

Abstract
Stroke and Alzheimer's disease, two diseases that disproportionately affect the aging population, share a subset of pathological findings and risk factors. The primary genetic risk factor after age for late-onset Alzheimer's disease, ApoE4, has also been shown to increase stroke risk and the incidence of post-stroke dementia. One mechanism by which ApoE4 contributes to disease is by inducing in neurons a resistance to Reelin, a neuromodulator that enhances synaptic function. Previous studies in Reelin knockout mice suggest a role for Reelin in protection against stroke; however, these studies were limited by the developmental requirement for Reelin in neuronal migration. To address the question of the effect of Reelin loss on stroke susceptibility in an architecturally normal brain, we utilized a novel mouse with induced genetic reduction of Reelin. We found that after transient middle cerebral artery occlusion, mice with complete adult loss of Reelin exhibited a similar level of functional deficit and extent of infarct as control mice. Together, these results suggest that physiological Reelin does not play a strong role in protection against stroke pathology.
Keywords: Reelin, Reelin conditional knockout, stroke, transient middle cerebral artery occlusion, ApoE4
Introduction
As the population ages and we implement better treatments for heart disease and cancer, the rates of neurological diseases, including Alzheimer's disease (AD) and stroke, are skyrocketing. Stroke now affects 800,000 Americans annually, and nearly 500,000 new AD cases are diagnosed in the U.S. each year.1,2 These diseases overlap to some extent, since dementia is a common long-term consequence of stroke,3,4 and after stroke there is an increase in amyloid (Aβ) and phosphorylated tau, the two characteristic markers of AD pathology.5,6 The shared pathological findings of AD and post-stroke recovery are mirrored by the identification of a common genetic risk factor, the ɛ4 isoform of Apolipoprotein E (ApoE4), which increases AD and cerebrovascular disease risk, as well as the likelihood of post-stroke dementia.7–9 Additionally, a role for ApoE4 in stroke susceptibility is supported by the finding that ApoE4-targeted replacement mice have larger infarct volumes and motor functional deficits following middle cerebral artery (MCA) inclusion.10
ApoE4 shares a common receptor, apolipoprotein receptor 2 (ApoEr2), with the neuromodulator Reelin. ApoE4 induces neuronal resistance to Reelin, which promotes AD pathogenesis by leaving neurons susceptible to Aβ toxicity.11 Though several studies have investigated the impact of ApoE4 in stroke, studies on the role of Reelin are limited. The microRNA miR-200c, which targets and eliminates Reelin, is transiently elevated post-stroke, and treatment with an antagomir of miR-200c increases Reelin levels slightly and reduces infarct size.12 Additionally, Reelin knockout (reeler) mice exhibit impaired recovery and larger infarct size following MCA occlusion.13 However, Reelin is required for neuronal migration, thus the severe disorganization of the reeler brain confounds these findings,14 and miR-200c has many targets in the brain, such as Zeb1.15 Consequently, the requirement for Reelin in stroke protection remains unresolved.
We recently established a novel Reelin conditional knockout mouse (cKO) mouse that allows us to bypass the developmental requirements for Reelin.16 To determine the effect of adult loss of Reelin on stroke susceptibility in the context of normal brain architecture, we subjected cKO mice to transient middle cerebral artery occlusion (tMCAo). In contrast to the previous findings in Reeler mice, Reelin cKO mice were no more susceptible to ischemic injury than control mice, as there was no difference between the two groups in either infarct size or post-stroke behavioral deficits. Taken together, these results suggest that Reelin plays a much smaller role in stroke pathology than previously thought.
Materials and methods
Animals
All animal care protocols were followed in accordance with the Institutional Animal Care and Use Committee of the University of Texas Southwestern Medical Center and according to the Association for Assessment and Accreditation of Laboratory Animal Care guidelines. B6.Cg-Tg(CAG-cre/Esr1)5Amc/J mice, referred to as CAG-CreERT2 mice in the text, were obtained from Jackson Laboratories.17 The Relnfl/fl mice were generated in our lab previously.16 The two lines were bred together to hemizygosity for CAG Cre and homozygosity for Relnfl/fl. Animals were group-housed in a standard 12-h light cycle and fed ad libitum standard mouse chow. Male littermates were separated into two groups of animals, whereby Reelin was conditionally knocked out or not, depending on the presence or absence of the Cre driver, respectively. All experiments and surgeries were performed by a blinded experimenter. Reporting of animal experiments below complies with ARRIVE guidelines.
