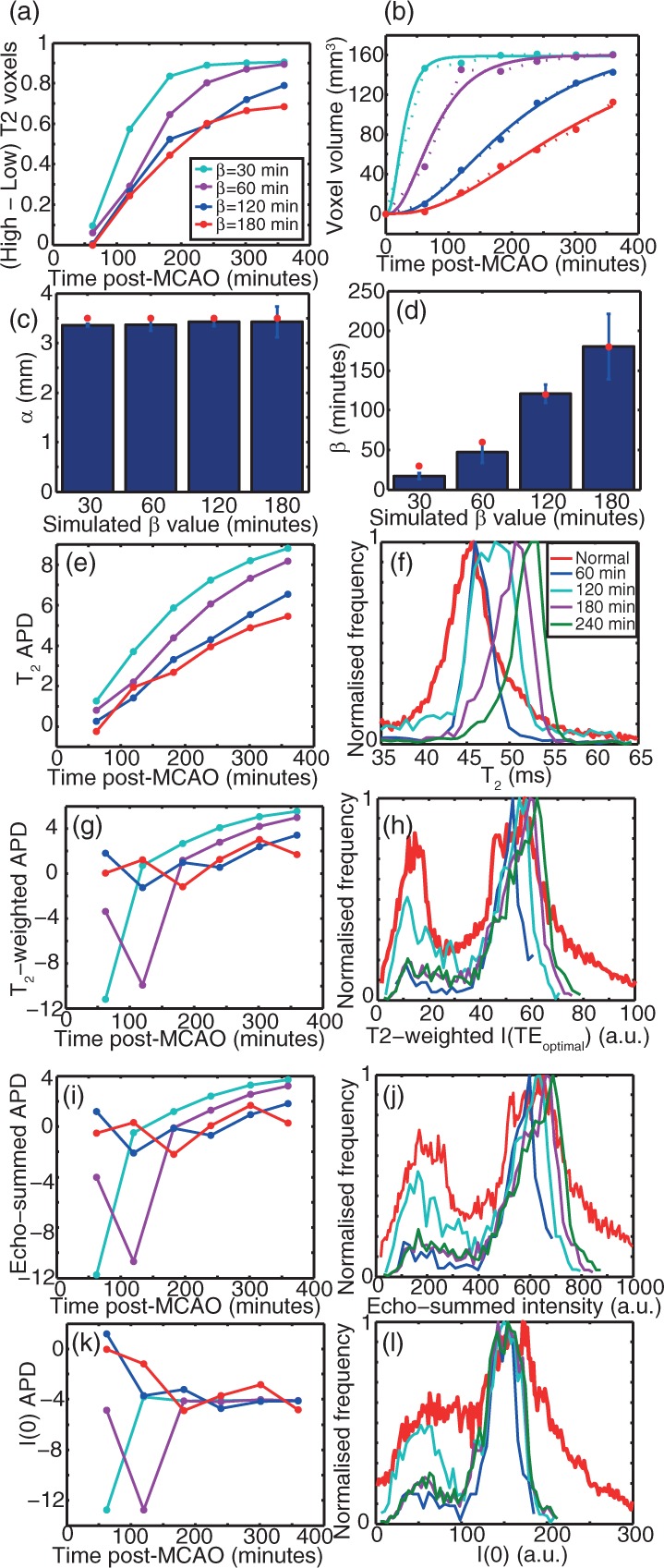
Time-dependence of MR parameters in lesions simulated in otherwise experimental data detected automatically using the criteria given in the Methods section, as a function of post-MCAO time and the lesion progression time coefficient β. (a): f2. (b) Lesion volume including the fit using equation (16) shown in solid lines. Dotted lines connect points as a visual guide. (c) and (d) α and β parameters, respectively, fitted to the extracted lesions from the simulated data using the automated procedure. (e) APD between the detected lesion and non-lesion T2. (f) Using β = 60 min, T2 histograms are plotted as a function of post-MCAO (onset) time. (g) and (h) APD between the detected lesion and non-lesion T2-weighted intensity at the optimum TE of 50 ms and T2-weighted intensity histograms for β = 60 min, respectively. (i) and (j) APD between the detected lesion and non-lesion echo-summed T2-weighted signal intensities and histograms of echo-summed T2-weighted signal intensities for β = 60 min, respectively. Panels (k) and (l) show the APD between the detected lesion and non-lesion I(0) (as determined from T2 fitting) and I(0) histograms for β = 60 min, respectively. Panels (a, b, c, e, g, i, k) share the legend shown in panel (a). Panels (f, h, j, l) share the legend shown in panel (f).
