

Abstract
There has been limited success in identifying causal variants underlying association signals observed in genome-wide association studies (GWAS). The use of 1000 Genomes Project (1KGP) allows the imputation to estimate the genetic information at untyped variants. However, long stretches of high linkage disequilibrium within the genome prevent us from differentiating between causal variants and perfect surrogates, thus limiting our ability to identify causal variants. Transethnic strategies have been proposed as a possible solution to mitigate this. However, these studies generally rely on imputing genotypes from multiple ancestries from 1KGP but not against population-specific reference panels. Here, we perform the first transethnic fine-mapping study across three Asian cohorts from diverse ancestries at the loci implicated with eye and blood lipid traits, using population-specific reference panels that have been generated by whole-genome sequencing samples from the same ancestry groups. Our study outlines several challenges faced in a fine-mapping exercise where one simply aims to meta-analyse existing GWAS that have been imputed against reference haplotypes from the 1KGP.
Introduction
Despite the success of genome-wide association studies (GWAS) in identifying genetic variants that correlate with complex diseases and human traits,1 the persistent problem of missing heritability signifies that the identified variants present, at best, a modest contribution to the phenotypic variance.2 A possible explanation is that the design of GWAS fundamentally relies on detecting proxy markers in the human genome, known as tagging single-nucleotide polymorphisms (SNPs), which are correlated to the biologically causal variants. The process of identifying the real causal variants is known as fine mapping. This usually requires complementing the patchy representation provided by genotyping microarrays in GWAS with denser sequence-level data, such as those from the 1000 Genomes Project (1KGP).3, 4 However, the perception and strategy towards fine mapping have evolved significantly.
An early study by Jallow et al.5 in localizing the hemoglobin S (HbS) variant in a malaria GWAS in The Gambia suggested that targeted sequencing of an implicated gene in a handful of population-specific individuals can provide a representative haplotype map that allows accurate imputation to isolate the protein-altering variant, which the use of an inappropriate haplotype map fail to achieve. Unfortunately, the presence of long-range linkage disequilibrium (LD) in most non-African populations meant there were numerous perfect surrogates that were virtually indistinguishable from the causal variants, thus compounding the quest to localize them.6, 7 Several reports then advocated the prospect of using different LD patterns intrinsic to multiple ancestries to overcome the challenge of long LD,8, 9, 10 although identifying causal variants with certainty proved elusive even with this strategy of transethnic fine mapping, as seen in a recent report for type 2 diabetes.11 This challenges the premise that a single causal variant will emerge with the strongest evidence of phenotypic association in transethnic fine mapping relative to all other neighboring markers.
The 1KGP supplies whole-genome sequence-level data for multiple populations from major ancestry groups globally. Statistical imputation against this reference panel produced in silico sequence-level information for the GWAS data at almost no additional cost,12, 13, 14, 15 and it was felt that this approach would provide greater resolution in our fine-mapping effort while mitigating the need for most populations to perform their own targeted or whole-genome sequencing. One concern in using these reference panels is the knowledge that the HbS variant was not identified as the causal variant associated with malaria when a non-population-specific reference panel was used. This raises a question whether a cosmopolitan reference panel such as 1KGP is adequate to impute populations that are not included in the cosmopolitan panel and if not, whether judicious use of population-specific reference panels will enhance our ability to localize protein-altering variants through transethnic fine mapping.
In this study, we aim to answer the two pressing questions through a series of simulation exercises as well as real GWAS data applications. To evaluate how fallacious the belief that transethnic fine mapping can identify the causal variant with certainty, we simulated 2000 collections of case–control data in three major ancestry groups from HapMap2. We mimicked the ideal situation where a common causal variant was present in all three populations and the imputation reference panel was perfectly matched to the case–control data in each population. We then investigated how often single-population fine-mapping or transethnic fine-mapping rediscovered the simulated causal variant as the SNP with the strongest evidence.
