

Abstract
Background
Upper respiratory infections (URI) and their complications are a major healthcare burden for pediatric populations. Although the microbiology of the nasopharynx is an important determinant of the complications of URI, little is known of the nasopharyngeal (NP) microbiota of children, the factors that affect its composition, and its precise relationship with URI.
Results
Healthy children (n = 47) aged 49–84 months from a prospective cohort study based in Wisconsin, USA, were examined. Demographic and clinical data and NP swab samples were obtained from participants upon entry to the study. All NP samples were profiled for bacterial microbiota using a phylogenetic microarray, and these data were related to demographic characteristics and upper respiratory health outcomes. The composition of the NP bacterial community of children was significantly related prior to the history of acute sinusitis (R2 = 0.070, P < 0.009). History of acute sinusitis was associated with significant depletion in relative abundance of taxa including Faecalibacterium prausnitzii and Akkermansia spp. and enrichment of Moraxella nonliquefaciens. Enrichment of M. nonliquefaciens was also a characteristic of baseline NP samples of children who subsequently developed acute sinusitis over the 1-year study period. Time to develop URI was significantly positively correlated with NP diversity, and children who experienced more frequent URIs exhibited significantly diminished NP microbiota diversity (P ≤ 0.05).
Conclusions
These preliminary data suggest that previous history of acute sinusitis influences the composition of the NP microbiota, characterized by a depletion in relative abundance of specific taxa. Diminished diversity was associated with more frequent URIs.
Electronic supplementary material
The online version of this article (doi:10.1186/s40168-016-0179-9) contains supplementary material, which is available to authorized users.
Keywords: Microbiota, Upper respiratory infection, URI, Acute sinusitis, Pediatrics, Children
Background
Upper respiratory infection (URI) is one of the most common reasons for children in the USA to seek medical care [1]. URIs frequently lead to bacterial complications, resulting in billions of dollars in health care expenditures [2]; 30 % of URIs are complicated by acute otitis media and 8 % by rhinosinusitis [2]. The presence of Streptococcus pneumoniae, Haemophilus influenzae, and/or Moraxalla catarrhalis in nasopharyngeal (NP) cultures from children with URIs is predictive of increased likelihood of progression to acute otitis media [2], and these species are also known to potentiate the effect of rhinovirus infection on the likelihood of asthma exacerbation [3].
A cross-sectional study of children and adults (n = 51 children, aged 1–4.5 years; n = 19 adults) in Canada demonstrated that, like the gastrointestinal microbiota [4], changes in NP bacterial microbiota occur with increasing age; less diverse communities are observed in younger children and more even, diverse communities are characteristic of adults [5]. The beta-diversity of bacterial communities was more similar in adults compared to that observed in children, indicating greater heterogeneity in upper airway microbiota in early life [5]. Teo et al. also recently demonstrated, using longitudinally obtained NP samples collected over the first year of life from a cohort of 234 Australian infants, that the NP microbiota undergoes compositional development during this period [6]. Subjects were grouped into one of six bacterial community states, each dominated by a distinct genus (Moraxella, Haemophilus, Streptococcus, Corynebacteria, Alloiococcus, or Staphylococcus) [6]. Infants whose NP microbiota were dominated by Moraxella, Haemophilus, or Streptococcus at the time of viral respiratory infection exhibited significantly higher risk for developing lower airway infection. In addition, early colonization with a Streptococcus-dominated community conferred an increased risk for the development of persistent asthma [6].
A separate study of the NP bacterial microbiota of 200 Dutch children sampled at 1.5, 6, 12, and 24 months identified eight distinct bacterial community profiles in this population [7]. While some of these community states exhibited compositional evolution over time, others remained stable over the 2-year study period. For example, Haemophilus-dominated communities never persisted for more than one time-point and exhibited a high magnitude of temporal compositional instability, while Moraxella or Corynebacterium/Dolosigram dominated communities were typically stable and also were associated with fewer parental reported URIs [7]. These observations indicate that the composition of the pediatric upper airway represents a critical factor that may either potentiate or protect against infection by respiratory pathogens. They also indicate that the interplay between the bacterial microbiota and respiratory pathogens associated with upper airway infection is important to consider. Both bacteria and viruses can influence each other’s pathogenicity [8] and a number of interactions between specific viruses and bacterial species have been reported in the airways [9, 10]. For example, human rhinovirus infection was found to significantly increase the binding of Staphylococcus aureus, S. pneumoniae, or H. influenzae to primary human nasal epithelial cells [11] and in an independent study of human bronchial epithelial cells, pre-incubation with S. pneumoniae significantly increased infection by human metapneumovirus while pre-incubation with H. influenzae, S. aureus, or M. catarrhalis had no effect [12].
The nuances of these relationships, especially in the context of the complex microbial communities of the upper airway (in both uncomplicated URI as well as acute sinusitis), are not completely understood. In this study, we sought to identify variables that influence the composition of the NP microbiota of healthy children and determine whether features of these communities are related to susceptibility to URI or acute sinusitis in a North American cohort of 49–84-month-old subjects.
Results
Variability in NP microbiota composition is significantly associated with a history of acute sinusitis
A total of 951 taxa were identified in baseline NP microbiota of participants (n = 47) in our cohort. These bacterial communities were variably composed of members of the Rickenellaceae, Lachnospiraceae, Verrucomicrobiaceae, Pseudomonadaceae, and Moraxellaceae as well as multiple unclassified members of the phylum Proteobacteria. To identify demographic features that might explain the observed variation in nasopharyngeal bacterial community composition (for demographic information see Additional file 1: Table S1), a pair-wise Canberra distance matrix was constructed and used in permutational multi-variate regression analyses against a total of 58 study variables (Additional file 1: Table S2). History of acute sinusitis (chronic sinusitis was an exclusion criterion) prior to entry into the study exhibited a strong significant relationship (Adonis, R2 = 0.070; P < 0.009) with bacterial community membership in our cohort (Fig. 1, Additional file 1: Table S2).
