

Abstract
Radical SAM enzymes use a [4Fe-4S] cluster to cleave S-adenosylmethionine (SAM) to initiate diverse radical reactions. These are thought to involve the 5′-deoxyadenosyl radical intermediate, which has not yet been detected. Here we rapid freeze-quench trap a catalytically competent intermediate in the reaction catalyzed by the radical SAM enzyme pyruvate formate-lyase activating enzyme. Characterization of the intermediate by electron paramagnetic resonance and 13C, 57Fe electron-nuclear double resonance spectroscopies reveals that it contains an organometallic center in which 5′-carbon of a SAM-derived deoxyadenosyl moiety forms a bond to the unique iron of the [4Fe-4S] cluster. Discovery of this intermediate extends the list of enzymatic bio-organometallic centers to the radical SAM enzymes, the largest enzyme superfamily known, and reveals intriguing parallels to B12 radical enzymes.
The radical SAM superfamily is the largest known, with over 100,000 functional domains found throughout all kingdoms of life.(1) These enzymes utilize a [4Fe-4S]+ cluster and S-adenosylmethionine (SAM) to catalyze a remarkably diverse array of finely-tuned (2) radical reactions that are central to key pathways, such as heme and chlorophyll biosynthesis, vitamin biosynthesis, DNA repair, and metal cluster assembly.(1) Spectroscopic studies of the radical SAM enzymes pyruvate formate-lyase activating enzyme (PFL-AE) and lysine 2,3-aminomutase (LAM), as confirmed by X-ray structure determinations, demonstrated that these enzymes bind SAM as a classical coordination complex in which the amino and carboxyl moieties of SAM chelate the unique iron of a [4Fe-4S] cluster.(3-6) One-electron reduction of SAM by the [4Fe-4S]+ cluster initiates SAM cleavage, generating the 5′-deoxyadenosyl (5′-dAdo•) radical intermediate which carries out subsequent radical chemistry. To date, neither the 5′-dAdo• radical nor any SAM- or 5′-dAdo- derived intermediate has been detected for any radical SAM enzyme. The use of the enzymatically active SAM analog 3′,4′-anhydro-S-adenosylmethionine (anSAM), however, has allowed characterization of the allylically stabilized anhydroadenosyl radical, a functional analog of the 5′-dAdo• radical, generated within the active site of LAM during the LAM-catalyzed reaction.(7-9)
PFL-AE is a radical SAM enzyme that catalyzes the formation of a catalytically essential glycyl (G•) radical on G734 of pyruvate formate-lyase (PFL), a central enzyme in anaerobic glucose metabolism (Fig. 1).(10) Here we use rapid freeze-quench (rfq) electron paramagnetic resonance (EPR) and electron-nuclear double resonance (ENDOR) spectroscopies to trap and characterize a catalytically competent intermediate species formed during the reaction catalyzed by PFL-AE. This approach was inspired by the hope of capturing the elusive 5′-dAdo• radical intermediate implicated as the central species in radical SAM mechanisms, but instead yielded a state with unanticipated properties.
Fig. 1.
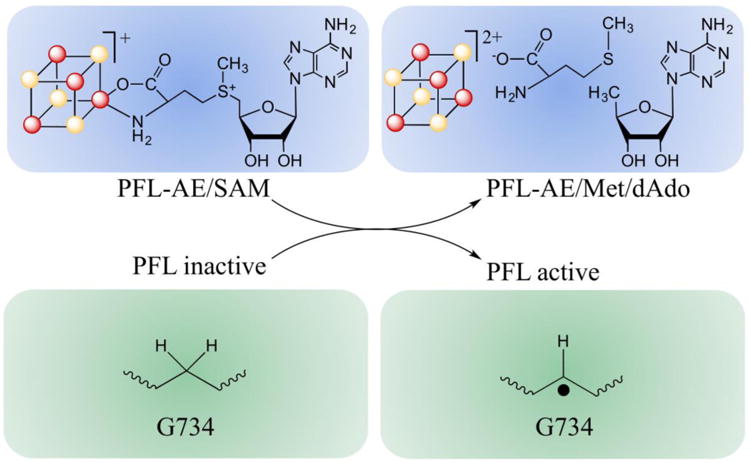
Activation of PFL by PFL-AE, with concomitant cleavage of SAM to methionine and 5′-deoxyadenosine.
