

Abstract
The etiology of diabetic peripheral neuropathy (DPN) involves an inter-related series of metabolic and vascular insults that ultimately contribute to sensory neuron degeneration. In the quest to pharmacologically manage DPN, small molecule inhibitors have targeted proteins and pathways regarded as “diabetes specific” as well as others whose activity are altered in numerous disease states. These efforts have not yielded any significant therapies, due in part to the complicating issue that the biochemical contribution of these targets/pathways to the progression of DPN does not occur with temporal and/or biochemical uniformity between individuals. In a complex, chronic neurodegenerative disease such as DPN, it is increasingly appreciated that effective disease management may not necessarily require targeting a pathway or protein considered to contribute to disease progression. Alternatively, it may prove sufficiently beneficial to pharmacologically enhance the activity of endogenous cytoprotective pathways to aid neuronal tolerance to and recovery from glucotoxic stress. In pursuing this paradigm shift, we have shown that modulating the activity and expression of molecular chaperones such as heat shock protein 70 (Hsp70) may provide translational potential for the effective medical management of insensate DPN. Considerable evidence supports that modulating Hsp70 has beneficial effects in improving inflammation, oxidative stress and glucose sensitivity. Given the emerging potential of modulating Hsp70 to manage DPN, the current review discusses efforts to characterize the cytoprotective effects of this protein and the benefits and limitations that may arise in drug development efforts that exploit its cytoprotective activity.
Keywords: diabetic neuropathy, molecular chaperones, heat shock proteins, Hsp70, novologues, inflammation, bioenergetics
1.0– Introduction
Over 300 million individuals worldwide are estimated to be affected by diabetes, with projections that exceed half a billion people by 2030 (Whiting et al., 2011). About half of these individuals will develop diabetic peripheral neuropathy (DPN). DPN symptoms can be quite varied and manifest in the extremities as weakness/numbness (insensate DPN) or painful tingling, burning or lancinating sensations (painful DPN). Though the painful aspects of DPN can be quite debilitating, the loss of sensation can lead to life altering amputations by exacerbating the seriousness of ulcerations that occur in 15% of patients. In fact, diabetes is the leading cause of non-accidental lower extremity amputations. Current therapies manage painful symptoms with anticonvulsants, antidepressants or opiates. However, pharmacologic approaches to manage insensate DPN have not realized any significant clinical success. To this point, management of glycemic control is our most effective option in ameliorating the onset and severity of DPN symptoms, but this approach is more effective for Type 1 than Type 2 diabetics (Callaghan et al., 2012).
The recurrent hyperglycemia central to the development of DPN results in a host of biochemical and metabolic changes. Elevated blood glucose levels can increase its flux through the polyol and hexosamine pathways, activate protein kinase C (PKC), increase poly(ADP-ribose) polymerase activity and enhance formation of advanced glycation endproducts (AGE), which activate AGE receptors (RAGE) to heighten oxidative stress (Brownlee, 2001; Edwards et al., 2008; Farmer et al., 2012). Though each pathway is likely to independently contribute to the metabolic alterations that underlie DPN, current theories suggest that the progression of DPN is strongly related to a convergence of these metabolic disturbances on promoting oxidative stress (Obrosova et al., 2005; Stavniichuk et al., 2011; Vareniuk et al., 2007) and mitochondrial dysfunction (Chowdhury et al., 2013; Farmer et al., 2012). Additionally, inflammatory signaling, a well-accepted component of Type 2 diabetes, is also fed by oxidative stress and can play a role in exacerbating DPN through cytokine production, immune cell infiltration and further elevation of oxidative stress (Wei et al., 2009; Wellen, 2005; Wright et al., 2006).
From the brief introduction above, it is quite evident that DPN has multiple pathogenetic mechanisms. Classic approaches to pharmacologically manage the disorder have assumed that targeting a single biochemical insult is sufficient to alter the interwoven molecular cascade that culminates in the onset of symptomatic sensory neuropathy and the eventual dying back of distal axons. If DPN arose from damage to a single cell population from a single biochemical insult, this approach may be highly fruitful. However, the natural history of the disorder develops over decades due to damage to vascular, glial and neuronal cell populations that are not necessarily affected synchronously by hyperglycemic stress. Indeed, despite the current focus on minimizing oxidative stress, antioxidant therapies have also met with limited success in treating DPN, though the use of α-lipoic acid may have some benefit (Garcia-Alcala et al., 2015; Ziegler et al., 2006; Ziegler et al., 2011). Thus, realigning our thinking toward pharmacologic approaches to address this problem may prove beneficial.
A rather unexplored and clinically unappreciated alternative approach for managing DPN is to de-emphasize that it is necessary to topple a biochemical kingpin to destroy the downstream network of molecular minions whose slaving’s contribute to disease progression. On the other hand, cells have evolved endogenous mechanisms to tolerate stress. Pharmacologic upregulation of these pathways may facilitate recovery from glucotoxicity and/or enhance the efficacy of agents that target direct pathogenetic mechanisms of DPN. Growing evidence suggests that modulating endogenous cytoprotective responses by targeting the activity and expression of molecular chaperones such as heat shock protein 90 (Hsp90) and Hsp70 may achieve this purpose.
2.0– Molecular Chaperones
Heat shock proteins are divided into families defined by their molecular size: Hsp110, Hsp100, Hsp90, Hsp70, Hsp60, Hsp40 and small Hsps (Kim et al., 2013; Saibil, 2013). This review will focus primarily on the Hsp90 and Hsp70 proteins that can be found in the endoplasmic reticulum, mitochondria, cytosol and extracellularly (Becker and Craig, 1994; Haas and Wabl, 1983; Hightower and Guidon, 1989; Leustek et al., 1989).
Both Hsp90 and Hsp70 have isoforms that are constitutively expressed or induced by thermal, ischemic, oxidative and metabolic stressors, as well as by exercise (Beckmann et al., 1992; Krause et al., 2007; Richard et al., 1996; Yang et al., 1996). Under physiologic conditions Hsp90 binds and inhibits the transcription factor heat shock factor 1 (HSF1). In response to the accumulation of damaged and/or unfolded proteins, HSF1 is released from this complex, undergoes trimerization and is phosphorylated before being translocated to the nucleus (Neef et al., 2011; Vihervaara and Sistonen, 2014). Upon binding to heat shock elements, HSF1 upregulates an array of inducible chaperones including forms of Hsp70 (Hsp70.1 and Hsp70.3) and Hsp90 (Hsp90α), as well as antioxidant proteins (Benarroch, 2011). Hsp70 can help refold unfolded proteins, prevent inappropriate protein interactions, manage protein aggregate formation and direct the degradation of damaged or dysfunctional proteins. Importantly, the efficacy of modulating chaperones is not limited to neurodegenerative diseases driven by the formation of protein aggregates. Hsp70 has also been shown to downregulate inflammatory signaling, decrease oxidative stress and improve mitochondrial function in DPN and other conditions whose etiology is not linked to the formation of protein aggregates (Ianaro et al., 2003; Jones et al., 2011; Li et al., 2012; Ma et al., 2015; Madden et al., 2008; Saibil, 2013; Zhang et al., 2012). However, an important aspect of Hsp70 biology that must be considered in moving forward in therapy development is how modulating the intra- versus extracellular pools of Hsp70 may affect disease progression.
Opposing effects have been demonstrated for intracellular Hsp70 (iHsp70) versus extracellular Hsp70 (eHsp70) in multiple studies (Krause et al., 2015; Rodrigues-Krause et al., 2012). Extracellular Hsp’s were first described as glia-axon transfer proteins when giant squid axons were subjected to heat stress (Tytell et al., 1986). Later studies specifically identified Hsp70 as one of the glia-axon transfer proteins (Hightower and Guidon, 1989). More recently, extracellular Hsp70 (eHsp70) has been shown in blood where it acts as a paracrine factor released from dendritic, neuronal and other cell types (De Maio, 2011). eHsp70 is associated with immune system activation and inflammatory/oxidative stress conditions (Whitham and Fortes, 2008) and has been hypothesized to act as a warning to the immune system for combatting infections (Campisi et al., 2003). Conversely, iHsp70 acts to suppress immune activation, in line with it also promoting anti-inflammatory and antioxidant actions (Ianaro et al., 2003; Johnson and Fleshner, 2006). Elevated eHsp70 expression and decreased iHsp70 expression are linked to diabetes, obesity and insulin resistance (Chung et al., 2008; Kurucz et al., 2002; Rodrigues-Krause et al., 2012). Considering these conflicting actions, this review will focus on differences in the mechanistic actions of extracellular versus intracellular Hsp70 proteins and what roles their pharmacological manipulation might have in therapies for DPN.
3.0– Extracellular Hsp70
3.1 – Import, export and neuronal support
Hsp70 export was unknowingly first characterized in heat-treated giant squid axon when protein levels of a heat shock-like transferrin protein increased in the axoplasm (Tytell et al., 1986). This transferrin protein was specifically identified as Hsp70 when heat-treated rat embryonic cell cultures (presumably fibroblasts) were shown to release Hsp70 into the media, independent of an increase in cell death (Hightower and Guidon, 1989). Glial eHsp70 secretion has also been confirmed in models of heat-treated glioblastoma cells (Guzhova et al., 2001).
The export mechanisms behind eHsp70 secretion are still not well understood since Hsp70 lacks a secretory signal sequence and inhibiting the secretory pathway has no effect on the release of eHsp70 (Hightower and Guidon, 1989). Some evidence has pointed toward an innate ability of the protein to traverse the cell membrane (Multhoff, 2007), lipid raft mediated lysosomal release (Hunter-Lavin et al., 2004) and secretory-like granule excretion (Evdonin et al., 2006). However, the largest collection of evidence points to exosome-dependent trafficking; heat shocked peripheral blood mononuclear cells (PBMC) increased eHsp70 levels and produced Hsp70 loaded exosomes (Lancaster and Febbraio, 2005). Though further study is needed to fully understand this Hsp70 export mechanism, exosomal eHsp70 release has been demonstrated in other models including colon and pancreatic cancer cell lines (Gastpar et al., 2005) as well as breast and leukemic cancer cells (Bausero et al., 2005).
Once excreted eHsp70 can be absorbed and utilized by neighboring cells, typically for the purpose of cell support (Guzhova et al., 2001). The neuroblastoma cell line LAN-5 showed lower levels of cytotoxicity from both heat shock and staurosporine treatment after internalizing exogenously administered Hsp70 (Guzhova et al., 2001). These cytoprotective effects can likewise translate to non-neoplastic neuronal models. Injection of recombinant human Hsp70 into G93A mutant Cu/Zn superoxide dismutase (SOD1) mice helped preserve motor neurons resulting in a delayed onset of amyotrophic lateral sclerosis (ALS) symptoms (Gifondorwa et al., 2007). In vitro and ex vivo experiments using a trophic factor deprived environment and sciatic nerve axotomy, respectively, confirm eHsp70 decreased motor neuron death (Robinson et al., 2005; Tidwell et al., 2004). Exogenously administered Hsp70 also preserved viability of dorsal root ganglia (DRG) sensory neurons after sciatic nerve axotomy (Tidwell et al., 2004). Though the exact mechanism for cellular protection is not known, eHsp70 may act as iHsp70 upon internalization (Turturici et al., 2011).