Fig. 1.
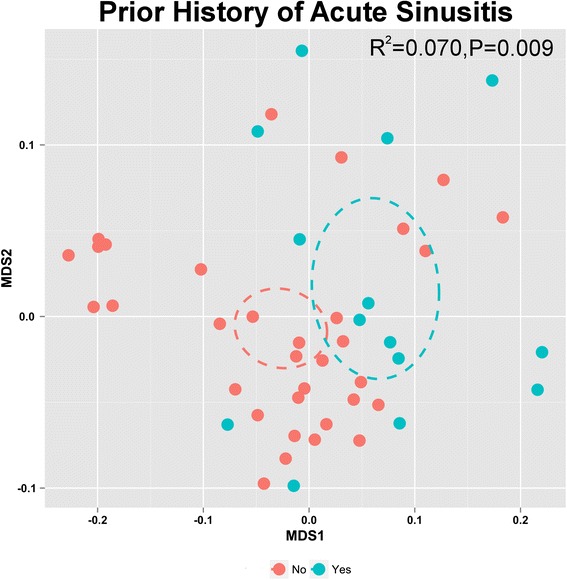
Permutation testing of non-metric dimensional scaling based on a Canberra distance matrix identifies acute sinusitis history (yes n = 14, no n = 33) as significantly (P ≤ 0.01) related to the observed variation in NP bacterial community composition upon entry to the study. Ellipses indicate 95 % confidence interval, stress = 0.1226
Our study used independent NP samples collected from individual participants over a 12-month study period that spanned all four seasons (see Additional file 2: Figure S1 for sampling schematic). Season of sample collection also demonstrated a relationship with bacterial beta-diversity (Adonis, R2 = 0.137, P < 0.006). However, samples were analyzed in batches as they were collected, and we noted that a significant relationship between batch and season existed (Fisher’s exact test P = 8.3 × 10−9) which confounded this observation. A similar analysis did not find a significant relationship between batch and on the history of acute sinusitis (Fisher’s exact test P > 0.12).
Compared with children who had no history of acute sinusitis (n = 33), those with a past history of acute sinusitis (n = 14) did not exhibit differences in α-diversity indices (Wilcoxon rank-sum test P > 0.5 for richness, Inverse Simpson, or Faith’s phylogenetic diversity), suggesting that differences in microbiota characterizing these groups may be due to the enrichment or depletion of a subset of taxa within these bacterial communities. A total of 309 taxa (representing 101 genera) exhibited significant differences in relative abundance between children with and without a history of acute sinusitis. NP samples from children with a prior history of acute sinusitis were characterized by significant depletion of 308 of the 309 taxa, including those represented by Akkermansia, Faecalibacterium prausnitzii, Clostridium, Lactobacillus, Prevotella, and Streptococcus species. The only taxon that exhibited a significant (Welch’s t test, P < 0.05, q < 0.05) increase in relative abundance in these subjects was represented by Moraxella nonliquefaciens (Fig. 2a; Additional file 1: Table S3; Additional file 3: Figure S2A).
Fig. 2.
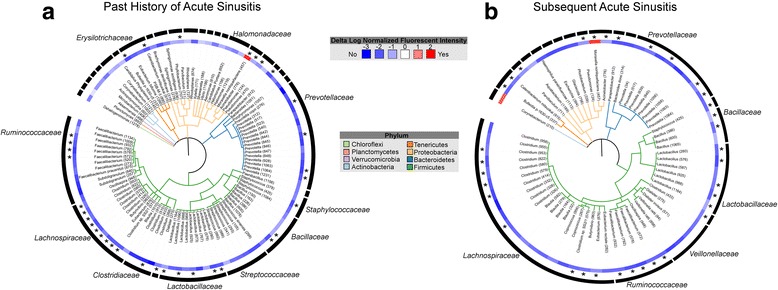
Phylogenetic trees displaying genera that exhibit significant differences in relative abundance across scomparison groups. a Children with a history of acute sinusitis (n = 14) have NP microbial communities significantly (Welch’s t test; P < 0.05, q < 0.05) enriched (red) for Moraxella nonliquefaciens and depleted (blue) of 100 genera compared with those without a history of sinusitis (n = 33). b Children who developed acute sinusitis during the 1-year period following baseline sampling (n = 7) have communities that are significantly (Welch’s t test; P ≤ 0.05, q < 0.25) enriched for Moraxella nonliquifaciens and Corynebacterium spp. (red) and depleted of 59 genera (blue) relative to children who did not develop acute sinusitis (n = 33). Branches are colored by phylum. Numbers specifying unique eOTUs within each genus are shown in parentheses; eOTUs consistently identified across both comparisons are indicated with an asterisk
Based on the relationship between a prior history of acute sinusitis and composition of the microbiota, we next examined differences in the relative abundance of baseline taxa associated with the development of acute sinusitis subsequent to baseline sampling (occurring during the 1 year clinical follow-up period; n = 33 healthy, n = 7 subsequent sinusitis). Subsequent acute sinusitis was associated with substantial taxon depletion (n = 143) and again included those belonging to Faecalibacterium, Clostridium, Lactobacillus, and Prevotella. Children who subsequently developed acute sinusitis displayed significant enrichment (Welch’s t test, P ≤ 0.05, q < 0.25) of Moraxella nonliquefaciens (Fig. 2b; Additional file 1: Table S4; Additional file 3: Figure S2B) and Corynebacterium spp. (Fig. 2b; Additional file 1: Table S4) in their baseline NP microbiota. It should be noted that this is a relatively high q value cut-off (q < 0.25) due to the small number of children (n = 7) who developed sinusitis during our study period, hence larger cohorts are needed to validate this finding. A large majority of the empiric Operational Taxonomic Units (eOTUs) depleted in subjects with a history of acute sinusitis were also depleted in children who developed acute sinusitis subsequent to baseline sampling, including multiple members of the Lactobacillus genus (Fig. 2b; Additional file 1: Table S4). Of note, a prior history of acute sinusitis was not significantly associated with the development of a subsequent episode of acute sinusitis following baseline sampling (Fisher’s exact test, P = 0.34), indicating that distinct groups of subjects were included in these analyses and that the taxonomic depletions and enrichments associated with acute sinusitis are consistent features of the disease.
