

Key Clinical Message
Rubinstein–Taybi syndrome is associated with intellectual and physical features. CREBBP and EP300 are causative. Few cases of EP300 mutations are reported. We report a case with mild features of RSTS and EP300 mutation on exome sequencing. This illustrates the utility of exome sequencing to expand every genetic phenotype.
Keywords: CREBBP, EP300, Rubinstein–Taybi syndrome, whole exome sequencing
Case Report
Rubinstein–Taybi syndrome (RSTS, OMIM #180849) was first described in 1963 and is known to be associated with mental impairment, growth deficiency, microcephaly, facial dysmorphia, and skeletal abnormalities 1. Two genes have been shown to result in this syndrome: CREBBP and EP300 2, 3. Mutations affecting CREBBP cause approximately 50–60% of cases, whereas mutations of EP300 are associated with only 3% of cases. Of note, a recent paper by Negri et al. suggests a higher incidence of EP300 mutations, closer to 8% of cases [2014].
Both CREBBP and EP300 are transcriptional coactivators and are highly conserved in the expression of many genes involved in embryology, cell growth and differentiation, and tumor suppression 3. The second function of these genes is to act as a histone acetyltransferase (HAT) which opens up the chromatin structure at the locus to be expressed, crucial for overall gene expression 4. While the sequences of EP300 and CREBBP are more than 70% similar, the two proteins have distinct functions and cannot replace one another 5.
To date, the literature reports 16 RSTS patients with EP300 mutations. In general, these patients have a clinical presentation milder than patients with CREBBP mutations 6. A recent paper expanded the phenotypic spectrum of EP300 mutations to include genitourinary anomalies and a myelomeningocele 7. Yet here, we report a case of a 11‐year‐old girl with very mild physical and cognitive features of RSTS and found to have an EP300 mutation on exome sequencing.
The patient is a Caucasian female presenting to our genetics office at 9 years of age for short stature, mild dysmorphic features, and mild cognitive impairment. She was the second pregnancy and second delivery for her healthy, nonconsanguineous parents. After an uncomplicated pregnancy, her birth was induced at 34 weeks gestation because of maternal hypertension. Birth weight was 1.8 kg (20th centile) and birth length was 40.6 cm (10th centile). She was hospitalized in the NICU for 2 weeks due to feeding difficulties.
Her past medical history was significant for an atrial septal defect that never required intervention. She was diagnosed as an infant with spina bifida occulta due to drainage from a sacral dimple. However, subsequent MRI imaging of the spine was normal. Her only surgical history was a tonsillectomy and adenoidectomy. She had one pilomatrixoma surgically removed from the upper arm. At the age of 8 years, her growth was noted to plateau and she crossed from the 50th–8th centile within 1 year. Evaluation showed hypothalamic‐pituitary‐adrenal axis suppression, thought to be secondary to exogenous glucocorticoid use in the form of inhaled steroids. She was prescribed growth hormone with good response. At 11 years 3 months, her growth parameters were: weight 38.8 kg (55th centile), height 145.1 cm (45th centile), and head circumference, 52 cm (25th centile).
With regard to development, walking and talking were delayed, both starting at 18 months of age. She received physical, occupational, and speech therapy by 2 years of age and improved. By the age of 11 years, she had undergone a full battery of neuropsychological testing on several occasions, at several hospitals. Each time, she was found to have overall intelligence in the low‐average range, with each subtest score in the low average to average ranges. Her lowest scores were on tasks with visual‐spatial or motor planning.
Her physical exam was significant for flat malar eminences with mild ptosis and down‐slanting palpebral fissures (Fig. 1). Her thumbs were not broad (Fig. 2). She also had evidence of hypertrophic scarring in several sites from minor trauma (Fig. 3).
Figure 1.