3.2 – Immunomodulation, inflammation and oxidative stress
In contrast to the effects of iHsp70, the actions of receptor-mediated eHsp70 are typically immune stimulating, pro-inflammatory and promote oxidative stress (Krause et al., 2015). For example, multiple studies have identified eHsp70 as mediating an initial inflammatory response after tissue injury (Kovalchin et al., 2006; Senf et al., 2013). Senf et al. (2013) reported that cardiotoxin injections into the tibialis anterior (TA) muscles of Hsp70 knockout mice induced severe muscle injury and delayed recovery when compared to control animals. Hsp70 expression vectors electroporated into TA muscles restored recovery time after injury by reinvigorating the initial post injury inflammatory response. Extracellular Hsp70 release after TA injury was then shown to be integral to restoring the early inflammatory response when cardiotoxin was injected into TA muscle with and without recombinant Hsp70. The exogenously administered Hsp70 increased macrophage and neutrophil infiltration and resultant cytokine production to control levels in almost all measures (Senf et al., 2013). This study summarizes eHsp70’s attributes as a chemoattractant, immune cell activator and pro-inflammatory mediator, which essentially allows it to act as an immunomodulatory agent.
Focusing first on chemoattraction, macrophages, neutrophils and natural killer (NK) cells directly respond to eHsp70 chemotaxic signaling (Horn et al., 2007; Ortega et al., 2009; Senf et al., 2013). However, eHsp70 can also indirectly increase chemotaxis of immune cells by stimulating T-cell mediated beta-chemokine release of Rantes and macrophage inflammatory protein-1β, which drive infiltration of numerous innate and adaptive immune components (Lehner et al., 2000). Once B- and T-lymphocytes have infiltrated the inflammatory site, they may use eHsp70 itself as a chemoattractant. In vitro experiments with and without heat shock have demonstrated lymphocyte export of eHsp70 (Hunter-Lavin et al., 2004). Similarly, increases in macrophage phagocytosis and neutrophil microbicide activity have been reported after exogenous Hsp70 administration (Kovalchin et al., 2006; Ortega et al., 2006). eHsp70 also prompted pro-inflammatory cytokine release from epithelial, innate immune and adaptive immune cells, which may explain why eHsp70 is associated with inflammatory cytokine production in multiple mouse and human studies (Asea et al., 2000; Asea et al., 2002; Basu et al., 2001; Chase et al., 2007; Dvoriantchikova et al., 2014; Dybdahl et al., 2005; Huang et al., 2013; Qiao et al., 2008). Collectively, these data support a strong capacity of eHsp70 to initiate and escalate an immune-mediated inflammatory response.
A growing list of receptors seem to mediate the immunomodulatory actions of eHsp70 by evoking a host of pro-inflammatory cytokines (IL-1β, IL-6, IL-8, IL-12,TNF-α) through a CD14-dependent, Toll-like receptor (TLR) 2/4 mechanism (Asea et al., 2000; Asea et al., 2002; Chase et al., 2007; Dvoriantchikova et al., 2014; Dybdahl, 2001). TLR2 has also been linked to eHsp70 stimulation of neutrophil chemotaxis (Ortega et al., 2009). Similarly, CD94, a C-type lectin receptor, has been demonstrated to mediate eHsp70 stimulation of IFN-γ release from NK cells (Gross et al., 2003). However, a separate report indicated that this response required co-culturing the NK cells with dendritic cells that produce NKG2D ligand after eHsp70 treatment (Qiao et al., 2008). The differences in these studies might be clarified by their use of patient derived NK cells versus the YT leukemic NK cell line. CD40 was determined to be necessary for mycobacterial-Hsp70 driven beta-chemokine release and human eHsp70 was demonstrated to interact with CD40 (Lehner et al., 2000). The CD40-eHsp70 dependent effects were expanded to include internalization of eHsp70-peptide complexes for antigen presentation in macrophages (Becker, 2002; Lehner et al., 2000; Wang et al., 2001). Surprisingly, an analogous study identified CD91, not CD40, as managing antigen presentation in macrophages and dendritic cells through the MHC class I molecule by an eHsp70-peptide complex (Basu et al., 2001). However, Theriault et al. (2005) demonstrated that Hsp70 has little capacity to directly bind any of the receptors mentioned above. Instead, they showed a robust ability for eHsp70 to bind LOX-1, a class E scavenger receptor that collects modified lipoproteins such as oxidized low-density lipoprotein. The authors hypothesize that eHsp70 interacts directly with LOX-1 and then indirectly utilizes CD14, CD40, CD91, CD94 and TRL2/4 to coordinate its effects (Theriault et al., 2005). This hypothesis is supported by eHsp70’s LOX-1 dependent ability to modulate antigen cross-presentation in dendritic cells (Delneste et al., 2002). With the exception of Threiault et al., (2005), one limitation of these studies is that they utilized antagonism and correlation to confirm the involvement of these receptors in eHsp70 signaling. Becker et al. (2002) and Wang et al. (2001) are the exceptions as both studies showed specific binding of fluorescently labeled eHsp70 after CD40 transfection in HEK293 and COS-7 cells, respectively. However, these data are in direct contradiction to Theriault et al. (2005) who showed a lack of fluorescent Hsp70 binding in HEK293 cells after CD40 transfection. Unfortunately, the authors were unable to reconcile the differences between these earlier results indicating more work is still needed to clarify how eHsp70 may interact with putative receptor proteins.
Downstream of eHsp70 binding, NF-κB has been implicated in neutrophil chemotaxis, bronchial epithelial cell mediated inflammation, LPS-induced liver injury, monocyte cytokine production and ischemia-reperfusion neurotoxic inflammation (Krause et al., 2015). Multiple lines of evidence support NF-κB involvement in these eHsp70 dependent processes: NF-κB nuclear translocation, Iκ-Bα phosphorylation, Bay 11-7082 and parthenolide antagonism and NF-κB activity measured by various reporter assays (Asea et al., 2000; Asea et al., 2002; Chase et al., 2007; Dvoriantchikova et al., 2014; Huang et al., 2013; Ortega et al., 2009). Protein kinase C (PKC) has also been implicated in the immunomodulatory action of eHsp70 based on antagonism by polymixin B (Dvoriantchikova et al., 2014). However, polymixin B has not been shown to inhibit eHsp70-dependent NF-κB activation, which suggests PKC and NF-κB may function as separate signaling pathways (Asea et al., 2000). One obvious gap in this research is that all evidence for eHsp70 activation of NF-κB signaling has evaluated CD14 and TLR 2/4 as the intermediary receptors. Work is still needed to investigate downstream eHsp70 signaling after LOX-1 binding and what role CD40, CD91 and CD94 might play in that process.
4.0– Intracellular Hsp70
4.1 – Chaperone functions
The molecular chaperone activity of intracellular heat shock proteins has been extensively characterized (De Maio, 2011). iHsp70 is an integral component of this chaperone system and the complex co-chaperone interactions and molecular mechanisms that facilitate this process have been described in numerous reviews (Becker and Craig, 1994; Kim et al., 2013; Saibil, 2013; Stetler et al., 2010). A major function of this chaperone activity is to maintain intracellular proteostasis by assisting nascent peptide folding, refolding of unfolded proteins, preventing inappropriate protein interactions, managing protein aggregate formation and degrading dysfunctional proteins (Saibil, 2013). These activities have particular relevance in neurodegenerative diseases, which are often associated with aggregates of mis-folded proteins, including Alzheimer’s disease, Parkinson’s disease, amyotrophic lateral sclerosis (ALS), Huntington disease and others (Turturici et al., 2011). It is believed that post-mitotic neurons affected in these pathologies are particularly vulnerable due to an inability to diminish mis-folded protein concentrations through cell division (Muchowski and Wacker, 2005). Consequently, enhancing iHsp70 action might prove a viable therapeutic approach to treating these pathologies. In vitro, iHsp70 increases the solubility and microtubule interaction of the tau protein, as well as early stage aggregation of Aβ that leads to amyloid plaque formation in Alzheimer’s disease (Dou et al., 2003; Evans et al., 2006). However, managing inappropriate protein interactions is only part of the solution to minimizing mis-folded protein stress. If a protein cannot be refolded to a functional state, then it must be degraded to prevent accumulation.
Superoxide dismutase 1 is a cytosolic free radical scavenger protein that is integral to managing oxidative stress and mutant SOD1 promotes mitochondrial dysfunction that contributes to the onset of familial ALS (Tan et al., 2014). CHIP is a C-terminal Hsp70 interacting protein that facilitates ubiquitination and degradation of mis-folded iHsp70 client proteins like mutant SOD1. Mutant SOD1 is also degraded by chaperone-mediated autophagy where regulator proteins such as Bcl-2-associated athanogene 3 (BAG-3) and Hsp22 direct iHsp70’s chaperone functions to identify, traffic and present mis-folded proteins for autophagic degradation (Kalmar et al., 2014). This orchestrated removal of dysfunctional protein is crucial for neurological homeostasis, as well as metabolic disorders such as diabetes mellitus. In particular, Type 2 diabetes has been specifically linked to endoplasmic reticulum (ER) stress, which results from mis-folded proteins accumulating in the ER. Glucose regulated protein 78, the ER specific Hsp70 family member, is a well-known mediator of the unfolded protein response (UPR) to manage ER stress (Kalmar et al., 2014; Lee and Ozcan, 2014). However, there is also evidence that iHsp70 mediated cytosolic protein degradation facilitates stabilization of ER stress in addition to the UPR (Goeckeler and Brodsky, 2010).
4.2 – Oxidative stress
A significant portion of mis-folded or dysfunctional proteins are caused by oxidative stress. Although the chaperone functions of iHsp70 are essential to managing intracellular damage directly, iHsp70 can also indirectly mitigate disturbances in proteostasis due to a potent ability to manage oxidative stress. In the human premonocytic U937 cell line, heat shock protects from H2O2-driven mitochondrial membrane depolarization and structural changes (Polla et al., 1996). Similarly, Hsp70 overexpressing transgenic animals have curtailed age-associated oxidative stress (Broome et al., 2006) and multiple cell lines are protected from ROS mediated cytotoxicity via heat treatment and/or iHsp70 overexpression (Bellmann et al., 1996; Bellmann et al., 1995; Jaattela and Wissing, 1993). Multiple mechanisms have been linked to mediating these protective effects. For example, the superoxide generating NADPH oxidases (NOX) are negatively regulated by iHsp70. In diabetic sensory neurons, NOX2 mRNA overexpression was reversed in an Hsp70-dependent manner by treatment with the C-terminal Hsp90 inhibitor, KU-596 and this correlated with an improvement in DPN (see Section 5.0) (Ma et al., 2015). Further, in vascular smooth muscle cells, co-immunoprecipitation, and Hsp70 knockdown studies demonstrate iHsp70 trafficking of NOX4 regulates degradation of the protein (Bocanegra et al., 2010; Gil Lorenzo et al., 2014). NOX isoforms are a major source of non-mitochondrial ROS and modulating Hsp70 in a mouse model of Type 2 diabetes decreased non-mitochondrial oxygen respiration, indirectly suggesting an effect on decreasing the activity of NOX isoforms (Ma et al., 2014).