When reduced PFL-AE was rapidly mixed with PFL and SAM followed by rfq of the reaction at times ranging from 25 ms to 1.0 s after mixing, the characteristic [4Fe-4S]+ EPR signal of reduced PFL-AE/SAM (g = 2.01, 1.88, 1.87)(11) diminishes in intensity concomitant with appearance of a new EPR signal, denoted Ω (g‖ = 2.035, g⊥ = 2.004), whose intensity reaches a maximum at 500 ms quench times (Fig. S1). At longer quench times the Ω signal is lost in parallel with the appearance of the G• radical signal characteristic of activated PFL, indicative that Ω is an intermediate in the formation of G• radical.
To explore the reactivity of Ω we cryo-annealed a frozen 500 ms quench sample for 1 or 3 minutes at progressively higher temperatures up to 220 K. During cryo-annealing the frozen solid, the Ω signal is lost in parallel with quantitative generation of the PFL G• g-2 radical signal, with its characteristic doublet from hyperfine coupling (A ∼ 15 G) to the α-proton (Fig. 2A).(12) This annealing progression confirms that Ω is a catalytically competent reaction intermediate that can generate the G• radical product by H-atom extraction from G734 of PFL, and can even do so in the frozen solid. As control, the rfq reaction was carried out using PFL G734A, where the G• radical site has been substituted with alanine. The rfq EPR spectrum at 12 K is identical to the one for PFL wild-type, indicating that Ω is formed by cleavage of SAM in these reactions (Fig. 2B), However, the EPR spectrum at 40 K shows that Ω does not proceed to produce a G• radical, at 500 ms quench time or longer, or during cryo-annealing.
Fig. 2.
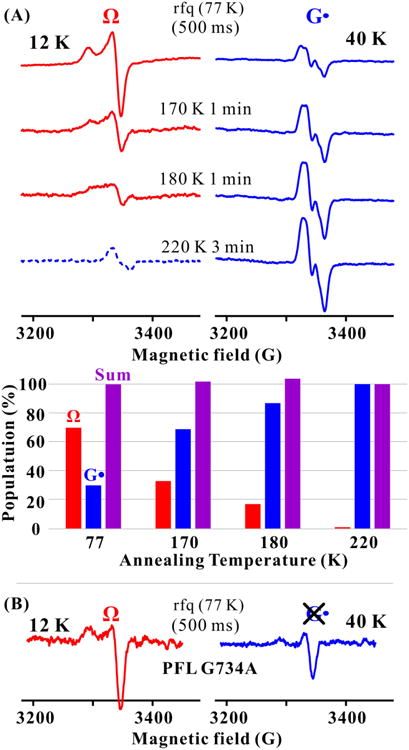
EPR spectra showing the formation of the PFL glycyl radical (G•) from Ω. (A, upper) EPR spectra of mixture of photoreduced PFL-AE and PFL/SAM freeze-quenched at ∼77K (500 ms) and stored at 77 K, and then annealed at progressively higher T for indicated times (See SI). At 12 K, the spectrum of G• radical is highly saturated and its amplitude diminished; at 40 K, signal from rapidly relaxing Ω is correspondingly diminished. The spectra here have had the residual intensities at both temperatures subtracted out (See SI, Fig. S2), with one exception. Ω is completely lost after annealing at 220 K; the dashed curve shows the residual signal from saturated G• radical. (A, lower) Populations of Ω and G• radical relative to the final (220 K) G• radical concentration taken as 100%, as derived from EPR spectra (See SI). (B) X-band EPR spectra for photoreduced PFL-AE freeze-quenched (77K) 500 ms after mixing with PFL G734A/SAM, with spectra collected at 12 and 40 K. Conditions: microwave frequency = 9.23 GHz, microwave power = 1 mW, 100 kHz modulation amplitude = 8 G, T, as indicated; the gain at a given T is fixed.
We conclude from these observations that (i) Ω is associated with PFL-AE and SAM or 5′-dAdo; (ii) that it is indeed a precursor in the formation of G• radical of the substrate protein PFL; and (iii) that in Ω the target G734 has been positioned so precisely that H-atom abstraction to generate the G• radical can take place without major conformational changes, which are quenched in the frozen solid.