Experimental timeline
Two-month-old Relnfl/fl mice with or without CAG Cre were given daily intra-peritoneal injections for five days with 135 mg/kg tamoxifen (Sigma-Aldrich, St. Louis, MO, USA) dissolved in sunflower oil. Ten days after the last tamoxifen injection, mice were trained on the Rotarod task. The following week, mice received a 45-min tMCAo. Two days later, the mice were assessed for motor impairment on rotarod. Later that afternoon, mice were sacrificed. The cerebella were removed and flash frozen in liquid nitrogen for analysis of Reelin levels, with the remaining brain subjected to triphenyltetrazolium chloride (TTC) staining.
Rotarod test
Mice were placed on a Rotamex rotarod apparatus (Columbus Instruments, Columbus, OH, USA), facing away from the experimenter. The rod initially rotated at 2 r/min and then accelerated at a rate of 1 r/min/5 s until the mice fell off the rod or 300 s passed. The time to falling off or the first “spin,” where the mouse completed a full rotation holding on to the rod, was considered the end of the trial. The mice received four trials per day with a 15-min inter-trial interval. Mice were trained until they reached ‘asymptote,’ which was defined as the point at which their learning no longer improved. For most mice, this was around day 4; 48 h after tMCAo, mice were tested again with four trials on the Rotarod to determine motor impairment.
Reelin analysis and Western blotting
Cerebella from experimental animals were analyzed to confirm Reelin knockout. After dissection of the cerebral hemispheres for TTC staining, cerebella were flash frozen in liquid nitrogen. The tissue was homogenized in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 0.1% SDS, 0.5% sodium deoxycholate, 1% NP40) with protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO, USA), then centrifuged at 14,000 r/min for 15 min at 4°C. Protein concentration was determined using the DC Protein Assay (Bio-Rad, Hercules, CA, USA); 15 μg of protein was resolved on 4–15% polyacrylamide gels (Bio-Rad, Hercules, CA, USA) and then transferred to nitrocellulose membranes. Blots were blocked in blocking buffer (LI-COR, Lincoln, NE, USA) and then incubated overnight in anti-Reelin primary antibody (G10,18 1:1000). Membranes were washed, incubated in donkey anti-mouse 800 secondary (LI-COR, Lincoln, NE, USA, 1:3000), and then imaged using the Odyssey CLx Infrared Imaging System (LI-COR, Lincoln, NE, USA). All conditional knockout animals used in this study had less than 5% residual Reelin, and thus none were excluded on the basis of incomplete knockout.
tMCAos
Animals were anesthetized with 2% isoflurane/70% NO2/30% O2 and kept warm on a heating pad at 37℃. A small incision was made to pull the muscle back from the skull and reveal the MCA for Doppler flowmetry. To induce a transient occlusion, an intra-luminal suture was inserted into the common carotid artery to block blood flow into the MCA. A transcranial Laser Doppler (Moor Instruments, Wilmington, DE, USA) detected successful MCA occlusion, defined as a greater than 80% reduction in blood flow compared to baseline value. The occlusion duration was 45 min, during which time mice were allowed to recover in a 37℃ incubator. Mice were then re-anesthetized and continued occlusion confirmed by Doppler. The occluding suture was then removed. A successfully reperfused MCA was defined as one that returned to more than 50% initial blood flow. Mice that did not meet both occlusion and reperfusion criteria were removed from the study. Out of a total group of 31 mice, 1 cKO mouse was not successfully occluded, 1 cKO and 1 control mouse died during tMCAo, 1 control mouse died the night after tMCAo, 1 cKO and 1 control mouse reperfused during the 45-min tMCAo, and 1 cKO mouse did not reperfuse after 45 min. These mice were removed from analysis, leaving a total of 13 control mice and 11 cKO mice. To evaluate stroke severity during occlusion and at two days post-stroke, mice were scored using a modified Neurological Severity Score.19 Mice received a score from 0 (no observable deficit) to 4 (unable to walk spontaneously), though 3 mice failed to have deficit scores recorded.