Much of total cellular ROS production does not require NOX activity and Hsp70 management of these non-NOX sources requires intervention with antioxidant enzymes such as superoxide dismutases. In Hsp70 knockout mice, a connection between Hsp70 and SOD1 expression was surmised since cerebrum homogenates from these animals showed a marked decrease in SOD1 protein levels and activity (Choi et al., 2005). Interestingly, this regulation may not be unidirectional, since SOD1 overexpressing transgenic mice have elevated levels of Hsp70 mRNA following cerebral ischemia (Kondo et al., 1996). Nevertheless, iHsp70 seems to have a capacity to modulate SOD1 expression. Evidence for Hsp70 regulation of SOD2 is not so clear-cut, however. Hsp70 knockout mice also have decreased SOD2 activity, but these mice do not show changes in SOD2 protein expression levels (Choi et al., 2005). This difference between SOD2 activity and expression may be explained by two observations. First, iHsp70 has been found to traffic SOD2 to the mitochondria (Afolayan et al., 2014) and second, SOD2 requires additional processing in the mitochondria before it becomes fully functional (Candas and Li, 2014). As a result, genetic deletion of iHsp70 would prevent SOD2 from being efficiently localized to the mitochondria for activation. Consistent with this premise, pharmacological induction of Hsp70 in hyperglycemically stressed rat embryonic sensory neurons increased mitochondrial SOD2 expression, as determined by quantitative mass spectrometry (Zhang et al., 2012).
Another major role iHsp70 plays in managing oxidative stress involves supporting proper mitochondrial function. Mitochondria are a major source of ROS production within the cell, having a concentration 5 to 10 fold higher than in the cytosol (Cadenas and Davies, 2000). Left unchecked, intracellular stress can overload or impair the mitochondrial machinery, decrease protein import and ultimately result in mitochondrial dysfunction and significant increases in oxidative stress (Baseler et al., 2011; Tomlinson and Gardiner, 2008). For instance, hyperglycemic stress of embryonic sensory neurons decreased the translation of numerous mitochondrial proteins and mitochondrial oxygen consumption while increasing superoxide levels. However, increasing iHsp70 levels with a C-terminal Hsp90 inhibitor decreased superoxide production and improved mitochondrial bioenergetics. (Zhang et al., 2012). Further studies from our lab have demonstrated that pharmacologically modulating Hsp70, has a beneficial effect on mitochondrial bioenergetics (Ma et al., 2014; Urban et al., 2012; Zhang et al., 2012). Interestingly, RNA-Seq analysis indicated that the mitochondrial transcriptome was unaltered by pharmacological modulation of Hsp70 in diabetic mice, despite an improvement in mitochondrial bioenergetics. Therefore iHsp70’s protection of mitochondrial bioenergetics may not be significantly mediated through gene transcription (Ma et al., 2015). iHsp70-dependent influx of mitochondrial proteins may offer an explanation for at least part of this mitochondrial bioenergetics protection (Afolayan et al., 2014).
Only 1% of mitochondrial proteins are encoded by mitochondrial DNA, the remaining 99% must be imported after nuclear transcription (Schmidt et al., 2010; Wiedemann et al., 2004). Hsp70 was first shown to bind and facilitate translocation of proteins to mitochondria in yeast (Deshaies et al., 1988; Murakami et al., 1988). Translocases of the outer mitochondrial membrane (Tom) form a complex that is composed of various receptors and Tom40, an import pore formed which most nuclear-transcribed proteins must traverse (Schmidt et al., 2010; Wiedemann et al., 2004). Tom70 acts as the mitochondrial receptor for iHsp70 to facilitate mitochondrial protein import (Young et al., 2003). After transit through the outer membrane, the preprotein that iHsp70 was transporting is pulled through the inner mitochondrial membrane using translocases of the inner mitochondrial membrane (Tim). Tim50 helps transition from the outer mitochondrial membrane to the inner, sending the preprotein to a channel formed by Tim23 and Tim17 (Schmidt et al., 2010; Wiedemann et al., 2004).
Mitochondrial Hsp70 is anchored to Tim44 and together they act as the major components of an ATP-dependent motor that pulls the preprotein into the inner mitochondrial space (Krimmer et al., 2000). Once inside, the preprotein is processed to facilitate proper mitochondrial function (Schmidt et al., 2010; Wiedemann et al., 2004). Mitochondrial dysfunction is only one mechanism by which intracellular stress can drive oxidative stress. Another can be seen in the interaction between hyperglycemia and thioredoxin interacting protein (TXNIP). It has been speculated that TXNIP plays a role in regulating insulin-independent glucose transport into tissues (Singh, 2013). This is largely based on an observation that TXNIP can regulate glucose transporter expression and localization (Wu et al., 2013). However, prolonged hyperglycemic stress can drive persistent activation of TXNIP, as was shown in the retina of Type 1 diabetic rats (Devi et al., 2012). TXNIP’s most well characterized function is as an inhibitor of the antioxidant and thiol reducing protein, thioredoxin (Trx), and extended inhibition of Trx by TXNIP drives oxidative stress (Schulze et al., 2004). Further exacerbating the situation, prolonged TXNIP activation can also increase inflammatory signaling, although it is unclear if these oxidative and inflammatory events occur sequentially or in parallel (Devi et al., 2012; Lane et al., 2013). Recent work from our lab has shown that Hsp70 may play a role in affecting the expression of TXNIP. RNA-seq analysis of mRNA from DRG of Type 1 diabetic mice showed a 2-fold elevated expression of TXNIP expression (Ma et al., 2015) and treating diabetic mice with the chaperone modulator, KU-596 (see section 5.0), suppressed TXNIP expression to control levels (Fig. 1A). Moreover, this suppression was Hsp70-dependent as TXNIP mRNA levels remained elevated (~1.4 fold) in DRG from diabetic Hsp70 KO mice that were treated with KU-596 (Fig. 1B). Though it remains unclear whether Hsp70 inhibited TXNIP overexpression directly or by alleviating the initial hyperglycemic/oxidative stress insult, these results suggest that Hsp70 attenuates intracellular oxidative stress induced by diabetes, which correlates with an improvement in clinically relevant measures of DPN.
Fig. 1. Modulating Hsp70 with KU-596 decreased the expression of txnip in diabetic DRG in an Hsp70-dependent manner.
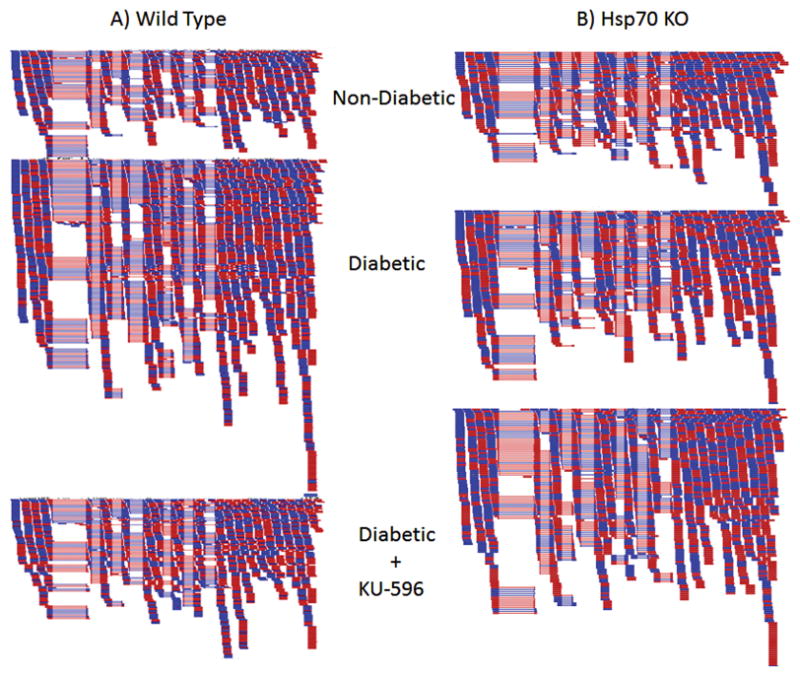
Wild type (A) and Hsp70 KO (B) mice were rendered diabetic with streptozotocin and after 12 weeks of diabetes, treated with vehicle or weekly doses of KU-596 at 20 mg/kg for 4 weeks. mRNA was harvested from the lumbar DRG and used for RNA-Seq analysis. Shown are representative alignments to the mouse txnip gene from non-diabetic, diabetic and diabetic animals that received KU-596 therapy. Dark blue bars indicate sequences that mapped to the forward strand while dark red bars are sequences that mapped to the reverse strand of the 8 exons of the txnip gene. See Ma et al., (2015) for additional details.
4.3 – Inflammation
Another major aspect of iHsp70 biology is its ability to regulate inflammatory signaling. Heat shock prevents TNF-α production after ischemic stress in the renal tubular epithelial cell line LLC-PK1 and in rat liver after induction of sepsis by cecal-ligation and puncture (CLP) (Chen et al., 2005; Meldrum, 2003). In vivo, Hsp70 overexpression suppresses increased expression of IL-6 and TNF-α induced by LPS, as well as elevation of IL-6 and cytokine-induced neutrophil chemoattractant-1 in rat blood after CLP (Dokladny et al., 2010; Weiss et al., 2007). Each of these studies reported that an Hsp70-dependent suppression of NF-κB activation mediated the anti-inflammatory action. Using DNA binding and nuclear localization, numerous studies have supported this claim with heat shock (Feinstein et al., 1996), liposomal Hsp70 delivery (Meldrum, 2003) and Hsp70 overexpression (Feinstein et al., 1997). Mechanistically, this effect may be due to Hsp70-dependent decrease in degradation of the NF-κB inhibitor (IκBα), which binds NF-κB to inhibit its nuclear localization (Dokladny et al., 2010; Feinstein et al., 1997; Wong et al., 1997). Precipitating this action is Hsp70’s ability to inhibit phosphorylation of the IκB kinase (IKK). IKK phosphorylates IκBα, causing it to dissociate from NF-κB and be degraded. In the absence of IKK phosphorylation, this process is blocked (Chan et al., 2004; Chung et al., 2008; Weiss et al., 2007). Immunoprecipitation studies demonstrate that Hsp70 associates with the IKK complex, which hinders the IKKβ subunit of the complex from being integrated (Weiss et al., 2007). Additional studies support Hsp70 binding to IKKγ (NEMO), which prevents its trimerization and function as a structural component of the IKK complex (Agou et al., 2002; Chen et al., 2005; Ran et al., 2004).