We also simulated T2-weighted images for a TE of 50 ms, which was calculated to be the optimal TE at 9.4 T for contrasting healthy and infarcted tissue (a TE of 50 ms is close to the T2 of the healthy brain parenchyma at 9.4 T). The resulting intensity distributions gave no clear separation of ischemic from non-ischemic tissue (Figure 6(g) and (h)). T2-weighted image intensity, even when optimised, is thus a far less robust experimental parameter to describe the ischaemic lesion than quantitative T2 (Figure 6). In these simulations, it was also observed that the use of an optimal TE gave near-identical results to using the sum of all echoes in a multi-contrast experiment, whereas the I(0) fitted during the computation of a T2 map (a proxy of proton density) gave different, and also rather uninformative, results.
Discussion
Predictions of the CED model and qualitative agreement with experiment
We have introduced a simple automated method for the rapid detection of cerebral ischaemia using quantitative ADC and T2 maps, as well a theoretical framework which explains many MRI observations and has enabled the validation of our lesion detection method. In particular, the CED model is able to provide a rational basis for the previously observed quasi-linear increase in T2 with time during the hyper-acute phase of ischaemia, the slower changes in T2 as compared to ADC, and identifies the rate of spread of ischaemia through rat brain parenchyma, encoded by the model parameter β, as the critical system parameter that directs the different form of the T2 time dependence in different areas of ischaemia. In addition, temporal changes in the spatial T2 distribution are accounted for, as well as the initial drop in T2, along with the basis for improved sensitivity of T2 to T2-weighted intensities to the time after onset.
The dependence of the T2 kinetics upon the rate of progression of ischaemia was also observed experimentally, and is now explicable with a single, measurable parameter. An easily accessible experimental parameter when assessing ischaemia is the width of the T2 distribution. In this regard, the CED model is broadly in agreement with experiment: the width of the lesion T2 distribution contracts relative to non-infarcted tissue around 1-h post-MCAo, before broadening around 2 h and contracting again (at β = 1 h). This time dependence in itself could in future be used as a restraint in determining parameters of clinical importance, being more informative than simply the average T2 over the lesion.17
An important finding of the simulations is that the T2-weighted signal intensity, even at optimal TE, is less sensitive to ischaemia, and apparently more so to tissue type, than T2. The latter can therefore be expected to be a more sensitive marker of ischaemia provided it can be experimentally determined with sufficient confidence.16 This may well be due to the broader dynamic range of T2-weighted intensity as a function of tissue type, such that ischaemia is a “second-order” perturbation, whilst the narrow dynamic range of absolute T2 as a function of tissue type make ischaemia the dominant “first-order” perturbation to its average value.
Rationalising the predictions of the CED model
The basis for the CED model’s predictions may be rationalised as follows: the fast drop in ADC and initial fast drop in T2 is driven by the shift of water from the extracellular space into the intracellular environment (i.e. cytotoxic oedema), before the slower dissociative terms take effect. However, the increased intracellular water content slowly shifts the equilibrium for supra-macromolecular structures, favouring states with fewer subunits. At the same time, associated with the ischaemic cascade is the activation of proteases.29 These two effects therefore decrease the average supra-macromolecular and macromolecular size intracellularly, so that bound intracellular T2 is substantially increased, and exchange with intracellular macromolecules no longer relaxes nuclear spins to such a degree. Nonetheless, ADC remains stable since the bound intracellular state contributes very little to its observed value; it is so very much slower than diffusion in either the free intra- or extracellular states, even with some dissociation.