The g-values for Ω are not compatible with assignment to a ‘free’ organic radical, such as the elusive 5′-dAdo• radical, which would exhibit a near-isotropic, g ∼ 2.0. Moreover, the EPR spectrum for the primary-carbon 5′-dAdo• radical would necessarily exhibit well-resolved splittings arising from the hyperfine coupling between the odd electron in the 2pπ orbital of C(5′) and the two equivalent protons of 1H2C(5′) (See SI, Fig. S3), but these are absent in the spectrum of Ω. The g-values do recall those of Compound ES of cytochrome c peroxidase, where they reflect a weak exchange coupling between an organic radical and an integer-spin metal-ion center.(13-15) The analogous situation here would involve the 5′-dAdo• radical spin-coupled to an integer-spin [4Fe-4S]2+ cluster. However, for multiple reasons this model also is unambiguously incapable of describing Ω, not least because the EPR spectrum for such a spin-coupled center would necessarily exhibit the same 1H hyperfine couplings to the 1H2C(5′) of 5′-dAdo• radical that are required for the ‘free’ radical, unchanged by the exchange interaction,(13-15) (See SI, Fig. S3), but that are absent in the spectrum of Ω.
To determine whether Ω nonetheless involves 5′-dAdo• radical in some fashion, we carried out rfq experiments, using 13C labeled SAM and 35 GHz CW/pulse ENDOR spectroscopies to examine Ω. When Ω was formed with SAM in which all adenosyl carbons, both of the base and ribose, are 13C ([adenosyl-13C10]-SAM), CW ENDOR spectra collected at g⊥ = 2.004 disclosed a strong coupling to 13C (Fig. 3A upper). This signal can be simulated by a hyperfine interaction with a single 13C with a large isotropic coupling, (aiso = 9.4 MHz) and an axial dipolar coupling (2T ≳ 5.3 MHz, depending on the extent to which the dipolar direction deviates from the g⊥ plane). A second, more weakly coupled 13C signal arising from 13C at other positions (A ∼ 0.7 MHz) is revealed by the Mims pulse technique (Fig. 3A inset).
Fig. 3.
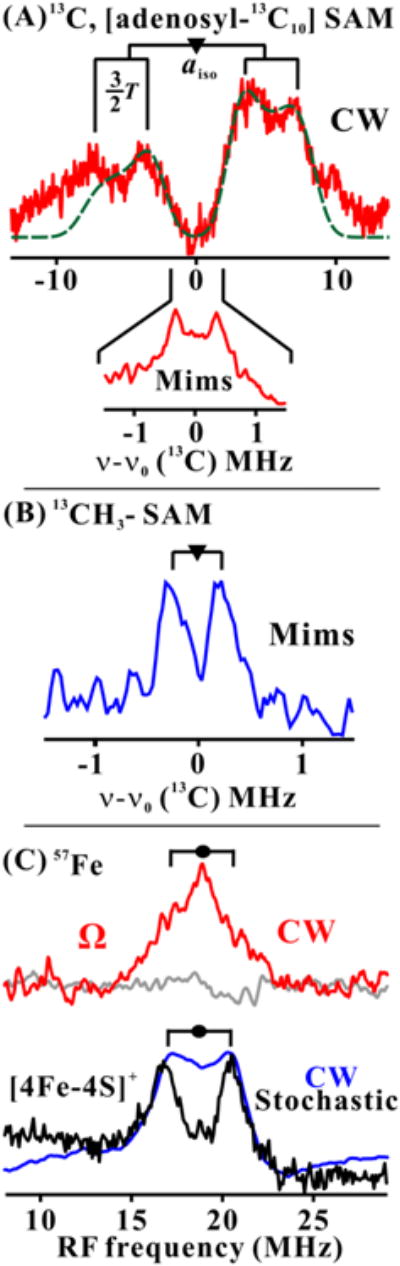
35 GHz ENDOR spectra at g⊥ for photoreduced PFL-AE freeze-quenched with PFL/SAM (See SI for details). To first order, an ENDOR spectrum of an I = 1/2 nucleus (N) in a frozen solution comprises a superposition of signals from different orientations, each signal a doublet at frequencies, ν± = |ν(N) ± A/2|, where ν(N) is the nuclear larmor frequency and A is the orientation-dependent hyperfine coupling.(23) For 13C, A/2 ≪ ν(13C) and it is convenient to plot spectra vs ν - ν(13C). For 57Fe, ν(57Fe) ≪ A/2 and spectra are plotted vs ν.