A second Hsp70 mechanism for hindering NF-κB activation has also been described since Hsp70 stabilized phosphorylated and ubiquitinated IκBα This interaction prevented the degradation of IκBα without affecting the IKK complex (Weiss et al., 2007). Additionally, a separate report suggests a third mechanism where Hsp70 directly upregulates IκBα expression to suppress NF-κB activation (Wong et al., 1999). Taken together, these studies outline a highly redundant system where iHsp70 can prevent NF-κB activation by manipulating IκB. Further, two studies also suggest that Hsp70 can also prevent NF-κB activation without altering IκBα concentrations. Overexpression of Hsp70 in C6 rat glioma cells did not prevent LPS induced IκBα degradation, despite suppression of p65 NF-κB subunit nuclear localization. The authors speculated that in certain situations, Hsp70 may instead prevent NF-κB nuclear localization by either competing for the nuclear pore or binding NF-κB after its initial dissociation from IκB (Feinstein et al., 1997). A later study replicated this finding using liposomal delivery of Hsp70 (Meldrum, 2003) and co-immunoprecipitation of Hsp70 with the p65 subunit of NF-κB, which supports the viability of this hypothesis (Chen et al., 2005). Clearly, this body of work demonstrates that NF-κB can be intimately regulated by iHsp70. However, as activation of the NF-κB pathway in sensory neurons has been shown to ameliorate DPN (Saleh et al., 2013a; Saleh et al., 2013b), it may be counterproductive to invoke therapies which may upregulate both the NF-κB pathway and iHsp70 in neurons.
4.4 – Insulin sensitivity, JNK and c-Jun
In Type 2 diabetes, iHsp70 may be able to improve insulin resistance and blood glucose levels. Mice fed a high fat diet show both insulin resistance and glucose intolerance compared to normal chow-fed animals. Transgenic animals overexpressing Hsp70 show no insulin resistance and markedly improved glucose tolerance when fed the high fat diet (Henstridge et al., 2014). The actions of Hsp70 overexpression were attributed to decreased phosphorylation od c-jun N-terminal kinase (JNK), a known regulator of insulin sensitivity. Heat treatment and pharmacological induction of Hsp70 also inhibited JNK phosphorylation (Chung et al., 2008; Henstridge et al., 2014a). Similarly, Hsp70 overexpression and heat shock suppressed JNK1 activity in NIH 3T3 cells after ultraviolet light stress. JNK1 and Hsp70 can physically interact and this prevented JNK1 phosphorylation and suppressed c-jun transactivating activity (Park et al., 2001). However, JNK regulation is not the only mechanism by which iHsp70 can modulate targets downstream of JNK.
C-jun is emerging as a key negative regulator of myelination (Arthur-Farraj et al., 2012; Hutton et al., 2011; Parkinson et al., 2008) and altering its expression via modulation of iHsp70 may offer an attractive approach to treat the segmental demyelination that occurs in human DPN, as well as in various inherited neuropathies. In myelinated DRG explants, induction of Hsp70 by treatment with the C-terminal Hsp90 modulator KU-32 inhibited c-jun/phospho-c-jun expression and decreased the extent of neuregulin-induced demyelination (Figs. 2A, 2C–E). Though KU-32 did not decrease the phosphorylation of JNK and ERK, inhibiting the proteasome with MG132 prevented the decrease in c-jun expression. These data support that iHsp70 facilitates the proteasomal degradation of c-jun to attenuate demyelination (Fig. 2) (Li et al., 2012). Importantly, the necessity of iHsp70 in preventing the increase in c-jun expression and demyelination was validated using myelinated DRG explants prepared from Hsp70 KO mice. Although neuregulin treatment promoted a robust induction of c-jun in these cultures, treatment with KU-32 was unable to block this increase and prevent demyelination, as was observed in myelinated neuronal cultures prepared from wild type mice (Figs. 2B, 2C–E). Similarly, adenovirus-mediated re-expression of Hsp70 in the Hsp70 KO neurons was sufficient to decrease c-jun induction following neuregulin treatment. Since transcriptomic analysis of mRNA from sural nerve of patients with progressive DPN showed an increase in a gene sub-network centered on c-jun (Hur et al., 2011), it is tempting to speculate that iHsp70 modulation of c-jun signaling may benefit later stages of progressive DPN. Unfortunately, diabetic rodent models do not recapitulate the extensive segmental demyelination that is observed in human DPN, but ongoing work using animal models of demyelinating neuropathies is examining whether decreasing c-jun expression by modulating iHsp70 may improve myelinated fiber function.
Fig. 2. Modulating Hsp70 with KU-32 decreased neuregulin-induced c-jun expression and demyelination in an Hsp70-dependent manner.
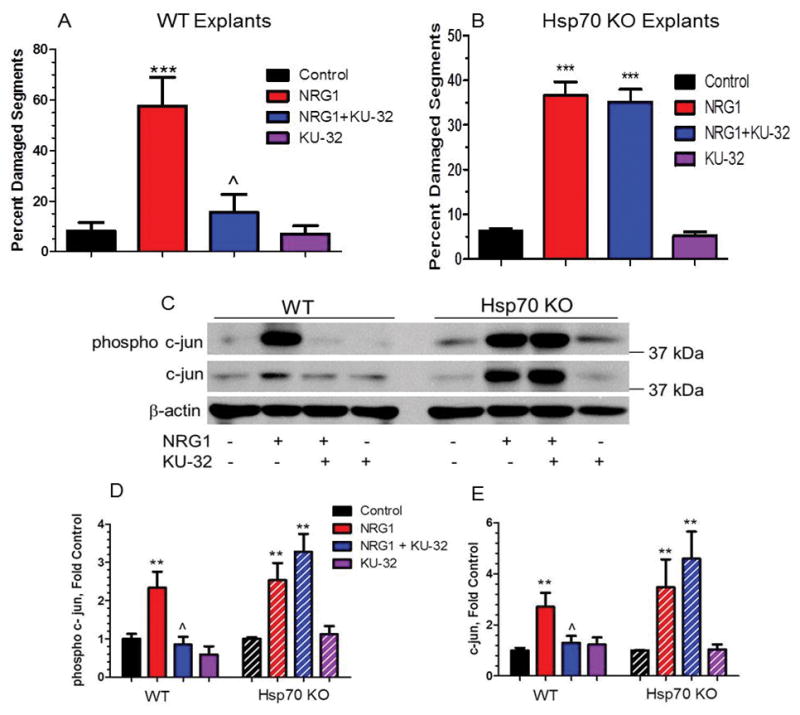
Myelinated DRG explants were prepared from wild type or Hsp70 KO mice. The cultures were treated with vehicle or 1 μM KU-32 overnight prior to inducing demyelination with 200 ng/ml neuregulin 1. Cultures were stained for myelin basic protein and the degeneration of the myelinated segments was quantified (A, B). Other cultures were prepared for immunoblot analysis of c-jun and the effects of the treatments on c-jun and phospho-c-jun expression quantified (C–E). Modified with permission from Li et al., (2012), ASN Neuro published by Portland Press.
5.0 – DPN and modulating Hsp70
Clearly, a large body of evidence supports the concept that modulating Hsp70 may have both beneficial and deleterious effects (Fig. 3). Indeed overexpression of iHsp70 is associated with various forms of cancer and its downregulation can increase sensitivity to chemotherapeutics (Lianos et al., 2015). eHsp70 may contribute to the development of experimental autoimmune encephalitis, an animal model of multiple sclerosis (Mansilla et al., 2014). Furthermore, elevated eHsp70 levels may contribute to inflammation in obese, Type 2 diabetic patients, ketoacidotic Type1 diabetics (Oglesbee et al., 2005; Rodrigues-Krause et al., 2012) and play a role in the progression of diabetes based on its ability to induce β-cell dysfunction and death (Krause et al., 2014). On the other hand, increasing iHsp70 expression in skeletal muscle improved insulin resistance and decreased metabolic parameters of Type 2 diabetes (Chung et al., 2008; Henstridge et al., 2014a). The chaperone balance hypothesis gives context to these observations by proposing that a high ratio of [eHsp70]:[iHsp70] is associated with promoting chronic inflammatory responses while a low [eHsp70]:[iHsp70] ratio is more anti-inflammatory (Krause et al., 2015). If this hypothesis proves to be true, then attempts to alter the progression of DPN by modulating Hsp70 may benefit from examining this relationship. This begets the question, what are our options for modulating Hsp70?
Fig. 3. Summary of potential impact of iHsp70 and eHsp70 on hyperglycemia-mediated dysfunction.

(I) Hyperglycemia manipulates multiple metabolic pathways to induce both intra- and extracellular stresses to the cell. These events converge at three points affecting mitochondrial dysfunction, oxidative stress and pro-inflammatory signaling to facilitate the progression of DPN. (II) iHsp70 inhibits hyperglycemia induced dysfunctions through multiple mechanisms including mitochondrial protein shuttling, up/downregulation of anti/pro-oxidant proteins respectively, repair or degradation of damaged and mis-folded protein, and inhibition of pro-inflammatory signaling via NF-κ B. (III) eHsp70 may play a complicated role since if internalized, it can bolster iHsp70 stores and actions while external binding to one of multiple purported receptors can antagonize the action of iHsp70 by initiating or exacerbating NF-κB mediated inflammatory signaling.
Though genetic approaches to downregulate or overexpress Hsp70 provide an avenue to modulate its activity in animal models of disease, these approaches have not been translated to humans. Similarly, no selective small molecule inhibitors (or activators) of inducible Hsp70 exist that can be translated to the clinic (Evans et al., 2010; Lianos et al., 2015). This difficulty is partially attributed to the fact that five cellular Hsp70 family members share 86–99% homology (Daugaard et al., 2007). However, there are viable small molecule therapies that can indirectly modulate Hsp70 levels by targeting Hsp90 or HSF1.
Hsp90 is required for the folding of many proteins into their final conformation and is the master regulator of the heat shock response. Numerous compounds have been developed that target either the N- or C-terminal of the protein to alter its enzymatic activity or interaction with co-chaperones (Miyata et al., 2013; Peterson and Blagg, 2009; Walton-Diaz et al., 2013). Since many Hsp90 client proteins are also oncoproteins, N-terminal inhibitors may serve as chemotherapeutics by promoting client protein degradation. As chemotherapeutics, one attribute of N-terminal Hsp90 inhibitors is their selectivity for inhibiting Hsp90 and promoting client protein degradation in cancerous versus non-cancerous cells (Chiosis et al., 2003; Kamal et al., 2003; Luo et al., 2008). However, induction of client protein degradation and cytotoxicity often occurs at drug concentrations that also induce the heat shock response. Unfortunately, an increase in chaperone expression can antagonize the compound’s cytotoxic potential by facilitating oncoprotein refolding, narrowing the drug’s effective therapeutic window (Peterson and Blagg, 2009). On the other hand, an increase in chaperone synthesis may be useful for treating neurodegenerative diseases since N-terminal Hsp90 inhibitors have been shown to limit or decrease tau aggregation in models of Alzheimer’s disease (Dickey et al., 2007; Luo et al., 2007), huntingtin protein in motor neurons (Waza et al., 2005), α-synuclein in Parkinson’s disease (Ebrahimi-Fakhari et al., 2013) and peripheral myelin protein 22 in Schwann cells from a mouse model of Charcot Marie Tooth 1A (Chittoor-Vinod et al., 2015). However, it is still necessary to dissociate the degradation of client proteins from stimulation of the heat shock response since client protein degradation could lessen the neuroprotective effectiveness of the drugs.