The inclusion of a third state in our model, the bound intracellular state, was largely to overcome the fact that two-state models of intra- and extracellular water fail to account fully for ADC kinetics23,24 and in our own pilot simulations fail comprehensively to explain T2 kinetics. However, we have not explored the possibility of a two-state model incorporating the effects of changes in tortuosity (either extra- or intracellular) to calculate T2. Models based on changes in extracellular tortuosity have been successfully applied to explain ADC changes when in conjunction with changes in water distribution (as mentioned in the Introduction), but the incorporation into a model for T2, no matter how interesting the prospect, lies beyond the scope of the current work. It is worth considering, however, that our approach to modelling the increase in intracellular ADC may be reformulated as a tortuosity model (commensurate with cytoskeletal remodelling in response to oedema), but in that case the calculation of T2 from the rotational correlation time from the ADC is not physically reasonable.
Shortcomings of the CED model
The proposed CED model is certainly incomplete, like any other three pool model is when trying to describe complex hydrodynamics in complex biological systems. First, it has been formulated only to model lesions with spherical, cylindrical or circular geometry. This can be extended, for example by modelling the lesion morphology using a polynomial expansion in three dimensions, such as a spherical harmonic series, but would require additional parameters (this is described in the Supplementary material). Second, we also have taken no account of blood oxygenation, which is likely to influence T2 at least very in early ischemia,20 through the BOLD effect. Vasogenic oedema also lies outside the scope of the model in its current incarnation, though again it could be incorporated into a more comprehensive formalism.
Our model includes a considerable number of parameters, not all of which yet have known values. Those that remain outstanding provide potential means to validate the model experimentally, and in principle could be made free parameters in the eventual case that the model is extended to such an extent as to be fitted to data. Our choice of the “healthy tissue” intracellular water fraction of 0.8 and “infarcted” intracellular water fraction rising to 0.95 were based on literature values.37 Likewise, we used water exchange timescales across the plasma membrane in the 0.1–1 s range (a lifetime of 550 ms has been measured38), but the model was not at all sensitive to this parameter, provided the lifetime was shorter than ∼1 s. Due to the total stability of the model in this case, we do not present simulations as a function of exchange rate constants. T2 for the “free” water in intra- and extracellular compartments have also to be chosen, both set at 65 ms in our simulations which was found to reproduce observed values at 9.4 T reasonably well.
When fitting to equation (4) to determine the model-dependent effective T2, deviations from mono-exponential behaviour can be expected, if there are slow-exchange terms in the kinetic matrix with large populations. Multi-exponential relaxation is therefore permitted within the CED model, as well as the model being arbitrarily extensible to any number of non-interacting (or slowly interacting) subspaces not currently accounted for, which may result in deviations from mono-exponential signal attenuation behaviour. However, as explained above, the exchange lifetimes of the components modelled argue for the CED model pertaining to a fast-exchange subspace of the total system. If there is a deviation from mono-exponential behaviour detectable in the experimental data (which in our data there was not, as shown in the Supplementary material, but in other systems may be the case), then one or more of the T2 “components” detected in experimental data belongs to a subspace not relevant to the CED model.
An explicit assumption of the model, and one that was necessary to describe the data, is that the ADC for extracellular water is higher than that for intracellular. These were set at in our simulations at 1.8 × 10−9 m2 s−1 and 0.45 × 10−9 m2 s−1 respectively, which placed the simulations in reasonable agreement with experiment, albeit without rigorous optimisation. These theoretical (essentially predicted) values are also in good agreement with experimental MRI observations for water ADC in the rat brain made using contrast agents to separate intra- and extracellular compartments.39
Summary and outlook
A major advantage of having a model that explains multiple MRI observations in ischemia is that the model can be applied to the design of future experiments or studies to maximise their impact and efficiency. Likewise, computerised means for the detection of ischemia and other applications in noisy data can be extensively tested using simulated data in advance of application to experimental data, reducing cost and animal burden, and ultimately with a potential translation into clinics. The purpose of our model was to understand the processes driving the time dependence of ischaemic stroke and serve as a means of testing the robustness of lesion detection approaches. Whilst we have not conducted a rigorous “system identification” analysis, it is clear that the rate of progression of ischemia is a parameter which drives the system. Realistically, in acute ischaemic stroke patients, symptoms will not allow comprehensive time-dependent data sets of the type possible in animal experimentation. Therefore, identification of parameters which facilitate estimation of system parameters from measurements at a single (and perhaps unknown) time after onset of symptoms is necessary. In that regard, our identification of the rate of lesion progression as a key system parameter as well as the width of the lesion T2 distribution having a characteristic time distribution may facilitate the formulation of an analytical or numerical model which does not require multiple time-point measurements. It is, however, certain that further theoretical and experimental work is required here.