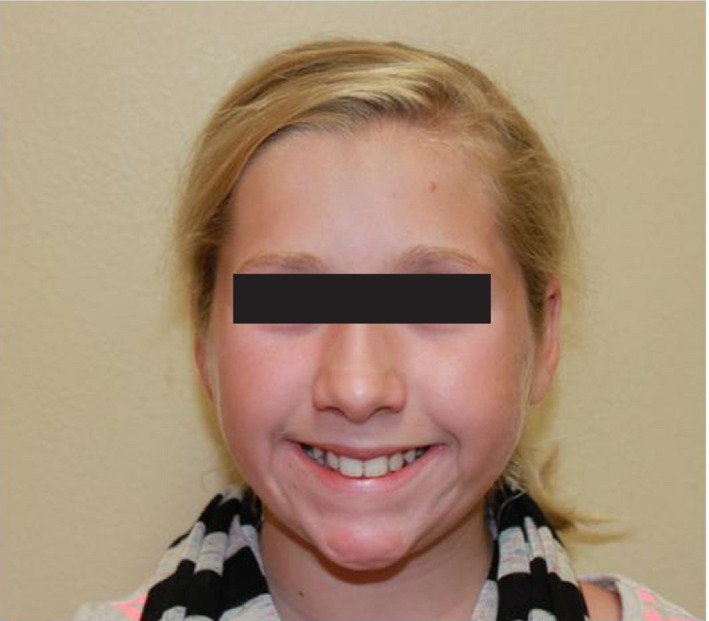
Picture taken at 10 years of age. Note the long columella of the nose and thin upper lip. The palpebral fissures are slightly down‐slanted. Additionally, while the eyebrows are slightly arched, there is no hirsutism.
Figure 2.
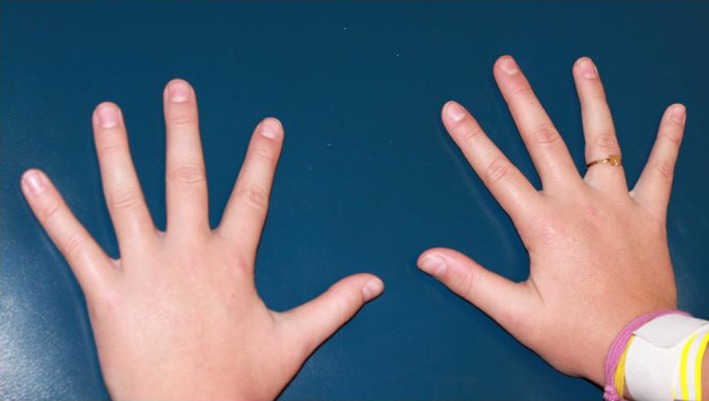
Picture of hands. No abnormalities noted.
Figure 3.
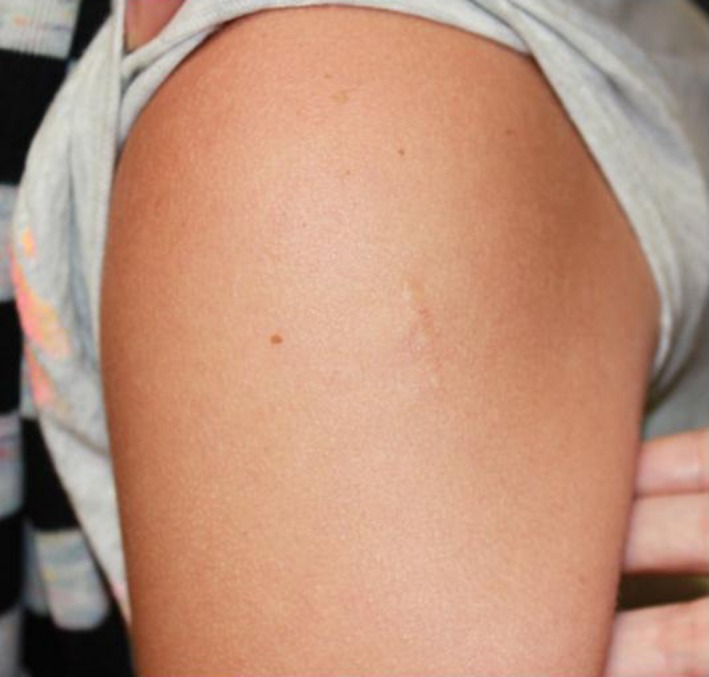
Note the hypertrophic scarring on the left upper arm from a prior simple laceration that did not require sutures.
The initial genetic workup included a karyotype and chromosomal microarray analysis (Affymetrix CytoScan HD microarray, ARUP©, Salt Lake City, UT). The karyotype and microarray both revealed a normal, female result. Our next evaluation was whole exome sequencing (WES) from a peripheral blood sample submitted to a reference laboratory (GeneDx©, Gaithersburg, MD) using their standard methodology. This test revealed that the patient was heterozygous for a c.1948delA mutation in the EP300 gene. This deletion in exon 10 results in a frameshift mutation and a premature stop codon. This mutation has not been previously reported but is predicted to cause a loss of normal protein function either through truncation or nonsense‐mediated mRNA decay. Both the patient's mother and brother were negative for this mutation. The patient's father was offered testing but declined.