Compositional differences based on URI frequency
We next examined whether features of the baseline composition of the NP microbiota were associated with susceptibility to URI by examining samples from 40 subjects (7 of 47 subjects were lost to follow-up) who reported a range of URI frequency (from 0–9; mean of 2.25) within the 12-month clinical follow-up period. Bacterial community richness, Shannon diversity, Inverse Simpson’s diversity, and Faith’s phylogenetic diversity indices were all significantly negatively associated with the frequency of URIs (negative binomial regression; P < 0.05; Table 1). Children who experienced at least one URI (n = 17) within 60 days of collection of the baseline sample had significantly lower phylogenetic diversity (Fig. 3a; Welch’s t test, P = 0.05; Shapiro-Wilk test P > 0.17) compared to those who had no URIs within that time frame (n = 23). Time to development of URI, defined as the number of days between the collection of the baseline sample and the first incidence of URI (a value of 365 days was assigned to those children who did not experience a URI during the year of monitoring), was also significantly correlated with phylogenetic diversity (Fig. 3b; Spearman Correlation, r = 0.421, P = 0.007). In addition, significantly decreased phylogenetic diversity (Fig. 3c; Wilcoxon rank-sum test, P = 0.05) was also observed among children who experienced a high frequency of URI (four or more URI, n = 13) relative to children who did not experience any URI (n = 8) over the 1-year study period following sample collection. Hence, these data indicate that diminished diversity of the NP microbiota is a precursor to URI in these children.
Table 1.
Negative binomial modeling indicates that NP bacterial alpha-diversity indices are inversely related to frequency of URI (n = 40)
| Diversity index | Estimate | P | Goodness of fit (X 2) |
|---|---|---|---|
| Shannon diversity index | −0.475 | 0.040 | 0.27 |
| Richness | −0.004 | 0.045 | 0.28 |
| Inverse Simpson’s index | −0.004 | 0.044 | 0.28 |
| Faith’s phylogenetic diversity index | −0.050 | 0.023 | 0.29 |
Fig. 3.
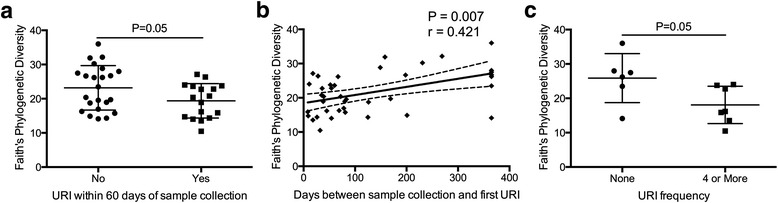
a Children who experience at least one URI within 60 days of sample collection (n = 17) exhibit significantly lower phylogenetic diversity than those who do not (n = 23) experience any URI within this time period (Welch’s t test, P = 0.05; Shapiro-Wilk test P > 0.17). b Increased phylogenetic diversity is significantly correlated with a greater number of days between sample collection and first subsequent URI (Spearman Correlation, r = 0.421, P = 0.007). Trendline and confidence intervals indicate that this relationship is also significant based on regression analysis (F test; R 2 = 0.233; P = 0.0016). c Children with four or more URIs (n = 13) exhibit significantly lower phylogenetic diversity than those who experienced no URIs (n = 8) over the 12-month clinical monitoring period (Wilcoxon rank-sum test, P = 0.05)
Discussion
Previous studies in a cohort of 234 Australian infants (up to 12 months of age) have demonstrated relationships between very early-life NP microbiota, characteristically dominated by specific bacterial genera, and respiratory illness [6]. Here we report similar relationships between acute sinusitis, URI, and the NP microbiota of children between 49 and 84 months of age in a US cohort using a distinct platform to generate bacterial microbiota profiles. Specifically, we identify a taxon represented by M. nonliquefaciens as significantly enriched both in children who report a past history of acute sinusitis and among those who developed sinusitis following baseline sampling, implicating this organism as potentially important in the pathogenesis of sinus infection. Teo and colleagues have previously demonstrated that Moraxella can stably colonize the microbiota of the infant NP, with 31 % of their cohort exhibiting this genus as the dominant member of the NP bacterial community [6]. In addition, the majority of infants colonized with Moraxella experienced a subsequent acute respiratory infection prior to the collection of additional samples within that study (samples were obtained at 2, 6, and 12 months of age) [6]. The authors also found that NP communities dominated by Moraxella (as well as those dominated by Streptococcus or Haemophilus) are also associated with significantly more frequent acute respiratory infections compared to healthy NP samples, even after adjusting for a large set of potential confounders (age, gender, season, number of prior infections, antibiotic intake, mother’s antibiotic intake, delivery mode, and breastfeeding). An independent study of children aged 4 to 12 years (n = 308) also demonstrated that those with upper airway microbiota significantly enriched for Moraxella catarrhalis experienced more severe and protracted symptoms of viral URI [3]. In contrast, Biesbroek and colleagues found that children with Moraxella-dominated NP bacterial communities suffered fewer URIs in 6 months following sample collection [7]. These apparently conflicting data underscore the limitations of biomarker gene sequencing approaches, which frequently fail to discriminate between discrete species or strains of a given genus, and provide no information on function of these organisms in the context of the specific microbial species with which they co-associate. In addition, previous studies have also demonstrated substantial variation in virulence capacity across members of the same species [13, 14] including Moraxella catarrhalis [15, 16], suggesting that distinct Moraxella species or strains may elicit dichotomous outcomes in pediatric patients. Hence, metagenomic studies of the functional genetic capacity as well as comparative genomic studies of Moraxella isolates are necessary to identify factors that potentiate the effect of upper airway infection and enhance or prolong respiratory symptoms. Such efforts may lead to improved prediction of susceptibility to infection.