TTC staining
Mice were sacrificed two days after tMCAo, shortly after rotarod testing. The brains were immediately sliced using a metal matrix into 1 mm coronal sections. Sections were immersed in 2% 2,3,5-triphenyltetrazolium chloride (TTC) (Sigma-Aldrich, St. Louis, MO, USA) in phosphate-buffered solution (PBS), which selectively stains active mitochondria. The sections were stained until they reached an appropriate color, and then the TTC solution was replaced with 4% paraformaldehyde (PFA). After 24 h in fixative, sections were arranged on glass slides and scanned. The sections were visualized using ImageJ (NIH, Bethesda, MD, USA), and the size of the infarct was quantified by a blinded observer. The indirect infarct was calculated as [contralateral hemisphere volume – (ipsilateral hemisphere volume – infarct volume)] and was used to account for swelling of the infarct.19 The percentage of edema present was calculated as [(ipsilateral volume – contralateral volume)/contralateral volume] × 100.
Cresyl violet and alkaline phosphatase staining
Mice were transcardially perfused with PBS, followed by fixative solution with 4% PFA in PBS. Brains were post-fixed in 4% PFA at 4℃ overnight and stored in PBS with 0.02% NaN3 at 4℃. Brains were embedded in 5% agarose (in PB), and 50 µm sections were cut with a Leica VT 1000S Vibratome. Slices were stored in PBS.
Brain sections rostral to the hippocampus were used for Cresyl Violet and Alkaline Phosphatase histological staining techniques. For Cresyl Violet staining, slices were arranged on slides and air-dried. Slices were rinsed with H2O prior incubation in 0.1% Cresyl Violet solution for 15 min. Excessive stain was removed by incubating the slices serially in 70% ethanol for 3 min, 95% ethanol for 2 min, and 100% ethanol for 2 min. The slices were cleared in Xylene for 5 min and mounted with Permount (Fisher Scientific). For Alkaline Phosphatase staining (VECTOR Laboratories), floating sections were incubated in the substrate working solution (according to manufacturer's protocol) until the desired staining intensity was obtained. Slices were rinsed in PBS, arranged on slides, and mounted with mounting medium (VectaMount). Stained slices were imaged with a NanoZoomer (Hamamatsu).
Statistical analysis
A priori power analysis was performed using G*Power software20 (Universitat Dusseldorf, Germany) to determine the sample size needed to yield 80% power for detecting an effect of Reelin loss on stroke severity. We first determined the average infarct volume based on prior studies of 45-min tMCAo in control mice, 102 mm3.21 We then assumed that Reelin loss would produce a 40% increase in infarct volume. We calculated an effect size of 1.75 (mean1-mean2)/s, where mean1 was 102 mm3, mean2 was mean1 times 1.4 (143 mm3), and s was defined as the SD of the mean of infarct volume, 23.2 mm3. This effect size was used in an a priori power analysis for a two-tailed t-test to detect a difference between two independent means. The results indicated that a sample size of seven mice per genotype would be required to achieve an 85% power for detecting an effect size of 1.75. Subsequent sample size exceeded this estimate.
Data were analyzed using GraphPad Prism software (version 6.0, GraphPad Software, San Diego, CA, USA). Statistical comparisons were evaluated using the two-tailed unpaired Student's t-test, except for the neurological deficit scores, which were evaluated using the Mann–Whitney rank-sum test, and the pre-stroke rotarod, which was evaluated by a two-way rmANOVA. Data are shown as the mean ± SEM, and statistical significance was set at p < 0.05.
Results
Confirmation of Reelin knockout
Reelin is required for normal brain development and thus Reelin knockout (reeler) mice have severely altered brain architecture and a variety of behavioral deficits.14 Recently generated CAG CreERT2; Relnfl/fl conditional Reelin knockout (cKO) mice allow for ubiquitous inducible loss of Reelin and bypasses the developmental requirement for Reelin.16 Experimental timeline for induction and tMCAo is shown in Figure 1(a). We confirmed that all conditional knockout (cKO) mice had efficient Reelin reduction following tamoxifen injection. All cKO mice exhibited a complete loss of Reelin at both the 380 kDa band, reflecting full length Reelin, and the 180 kDa band cleavage product (Figure 1(b)) in cerebellar tissue, a region unaffected by the tMCAo.