(A) 13C CW ENDOR for [adenosyl-13C10] SAM. Best match simulation to axial hyperfine tensor (See SI), green dash line. Simulation parameters, aiso = 9.4 MHz, 2T = 5.3 MHz and β = 90°. Conditions: microwave frequency = 35.39 GHz, microwave power = 1 mW, 100 kHz modulation amplitude = 1.3 G, rf sweep rate = 1 MHz/s and T = 2 K. Inset: Mims ENDOR spectrum. Conditions: microwave frequency = 35.20 GHz, MW pulse length, (π/2) = 50 ns, τ = 500 ns and T = 2 K. (B) Mims ENDOR spectrum from [methyl-13C] SAM. Conditions: microwave frequency = 35.08 GHz, MW pulse length, (π/2) = 50 ns, τ = 500 ns and T = 2 K. (C) 57Fe CW ENDOR for 57Fe enriched Ω (prepared using [adenosyl-13C10] SAM) and photoreduced PFL-AE. Upper: CW ENDOR spectra for 57Fe-enriched (red) and natural abundance (gray) rfq samples. Lower: Frequency sweep and randomly hopped stochastic CW ENDOR spectra (23) for 57Fe-enriched reduced PFL-AE. Conditions: microwave frequency = 35.45 GHz and 35.07GHz for rfq and 57Fe-enriched reduced PFL-AE, respectively, microwave power = 1 mW, 100 kHz modulation amplitude = 1.3 G, rf sweep rate = 1 MHz/s, stochastic CW ENDOR cycle; rf-on = 3 ms, rf-off = 1 ms, sample collection time = 3 ms, and T = 2 K.
The strong isotropic coupling to one 13C of SAM reveals that this carbon is covalently integrated into the paramagnetic center of Ω. However, in addition to the absence of a large 1H2C(5′) hyperfine coupling (vide supra), the magnitude of aiso is ∼ ten-fold too low (16, 17) to be attributed to the sought-for 5′-dAdo• primary-carbon radical, either ‘free’ or weakly-coupled to the cluster spin. An alternative possibility is that the unpaired electron of Ω is on the sulfur of an intact SAM neutral radical, but in this case we would expect that each of the three carbons bound to the sulfur would exhibit a similarly strong hyperfine coupling. Instead, experiments using [methyl-13C]-SAM showed the methyl-13C is only weakly-coupled (A ∼ 0.5 MHz) (Fig. 3B), ruling out this alternative.
These considerations lead to the idea that Ω involves a SAM fragment associated with an S = 1/2 form of the [4Fe-4S] cluster. To test this idea we used 57Fe-enriched PFL-AE to prepare Ω. ENDOR of this sample revealed an 57Fe signal centered at A/2 = ∼17 MHz, essentially the same as the much better-resolved signal for the 57Fe-labeled protein in its paramagnetic [4Fe-4S]+ state (Fig. 3C). This establishes that the electron spin giving rise to the Ω EPR signal resides on the iron-sulfur cluster, while the isotropic coupling to a single carbon of dAdo requires that carbon of the 5′-dAdo fragment to be covalently bound to the paramagnetic cluster. Although all carbons of the adenosyl moiety of SAM were 13C in this sample, the only reasonable interpretation is that the 5′-C of the 5′-dAdo• radical created by reductive cleavage of SAM has formed an Fe-C bond with the unique cluster iron, and gives rise to the stronger coupling observed in the 13C ENDOR spectrum of Ω (Fig. 4). The weakly-coupled carbon observed in the Mims ENDOR (Fig. 3B) would then be assigned to 4′-C of dAdo.
Fig. 4.