We were surprised our testing revealed an EP300 mutation as RSTS was never considered in the differential diagnosis. Since the father was not tested and this mutation has never been reported, the causative nature of this mutation for her features is largely speculative. However, in retrospect, her short stature, mild cognitive delays, history of pilomatrixoma, and minor facial features fit some aspects of the diagnosis. Even the more subtle findings of maternal hypertension at the end of pregnancy and skin scarring have been reported previously 4, 6, 8, 9. Like so many have appreciated, mutations in the EP300 can lead to a milder phenotype. Our patient has no notable skeletal findings, lacks hirsutism and microcephaly, and the cognitive delays are quite subtle such that she has a normal IQ on repeated testing.
Negri et al. hypothesized that a truncated EP300 protein leads to varying phenotypes depending on how severely the protein product is affected 6. Our patient's EP300 mutation in exon 10 preserves the HAT domain. Alterations in the HAT domain are proposed to cause the classic RSTS phenotype and the cognitive impairment 10. Our patient's mild phenotype further confirms this idea that as more of the EP300 protein is preserved, and particularly the HAT domain, the milder the phenotype. Again, this case also illustrates that as the prevalence of WES increases, we will likely find that EP300 mutations are more common, and therefore better understand the full range of associated phenotypes.
Conflict of Interest
None declared.
Clinical Case Reports 2016; 4(7): 696–698
Elizabeth A. Sellars and Bonnie R. Sullivan contributed equally to the paper.
References
- 1. Rubinstein, J. H. , and Taybi H.. 1963. Broad thumbs and toes and facial abnormalities: a possible mental retardation syndrome. Am. J. Dis. Child. 105:588–608. [DOI] [PubMed] [Google Scholar]
- 2. Roelfsema, J. H. , White S. J., Ariyürek Y., Bartholdi D., Niedrist D., Papadia F., et al. 2005. Genetic heterogeneity in Rubinstein‐Taybi sydrome: mutations in both the CBP and EP300 genes cause disease. Am. J. Hum. Genet. 76:572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zimmermann, N. , Acosta A. M., Kohlhase J., and Bartsch O.. 2007. Confirmation of EP300 gene mutations as a rare cause of Rubinstein‐Taybi syndrome. Eur. J. Hum. Genet. 15:837–842. [DOI] [PubMed] [Google Scholar]
- 4. Tsai, A. C. H. , Dossett C. J., Walton C. S., Cramer A. E., Eng P. A., Nowakowska B. A., et al. 2011. Exon deletions of the EP300 and CREBBP genes in two children with Rubinstein‐Taybi syndrome detected by aCGH. Eur. J. Hum. Genet. 19:43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Viosca, J. , Lopez‐Atalaya J., Olivares R., Eckner R., and Barco A.. 2010. Syndromic features and mild cognitive impairment in mice with genetic reduction on p300 activity: differential contribution of p300 and CBP to Rubinstein‐Taybi syndrome etiology. Neurobiol. Dis. 37:186–194. [DOI] [PubMed] [Google Scholar]
- 6. Negri, G. , Milani D., Colapietro P., Forzano F., Della Monica M., Rusconi D., et al. 2014. Clinical and molecular characterization of Rubinstein‐Taybi syndrome patients carrying distinct novel mutations of the EP300 gene. Clin. Genet. 87:148–154. [DOI] [PubMed] [Google Scholar]
- 7. Solomon, B. D. , Bodian D. L., Khromykh A., Mora G. G., Lanpher B. C., Iyer R. K., et al. 2015. Expanding the phenotypic spectrum in EP300‐related Rubinstein‐Taybi syndrome. Am. J. Hum. Genet. A 167A:1111–1116. [DOI] [PubMed] [Google Scholar]
- 8. Bartholdi, D. , Roelfsema J. H., Papadia F., Breuning M. H., Niedrist D., Hennekam R. C., et al. 2007. Genetic heterogeneity in Rubinstein‐Taybi syndrome: delineation of the phenotype of the first patients carrying mutations in EP300. J. Med. Genet. 44:327–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Foley, P. , Bunyan D., Stratton J., Dillon M., and Lynch S. A.. 2009. Further case of Rubinstein‐Taybi syndrome due to a deletion in EP300. Am. J. Med. Genet. A 149A:997–1000. [DOI] [PubMed] [Google Scholar]
- 10. Roelfsema, J. H. , and Peters D. J.. 2007. Rubinstein‐Taybi syndrome: clinical and molecular overview. Expert Rev. Mol. Med. 9:1–16. [DOI] [PubMed] [Google Scholar]