Figure 1.
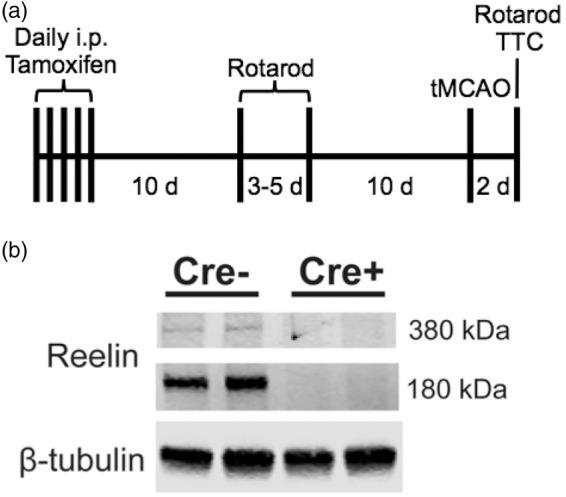
tMCAo timeline and confirmation of Reelin knockout. (a) Experimental timeline. All mice were injected with tamoxifen for five days; 16 days later the mice were trained on the rotarod. A 45-min tMCAo was induced after an additional 10 days. Two days later, mice were tested on the rotarod prior to sacrifice. (b) Reelin levels are negligible in Reelin cKO (Cre+) mice (n = 13 Cre− and n = 11 Cre + animals) in post-stroke cerebella.
Reelin cKO mice show similar levels of acute post-stroke injury
To determine the effect of adult Reelin loss on post-stroke infarct damage, we performed TTC staining on acutely prepared brain tissues. tMCAo-induced infarct volumes were similar between the cKO (36.00 ± 7.80 mm3; n = 11) and control (44.59 ± 6.86 mm3; n = 13) groups. While tMCAo infarct size was variable, and potentially confounded by the modest impact of the tMCAo, there was no statistically significant effect of Reelin loss on indirect infarct size (p = 0.4157) (Figure 2(a)). Conditional loss of Reelin also did not significantly affect ipsilateral hemispheric swelling between groups (cKO: 0.75 ± 1.71% vs. control: −1.49 ± 1.32 %; p = 0.3046; Figure 2(b)).
Figure 2.

Reelin loss has no significant effect on size of infarct post-tMCAo. (a) Representative brain sections from control (Cre−) and cKO (Cre+) mice two days after tMCAo. (b) Quantification of indirect stroke volume (control: 44.59 ± 6.86 mm3, n = 13; cKO: 36.00 ± 7.80 mm3, n = 11; p = 0.4157) and edema (control: −1.49 ± 1.32%, n = 13; cKO: 0.75 ± 1.71% n = 11; p = 0.3046). Data are represented as mean ± S.E.M.
We previously found that adult loss of Reelin does not accelerate AD-related amyloid pathology, but does increase synaptic susceptibility to amyloid toxicity, resulting in cognitive impairment in the presence of elevated Aβ.16 Here, it similarly could be possible that while Reelin loss does not accelerate stroke pathology, it does leave mice more susceptible to functional deficits. cKO mice exhibited between a mild to moderate impairment (average score 1.91 ± 0.11) immediately following tMCAo induction (Figure 3(a)) using a standard 0–5 deficit scale, with some improvement at two days post-tMCAo (average score 1.46 ± 0.13; Figure 3(b)). Importantly, there was no significant difference compared to control mice at either initial impairment (average score 1.95 ± 0.14; p = 0.99) or recovery (average score 1.29 ± 0.07; p = 0.37).
Figure 3.

Reelin cKO mice do not have significantly increased functional deficit over control mice following tMCAo. (a) Control and cKO mice showed a similar impairment in motor function during the tMCAO (control: 1.95 ± 0.138, n = 10; cKO 1.91 ± 0.375; Mann–Whitney p = 0.9936). (b) Both genotypes also had a similar impairment two days following tMCAo (control: 1.29 ± 0.258, n = 12; cKO 1.46 ± 0.416; Mann–Whitney p = 0.3667). (c) Control and cKO mice exhibited a similar rotarod ability two days post-tMCAo (control: 57.07 ± 9.51 s, n = 13; cKO: 47.96 ± 10.30 s, n = 11; unpaired t-test p = 0.9703). Data are represented as mean ± S.E.M.