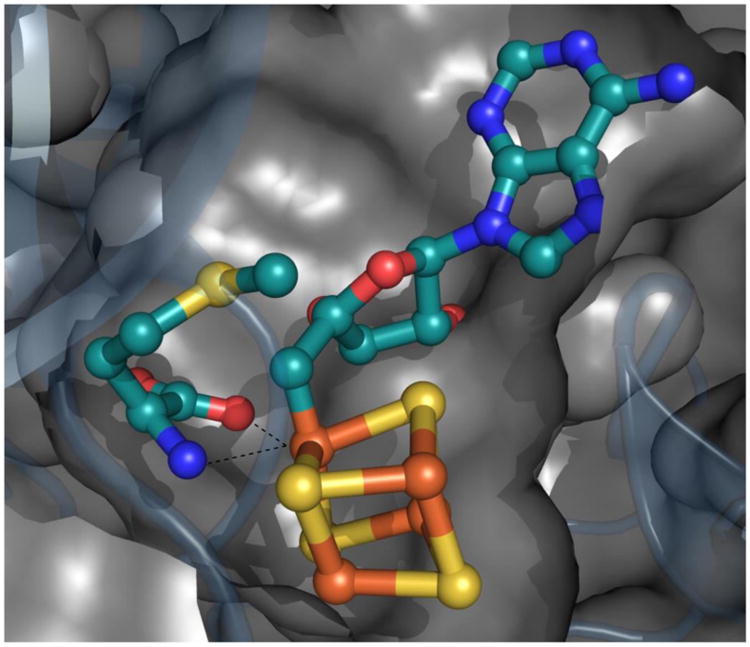
Model for bio-organometallic intermediate, Ω. Whether methionine remains coordinated to the unique iron is not currently known.
In the commonly proposed mechanism of radical SAM enzymes, an electron from a [4Fe-4S]+ cluster is transferred to the sulfonium of SAM, thereby promoting S-[5′-C] bond cleavage to generate the 5′-dAdo• radical intermediate.(1) If this is indeed the initial step in catalysis, then our current results would indicate that in PFL-AE, the 5′-dAdo• radical thus formed by SAM reduction adds to the [4Fe-4S]2+ cluster product of the electron transfer, to generate Ω, an organometallic intermediate in which the S = 1/2 cluster, formally [4Fe-4S]3+, is covalently linked to 5′-dAdo through an Fe-C bond between [5′-C] and the unique iron of the cluster. As a plausible alternative mechanism, nucleophilic attack of the unique iron of the [4Fe-4S]+ cluster on the 5′-C of SAM initiates SAM cleavage, to release methionine and generate the [4Fe-4S]3+-adenosyl intermediate, Ω. In this case, 5′-dAdo• radical truly is ‘never free’.(9)
Regardless of the mechanism for its formation, the organometallic intermediate Ω proposed here for PFL-AE provides intriguing parallels to the adenosylcobalamin cofactor, which is used by B12 radical enzymes to initiate radical catalysis by homolytic cleavage of the Co(III)-[5′-C]-deoxyadenosyl bond to generate a 5′-dAdo• radical and Co(II) cobalamin.(18, 19) In analogy, the H-atom abstraction from G734 by Ω would begin with homolytic cleavage of the [4Fe-4S]3+ Fe-[5′-C]-adenosyl bond to generate [4Fe-4S]2+ and a 5′-dAdo• radical, which then abstracts a precisely positioned hydrogen atom from G734 of PFL.
The results reported here provide evidence, that, contrary to expectation, cleavage of SAM by PFL-AE generates a catalytically competent bio-organometallic intermediate Ω in which the 5′-dAdo moiety derived from SAM is covalently bound through 5′-C to the [4Fe-4S] cluster (Fig. 4). Remarkably, this intermediate generates the G• product radical on PFL during annealing of the activated protein-protein complex in the frozen solid at temperatures at least as low as 170 K (Fig. 2). This confirms that in the ternary complex the structures of both PFL and PFL-AE have rearranged so that the target G734 of PFL is proximate to the [4Fe-4S]/SAM radical-generating construct of PFL-AE. This proximity, which was predicted based on structural studies of PFL-AE in complex with a heptamer peptide of PFL,(5) requires that formation of the PFL-AE/PFL complex involves substantial conformational changes in PFL, whose G734 residue is normally buried 8 Å from the protein surface.(20, 21)
If Ω is indeed formed through nucleophilic attack of the unique Fe on the 5′-C of SAM as a means to cleave the S-[5′-C] bond of SAM and generate a “radical initiator” Fe-[5′-C] bond, then such an intermediate may well be common to many or even all radical-SAM enzymes. In particular, it is interesting to consider whether the formation of Ω might represent a mechanistic difference between those radical SAM enzymes that, like PFL-AE, use SAM as a co-substrate, and those that use SAM as a cofactor; further studies will be required to address this possibility. The mechanistic importance of such an intermediate may lie in providing a means to control and store the reactive 5′-dAdo• radical intermediate until the target hydrogen atom of substrate is bound appropriately for abstraction. Given the complexity of the 170 kDa substrate PFL, and the requirement for large protein conformational changes during its activation by PFL-AE,(5, 21) the need for such storage and control seems plausible. In either case, the discovery of this organometallic ‘radical-initiator’ moiety in a radical-SAM enzyme provides an additional intriguing parallel to the B12 radical enzymes: not only do both classes utilize a 5′-dAdo• radical in catalytic H-atom abstraction, but both employ a precursor complex with a direct metal-[5′-C] bond.