Our data did not find a relationship between URI frequency and Moraxella enrichment; what was apparent, however, was an association between this genus and acute sinusitis. Since the study by Biesbroek and colleagues used retrospective parent-reported mild respiratory tract infections as outcomes, the role of Moraxella in a more severe infection or sinusitis outcomes cannot be assessed in that study [7].
In addition to Moraxella, a Corynebacterium was enriched in relative abundance in the NP microbiota of children who experienced acute sinusitis subsequent to baseline sample collection during the study period. The small numbers in this study and borderline false discovery correction values mandate caution in the interpretation of these findings. However, Abreu et al. previously found Corynebacterium tuberculostearicum to be significantly enriched in the maxillary sinuses of adults with chronic rhinosinusitis compared to healthy control subjects [17]. The authors subsequently confirmed the ability of C. tuberculostearicum to induce acute sinusitis in the context of an antimicrobial-depleted murine model of sinus infection. Moreover co-installation of Lactobacillus sakei (one of a number of taxa acutely depleted in relative abundance among chronic rhinosinusitis patients) protected animals against C. tuberculostearicum infection [17]. Our pediatric data exhibits similarity with these murine studies, in that six members of the Lactobacillus genus were among those taxa most significantly depleted in relative abundance in the NP bacterial communities of children who developed sinusitis during our study. Five of these same taxa were also depleted in relative abundance in the NP microbial communities of children with a prior history of sinusitis. In addition to Lactobacillus, many other bacterial taxa including Akkermansia, Faecalibacterium prausnitzii, Clostridium, Prevotella, and Streptococcus species were depleted in relative abundance among children with a prior history of acute sinusitis. Though traditionally associated with gut microbiota, anaerobic bacterial species can exist in biofilms in the upper respiratory tract [18] and Akkermansia and Faecalibacterium have previously been detected in the nasopharynx of children [19, 20]. While its role in the airway is unknown, gastrointestinal Akkermansia muciniphilia metabolizes mucin and has been shown to activate immune homeostasis, increasing host expression of antimicrobial peptides such as RegIIIγ and improving barrier function via an increase in 2-oleoylgylcercerol [21–23]. However, whether such mechanisms play a role at the airway mucosal surface remains to be determined. Mechanisms by which Lactobacillus and other bacterial species depleted in the NP microbiota of sinusitis patients may prevent the development of disease include competitive exclusion of pathogenic species. A previous murine study indicated that intra-nasal inoculation of mice with L. fermentum decreased S. pneumoniae burden throughout the respiratory tract and increased the number of activated macrophages in the lung and lymphocytes in the tracheal lamina propria [24]. Hence, it is plausible that the absence of NP genera with known competitive exclusion and immunomodulatory capabilities leads to pathogen expansion and associated clinical manifestations of upper airway infection.
Permutational analyses indicated that the composition of the NP microbiota is significantly associated with the season of sample collection. While this observation in our study was confounded by a batch effect, our finding corroborates those of an Australian infant cohort, indicating that the composition of the NP microbiota in the first year of life is related to season [6]. Seasonality has previously been related to respiratory illness [25], and in our study, we demonstrate that increased NP diversity is associated with protection against URI. These data raise the intriguing possibility that the composition of the microbiota of the upper airway is related both to local climactic conditions and seasonal susceptibility to upper airway infection. However, studies involving repeated measures from individual participants are necessary to corroborate our observations and to determine composition dynamics of the NP microbiota that contribute to this phenomenon.