Since the tMCAo injury often spans motor cortex and striatum, we wanted to see if there was a specific effect on motor function. Prior to tMCAo, mice were trained daily on the rotarod task until they hit a ceiling on their learning, which took three to five days. We confirmed our prior results that adult loss of Reelin does not significantly affect acquisition of the rotarod task in uninjured mice16 (Figure 3(c)). Two days after tMCAo, mice exhibited a significant reduction in motor function compared to pre-stroke baseline for both the cKO (pre-stroke: 85.11 ± 6.87 s vs. post-tMCAo: 47.96 ± 10.31 s; p = 0.0031) and control (pre-stroke: 76.76 ± 6.73 s vs. post-tMCAo: 57.07 ± 9.51 s; p = 0.038) groups. There was, however, no significant difference in the magnitude of deficit between the two genotypes (two-way repeated measures ANOVA, pTime < 0.003, pGenotype n.s. pInteraction n.s.; Figure 3(c)). Taken together, these behavioral results suggest that adult loss of Reelin causes no additional functional impairment during the acute stage of recovery following tMCAo.
Finally, to confirm that our results were not affected by physiological differences in brain structure or vasculature between control and cKO mice, we performed Cresyl Violet and Alkaline Phosphatase staining. Between the two genotypes, no qualitative differences were observed in the anatomy (Figure 4(a) and (b)) or the brain vasculature (Figure 4(c) and (d)). Additionally, we evaluated the difference in cerebral blood flow (CBF) by Doppler flowmetry at multiple time points during the procedure. There was no significant difference between CBF in cKO and control mice before induction of MCAo (p = 0.243), immediately after induction of tMCAo (p = 0.922), just prior to restoration of flow (p = 0.906), and immediately after removal of suture and restoration of flow (p = 0.5133 Figure 4(e) and (h)).
Figure 4.

No obvious difference in brain anatomy, vasculature, and intrinsic pathophysiology between control and cKO mice. (a) and (c) cresyl violet staining; (b) and (d) alkaline phosphatase staining of control (a and c) and cKO (b and d) brain sections. (e–h) control and cKO mice had similar cerebral flow by Doppler flowmetry before occlusion (e) control: 692.1 a.u., cKO: 761.6 a.u., p = 0.243), immediately after occlusion (f) control: 72.3 a.u., cKO: 78.1 a.u., p = 0.721), prior to suture removal (g) control: 56.0 a.u., cKO: 63.0 a.u., p = 0.612), and after restoration of MCA flow (h) control: 651.5 a.u., cKO: 690.3 a.u., p = 0.666). Data are represented as mean ± S.E.M.
Discussion
In this study, we examined the physiological role Reelin plays in protection against ischemic injury. We found that mice with induced genetic reduction of Reelin during adulthood exhibited similar injury following transient stroke compared to control mice for both infarct size and behavioral impairment two days after stroke. These results suggest that endogenous Reelin does not play a large role in short-term stroke recovery. Previous studies into the role of Reelin in stroke pathology have been limited. Reelin levels are transiently reduced post-stroke, and Reeler mice have increased susceptibility to tMCAo.12,13 Though Reeler mice have grossly normal cerebral vasculature, which permits induction of tMCAo, Reeler mice have severe architectural deficits, including complete disruption of cortical and hippocampal layering, and extreme hypofoliation of the cerebellum, which leads to a characteristic ataxia.14 Moreover, Reeler mice have a reduction in spine density and impaired neurogenesis.22 It is possible that it is these developmentally regulated structural changes, and not loss of endogenous adult Reelin, that lead to increased stroke size in Reeler mice. Our finding that adult cKO mice – which have normal brain architecture but undetectable Reelin – have no increased susceptibility to tMCAo supports this hypothesis.