The work presented here reveals a catalytically competent [4Fe-4S]-cluster-bound 5′-deoxyadenosyl species in PFL-AE, a 5′-deoxyadenosyl-derived intermediate never before observed among the vast radical SAM enzyme superfamily. This radical initiator Fe-[5′-C]-adenosyl complex expands the growing list of enzymatic bio-organometallic centers, whose first entry was coenzyme B12,(18, 19) but now includes the active-site metal clusters of hydrogenases,(22) nitrogenase,(23, 24) and a catalytic intermediate of IspH.(25, 26) Our study shows that radical SAM enzymes, the largest enzyme superfamily currently known, also can function through an organometallic center.
Supplementary Material
Acknowledgments
This work was funded by the NIH (GM 111097, BMH; GM 54608, JBB). The authors gratefully acknowledge the contributions of Boi Hanh (Vincent) Huynh and Carsten Krebs in the early stages of this study.
Footnotes
Supplementary Materials: Materials and Methods
References and Notes
- 1.Broderick JB, Duffus BR, Duschene KS, Shepard EM. Radical S-Adenosylmethionine Enzymes. Chem Rev. 2014;114:4229. doi: 10.1021/cr4004709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sicoli G, et al. Fine-tuning of a radical-based reaction by radical S-adenosyl-L-methionine tryptophan lyase. Science. 2016;351:1320. doi: 10.1126/science.aad8995. [DOI] [PubMed] [Google Scholar]
- 3.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. An anchoring role for FeS Clusters: Chelation of the amino acid moiety of S-adenosylmethionine to the unique iron site of the [4Fe-4S] cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:11270. doi: 10.1021/ja027078v. [DOI] [PubMed] [Google Scholar]
- 4.Chen D, Walsby C, Hoffman BM, Frey PA. Coordination and mechanism of reversible cleavage of S-adenosylmethionine by the [4Fe-4S] center in lysine 2,3-aminomutase. J Am Chem Soc. 2003;125:11788. doi: 10.1021/ja036120z. [DOI] [PubMed] [Google Scholar]
- 5.Vey JL, et al. Structural basis for glycyl radical formation by pyruvate formate-lyase activating enzyme. Proc Natl Acad Sci USA. 2008;105:16137. doi: 10.1073/pnas.0806640105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lepore BW, Ruzicka FJ, Frey PA, Ringe D. The x-ray crystal structure of lysine-2,3-aminomutase from Clostridium subterminale. Proc Natl Acad Sci USA. 2005;102:13819. doi: 10.1073/pnas.0505726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magnusson OT, Reed GH, Frey PA. Spectroscopic Evidence for the participation of an allylic analogue of the 5′-deoxyadenosyl radical in the reaction of lysine 2,3-aminomutase. J Am Chem Soc. 1999;121:9764. [Google Scholar]
- 8.Magnusson OT, Reed GH, Frey PA. Characterization of an Allylic Analogue of the 5′-Deoxyadenosyl Radical: An Intermediate in the Reaction of Lysine 2,3-Aminomutase. Biochemistry. 2001;40:7773. doi: 10.1021/bi0104569. [DOI] [PubMed] [Google Scholar]
- 9.Horitani M, et al. Why Nature Uses Radical SAM Enzymes so Widely: Electron Nuclear Double Resonance Studies of Lysine 2,3-Aminomutase Show the 5′-dAdo• “Free Radical” is Never Free. J Am Chem Soc. 2015;137:7111. doi: 10.1021/jacs.5b00498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shisler KA, Broderick JB. Glycyl Radical Activating Enzymes: Structure, Mechanism, and Substrate Interactions. Archives of biochemistry and biophysics. 2014;546:64. doi: 10.1016/j.abb.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Walsby CJ, et al. Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe-4S]+ cluster of pyruvate formate-lyase activating enzyme. J Am Chem Soc. 2002;124:3143. doi: 10.1021/ja012034s. [DOI] [PubMed] [Google Scholar]