In summary, we have introduced a simple empirical parameter to describe T2 kinetics in ischemia which is numerically stable and obtained experimental data in a rat MCAo model. We have introduced a mathematical model based on hydrodynamics to explain ADC and T2 from unified biophysics and used it to explain many of our findings, as well as others in the literature. Although the excellent agreement between the model’s predictions is necessary, but not sufficient to validate its hypotheses, we believe that such a model is of great value in understanding ischaemia and designing MRI experiments for its detection. With an understanding of the parameters which drive ADC and T2 kinetics in ischaemia, a simplified model may be derived which could be used to extract the most relevant parameters describing ischemia from MRI data, such as time of onset,15,17,18 which would be of great value in clinical MRI.
Supplementary Material
Acknowledgement
The skilful technical assistance by Mrs Maarit Pulkkinen is greatly acknowledged.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: MJK is funded by grants from ARUK and BRACE Charity. BLM and HJR received a travel grant to University of Eastern Finland from the School of Experimental Psychology, University of Bristol. OG and KJ are funded by Academy of Finland, UEF-Brain strategic funding from University of Eastern Finland and by Biocenter Finland
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MJK created the model, performed the simulations, fitted the experimental data into the model and wrote the first draft of the paper; BLM and HJR performed the MRI scans and initial analyses of experimental data; KJ and OHJG designed the MRI protocol, supervised animal experiments and MRI scans; RAK initiated and supervised the study, contributed to the creation of the model and finalised the paper. All authors reviewed the paper and approved it for submission.
Supplementary material
Supplementary material for this paper can be found at http://jcbfm.sagepub.com/content/by/supplemental-data
References
- 1.Kim BJ, Kang HG, Kim HJ, et al. Magnetic resonance imaging in acute ischemic stroke treatment. J Stroke 2014; 16: 131–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heiss WD, Kidwell CS. Imaging for prediction of functional outcome and assessment of recovery in ischemic stroke. Stroke 2014; 45: 1195–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hacke W, Donnan G, Fieschi C, et al. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 2004; 363: 768–774. [DOI] [PubMed] [Google Scholar]
- 4.Wardlaw JM, Murray V, Berge E, et al. Alteplase for ischaemic stroke–responses. Lancet 2014; 384: 661–662. [DOI] [PubMed] [Google Scholar]
- 5.Wester P, Radberg J, Lundgren B, et al. Factors associated with delayed admission to hospital and in-hospital delays in acute stroke and TIA: A prospective, multicenter study.Seek- Medical-Attention-in-Time Study Group. Stroke 1999; 30: 40–48. [DOI] [PubMed] [Google Scholar]
- 6.Knight RA, Dereski MO, Helpern JA, et al. Magnetic resonance imaging assessment of evolving focal cerebral ischemia. Comparison with histopathology in rats. Stroke 1994; 25: 1252–1261. [DOI] [PubMed] [Google Scholar]
- 7.Welch KM, Windham J, Knight RA, et al. A model to predict the histopathology of human stroke using diffusion and T2-weighted magnetic resonance imaging. Stroke 1995; 26: 1983–1989. [DOI] [PubMed] [Google Scholar]