The modest reduction of Reelin post-stroke is due to upregulation of miR200-c shortly after ischemia, which has Reelin as one of its targets.12,23 Antagonism of miR200-c protects against tMCAo; however, whether this effect is mediated by Reelin was previously unclear. Here, we show that Reelin loss does not affect immediate tMCAo recovery, in contrast to miR200-c, the inhibition of which is protective 24 h post-tMCAo.12 miR200-c has numerous targets that have been primarily investigated for cancer-related mechanisms,24 as well as several targets implicated in brain function, such as very low-density lipoprotein receptor (Vldlr) and zinc finger E-box-binding homeobox 1 (Zeb1).15,25 Vldlr, as one of the two canonical receptors for Reelin, is unlikely to be the main target of miR200-c in stroke, given our finding that loss of Reelin has no significant effect on tMCAo. However, there are Reelin-independent functions of Vldlr26 that cannot be excluded. Conversely, Zeb1 enacts part of the protective response to ischemia by repressing transcription of a pro-apoptotic isoform of p73,27 and thus its reduction by miR200-c may mediate the negative effect of miR200-c on post-stroke recovery. Given our negative findings in the cKO mice, Zeb1 is a strong candidate to mediate the effect of miR200-c.
The data presented here reflect an acute effect following a transient, relatively mild, stroke. Though we have shown endogenous Reelin is not required for the immediate recovery two days after stroke, this does not preclude a role for Reelin in long-term recovery after ischemia. A subtle deficit may be present if the post-stroke recovery time was extended to cover the course of weeks, rather than days. Moreover, the stroke model used in this study was a transient ischemia lasting only 45 min, which induces a relatively moderate infarct and motor impairment. It is possible that Reelin is more important for recovery from more severe strokes. To investigate this possibility, we initiated studies using permanent MCAOs; however, since this model results in a high mortality rate and our preliminary results indicated there was no significant effect of Reelin loss after transient stroke, we did not continue for ethical considerations.
Additionally, while our data indicate that endogenous Reelin is not required for post-stroke protection, this does not preclude a protective effect of exogenous or augmented Reelin. Exogenous application of Reelin enhances learning and memory in vivo, and overexpression of Reelin enhances cognitive function in a mouse model of AD.28,29 Moreover, exogenous application of Reelin enhances learning in a mouse model of Angelman disease,30 which does not involve Aβ toxicity, suggesting the ability of Reelin to strengthen synapses in ailments outside of Alzheimer's. It is possible that treatment with Reelin supplementation or a small molecule that increases Reelin levels could have a therapeutic effect on stroke; however, more studies are required to determine the effectiveness of raising Reelin levels in stroke protection.
We previously found that ApoE4 induces Reelin resistance due to alterations in vesicle trafficking, causing neurons to be more susceptible to damage by Aβ.11 ApoE4 in humans may induce a partial Reelin deficiency, contributing to disease pathogenesis. Since ApoE4 individuals have more negative outcomes following stroke,9 we wanted to investigate the contribution of loss of Reelin signaling to stroke susceptibility. Our current studies show that loss of Reelin signaling is not likely to be the driver of increased stroke susceptibility in ApoE4 individuals, thus other mechanisms by which ApoE4 could impact stroke outcome must be considered. In AD, ApoE4 accelerates the deposition of Aβ.31 Patients with post-stroke dementia have high levels of Aβ, and mouse models of AD that overproduce Aβ are more susceptible to ischemic brain damage.32,33 In addition to its effect on Aβ levels, possession of the ApoE4 allele causes patients to be more susceptible to cerebral amyloid angiopathy (CAA) and cerebral hemorrhage, a finding mirrored in mouse models of AD.34,35 CAA also independently increases susceptibility to stroke in both patients and mice.36,37 Thus, ApoE4 may mediate its effect on stroke outcomes both through Aβ deposition and development of CAA. Moreover, since Reelin protects against Aβ toxicity,38 it is possible that the reason we observed no significant effect of Reelin loss on stroke recovery is that mice do not express human Aβ. Thus, repeating the tMCAo experiments in an Aβ-overproducing mouse model may reveal a role for Reelin in stroke recovery. However, it is important to note that in the absence of human APP and Aβ, mice expressing ApoE4 are more susceptible to stroke than ApoE3 mice, thus ApoE4 must have some effect independent of its effect on Aβ.10 One hypothesis could be the effect of ApoE4 on glutamate receptors and glutamate reuptake, which may lead to increased susceptibility to excitotoxicity.39
Taken together, our results show that complete loss of Reelin expression in the adult results in no exacerbation of acute deficits caused by ischemic insult. While this does not preclude a role for Reelin in long-term ischemic injury, or even a protective effect of exogenous Reelin, it does suggest that the mechanism by which ApoE4 impedes stroke recovery is Reelin-independent. Future studies will have to investigate the alternative means by which ApoE4 contributes to stroke pathology.