Over the past 5 years we have identified novologues as a class of C-terminal Hsp90 inhibitors derived from novobiocin that induce Hsp70 in the absence of significant client protein degradation (Kusuma et al., 2012; Urban et al., 2010). KU-32 and KU-596 are two such compounds that were mentioned above and these drugs improved multiple clinical indices of DPN in models of both Type 1 and Type 2 diabetes, in an Hsp70-dependent manner (Ma et al., 2014; Ma et al., 2015; Urban et al., 2010; Urban et al., 2012). Similarly, an N-terminal Hsp90 inhibitor has proven beneficial in treating diabetic nephropathy and the physiologic improvement in renal function may also be Hsp70-dependent (Lazaro et al., 2015). As neither of these diabetic complications has an etiology that centers on formation of any one protein aggregate, clearly modulating Hsp70 may prove efficacious toward treating complex metabolic complications of diabetes. Although all these compounds were shown to increase iHsp70, their effects on eHsp70 levels were not examined. Consistent with many of the studies described in section 4.0, novologues diminished the level of oxidative stress, improved mitochondrial bioenergetics and decreased an array of genes in DRG that were associated with inflammation (Ma et al., 2014; Ma et al., 2015; Urban et al., 2012; Zhang et al., 2012). The improvement of DPN and decrease in inflammation by KU-596 therapy was also associated with a transcriptomic profile that was predicted to inhibit the NF-κB pathway in DRG of diabetic wild type mice but not diabetic Hsp70 KO mice (Ma et al., 2015). These actions would be inconsistent with enhancing systemic levels of eHsp70, but it is possible that a more localized exchange of eHsp70 between Schwann cells and sensory neurons may contribute to the dependence of drug effectiveness on Hsp70.
It is interesting that increasing Hsp70 in skeletal muscle using the HSF1 activator, BGP-15, improved fasting glucose and insulin signaling in obese mice (Henstridge et al., 2014a; Henstridge et al., 2014b). However, novologues improved DPN in an Hsp70-dependent manner without correcting any metabolic parameters in models of both Type 1 and Type 2 diabetes. Similarly, the N-terminal Hsp90 inhibitor and geldanamycin derivative (DMAG) improved diabetic nephropathy without effecting metabolic measures of diabetes; weight loss, blood glucose and HbA1c (Lazaro et al., 2015). The reason for the difference between these compounds is curious, but may lie in BGP-15 being a more direct activator of HSF-1. It is encouraging that no adverse effects were reported from a short 28 day clinical trial in patients given 200 or 400 mg BGP-15 daily, which may be consistent with minimal drug effect on increasing eHsp70 activity (Literáti-Nagy et al., 2009). As treatment of diabetic complications such as DPN will require prolonged drug administration, it will be important to assess any effects on eHsp70 as novel modulators of Hsp70 move forward clinically. Fortunately, eHsp70 should be easy to monitor in a clinical trial and this will provide some insight into whether this pool of the protein correlates with adverse, presumably inflammatory, side effects.
6.0 – Concluding remarks
In summary, results in animal models and small clinical trials clearly provide compelling proof-of-concept that modulating molecular chaperones and particularly iHsp70 provides a viable approach to improve insulin resistance and diabetic complications. Although we are not putting forth the premise that directly targeting proteins and pathways that are linked to the pathophysiological progression of DPN is a less than fruitful approach to disease management, it seems that altering any one pathway may be insufficient to treat the human disease. A similar fate may befall an approach that only modulates iHsp70 to treat DPN. However, since modulating Hsp70 improves oxidative stress, mitochondrial function and to some extent inflammation, increasing the ability of neurons and Schwann cells to tolerate ongoing diabetic stress may facilitate the action of additional agents or improve the ability of glycemic control to slow the rate of onset and diminish the severity of DPN, especially in Type 2 diabetic patients.
Acknowledgments
This work was supported by grant DK095911 from the National Institute of Diabetes, Digestive and Kidney Diseases to RTD and a post-doctoral fellowship from the Pharmaceutical Research and Manufacturers of America Foundation to SME.
References
- Afolayan AJ, Teng RJ, Eis A, Rana U, Broniowska KA, Corbett JA, et al. Inducible Hsp70 regulates superoxide dismutase-2 and mitochondrial oxidative stress in the endothelial cells from developing lungs. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2014;306:L351–360. doi: 10.1152/ajplung.00264.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agou F, Ye F, Goffinont S, Courtois G, Yamaoka S, Israel A, et al. Nemo trimerizes through its coiled-coil C-terminal domain. Journal of Biological Chemistry. 2002;277:17464–17475. doi: 10.1074/jbc.M201964200. [DOI] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, et al. C-jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron. 2012;75:633–647. doi: 10.1016/j.neuron.2012.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asea A, Kraeft SK, Kurt-Jones EA, Stevenson MA, Chen LB, Finberg RW, et al. Hsp70 stimulates cytokine production through a CD14-dependent pathway, demonstrating its dual role as a chaperone and cytokine. Nature Medicine. 2000;6:435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, et al. Novel signal transduction pathway utilized by extracellular Hsp70: Role of toll-like receptor (TLR) 2 and TLR4. Jornal of Biological Chemistry. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D, Powell MJ, et al. Proteomic alterations of distinct mitochondrial subpopulations in the Type 1 diabetic heart: Contribution of protein import dysfunction. American Journal of Physiology: Regulatory, Integrative and Comparative Physiology. 2011;300:R186–200. doi: 10.1152/ajpregu.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Binder RJ, Ramalingam T, Srivastava PK. CD91 is a common receptor for heat shock proteins GRP96, Hsp90, Hsp70, and calreticulin. Immunity. 2001;14:303–313. doi: 10.1016/s1074-7613(01)00111-x. [DOI] [PubMed] [Google Scholar]
- Bausero MA, Gastpar R, Multhoff G, Asea A. Alternative mechanism by which IFN-γ enhances tumor recognition: Active release of heat shock protein 72. Journal of Immunology. 2005;175:2900–2912. doi: 10.4049/jimmunol.175.5.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker J, Craig EA. Heat-shock proteins as molecular chaperones. European Journal of Biochemistry. 1994;219:11–23. doi: 10.1007/978-3-642-79502-2_2. [DOI] [PubMed] [Google Scholar]
- Becker T. CD40, an extracellular receptor for binding and uptake of Hsp70-peptide complexes. Journal of Cell Biology. 2002;158:1277–1285. doi: 10.1083/jcb.200208083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann RP, Lovett M, Welch WJ. Examining the function and regulation of Hsp 70 in cells subjected to metabolic stress. Journal of Cell Biology. 1992;117:1137–1150. doi: 10.1083/jcb.117.6.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmann K, Jaattela M, Wissing D, Burkart V, Kolb H. Heat shock protein Hsp70 overexpression confers resistance against nitric oxide. FEBS Letters. 1996;391:185–188. doi: 10.1016/0014-5793(96)00730-2. [DOI] [PubMed] [Google Scholar]
- Bellmann K, Wenz A, Radons J, Burkart V, Kleemann R, Kolb H. Heat shock induces resistance in rat pancreatic islet cells against nitric oxide, oxygen radicals and streptozotocin toxicity in vitro. Journal of Clinical Investigation. 1995;95:2840–2845. doi: 10.1172/JCI117989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benarroch EE. Heat shock proteins: Multiple neuroprotective functions and implications for neurologic disease. Neurology. 2011;76:660–667. doi: 10.1212/WNL.0b013e31820c3119. [DOI] [PubMed] [Google Scholar]
- Bocanegra V, Manucha W, Pena MR, Cacciamani V, Valles PG. Caveolin-1 and Hsp70 interaction in microdissected proximal tubules from spontaneously hypertensive rats as an effect of losartan. Journal of Hypertension. 2010;28:143–155. doi: 10.1097/HJH.0b013e328332b778. [DOI] [PubMed] [Google Scholar]
- Broome CS, Kayani AC, Palomero J, Dillmann WH, Mestril R, Jackson MJ, et al. Effect of lifelong overexpression of Hsp70 in skeletal muscle on age-related oxidative stress and adaptation after nondamaging contractile activity. FASEB Journal. 2006;20:1549–1551. doi: 10.1096/fj.05-4935fje. [DOI] [PubMed] [Google Scholar]
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- Cadenas E, Davies KJ. Mitochondrial free radical generation, oxidative stress, and aging. Free Radicals in Biology and Medicine. 2000;29:222–230. doi: 10.1016/s0891-5849(00)00317-8. [DOI] [PubMed] [Google Scholar]
- Callaghan BC, Cheng HT, Stables CL, Smith AL, Feldman EL. Diabetic neuropathy: Clinical manifestations and current treatments. Lancet Neurology. 2012;11:521–534. doi: 10.1016/S1474-4422(12)70065-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, Leem TH, Fleshner M. Stress-induced extracellular Hsp72 is a functionally significant danger signal to the immune system. Cell Stress and Chaperones. 2003;8:272–286. doi: 10.1379/1466-1268(2003)008<0272:sehiaf>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candas D, Li JJ. MnSOD in oxidative stress response-potential regulation via mitochondrial protein influx. Antioxidant and Redox Signaling. 2014;20:1599–1617. doi: 10.1089/ars.2013.5305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Ou CC, Wang LL, Chan SH. Heat shock protein 70 confers cardiovascular protection during endotoxemia via inhibition of Nuclear Factor-KappaB activation and inducible nitric oxide synthase expression in the rostral ventrolateral medulla. Circulation. 2004;110:3560–3566. doi: 10.1161/01.CIR.0000143082.63063.33. [DOI] [PubMed] [Google Scholar]
- Chase MA, Wheeler DS, Lierl KM, Hughes VS, Wong HR, Page K. Hsp72 induces inflammation and regulates cytokine production in airway epithelium through a TLR- and NF-κB-dependent mechanism. Journal of Immunology. 2007;179:6318–6324. doi: 10.4049/jimmunol.179.9.6318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HW, Kuo HT, Wang SJ, Lu TS, Yang RC. In vivo heat shock protein assembles with septic liver NF-κB/I-κB complex regulating NF-κB activity. Shock. 2005;24:232–238. doi: 10.1097/01.shk.0000174020.87439.f2. [DOI] [PubMed] [Google Scholar]
- Chiosis G, Huezo H, Rosen N, Mimnaugh E, Whitesell L, Neckers L. 17AAG: Low target binding affinity and potent cell activity--finding an explanation. Molecular Cancer Therapy. 2003;2:123–129. [PubMed] [Google Scholar]