- 8.Hoehn-Berlage M, Eis M, Back T, et al. Changes of relaxation times (T1, T2) and apparent diffusion coefficient after permanent middle cerebral artery occlusion in the rat: temporal evolution, regional extent, and comparison with histology. Magn Reson Med 1995; 34: 824–834. [DOI] [PubMed] [Google Scholar]
- 9.Kauppinen RA. Multiparametric magnetic resonance imaging of acute experimental brain ischaemia. Prog Nucl Magn Reson Spectr 2014; 80: 12–25. [DOI] [PubMed] [Google Scholar]
- 10.Schaefer PW, Grant PE, Gonzalez RG. Diffusion-weighted MR Imaging of the Brain. Radiology 2000; 217: 331–345. [DOI] [PubMed] [Google Scholar]
- 11.Roberts TP, Rowley HA. Diffusion weighted magnetic resonance imaging in stroke. Eur J Radiol 2003; 45: 185–194. [DOI] [PubMed] [Google Scholar]
- 12.van der Toorn A, Sykova E, Dijkhuizen RM, et al. Dynamic changes in water ADC, energy metabolism, extracellular space volume, and tortuosity in neonatal rat brain during global ischemia. Magn Reson Med 1996; 36: 52–60. [DOI] [PubMed] [Google Scholar]
- 13.van Gelderen P, de Vleeschouwer MHM, DesPres D, et al. Water diffusion and acute stroke. Magn Reson Med 1994; 31: 154–163. [DOI] [PubMed] [Google Scholar]
- 14.Grohn OHJ, Lukkarinen JA, Oja JME, et al. Noninvasive Detection of Cerebral Hypoperfusion and Reversible Ischemia From Reductions in the Magnetic Resonance Imaging Relaxation Time, T2. J Cereb Blood Flow Metab 1998; 18: 911–920. [DOI] [PubMed] [Google Scholar]
- 15.Jokivarsi KT, Hiltunen Y, Grohn H, et al. Estimation of the onset time of cerebral ischemia using T1rho and T2 MRI in rats. Stroke 2010; 41: 2335–2340. [DOI] [PubMed] [Google Scholar]
- 16.Rogers HJ, McGarry BL, Knight MJ, et al. Timing the ischaemic stroke by 1H-MRI: improved accuracy using absolute relaxation times over signal intensities. Neuroreport 2014; 25: 1180–1185. [DOI] [PubMed] [Google Scholar]
- 17.Siemonsen S, Mouridsen K, Holst B, et al. Quantitative T2 values predict time from symptom onset in acute stroke patients. Stroke 2009; 40: 1612–1616. [DOI] [PubMed] [Google Scholar]
- 18.Thomalla G, Cheng B, Ebinger M, et al. DWI-FLAIR mismatch for the identification of patients with acute ischaemic stroke within 4.5 h of symptom onset (PRE-FLAIR): A multicentre observational study. Lancet Neurol 2011; 10: 978–986. [DOI] [PubMed] [Google Scholar]
- 19.Cheng B, Brinkmann M, Forkert ND, et al. Quantitative measurements of relative fluid-attenuated inversion recovery (FLAIR) signal intensities in acute stroke for the prediction of time from symptom onset. J Cereb Blood Flow Metab 2013; 33: 76–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grohn OHJ, Kettunen MI, Penttonen M, et al. Graded reduction of cerebral blood flow in rat as detected by the nuclear magnetic resonance relaxation time T2: A theoretical and experimental approach. J Cereb Blood Flow Metab 2000; 20: 316–326. [DOI] [PubMed] [Google Scholar]
- 21.Rekik I, Allassonnière S, Durrleman S, et al. Spatiotemporal dynamic simulation of acute perfusion/diffusion ischemic stroke lesions evolution: A pilot study derived from longitudinal MR patient data. Comput Math Meth Med 2013; 2013: 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dronne MA, Boissel JP, Grenier E, et al. Mathematical modelling of an ischemic stroke: An integrative approach. Acta Biotheor 2004; 52: 255–272. [DOI] [PubMed] [Google Scholar]
- 23.Gass A, Niendorf T, Hirsch JG. Acute and chronic changes of the apparent diffusion coefficient in neurological disorders—biophysical mechanisms and possible underlying histopathology. J Neurol Sci 2001; 186: S15–S23. [DOI] [PubMed] [Google Scholar]