- 12.Wagner AFV, Frey M, Neugebauer FA, Schäfer W, Knappe J. The Free Radical in Pyruvate Formate-Lyase is Located on Glycine-734. Proc Natl Acad Sci USA. 1992;89:996. doi: 10.1073/pnas.89.3.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sivaraja M, Goodin DB, Smith M, Hoffman BM. Identification by ENDOR of Trp191 as the free-radical site in cytochrome C peroxidase compounds ES. Science. 1989;245:738. doi: 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- 14.Houseman ALP, Doan PE, Goodin DB, Hoffman BM. Comprehensive explanation of the anomolous EPR spectra of wild-type and mutant cytochrome C peroxidase compound ES. Biochemistry. 1993;32:4430. doi: 10.1021/bi00067a036. [DOI] [PubMed] [Google Scholar]
- 15.Huyett JE, et al. Compound ES of cytochrome C peroxidase contains a Trp Pi-Cation radical: Characterization by CW and pulsed Q-band ENDOR. J Am Chem Soc. 1995;117:9033. [Google Scholar]
- 16.McConnell HM, Fressenden RW. Carbon-13 Hyperfine Splitting in CH(COOH)2. J Chem Phys. 1959;31:1688. [Google Scholar]
- 17.Cole T, Heller C. Hyperfine Splittings in the (HOOC)C13H(COOH) Radical. J Chem Phys. 1961;34:1085. [Google Scholar]
- 18.Banerjee R. Radical carbon skeleton rearrangements: catalysis by coenzyme B12-dependent mutases. Chem Rev. 2003;103:2083. doi: 10.1021/cr0204395. [DOI] [PubMed] [Google Scholar]
- 19.Brown KL. Chemistry and Enzymology of Vitamin B12. Chem Rev. 2005;105:2075. doi: 10.1021/cr030720z. [DOI] [PubMed] [Google Scholar]
- 20.Becker A, et al. Structure and mechanism of the glycyl radical enzyme pyruvate formate-lyase. Nat Struct Biol. 1999;6:969. doi: 10.1038/13341. [DOI] [PubMed] [Google Scholar]
- 21.Peng Y, Veneziano SE, Gillispie GD, Broderick JB. Pyruvate formate-lyase, Evidence for an open conformation favored in the presence of its activating enzyme. J Biol Chem. 2010;285:27224. doi: 10.1074/jbc.M109.096875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lubitz W, Ogata H, Rudiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 23.Cutsail GE, III, Telser J, Hoffman BM. Advanced paramagnetic resonance spectroscopies of iron-sulfur proteins: Electron nuclear double resonance (ENDOR) and electron spin echo envelope modulation (ESEEM) Biochim Biophys Acta. 2015;1853:1370. doi: 10.1016/j.bbamcr.2015.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee HI, et al. An organometallic intermediate during alkyne reduction by nitrogenase. J Am Chem Soc. 2004;126:9563. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 25.Yu W, Lees NS, Hall D, Welideniya D, Hoffman BM. A closer look at the spectroscopic properties of possible reaction intermediates in wild-type and mutant (E)-4-hydroxy-3-methylbut-2-enyl diphosphate reductase. Biochemistry. 2012;31:4835. doi: 10.1021/bi3001215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Span I, et al. Insights into the binding of pyridines to the iron-sulfur enzyme IspH. J Am Chem Soc. 2014;136:7926. doi: 10.1021/ja501127j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Broderick JB, et al. Pyruvate formate-lyase activating enzyme: strictly anaerobic isolation yields active enzyme containing a [3Fe-4S]+ cluster. Biochem Biophys Res Commun. 2000;269:451. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 28.Krebs C, Henshaw TF, Cheek J, Huynh B, Broderick JB. Conversion of 3Fe-4S to 4Fe-4S Clusters in Native Pyruvate Formate-Lyase Activating Enzyme: Mössbauer Characterization and Implications for Mechanism. J Am Chem Soc. 2000 Dec 01;122:12497. 2000. [Google Scholar]