- Chittoor-Vinod VG, Lee S, Judge SM, Notterpek L. Inducible Hsp70 is critical in preventing the aggregation and enhancing the processing of PMP22. ASN NEURO. 2015;7:1–17. doi: 10.1177/1759091415569909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S, Park KA, Lee HJ, Park MS, Lee JH, Park KC, et al. Expression of Cu/Zn SOD protein is suppressed in Hsp 70.1 knockout mice. Journal of Biochemistry and Molecular Biology. 2005;38:111–114. doi: 10.5483/bmbrep.2005.38.1.111. [DOI] [PubMed] [Google Scholar]
- Chowdhury SK, Smith DR, Fernyhough P. The role of aberrant mitochondrial bioenergetics in diabetic neuropathy. Neurobiology of Disease. 2013;51:56–65. doi: 10.1016/j.nbd.2012.03.016. [DOI] [PubMed] [Google Scholar]
- Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, et al. Hsp72 protects against obesity-induced insulin resistance. Proceedings of the National Academy of Science. 2008;105:1739–1744. doi: 10.1073/pnas.0705799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Letters. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- De Maio A. Extracellular heat shock proteins, cellular export vesicles, and the stress observation system: A form of communication during injury, infection, and cell damage. It is never known how far a controversial finding will go! Dedicated to Ferruccio Ritossa. Cell Stress and Chaperones. 2011;16:235–249. doi: 10.1007/s12192-010-0236-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delneste Y, Magistrelli G, Gauchat J, Haeuw J, Aubry J, Nakamura K, et al. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity. 2002;17:353–362. doi: 10.1016/s1074-7613(02)00388-6. [DOI] [PubMed] [Google Scholar]
- Deshaies RJ, Koch BD, Werner-Washburne M, Craig EA, Schekman R. A subfamily of stress proteins facilitates translocation of secretory and mitochondrial precursor polypeptides. Nature. 1988;332:800–805. doi: 10.1038/332800a0. [DOI] [PubMed] [Google Scholar]
- Devi TS, Lee I, Huttemann M, Kumar A, Nantwi KD, Singh LP. TXNIP links innate host defense mechanisms to oxidative stress and inflammation in retinal Muller glia under chronic hyperglycemia: Implications for diabetic retinopathy. Experimental Diabetes Research. 2012;2012:438238. doi: 10.1155/2012/438238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickey CA, Kamal A, Lundgren K, Klosak N, Bailey RM, Dunmore J, et al. The high-affinity Hsp90-Chip complex recognizes and selectively degrades phosphorylated tau client proteins. Journal of Clinical Investigation. 2007;117:648–658. doi: 10.1172/JCI29715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokladny K, Lobb R, Wharton W, Ma TY, Moseley PL. LPS-induced cytokine levels are repressed by elevated expression of Hsp70 in rats: Possible role of NF-kappaB. Cell Stress and Chaperones. 2010;15:153–163. doi: 10.1007/s12192-009-0129-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou F, Netzer WJ, Tanemura K, Li F, Hartl FU, Takashima A, et al. Chaperones increase association of tau protein with microtubules. Proceedings of the National Academy of Science. 2003;100:721–726. doi: 10.1073/pnas.242720499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvoriantchikova G, Santos AR, Saeed AM, Dvoriantchikova X, Ivanov D. Putative role of protein kinase C in neurotoxic inflammation mediated by extracellular heat shock protein 70 after ischemia-reperfusion. Journal of Neuroinflammation. 2014;11:81. doi: 10.1186/1742-2094-11-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dybdahl B. Inflammatory response after open heart surgery: Release of heat-shock protein 70 and signaling through Toll-like receptor-4. Circulation. 2001;105:685–690. doi: 10.1161/hc0602.103617. [DOI] [PubMed] [Google Scholar]
- Dybdahl B, Slordahl SA, Waage A, Kierulf P, Espevik T, Sundan A. Myocardial ischaemia and the inflammatory response: Release of heat shock protein 70 after myocardial infarction. Heart. 2005;91:299–304. doi: 10.1136/hrt.2003.028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi-Fakhari D, Saidi LJ, Wahlster L. Molecular chaperones and protein folding as therapeutic targets in Parkinson's disease and other synucleinopathies. Acta Neuropathology Communications. 2013;1:79. doi: 10.1186/2051-5960-1-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards JL, Vincent AM, Cheng HT, Feldman EL. Diabetic neuropathy: Mechanisms to management. Pharmacology and Therapeutics. 2008;120:1–34. doi: 10.1016/j.pharmthera.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (Hsp70) as an emerging drug target. Journal of Medicinal Chemistry. 2010;53:4585–4602. doi: 10.1021/jm100054f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans CG, Wisen S, Gestwicki JE. Heat shock proteins 70 and 90 inhibit early stages of amyloid beta-(1–42) aggregation in vitro. Journal of Biological Chemistry. 2006;281:33182–33191. doi: 10.1074/jbc.M606192200. [DOI] [PubMed] [Google Scholar]
- Evdonin AL, Martynova MG, Bystrova OA, Guzhova IV, Margulis BA, Medvedeva ND. The release of Hsp70 from A431 carcinoma cells is mediated by secretory-like granules. European Journal of Cell Biology. 2006;85:443–455. doi: 10.1016/j.ejcb.2006.02.008. [DOI] [PubMed] [Google Scholar]
- Farmer KL, Li C, Dobrowsky RT. Diabetic peripheral neuropathy: Should a chaperone accompany our therapeutic approach? Pharmacologic Reviews. 2012;64:880–900. doi: 10.1124/pr.111.005314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Aquino DA, Li GC, Xu H, Reis DJ. Heat shock protein 70 suppresses astroglial-inducible nitric-oxide synthase expression by decreasing NFkappaB activation. Journal of Biological Chemistry. 1996;271:17724–17732. doi: 10.1074/jbc.271.30.17724. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Galea E, Reis DJ. Suppression of glial nitric oxide synthase induction by heat shock: Effects on proteolytic degradation of IkappaB-alpha. Nitric Oxide. 1997;1:167–176. doi: 10.1006/niox.1997.0117. [DOI] [PubMed] [Google Scholar]
- Garcia-Alcala H, Santos Vichido CI, Islas Macedo S, Genestier-Tamborero CN, Minutti-Palacios M, Hirales Tamez O, et al. Treatment with alpha-lipoic acid over 16 weeks in Type 2 diabetic patients with symptomatic polyneuropathy who responded to initial 4-week high-dose loading. Journal of Diabetes Research. 2015;2015:189857. doi: 10.1155/2015/189857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gastpar R, Gehrmann M, Bausero MA, Asea A, Gross C, Schroeder JA, et al. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Research. 2005;65:5238–5247. doi: 10.1158/0008-5472.CAN-04-3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifondorwa DJ, Robinson MB, Hayes CD, Taylor AR, Prevette DM, Oppenheim RW, et al. Exogenous delivery of heat shock protein 70 increases lifespan in a mouse model of amyotrophic lateral sclerosis. Journal of Neuroscience. 2007;27:13173–13180. doi: 10.1523/JNEUROSCI.4057-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil Lorenzo AF, Bocanegra V, Benardon ME, Cacciamani V, Valles PG. Hsp70 regulation on NOX4/p22Phox and cytoskeletal integrity as an effect of losartan in vascular smooth muscle cells. Cell Stress and Chaperones. 2014;19:115–134. doi: 10.1007/s12192-013-0439-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goeckeler JL, Brodsky JL. Molecular chaperones and substrate ubiquitination control the efficiency of endoplasmic reticulum-associated degradation. Diabetes Obesity and Metabolism. 2010;12(Suppl 2):32–38. doi: 10.1111/j.1463-1326.2010.01273.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Hansch D, Gastpar R, Multhoff G. Interaction of heat shock protein 70 peptide with NK cells involves the NK receptor CD94. Biological Chemistry. 2003;384:267–279. doi: 10.1515/BC.2003.030. [DOI] [PubMed] [Google Scholar]
- Guzhova I, Kislyakova K, Moskaliova O, Fridlanskaya I, Tytell M, Cheetham M, et al. In vitro studies show that Hsp70 can be released by glia and that exogenous Hsp70 can enhance neuronal stress tolerance. Brain Research. 2001;914:66–73. doi: 10.1016/s0006-8993(01)02774-3. [DOI] [PubMed] [Google Scholar]
- Haas IG, Wabl M. Immunoglobulin heavy chain binding protein. Nature. 1983;306:387–389. doi: 10.1038/306387a0. [DOI] [PubMed] [Google Scholar]
- Henstridge DC, Bruce CR, Drew BG, Tory K, Kolonics A, Estevez E, et al. Activating Hsp72 in rodent skeletal muscle increases mitochondrial number and oxidative capacity and decreases insulin resistance. Diabetes. 2014a;63:1881–1894. doi: 10.2337/db13-0967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henstridge DC, Whitham M, Febbraio MA. Chaperoning to the metabolic party: The emerging therapeutic role of heat-shock proteins in obesity and Type 2 diabetes. Molecular Metabolism. 2014b;3:781–793. doi: 10.1016/j.molmet.2014.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hightower LE, Guidon PT., Jr Selective release from cultured mammalian cells of heat-shock (stress) proteins that resemble glia-axon transfer proteins. Journal of Cell Physiology. 1989;138:257–266. doi: 10.1002/jcp.1041380206. [DOI] [PubMed] [Google Scholar]
- Horn P, Kalz A, Lim CL, Pyne D, Saunders P, Mackinnon L, et al. Exercise-recruited NK cells display exercise-associated eHsp-70. Exercise and Immunology Reviews. 2007;13:100–111. [PubMed] [Google Scholar]
- Huang C, Wang J, Chen Z, Wang Y, Zhang W. 2-Phenylethynesulfonamide prevents induction of pro-inflammatory factors and attenuates LPS-induced liver injury by targeting NHE1-Hsp70 complex in mice. PLoS One. 2013;8:e67582. doi: 10.1371/journal.pone.0067582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter-Lavin C, Davies EL, Bacelar MM, Marshall MJ, Andrew SM, Williams JH. Hsp70 release from peripheral blood mononuclear cells. Biochemical and Biophysical Research Communications. 2004;324:511–517. doi: 10.1016/j.bbrc.2004.09.075. [DOI] [PubMed] [Google Scholar]
- Hur J, Sullivan KA, Pande M, Hong Y, Sima AAF, Jagadish HV, et al. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain. 2011;134:3222–3235. doi: 10.1093/brain/awr228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutton EJ, Carty L, Laurá M, Houlden H, Lunn MPT, Brandner S, et al. C-jun expression in human neuropathies: A pilot study. Journal of Peripheral Nervous System. 2011;16:295–303. doi: 10.1111/j.1529-8027.2011.00360.x. [DOI] [PubMed] [Google Scholar]
- Ianaro A, Lalenti A, Maffia P, Di Meglio P, Di Rosa M, Santoro MG. Anti-inflammatory activity of 15-deoxy-delta 12,14-PGJ2 and 2-cyclopenten-1-one: Role of the heat shock response. Molecular Pharmacology. 2003;64:85–93. doi: 10.1124/mol.64.1.85. [DOI] [PubMed] [Google Scholar]
- Jaattela M, Wissing D. Heat-shock proteins protect cells from monocyte cytotoxicity: Possible mechanism of self-protection. Journal of Experimental Medicine. 1993;177:231–236. doi: 10.1084/jem.177.1.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Fleshner M. Releasing signals, secretory pathways, and immune function of endogenous extracellular heat shock protein 72. Journal of Leukocyte Biology. 2006;79:425–434. doi: 10.1189/jlb.0905523. [DOI] [PubMed] [Google Scholar]