Limitations of our study include limited ability to discern causal connections between enriched Operational Taxonomic Units (OTUs) and sinusitis history. We do show that a history of sinusitis, its pathophysiology or treatment, may shape the NP microbiota—which may inform future studies and their design. Additionally, though we recognize that the composition of the microbiota in the upper airways is likely highly influenced by antibiotic administration, obtaining a complete history of previous antibiotic administration is difficult and often imprecise due to poor adherence and transition across multiple medical providers and systems and thus was not examined in this study. The pervasive effects of antimicrobials on the human microbiota are well-described [26, 27], and it is likely that lifetime antibiotic use plays an important role in shaping the baseline NP microbial community. However, such studies are best suited to birth cohorts, where this information is collected prospectively and can be related retrospectively both to microbiota composition and health outcomes. This study used the 16S rRNA PhyloChip, rather than the next generation 16S rRNA gene sequencing. The microarray platform offers the advantage of detecting OTUs in parallel, allowing for the detection of rare, low abundance organisms that may not be detected by shallower sequencing efforts [28]. However, the PhyloChip is limited in only determining differences in relative abundance within each OTU and cannot provide insight into community structure, absolute abundance of OTUs, or discover novel OTUs. Several studies have, however, validated the application of this assay, demonstrating consistency in findings generated by this approach and parallel sequencing of the same samples [28, 29]
Conclusions
The composition of the NP microbiota in healthy children between 49 and 84 months of age is associated with past and subsequent history of acute sinusitis and frequency of URI. Widespread bacterial taxon depletion and enrichment of M. liquefaciens and C. tuberculostearicum are associated with upper airway infection and the development of acute sinusitis. Collectively, these findings provide evidence of close connections between microbial colonization of the airways and susceptibility to upper respiratory illnesses in early childhood and raise the possibility that the manipulation of the airway microbiota could be applied to the prevention of childhood respiratory illnesses.
Methods
Study population
Forty-seven healthy children aged 49–84 months were recruited during well-child visits from two primary care clinics in Madison, WI, for a prospective, longitudinal cohort study. This study was approved by the University of Wisconsin Institutional Review Board. Informed parental (or legal guardian) written consent was obtained as well as assent from older children (signed consent documentation was retained as part of the patient’s file). Information on demographic variables, pets in the household, medical history, and various exposures (e.g., farm animals, medications; Additional file 1: Table S1) was collected. Children were enrolled between February 2012 and February 2013 and followed for 1 year (Additional file 2: Figure S1). Seven children discontinued participation before completing a full year of the study due to discomfort with the sampling procedure. NP surveillance samples were obtained from the children at entry to the study (n = 47); enrollment samples were collected from participants across all four seasons (defined by equinox and solstice dates: spring commencing on March 20th, summer, June 20th, fall, September 22nd and winter, December 21st) [30]. Longitudinal clinical follow-up was conducted for 12 months following entry into the study and baseline sample collection. To monitor URI frequency, parents were instructed to call the study nurse when respiratory symptoms persisted for at least 48 h and record their children’s respiratory symptoms. Parents were also contacted monthly by the study team to determine the occurrence of episodes of infection, which they may have previously failed to report. URI was defined as nasal congestion, nasal discharge, or cough (with or without fever) lasting 48 hours or more. Sinusitis was defined in one of three ways: (1) respiratory symptoms (including nasal discharge or cough or both) having lasted more than 10 days and not improving, (2) a combination of purulent (thick, colored, and opaque) nasal discharge plus temperature >39 °C for at least 72 h, or (3) sudden worsening of a viral URI after apparent improvement usually beyond the sixth day of illness [31]. The medical record was investigated to determine if there were unreported instances of antimicrobial use. Children were excluded from the study if they had a congenital or acquired immunodeficiency, craniofacial abnormalities, cystic fibrosis, allergic rhinitis, received antibiotics in the past 30 days, developed a URI within 1 week of baseline sampling, or a previous diagnosis of chronic sinusitis.
Sample processing
A flocked swab was placed into the nasopharynx and rotated. The swab was cut off with sterile scissors and placed into sterile DNAase/RNase-free cryovials containing 2 ml of RNALater (Ambion). Samples were collected at the University of Wisconsin, Madison, and stored at 4 °C for 24 h to permit preservative to penetrate cells, prior to freezing at −80 °C and then shipped in batches to the University of California San Francisco for microbiota analysis. DNA was extracted using a combination of bead beating and the AllPrep kit (Qiagen) as previously described [17]. Extracted DNA was used for high-resolution microbiota profiling using the 16S rRNA PhyloChip (Second Genome) and TaqMan qPCR. To assess relationships between environmental exposures or URI and NP microbiota, a single baseline sample from each child (n = 47) collected during a healthy (non-URI) period was used.
Microbiota profiling
Microbiota profiling with 16S rRNA PhyloChip (version G3) which can detect up to 60,000 bacterial and archeal taxa was performed as previously described [32, 33]. Briefly, extracted DNA was used as template (30 ng) in eight PCR reactions which run across a gradient of annealing temperatures (48.7–57.1 °C). Amplicons were pooled per sample, gel purified, and quantified with Alpha Ease software version 4.1.0 (Alpha Innotech Corporation) using E-gels (Invitrogen). A total of 250 ng of fragmented, biotin-labeled 16S rRNA product per sample was applied to each PhyloChip. Detection and quantification criteria for each OTU detected by PhyloChip were defined using the empiric Operational Taxonomic Unit approach as previously described [34]. Briefly, groups of probes are assigned to a specific eOTU based on taxonomic relatedness as well as their correlation in fluorescent intensity throughout a given experiment and then taxonomically annotated by a Bayesian method based on the sequence of the probes within each eOTU probe grouping. An 80 % certainty cut-off was used for taxonomic annotation. A negative control sample was carried through from extraction to microbiota profiling on the PhyloChip; a total of seven eOTUs were detected in this sample (see Additional file 1: Table S5). These eOTUs were removed from the dataset and not considered in data analyses.
Viral detection
Viral detection by the PCR-based Respiratory Multicode Assay (EraGen Biosciences) [35] as well as human rhinovirus detection by PCR [36] was performed as previously described.