- 29.Broderick JB, et al. Pyruvate formate-lyase activating enzyme: strictly anaerobic isolation yields active enzyme containing a [3Fe-4S]+ cluster. Biochem Biophys Res Comm. 2000;269:451. doi: 10.1006/bbrc.2000.2313. [DOI] [PubMed] [Google Scholar]
- 30.Nnyepi MR, Peng Y, Broderick JB. Inactivation of E. coli pyruvate formate-lyase: Role of AdhE and small molecules. Arch Biochem Biophys. 2007;459:1. doi: 10.1016/j.abb.2006.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Walsby JC, et al. Electron-Nuclear Double Resonance Spectroscopic Evidence That S-Adenosylmethionine Binds in Contact with the Catalytically Active [4Fe–4S]+ Cluster of Pyruvate Formate-Lyase Activating Enzyme. J Am Chem Soc. 2002 Mar 01;124:3143. doi: 10.1021/ja012034s. 2002. [DOI] [PubMed] [Google Scholar]
- 32.Walsby CJ, Ortillo D, Broderick WE, Broderick JB, Hoffman BM. An Anchoring Role for FeS Clusters: Chelation of the Amino Acid Moiety of S-Adenosylmethionine to the Unique Iron Site of the [4Fe–4S] Cluster of Pyruvate Formate-Lyase Activating Enzyme. J Am Chem Soc. 2002 Sep 01;124:11270. doi: 10.1021/ja027078v. 2002. [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Perera R, Hager LP, Dawson JH, Hoffman BM. Rapid Freeze-Quench ENDOR Study of Chloroperoxidase Compound I: The Site of the Radical. J Am Chem Soc. 2006 May 01;128:5598. doi: 10.1021/ja060776l. 2006. [DOI] [PubMed] [Google Scholar]
- 34.Lin Y, Gerfen GJ, Rousseau DL, Yeh SR. Ultrafast Microfluidic Mixer and Freeze-Quenching Device. Analytical Chemistry. 2003 Oct 01;75:5381. doi: 10.1021/ac0346205. 2003. [DOI] [PubMed] [Google Scholar]
- 35.Aitha M, et al. Investigating the position of the hairpin loop in New Delhi metallo-beta-lactamase, NDM-1, during catalysis and inhibitor binding. J Inorg Biochem. 2016;156:35. doi: 10.1016/j.jinorgbio.2015.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Werst MM, Davoust CE, Hoffman BM. Ligand Spin Densities in Blue Copper Proteins by Q-band 1H and 14N ENDOR Spectroscopy. J Am Chem Soc. 1991;113:1533. [Google Scholar]
- 37.Davoust CE, Doan PE, Hoffman BM. Q-Band Pulsed Electron Spin-Echo Spectrometer and Its Application to ENDOR and ESEEM. Journal of Magnetic Resonance. 1996;119:38. [Google Scholar]
- 38.Doan PE, Hoffman BM. Making hyperfine selection in Mims ENDOR independent of deadtime. Chemical Physics Letters. 1997;269:208. [Google Scholar]
- 39.Stoll S, Schweiger A. EasySpin a comprehensive software package for spectral simulation and analysis in EPR. J Magn Res. 2006;178:42. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 40.Doan PE. In: Paramagnetic Resonance of Metallobiomolecules. Telser J, editor. American Chemical Society; 2003. pp. 55–81. [Google Scholar]
- 41.McConnell HM, Heller C, Cole T, Fessenden RW. Radiation damage in organic crystals. I. CH(COOH)2 in malonic acid. J Am Chem Soc. 1960;82:766. [Google Scholar]
- 42.Horsfield A, Morton JR, Whiffen DH. Electron-spin resonance of gamma-irradiated malonic acid. Mol Phys. 1961;4:327. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.