Funding
This work was supported by NIH grants F30AG047799 (to CLD), R37HL63762 (to JH), R01NS093382 (to JH), and R01NS088555 (to AMS), the American Health Assistance Foundation, the Consortium for Frontotemporal Dementia Research, the Bright Focus Foundation, the Lupe Murchison Foundation, the Ted Nash Long Life Foundation, the Deutsche Forschungsgemeinschaft (DFG) (grant numbers SFB 780/TP5 to JH), the American Heart Association (14SDG18410020 to AMS), and the Haggerty Center for Brain Injury and Repair (UTSW, to AMS).
Acknowledgement
The authors thank the Neuromodels Core at UT Southwestern for technical assistance.
Footnotes
*These authors contributed equally to this work.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
CLD, JH, and AMS designed experiments. CLD, CD, EP, TP, and AMS performed experiments. CLD, TP, JH, and AMS prepared the manuscript.
References
- 1.Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics – 2014 update: a report from the American Heart Association. Circulation 2014; 129: e28–e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alzheimer's A. 2014 Alzheimer's disease facts and figures. Alzheimers Dement 2014; 10: e47–e92. [DOI] [PubMed] [Google Scholar]
- 3.Snowdon DA, Greiner LH, Mortimer JA, et al. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA 1997; 277: 813–817. [PubMed] [Google Scholar]
- 4.Pendlebury ST, Rothwell PM. Prevalence, incidence, and factors associated with pre-stroke and post-stroke dementia: a systematic review and meta-analysis. Lancet Neurol 2009; 8: 1006–1018. [DOI] [PubMed] [Google Scholar]
- 5.Hiltunen M, Makinen P, Peraniemi S, et al. Focal cerebral ischemia in rats alters APP processing and expression of Abeta peptide degrading enzymes in the thalamus. Neurobiol Dis 2009; 35: 103–1. [DOI] [PubMed] [Google Scholar]
- 6.Uchihara T, Tsuchiya K, Kondo H, et al. Widespread appearance of Alz-50 immunoreactive neurons in the human brain with cerebral infarction. Stroke 1995; 26: 2145–2148. [DOI] [PubMed] [Google Scholar]
- 7.Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993; 261: 921–923. [DOI] [PubMed] [Google Scholar]
- 8.McCarron MO, Delong D, Alberts MJ. APOE genotype as a risk factor for ischemic cerebrovascular disease: a meta-analysis. Neurology 1999; 53: 1308–1311. [DOI] [PubMed] [Google Scholar]
- 9.Wagle J, Farner L, Flekkoy K, et al. Association between ApoE epsilon4 and cognitive impairment after stroke. Dement Geriatr Cogn Disord 2009; 27: 525–533. [DOI] [PubMed] [Google Scholar]
- 10.Sheng H, Laskowitz DT, Bennett E, et al. Apolipoprotein E isoform-specific differences in outcome from focal ischemia in transgenic mice. J Cereb Blood Flow Metabol 1998; 18: 361–366. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, Durakoglugil MS, Xian X, et al. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. Proc Natl Acad Sci U S A 2010; 107: 12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stary CM, Xu L, Sun X, et al. MicroRNA-200c contributes to injury from transient focal cerebral ischemia by targeting reelin. Stroke 2015; 46: 551–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Won SJ, Kim SH, Xie L, et al. Reelin-deficient mice show impaired neurogenesis and increased stroke size. Exper Neurol 2006; 198: 250–259. [DOI] [PubMed] [Google Scholar]
- 14.Falconer DS. Two new mutants, ‘trembler’ and ‘reeler’, with neurological actions in the house mouse (Mus musculus L.). J Genet 1951; 50: 192–201. [DOI] [PubMed] [Google Scholar]
- 15.Hurteau GJ, Carlson JA, Spivack SD, et al. Overexpression of the microRNA hsa-miR-200c leads to reduced expression of transcription factor 8 and increased expression of E-cadherin. Cancer Res 2007; 67: 7972–7976. [DOI] [PubMed] [Google Scholar]
- 16.Lane-Donovan C, Philips GT, Wasser CR, et al. Reelin protects against amyloid beta toxicity in vivo. Sci Signal 2015; 8: ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hayashi S, McMahon AP. Efficient recombination in diverse tissues by a tamoxifen-inducible form of Cre: a tool for temporally regulated gene activation/inactivation in the mouse. Dev Biol 2002; 244: 305–318. [DOI] [PubMed] [Google Scholar]