Bacterial burden
Total 16S copy number was determined as previously described using TaqMan universal PCR Master mix (Applied Biosystems) with the following primers: P981F, 5′-TGGAGCATGTGGTTTAATTCGA; P1033R, 5′-TGCGGGACTTAACCCAACA; UniProbe, 5′-CACGAGCTGACGACARCCATGCA [37]. To determine if bacterial burden had an effect on the observed variation in the bacterial community composition of the nasopharynx, a pair-wise Canberra distance matrix was constructed and used in permutational multi-variate regression analyses and no significant relationship was found (Adonis, P = 0.529, R2 = 0.050).
Statistical analysis
Much of the statistical analyses were performed using custom scripts and packages in the R statistical environment as we have previously described [33]. Faith’s phylogenetic diversity is defined as “the sum of the lengths of all those branches that are members of the corresponding minimum spanning path”; the tree referring to a bacterial phylogenetic tree was calculated using QIIME [38, 39]. All other diversity indices were calculated using the Vegan (http://vegan.r-forge.r-project.org/) package in the R statistical environment. The indices used are defined as follows: Richness is the total number of unique OTUs; Inverse Simpson’s index is 1/D = ∑ (n/N)2, where D = Simpson’s index, n = the total number of organisms of a particular OTU, and N = the total number of organisms of all OTUs; Shannon’s diversity is calculated by the equation H = −∑*(pi ln(pi)), where H = Shannon’s index and p is the proportional abundance of OTU i.
Fluorescence intensities were used to calculate weighted, pair-wise Canberra distances with the following equation: Djk = (1/NZ) * ΣAll Non-Zero OTUs(|xij − xik|/(xij + xik)) where j = sample 1, k = sample 2, NZ = the number of OTUs with fluorescence intensities greater than 0 in at least one sample, x = fluorescence intensity, and i = OTU. Canberra distance calculates pair-wise distances, based on the average absolute difference in abundance between OTUs present normalized to the combined OTU abundance. Canberra distances were visualized using non-metric multi-dimensional scaling to illustrate community dissimilarity. Negative binomial regression was used to assess trends in microbiota alpha-diversity relative to URI frequency [40]. As previously described, two tailed Welch’s t tests adjusted for false discovery with q values were used on log-normalized fluorescence data to assess differences in taxon relative abundance between specific groups (an underlying normal distribution of probe set fluorescent intensities is assumed) [17, 33, 41]. Because each probe set (corresponding to a given eOTU) is composed of a group of oligonucleotides with different sequences and therefore different hybridization affinity with their target 16S rRNA, comparisons in relative abundance cannot be made across eOTUs, within a bacterial community. Prism version 6.0 (GraphPad Software, www.graphpad.com) was used to construct figures and perform statistical tests. The Interactive Tree of Life (iTOL, http://itol.embl.de) was used to construct and visualize bacterial phylogenetic trees [42].
Abbreviations
eOTU, empiric operational taxonomic unit; NP, nasopharyngeal; OTU, operational taxonomic unit; URI, upper respiratory infection
Acknowledgements
We thank the subjects and their parents whose participation made this study possible.
Funding
This work was supported by funding from the National Institutes of Health, award number 5RO1AI097172-03. The National Institutes of Health played no role in the design of the study and the collection, analysis, and interpretation of the data or in writing the manuscript.
Availability of data
Our microarray data are publicly available at http://greengenes.secondgenome.com/downloads/phylochip_datasets under the file name UCSF_CASantee_2016_Microbiome.tgz
Authors’ contributions
CAS performed the statistical analyses, prepared the figures, and drafted the manuscript. NAN performed the G3 PhyloChip 16S rRNA profiling. AAF generated a custom script for the negative binomial distribution analysis used in this study. GPD collected the samples and patient data and helped draft the manuscript. JEG performed the viral detection assays and contributed to the manuscript preparation. ERW conceived the study, participated in its design and coordination, and contributed to the manuscript preparation. SVL conceived the study, participated in its design and coordination, and constructed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Ethics approval and consent to participate
This study was approved by the University of Wisconsin Institutional Review Board. Informed parental (or legal guardian) written consent was obtained as well as assent from older children (signed consent documentation was retained as part of the patient’s file).