- Jones Q, Voegeli TS, Li G, Chen Y, William Currie R. Heat shock proteins protect against ischemia and inflammation through multiple mechanisms. Inflammation & Allergy – Drug Targets. 2011;10:247–259. doi: 10.2174/187152811796117726. [DOI] [PubMed] [Google Scholar]
- Kalmar B, Lu CH, Greensmith L. The role of heat shock proteins in amyotrophic lateral sclerosis: The therapeutic potential of arimoclomol. Pharmacology and Therapeutics. 2014;141:40–54. doi: 10.1016/j.pharmthera.2013.08.003. [DOI] [PubMed] [Google Scholar]
- Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, et al. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407–410. doi: 10.1038/nature01913. [DOI] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, Hartl FU. Molecular chaperone functions in protein folding and proteostasis. Annual Review of Biochemistry. 2013;82:323–355. doi: 10.1146/annurev-biochem-060208-092442. [DOI] [PubMed] [Google Scholar]
- Kondo T, Murakami K, Honkaniemi J, Sharp FR, Epstein CJ, Chan PH. Expression of Hsp70 mRNA is induced in the brain of transgenic mice overexpressing human CuZn-superoxide dismutase following transient global cerebral ischemia. Brain Research. 1996;737:321–326. doi: 10.1016/0006-8993(96)00949-3. [DOI] [PubMed] [Google Scholar]
- Kovalchin JT, Wang R, Wagh MS, Azoulay J, Sanders M, Chandawarkar RY. In vivo delivery of heat shock protein 70 accelerates wound healing by up-regulating macrophage-mediated phagocytosis. Wound Repair and Regeneration. 2006;14:129–137. doi: 10.1111/j.1743-6109.2006.00102.x. [DOI] [PubMed] [Google Scholar]
- Krause M, Heck TG, Bittencourt A, Scomazzon SP, Newsholme P, Curi R, et al. The chaperone balance hypothesis: The importance of the extracellular to intracellular Hsp70 ratio to inflammation-driven Type 2 diabetes, the effect of exercise, and the implications for clinical management. Mediators and Inflammation. 2015;2015:249205. doi: 10.1155/2015/249205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause M, Keane K, Rodrigues-Krause J, Crognale D, Egan B, De Vito G, et al. Elevated levels of extracellular heat-shock protein 72 (eHsp72) are positively correlated with insulin resistance in vivo and cause pancreatic beta-cell dysfunction and death in vitro. Clinical Science (London) 2014;126:739–752. doi: 10.1042/CS20130678. [DOI] [PubMed] [Google Scholar]
- Krause MS, Oliveira LP, Jr, Silveira EM, Vianna DR, Rossato JS, Almeida BS, et al. MRP1/GS-X pump ATPase expression: Is this the explanation for the cytoprotection of the heart against oxidative stress-induced redox imbalance in comparison to skeletal muscle cells? Cell Biochemistry and Function. 2007;25:23–32. doi: 10.1002/cbf.1343. [DOI] [PubMed] [Google Scholar]
- Krimmer T, Rassow J, Kunau WH, Voos W, Pfanner N. Mitochondrial protein import motor: The ATPase domain of matrix Hsp70 is crucial for binding to TIM44, while the peptide binding domain and the carboxy-terminal segment play a stimulatory role. Molecular and Cellular Biology. 2000;20:5879–5887. doi: 10.1128/mcb.20.16.5879-5887.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurucz I, Morva A, Vaag A, Eriksson KF, Huang X, Groop L, et al. Decreased expression of heat shock protein 72 in skeletal muscle of patients with Type 2 diabetes correlates with insulin resistance. Diabetes. 2002;51:1102–1109. doi: 10.2337/diabetes.51.4.1102. [DOI] [PubMed] [Google Scholar]
- Kusuma BR, Zhang L, Sundstrom T, Peterson LB, Dobrowsky RT, Blagg BSJ. Synthesis and evaluation of novologues as C-terminal Hsp90 inhibitors with cytoprotective activity against sensory neuron glucotoxicity. Journal of Medicinal Chemistry. 2012;55:5797–5812. doi: 10.1021/jm300544c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster GI, Febbraio MA. Exosome-dependent trafficking of Hsp70: A novel secretory pathway for cellular stress proteins. Journal of Biological Chemistry. 2005;280:23349–23355. doi: 10.1074/jbc.M502017200. [DOI] [PubMed] [Google Scholar]
- Lane T, Flam B, Lockey R, Kolliputi N. Txnip shuttling: Missing link between oxidative stress and inflammasome activation. Frontiers in Physiology. 2013;4:50. doi: 10.3389/fphys.2013.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazaro I, Oguiza A, Recio C, Mallavia B, Madrigal-Matute J, Blanco J, et al. Targeting Hsp90 ameliorates nephropathy and atherosclerosis through suppression of NF-kappaB and STAT signaling pathways in diabetic mice. Diabetes. 2015;64:3600–3613. doi: 10.2337/db14-1926. [DOI] [PubMed] [Google Scholar]
- Lee J, Ozcan U. Unfolded protein response signaling and metabolic diseases. Journal of Biological Chemistry. 2014;289:1203–1211. doi: 10.1074/jbc.R113.534743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehner T, Bergmeier LA, Wang Y, Tao L, Sing M, Spallek R, et al. Heat shock proteins generate beta-chemokines which function as innate adjuvants enhancing adaptive immunity. European Journal of Immunology. 2000;30:594–603. doi: 10.1002/1521-4141(200002)30:2<594::AID-IMMU594>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Leustek T, Dalie B, Amir-Shapira D, Brot N, Weissbach H. A member of the Hsp70 family is localized in mitochondria and resembles Escherichia coli DNAK. Proceedings of the National Academy of Science. 1989;86:7805–7808. doi: 10.1073/pnas.86.20.7805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Ma J, Zhao H, Blagg BS, Dobrowsky RT. Induction of heat shock protein 70 (Hsp70) prevents neuregulin-induced demyelination by enhancing the proteasomal clearance of c-jun. ASN Neuro. 2012;4:e00102. doi: 10.1042/20120047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lianos GD, Alexiou GA, Mangano A, Mangano A, Rausei S, Boni L, et al. The role of heat shock proteins in cancer. Cancer Letters. 2015;360:114–118. doi: 10.1016/j.canlet.2015.02.026. [DOI] [PubMed] [Google Scholar]
- Literáti-Nagy B, Kulcsár E, Literáti-Nagy Z, Buday B, Péterfai É, Horváth T, et al. Improvement of insulin sensitivity by a novel drug, BGP-15, in insulin-resistant patients: A proof of concept randomized double-blind clinical trial. Hormone and Metabolism Research. 2009;41:374–380. doi: 10.1055/s-0028-1128142. [DOI] [PubMed] [Google Scholar]
- Luo W, Dou F, Rodina A, Chip S, Kim J, Zhao Q, et al. Roles of heat-shock protein 90 in maintaining and facilitating the neurodegenerative phenotype in tauopathies. Proceedings of the National Academy of Science. 2007;104:9511–9516. doi: 10.1073/pnas.0701055104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Rodina A, Chiosis G. Heat shock protein 90: Translation from cancer to Alzheimer's disease treatment? BMC Neuroscience. 2008;9(Suppl 2):S7. doi: 10.1186/1471-2202-9-S2-S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Farmer KL, Pan P, Urban MJ, Zhao H, Blagg BS, et al. Heat shock protein 70 is necessary to improve mitochondrial bioenergetics and reverse diabetic sensory neuropathy following KU-32 therapy. Journal of Pharmacology and Experimental Therapeutics. 2014;348:281–292. doi: 10.1124/jpet.113.210435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Pan P, Anyika M, Blagg BS, Dobrowsky RT. Modulating molecular chaperones improves mitochondrial bioenergetics and decreases the inflammatory transcriptome in diabetic sensory neurons. ACS Chemical Neuroscience. 2015;6:1637–1648. doi: 10.1021/acschemneuro.5b00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden LA, Sandstrom ME, Lovell RJ, McNaughton L. Inducible heat shock protein 70 and its role in preconditioning and exercise. Amino Acids. 2008;34:511–516. doi: 10.1007/s00726-007-0004-7. [DOI] [PubMed] [Google Scholar]
- Mansilla MJ, Costa C, Eixarch H, Tepavcevic V, Castillo M, Martin R, et al. Hsp70 regulates immune response in experimental autoimmune encephalomyelitis. PLoS One. 2014;9:e105737. doi: 10.1371/journal.pone.0105737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum KK. Liposomal delivery of heat shock protein 72 into renal tubular cells blocks Nuclear Factor-kappaB activation, Tumor Necrosis Factor-alpha production, and subsequent ischemia-induced apoptosis. Circulation Research. 2003;92:293–299. doi: 10.1161/01.res.0000057754.35180.99. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Nakamoto H, Neckers L. The therapeutic target Hsp90 and cancer hallmarks. Current Pharmaceutical Design. 2013;19:347–365. doi: 10.2174/138161213804143725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nature Reviews Neuroscience. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Multhoff G. Heat shock protein 70 (Hsp70): Membrane location, export and immunological relevance. Methods. 2007;43:229–237. doi: 10.1016/j.ymeth.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Murakami H, Pain D, Blobel G. 70-kd heat shock-related protein is one of at least two distinct cytosolic factors stimulating protein import into mitochondria. Journal of Cell Biology. 1988;107:2051–2057. doi: 10.1083/jcb.107.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neef DW, Jaeger AM, Thiele DJ. Heat shock transcription factor 1 as a therapeutic target in neurodegenerative diseases. Nature Reviews Drug Discovery. 2011;10:930–944. doi: 10.1038/nrd3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obrosova IG, V, Drel R, Pacher P, Ilnytska O, Wang ZQ, Stevens MJ, et al. Oxidative-nitrosative stress and poly(ADP-ribose) polymerase (PARP) activation in experimental diabetic neuropathy: The relation is revisited. Diabetes. 2005;54:3435–3441. doi: 10.2337/diabetes.54.12.3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oglesbee MJ, Herdman AV, Passmore GG, Hoffman WH. Diabetic ketoacidosis increases extracellular levels of the major inducible 70-kDa heat shock protein. Clinical Biochemistry. 2005;38:900–904. doi: 10.1016/j.clinbiochem.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Ortega E, Giraldo E, Hinchado MD, Martinez M, Ibanez S, Cidoncha A, et al. Role of Hsp72 and norepinephrine in the moderate exercise-induced stimulation of neutrophils' microbicide capacity. European Journal of Applied Physiology. 2006;98:250–255. doi: 10.1007/s00421-006-0269-7. [DOI] [PubMed] [Google Scholar]
- Ortega E, Hinchado MD, Martin-Cordero L, Asea A. The effect of stress-inducible extracellular Hsp72 on human neutrophil chemotaxis: A role during acute intense exercise. Stress. 2009;12:240–249. doi: 10.1080/10253890802309853. [DOI] [PubMed] [Google Scholar]