- 18.Beffert U, Durudas A, Weeber EJ, et al. Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J Neurosci 2006; 26: 2041–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stowe AM, Altay T, Freie AB, et al. Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol 2011; 69: 975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Faul F, Erdfelder E, Lang AG, et al. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Meth 2007; 39: 175–191. [DOI] [PubMed] [Google Scholar]
- 21.Stowe AM, Wacker BK, Cravens PD, et al. CCL2 upregulation triggers hypoxic preconditioning-induced protection from stroke. J Neuroinflammation 2012; 9: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao S, Chai X, Frotscher M. Balance between neurogenesis and gliogenesis in the adult hippocampus: role for reelin. Dev Neurosci 2007; 29: 84–90. [DOI] [PubMed] [Google Scholar]
- 23.Lee ST, Chu K, Jung KH, et al. MicroRNAs induced during ischemic preconditioning. Stroke 2010; 41: 1646–1651. [DOI] [PubMed] [Google Scholar]
- 24.Hilmarsdottir B, Briem E, Bergthorsson JT, et al. Functional role of the microRNA-200 family in breast morphogenesis and neoplasia. Genes 2014; 5: 804–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rebustini IT, Hayashi T, Reynolds AD, et al. miR-200c regulates FGFR-dependent epithelial proliferation via Vldlr during submandibular gland branching morphogenesis. Development 2012; 139: 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su J, Klemm MA, Josephson AM, et al. Contributions of VLDLR and LRP8 in the establishment of retinogeniculate projections. Neural Dev 2013; 8: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bui T, Sequeira J, Wen TC, et al. ZEB1 links p63 and p73 in a novel neuronal survival pathway rapidly induced in response to cortical ischemia. PloS One 2009; 4: e4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pujadas L, Rossi D, Andres R, et al. Reelin delays amyloid-beta fibril formation and rescues cognitive deficits in a model of Alzheimer's disease. Nat Commun 2014; 5: 3443. [DOI] [PubMed] [Google Scholar]
- 29.Rogers JT, Rusiana I, Trotter J, et al. Reelin supplementation enhances cognitive ability, synaptic plasticity, and dendritic spine density. Learn Mem 2011; 18: 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hethorn WR, Ciarlone SL, Filonova I, et al. Reelin supplementation recovers synaptic plasticity and cognitive deficits in a mouse model for Angelman syndrome. Eur J Neurosc 2015; 41: 1372–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Holtzman DM, Bales KR, Tenkova T, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 2000; 97: 2892–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang F, Eckman C, Younkin S, et al. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci 1997; 17: 7655–7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koistinaho M, Kettunen MI, Goldsteins G, et al. Beta-amyloid precursor protein transgenic mice that harbor diffuse A beta deposits but do not form plaques show increased ischemic vulnerability: role of inflammation. Proc Natl Acad Sci U S A 2002; 99: 1610–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greenberg SM, Rebeck GW, Vonsattel JP, et al. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann Neurol 1995; 38: 254–259. [DOI] [PubMed] [Google Scholar]
- 35.Fryer JD, Simmons K, Parsadanian M, et al. Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci 2005; 25: 2803–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okazaki H, Reagan TJ, Campbell RJ. Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc 1979; 54: 22–31. [PubMed] [Google Scholar]
- 37.Milner E, Zhou ML, Johnson AW, et al. Cerebral amyloid angiopathy increases susceptibility to infarction after focal cerebral ischemia in Tg2576 mice. Stroke 2014; 45: 3064–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grayson DR, Jia X, Chen Y, et al. Reelin promoter hypermethylation in schizophrenia. Proc Natl Acad Sci U S A 2005; 102: 9341–9346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lane-Donovan C, Philips GT, Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron 2014; 83: 771–787. [DOI] [PMC free article] [PubMed] [Google Scholar]