Additional files
Summary table of demographic characteristics, health history, and environmental exposures of study subjects. Table S2. Relationship between community composition variance using Adonis (PermANOVA) and a Canberra distance matrix of surveillance (healthy) samples (n = 47) and 58 measured variables detailed below. Note, only one individual was exposed to a cat at school and that same individual was the only subject with a positive corona virus (CoV) result in the Respiratory MultiCode Assay (RMA) viral screen, so the findings for these variables are inconclusive. Findings ranked by P value. Table S3. Genera enriched in children with a history of acute sinusitis (n = 14 shown in gray and those enriched in children without (n = 33) shown in white by Welch’s t test (P < 0.05, q < 0.05, ranked by P). Table S4. Taxa enriched in children with at least one episode of acute sinusitis during 1 year after sample collection (n = 7), shown in gray and those enriched in children who did not experience acute sinusitis (n = 33) during 1 year after sample collection by Welch’s t test, shown in white (P ≤ 0.05, q < 0.25, ranked by P). Table S5. Taxa detected on negative control PhyloChip and removed from further data analysis. (DOCX 76 kb)
Schematic of sampling and clinical follow-up time periods. Number in circle indicates the number of independent samples collected during that month, and the line indicates the time span of subsequent clinical monitoring. Colors indicate season. (TIFF 552 kb)
Normalized fluorescent intensity for Moraxella nonliquefaciens (eOTU 457) plotted for each subject showing significant differences (Welch’s t test, P ≤ 0.05) between (A) children with a prior history of acute sinusitis and those without a prior history of acute sinusitis as well as (B) children who developed acute sinusitis during the year following sample collection and those who did not. (TIFF 340 kb)
References
- 1.Ball TM, Holberg CJ, Aldous MB, Martinez FD, Wright AL. Influence of attendance at day care on the common cold from birth through 13 years of age. Arch Pediatr Adolesc Med. 2002;156:121–6. doi: 10.1001/archpedi.156.2.121. [DOI] [PubMed] [Google Scholar]
- 2.Wald ER. Acute otitis media and acute bacterial sinusitis. Clin Infect Dis. 2011;52(Suppl 4):S277–83. doi: 10.1093/cid/cir042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kloepfer KM, Lee WM, Pappas TE, Kang TJ, Vrtis RF, Evans MD, et al. Detection of pathogenic bacteria during rhinovirus infection is associated with increased respiratory symptoms and asthma exacerbations. J Allergy Clin Immunol. 2014;133(5):1301–7. doi: 10.1016/j.jaci.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Backhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, et al. Dynamics and stabilization of the human gut microbiome during the first year of life. Cell Host Microbe. 2015;17(5):690–703. doi: 10.1016/j.chom.2015.04.004. [DOI] [PubMed] [Google Scholar]
- 5.Stearns JC, Davidson CJ, McKeon S, Whelan FJ, Fontes ME, Schryvers AB, et al. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015;9(5):1268. doi: 10.1038/ismej.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teo Shu M, Mok D, Pham K, Kusel M, Serralha M, Troy N, et al. The infant nasopharyngeal microbiome impacts severity of lower respiratory infection and risk of asthma development. Cell Host Microbe. 2015;17(5):704–15. doi: 10.1016/j.chom.2015.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biesbroek G, Tsivtsivadze E, Sanders EA, Montijn R, Veenhoven RH, Keijser BJ, et al. Early respiratory microbiota composition determines bacterial succession patterns and respiratory health in children. Am J Respir Crit Care Med. 2014;190(11):1283–92. doi: 10.1164/rccm.201407-1240OC. [DOI] [PubMed] [Google Scholar]
- 8.Kane M, Case LK, Kopaskie K, Kozlova A, MacDearmid C, Chervonsky AV, et al. Successful transmission of a retrovirus depends on the commensal microbiota. Science. 2011;334(6053):245–9. doi: 10.1126/science.1210718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bosch AA, Biesbroek G, Trzcinski K, Sanders EA, Bogaert D. Viral and bacterial interactions in the upper respiratory tract. PLoS Pathog. 2013;9(1):e1003057. doi: 10.1371/journal.ppat.1003057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lynch SV. Viruses and microbiome alterations. Ann Am Thorac Soc. 2014;11(Suppl 1):S57–60. doi: 10.1513/AnnalsATS.201306-158MG. [DOI] [PubMed] [Google Scholar]
- 11.Wang JH, Kwon HJ, Jang YJ. Rhinovirus enhances various bacterial adhesions to nasal epithelial cells simultaneously. Laryngoscope. 2009;119(7):1406–11. doi: 10.1002/lary.20498. [DOI] [PubMed] [Google Scholar]
- 12.Verkaik NJ, Nguyen DT, de Vogel CP, Moll HA, Verbrugh HA, Jaddoe VW, et al. Streptococcus pneumoniae exposure is associated with human metapneumovirus seroconversion and increased susceptibility to in vitro HMPV infection. Clin Microbiol Infect. 2011;17(12):1840–4. doi: 10.1111/j.1469-0691.2011.03480.x. [DOI] [PubMed] [Google Scholar]
- 13.Eveillard M, Soltner C, Kempf M, Saint-Andre JP, Lemarie C, Randrianarivelo C, et al. The virulence variability of different Acinetobacter baumannii strains in experimental pneumonia. J Infect. 2010;60(2):154–61. doi: 10.1016/j.jinf.2009.09.004. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez-Pando R, Marquina-Castillo B, Barrios-Payan J, Mata-Espinosa D. Use of mouse models to study the variability in virulence associated with specific genotypic lineages of Mycobacterium tuberculosis. Infect Genet Evol. 2012;12(4):725–31. doi: 10.1016/j.meegid.2012.02.013. [DOI] [PubMed] [Google Scholar]
- 15.Verduin CM, Kools-Sijmons M, van der Plas J, Vlooswijk J, Tromp M, van Dijk H, et al. Complement-resistant Moraxella catarrhalis forms a genetically distinct lineage within the species. FEMS Microbiol Lett. 2000;184(1):1–8. doi: 10.1111/j.1574-6968.2000.tb08981.x. [DOI] [PubMed] [Google Scholar]