- 24.Anderson AW, Xie J, Pizzonia J, et al. Effects of cell volume fraction changes on apparent diffusion in human cells. Magn Reson Imaging 2000; 18: 689–695. [DOI] [PubMed] [Google Scholar]
- 25.Latour LL, Svoboda K, Mitra PP, et al. Time-dependent diffusion of water in a biological model system. Proc Natl Acad Sci USA 1994; 91: 1229–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moseley ME, Cohen Y, Mintorovitch J, et al. Early detection of regional cerebral ischemia in cats: Comparison of diffusion- and T2-weighted MRI and spectroscopy. Magn Reson Med 1990; 14: 330–346. [DOI] [PubMed] [Google Scholar]
- 27.Mintorovitch J, Moseley ME, Chileuitt L, et al. Comparison of diffusion- and T2-weighted MRI for the early detection of cerebral ischemia and reperfusion in rats. Magn Reson Med 1991; 18: 39–50. [DOI] [PubMed] [Google Scholar]
- 28.Benveniste H, Hedlund LW, Johnson GA. Mechanism of detection of acute cerebral ischemia in rats by diffusion-weighted magnetic resonance microscopy. Stroke 1992; 23: 746–754. [DOI] [PubMed] [Google Scholar]
- 29.Sotak CH. Nuclear magnetic resonance (NMR) measurement of the apparent diffusion coefficient (ADC) of tissue water and its relationship to cell volume changes in pathological states. Neurochem Internatl 2004; 45: 569–582. [DOI] [PubMed] [Google Scholar]
- 30.Szafer A, Zhong J, Gore JC. Theoretical model for water diffusion in tissues. Magn Reson Med 1995; 33: 697–712. [DOI] [PubMed] [Google Scholar]
- 31.Hakumaki JM, Pirttila TRM, Kauppinen RA. Reduction in water and metabolite apparent diffusion coefficients during energy failure involves cation-dependent mechanisms. A proton nuclear magnetic resonance study of rat cortical brain slices. J Cereb Blood Flow Metab 2000; 20: 405–411. [DOI] [PubMed] [Google Scholar]
- 32.Norris DG, Niendorf T, Leibfritz D. Health and infarcted brain tissues studied at short diffusion times: the origins of apparent restriction and the reduction in apparent diffusion coefficient. NMR Biomed 1994; 7: 304–310. [DOI] [PubMed] [Google Scholar]
- 33.Niendorf T, Norris DG, Leibfritz D. Detection of apparent restricted diffusion in healthy rat brain at short diffusion times. Magn Reson Med 1994; 32: 672–677. [DOI] [PubMed] [Google Scholar]
- 34.Norris DG, Niendorf T, Hoehn-Berlage M, et al. Incidence of apparent restricted diffusion in three different models of cerebral infarction. Magn Reson Imag 1994; 12: 1175–1182. [DOI] [PubMed] [Google Scholar]
- 35.Sykova E, Svoboda J, Polak J, et al. Extracellular volume fraction and diffusion characteristics during progressive ischemia and terminal anoxia in the spinal cord of the rat. J Cereb Blood Flow Metab 1994; 14: 301–311. [DOI] [PubMed] [Google Scholar]
- 36.Longa EZ, Weinstein PR, Carlson S, et al. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989; 20: 84–91. [DOI] [PubMed] [Google Scholar]
- 37.Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiol Rev 1985; 65: 101–148. [DOI] [PubMed] [Google Scholar]
- 38.Quirk JD, Bretthorst GL, Duong TQ, et al. Equilibrium water exchange between the intra- and extracellular spaces of mammalian brain. Magn Reson Med 2003; 50: 493–499. [DOI] [PubMed] [Google Scholar]
- 39.Silva MD, Omae T, Helmer KG, et al. Separating changes in the intra- and extracellular water apparent diffusion coefficient following focal cerebral ischemia in the rat brain. Magn Reson Med 2002; 48: 826–837. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.