- Park HS, Lee JS, Huh SH, Seo JS, Choi EJ. Hsp72 functions as a natural inhibitory protein of c-jun N-terminal kinase. EMBO Journal. 2001;20:446–456. doi: 10.1093/emboj/20.3.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, et al. C-jun is a negative regulator of myelination. Journal of Cell Biology. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LB, Blagg BS. To fold or not to fold: Modulation and consequences of Hsp90 inhibition. Future Medicinal Chemistry. 2009;1:267–283. doi: 10.4155/fmc.09.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polla BS, Kantengwa S, Francois D, Salvioli S, Franceschi C, Marsac C, et al. Mitochondria are selective targets for the protective effects of heat shock against oxidative injury. Proceedings of the National Academy of Science. 1996;93:6458–6463. doi: 10.1073/pnas.93.13.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Y, Liu B, Li Z. Activation of NK cells by extracellular heat shock protein 70 through induction of NKG2D ligands on dendritic cells. Cancer Immunity. 2008;8:12. [PMC free article] [PubMed] [Google Scholar]
- Ran R, Lu A, Zhang L, Tang Y, Zhu H, Xu H, et al. Hsp70 promotes TNF-mediated apoptosis by binding IKK gamma and impairing NF-kappaB survival signaling. Genes and Development. 2004;18:1466–1481. doi: 10.1101/gad.1188204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard V, Kaeffer N, Thuillez C. Delayed protection of the ischemic heart--from pathophysiology to therapeutic applications. Fundamental and Clinical Pharmacology. 1996;10:409–415. doi: 10.1111/j.1472-8206.1996.tb00595.x. [DOI] [PubMed] [Google Scholar]
- Robinson MB, Tidwell JL, Gould T, Taylor AR, Newbern JM, Graves J, et al. Extracellular heat shock protein 70: A critical component for motoneuron survival. Journal of Neuroscience. 2005;25:9735–9745. doi: 10.1523/JNEUROSCI.1912-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues-Krause J, Krause M, O'Hagan C, De Vito G, Boreham C, Murphy C, et al. Divergence of intracellular and extracellular Hsp72 in Type 2 diabetes: Does fat matter? Cell Stress and Chaperones. 2012;17:293–302. doi: 10.1007/s12192-011-0319-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saibil H. Chaperone machines for protein folding, unfolding and disaggregation. Nature Reviews Molecular Cell Biology. 2013;14:630–642. doi: 10.1038/nrm3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Roy Chowdhury SK, Smith DR, Balakrishnan S, Tessler L, Martens C, et al. Ciliary neurotrophic factor activates NF-kappaB to enhance mitochondrial bioenergetics and prevent neuropathy in sensory neurons of streptozotocin-induced diabetic rodents. Neuropharmacology. 2013a;65:65–73. doi: 10.1016/j.neuropharm.2012.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh A, Schapansky J, Smith DR, Young N, Odero GL, Aulston B, et al. Normalization of NF-kappaB activity in dorsal root ganglia neurons cultured from diabetic rats reverses neuropathy-linked markers of cellular pathology. Experimental Neurology. 2013b;241:169–178. doi: 10.1016/j.expneurol.2012.11.009. [DOI] [PubMed] [Google Scholar]
- Schmidt O, Pfanner N, Meisinger C. Mitochondrial protein import: From proteomics to functional mechanisms. Nature Reviews Molecular Cell Biology. 2010;11:655–667. doi: 10.1038/nrm2959. [DOI] [PubMed] [Google Scholar]
- Schulze PC, Yoshioka J, Takahashi T, He Z, King GL, Lee RT. Hyperglycemia promotes oxidative stress through inhibition of thioredoxin function by thioredoxin-interacting protein. Journal of Biological Chemistry. 2004;279:30369–30374. doi: 10.1074/jbc.M400549200. [DOI] [PubMed] [Google Scholar]
- Senf SM, Howard TM, Ahn B, Ferreira LF, Judge AR. Loss of the inducible Hsp70 delays the inflammatory response to skeletal muscle injury and severely impairs muscle regeneration. PLoS One. 2013;8:e62687. doi: 10.1371/journal.pone.0062687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh LP. Thioredoxin interacting protein (TXNIP) and pathogenesis of diabetic retinopathy. Journal of Clinical and Experimental Ophthamology. 2013;4 doi: 10.4172/2155-9570.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavniichuk R, V, Drel R, Shevalye H, Maksimchyk Y, Kuchmerovska TM, Nadler JL, et al. Baicalein alleviates diabetic peripheral neuropathy through inhibition of oxidative–nitrosative stress and p38 MAPK activation. Experimental Neurology. 2011;230:106–113. doi: 10.1016/j.expneurol.2011.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetler RA, Gan Y, Zhang W, Liou AK, Gao Y, Cao G, et al. Heat shock proteins: Cellular and molecular mechanisms in the central nervous system. Progress in Neurobiology. 2010;92:184–211. doi: 10.1016/j.pneurobio.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan W, Pasinelli P, Trotti D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochimica Biophysica Acta. 2014;1842:1295–1301. doi: 10.1016/j.bbadis.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriault JR, Mambula SS, Sawamura T, Stevenson MA, Calderwood SK. Extracellular Hsp70 binding to surface receptors present on antigen presenting cells and endothelial/epithelial cells. FEBS Letters. 2005;579:1951–1960. doi: 10.1016/j.febslet.2005.02.046. [DOI] [PubMed] [Google Scholar]
- Tidwell JL, Houenou LJ, Tytell M. Administration of Hsp70 in vivo inhibits motor and sensory neuron degeneration. Cell Stress and Chaperones. 2004;9:88–98. doi: 10.1379/CSC-9R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomlinson DR, Gardiner NJ. Glucose neurotoxicity. Nature Reviews Neuroscience. 2008;9:36–45. doi: 10.1038/nrn2294. [DOI] [PubMed] [Google Scholar]
- Turturici G, Sconzo G, Geraci F. Hsp70 and its molecular role in nervous system diseases. Biochemical Research International. 2011;2011:618127. doi: 10.1155/2011/618127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytell M, Greenberg SG, Lasek RJ. Heat shock-like protein is transferred from glia to axon. Brain Research. 1986;363:161–164. doi: 10.1016/0006-8993(86)90671-2. [DOI] [PubMed] [Google Scholar]
- Urban MJ, Li C, Yu C, Lu Y, Krise JM, McIntosh MP, et al. Inhibiting heat shock protein 90 reverses sensory hypoalgesia in diabetic mice. ASN NEURO. 2010;2:189–199. doi: 10.1042/AN20100015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MJ, Pan P, Farmer KL, Zhao H, Blagg BS, Dobrowsky RT. Modulating molecular chaperones improves sensory fiber recovery and mitochondrial function in diabetic peripheral neuropathy. Experimental Neurology. 2012;235:388–396. doi: 10.1016/j.expneurol.2012.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vareniuk I, I, Pavlov A, Drel VR, Lyzogubov VV, Ilnytska O, Bell SR, et al. Nitrosative stress and peripheral diabetic neuropathy in leptin-deficient (ob/ob) mice. Experimental Neurology. 2007;205:425–436. doi: 10.1016/j.expneurol.2007.03.019. [DOI] [PubMed] [Google Scholar]
- Vihervaara A, Sistonen L. HSF1 at a glance. Journal of Cell Science. 2014;127:261–266. doi: 10.1242/jcs.132605. [DOI] [PubMed] [Google Scholar]
- Walton-Diaz A, Khan S, Bourboulia D, Trepel JB, Neckers L, Mollapour M. Contributions of co-chaperones and post-translational modifications towards Hsp90 drug sensitivity. Future Medicinal Chemistry. 2013;5:1059–1071. doi: 10.4155/fmc.13.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Kelly CG, Karttunen JT, Whittall T, Lehner PJ, Duncan L, et al. CD40 is a cellular receptor mediating mycobacterial heat shock protein 70 stimulation of CC-chemokines. Immunity. 2001;15:971–983. doi: 10.1016/s1074-7613(01)00242-4. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, et al. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nature Medicine. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Wei W, Liu Q, Tan Y, Liu L, Li X, Cai L. Oxidative stress, diabetes, and diabetic complications. Hemoglobin. 2009;33:370–377. doi: 10.3109/03630260903212175. [DOI] [PubMed] [Google Scholar]
- Weiss YG, Bromberg Z, Raj N, Raphael J, Goloubinoff P, Ben-Neriah Y, et al. Enhanced heat shock protein 70 expression alters proteasomal degradation of IκB kinase in experimental acute respiratory distress syndrome. Critical Care Medicine. 2007;35:2128–2138. doi: 10.1097/01.ccm.0000278915.78030.74. [DOI] [PubMed] [Google Scholar]
- Wellen KE. Inflammation, stress, and diabetes. Journal of Clinical Investigation. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitham M, Fortes MB. Heat shock protein 72: Release and biological significance during exercise. Frontiers in Bioscience. 2008;13:1328–1339. doi: 10.2741/2765. [DOI] [PubMed] [Google Scholar]
- Whiting DR, Guariguata L, Weil C, Shaw J. IDF Diabetes Atlas: Global estimates of the prevalence of diabetes for 2011 and 2030. Diabetes Research and Clinical Practice. 2011;94:311–321. doi: 10.1016/j.diabres.2011.10.029. [DOI] [PubMed] [Google Scholar]
- Wiedemann N, Frazier AE, Pfanner N. The protein import machinery of mitochondria. Journal of Biological Chemistry. 2004;279:14473–14476. doi: 10.1074/jbc.R400003200. [DOI] [PubMed] [Google Scholar]
- Wong HR, Ryan M, Wispe JR. The heat shock response inhibits inducible nitric oxide synthase gene expression by blocking I kappa-B degradation and NF-kappa B nuclear translocation. Biochemical and Biophysical Research Communications. 1997;231:257–263. doi: 10.1006/bbrc.1997.6076. [DOI] [PubMed] [Google Scholar]
- Wong HR, Ryan MA, Menendez IY, Wispe JR. Heat shock activates the I-kappaBalpha promoter and increases I-kappaBalpha mRNA expression. Cell Stress and Chaperones. 1999;4:1–7. doi: 10.1006/csac.1998.0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright E, Jr, Scism-Bacon JL, Glass LC. Oxidative stress in type 2 diabetes: The role of fasting and postprandial glycaemia. Internation Journal of Clinical Practice. 2006;60:308–314. doi: 10.1111/j.1368-5031.2006.00825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Zheng B, Shaywitz A, Dagon Y, Tower C, Bellinger G, et al. AMPK-dependent degradation of TXNIP upon energy stress leads to enhanced glucose uptake via Glut1. Molecular Cell. 2013;49:1167–1175. doi: 10.1016/j.molcel.2013.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XM, Baxter GF, Heads RJ, Yellon DM, Downey JM, Cohen MV. Infarct limitation of the second window of protection in a conscious rabbit model. Cardiovascular Research. 1996;31:777–783. doi: 10.1016/0008-6363(96)00026-0. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU. Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell. 2003;112:41–50. doi: 10.1016/s0092-8674(02)01250-3. [DOI] [PubMed] [Google Scholar]
- Zhang L, Zhao H, Blagg BS, Dobrowsky RT. A C-terminal heat shock protein 90 inhibitor decreases hyperglycemia-induced oxidative stress and improves mitochondrial bioenergetics in sensory neurons. Journal of Proteome Research. 2012;11:129–137. doi: 10.1021/pr300056m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler D, Ametov A, Barinov A, Dyck PJ, Gurieva I, Low PA, et al. Oral treatment with α-lipoic acid improves symptomatic diabetic polyneuropathy. Diabetes Care. 2006;29:2365–2370. doi: 10.2337/dc06-1216. [DOI] [PubMed] [Google Scholar]
- Ziegler D, Low PA, Litchy WJ, Boulton AJ, Vinik AI, Freeman R, et al. Efficacy and safety of antioxidant treatment with alpha-lipoic acid over 4 years in diabetic polyneuropathy: The Nathan 1 Trial. Diabetes Care. 2011;34:2054–2060. doi: 10.2337/dc11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]