- 16.de Vries SP, Bootsma HJ, Hays JP, Hermans PW. Molecular aspects of Moraxella catarrhalis pathogenesis. Microbiol Mol Biol Rev. 2009;73(3):389–406. doi: 10.1128/MMBR.00007-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abreu NA, Nagalingam NA, Song Y, Roediger FC, Pletcher SD, Goldberg AN, et al. Sinus microbiome diversity depletion and Corynebacterium tuberculostearicum enrichment mediates rhinosinusitis. Sci Transl Med. 2012;4:151ra24. doi: 10.1126/scitranslmed.3003783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YC, Post JC. Biofilms in pediatric respiratory and related infections. Curr Allergy Asthma Rep. 2009;9(6):449–55. doi: 10.1007/s11882-009-0066-6. [DOI] [PubMed] [Google Scholar]
- 19.Bogaert D, Keijser B, Huse S, Rossen J, Veenhoven R, van Gils E, et al. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One. 2011;6(2):e17035. doi: 10.1371/journal.pone.0017035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sakwinska O, Schmid VB, Berger B, Bruttin A, Keitel K, Lepage M, et al. Nasopharyngeal microbiota in healthy children and pneumonia patients. J Clin Microbiol. 2014;52(5):1590–4. doi: 10.1128/JCM.03280-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Derrien M, Van Baarlen P, Hooiveld G, Norin E, Müller M, de Vos WM. Modulation of mucosal immune response, tolerance, and proliferation in mice colonized by the mucin-degrader Akkermansia muciniphila. Front Microbiol. 2011;2:166. doi: 10.3389/fmicb.2011.00166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol. 2004;54:1469–76. doi: 10.1099/ijs.0.02873-0. [DOI] [PubMed] [Google Scholar]
- 23.Everard A, Belzer C, Geurts L, Ouwerkerk JP, Druart C, Bindels LB, et al. Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci U S A. 2013;110(22):9066–71. doi: 10.1073/pnas.1219451110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Gutierrez RC, Santos V, Nader-Macías ME. Protective effect of intranasally inoculated Lactobacillus fermentum against Streptococcus pneumoniae challenge on the mouse respiratory tract. FEMS Immunol Med Microbiol. 2001;31(3):187–95. doi: 10.1111/j.1574-695X.2001.tb00519.x. [DOI] [PubMed] [Google Scholar]
- 25.Paynter S, Ware RS, Sly PD, Williams G, Weinstein P. Seasonal immune modulation in humans: observed patterns and potential environmental drivers. J Infect. 2015;70(1):1–10. doi: 10.1016/j.jinf.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 26.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4554–61. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saulnier DM, Riehle K, Mistretta TA, Diaz MA, Mandal D, Raza S, et al. Gastrointestinal microbiome signatures of pediatric patients with irritable bowel syndrome. Gastroenterology. 2011;141(5):1782–91. doi: 10.1053/j.gastro.2011.06.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fujimura KE, Rauch M, Matsui E, Iwai S, Calatroni A, Lynn H, et al. Development of a standardized approach for environmental microbiota investigations related to asthma development in children. J Microbiol Methods. 2012;91(2):231–9. doi: 10.1016/j.mimet.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.United States Naval Observatory. Earth’s seasons: equinoxes, solstices, perphelion, and aphelion, 2000-2020. United States Naval Observatory. 2013. http://aa.usno.navy.mil/data/docs/EarthSeasons.php. Accessed 22 Sept 2014.
- 31.Wald ER, Applegate KE, Bordley C, Darrow DH, Glode MP, Marcy SM, et al. Clinical practice guideline for the diagnosis and management of acute bacterial sinusitis in children aged 1 to 18 years. Pediatrics. 2013;132(1):e262–e80. doi: 10.1542/peds.2013-1071. [DOI] [PubMed] [Google Scholar]
- 32.Hazen TC, Dubinsky EA, DeSantis TZ, Andersen GL, Piceno YM, Singh N, et al. Deep-sea oil plume enriches indigenous oil-degrading bacteria. Science. 2010;330(6001):204–8. doi: 10.1126/science.1195979. [DOI] [PubMed] [Google Scholar]
- 33.Cox MJ, Huang YJ, Fujimura KE, Liu JT, McKean M, Boushey HA, et al. Lactobacillus casei abundance is associated with profound shifts in the infant gut microbiome. PLoS One. 2010;5(1):e8745. doi: 10.1371/journal.pone.0008745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miezeiewski M, Schnaufer T, Muravsky M, Wang S, Caro-Aguilar I, Secore S et al. An in vitro culture model to study the dynamics of colonic microbiota in Syrian golden hamsters and their susceptibility to infection with Clostridium difficile. ISME J. 2014:1–12. doi:10.1038/ismej.2014.127. [DOI] [PMC free article] [PubMed]
- 35.Lee W-M, Grindle K, Pappas T, Marshall DJ, Moser MJ, Beaty EL, et al. High-throughput, sensitive, and accurate multiplex PCR-microsphere flow cytometry system for large-scale comprehensive detection of respiratory viruses. J Clin Microbiol. 2007;45(8):2626–34. doi: 10.1128/JCM.02501-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bochkov YA, Grindle K, Vang F, Evans MD, Gern JE. Improved molecular typing assay for rhinovirus species A, B, and C. J Clin Microbiol. 2014;52(7):2461–71. doi: 10.1128/JCM.00075-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang S, Lin S, Kelen GD, Quinn TC, Dick JD, Gaydos CA, et al. Quantitative multiprobe PCR assay for simultaneous detection and identification to species level of bacterial pathogens. J Clin Microbiol. 2002;40(9):3449–54. doi: 10.1128/JCM.40.9.3449-3454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, et al. CorrespondEnce QIIME allows analysis of high-throughput community sequencing data intensity normalization improves color calling in SOLiD sequencing. Nat Publishing Group. 2010;7:335–6. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Faith DP. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. doi: 10.1016/0006-3207(92)91201-3. [DOI] [Google Scholar]
- 40.Hilbe J. Negative binomial regression. 2. Cambridge: Cambridge University Press; 2011. [Google Scholar]
- 41.Fujimura KE, Demoor T, Rauch M, Faruqi A, Jang S, Johnson CC, et al. House dust exposure mediates gut microbiome Lactobacillus enrichment and airway immune defense against allergens and virus infection. Proc Natl Acad Sci U S A. 2014;111:805–10. doi: 10.1073/pnas.1310750111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Letunic I, Bork P. Interactive tree of life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39(Web Server issue):W475–8. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]