To evaluate the effect of the two types of reference panels in imputing populations absent from the cosmopolitan panel, we performed transethnic fine mapping with GWAS data from three ancestry groups in Asia, consisting of East Asian Han Chinese, Southeast Asian Malays and South Asian Indians residing in Singapore. Besides the cosmopolitan reference panel from 1000G project phase I, we possessed population-specific reference panels for Southeast Asian Malays16 and South Asian Indians17 that were generated from high-coverage whole-genome sequencing. The intention is to locate the potential protein-altering variants underpinning the association between 176 genetic loci sieved from the NIH GWAS catalog (http://www.genome.gov/gwastudies)18 with either eye-related traits such as corneal curvature (CC), central corneal thickness (CCT), corneal astigmatism (CA) and optic disk area (ODA)19, 20, 21, 22, 23, 24, 25 or blood lipid measurements such as triglycerides (TGs), high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C).26, 27, 28, 29, 30
Materials and methods
Simulation setup
We used the HAPGEN program31 with haplotype and recombination data from phase 2 of the International HapMap Project32 to simulate case–control data with preselected SNPs as the causal variants, to evaluate whether these variants necessarily exhibit the strongest association evidence. A total of 2000 SNPs that were polymorphic in all three HapMap panels (CEU from European ancestry, JPT+CHB from Asian ancestry and YRI from African ancestry) but were not found on commercial microarrays were chosen as causal variants. For each SNP, 1000 cases and 1000 controls were simulated for each ancestry panel assuming a multiplicative model with an allelic relative risk of 1.5. The simulated data was thinned to retain only the SNPs that were located on commercial microarrays, before being recovered by imputation with IMPUTE14 against the respective HapMap reference panel. This allowed the association evidence at each causal variant to be ranked against neighboring markers located within 750 kb. A logistic regression assuming additive model was performed on the simulated data. A detailed description of the simulation setup can be found in the Supplementary Material.
GWAS cohorts
Our study considered data from three independent genome-wide studies involving 1889 Chinese from the Singapore Chinese Eye Study (SCES), 2542 Malays from the Singapore Malay Eye Study (SiMES) and 2538 Asian Indians from the Singapore Indian Eye Study (SINDI).33, 34 All samples have been genotyped on the Illumina HumanHap 610-Quad BeadChip. The cohorts were imputed against two haplotype reference panels: (i) each cohort was imputed to the combined 1KGP phase 1 panel with 1092 individuals from 14 populations; and (ii) a population-specific panel from whole-genome sequencing 286 East Asian samples (from 1KGP), 96 Southeast Asian Malays (from Singapore Sequence Malay Project (SSMP); http://www.statgen.nus.edu.sg/~SSMP/download/vcf/) and 38 South Asian Indians (from Singapore Sequence Indian Project (SSIP); http://www.statgen.nus.edu.sg/~SSIP/download/vcf/) was used to impute the Chinese, Malay and Indian cohort, respectively. Imputed SNPs with information ≥0.50 (estimated by IMPUTE2) were retained for analysis.35, 36 Details of the quality control criteria for genotyping and imputation for each cohort, including correction for covariates, Hardy–Weinberg equilibrium, minor allele frequency threshold and SNP/sample call rate are outlined in Supplementary Table S1.
Identification of trait-associated loci
We identified 176 unique loci from the NIH GWAS catalog that were associated with either eye-related traits or blood lipid. Several features of interest were recorded for each study: first author, journal, year of publication, genetic ancestry, sample size in GWAS stage, total sample size in replication stage, array genotyped, genomic control factor in GWAS stage (if available), use of imputed SNPs (Y/N) and the number of genomic regions achieving genome-wide significance in the initial and final stage (Supplementary Table S2). There was a GWAS for CCT that had been published using the cohorts from Singapore,37 which was absent from the GWAS catalog, and we appended the loci reported in this study that achieved genome-wide significance.
Statistical analyses
The analysis at each locus included SNPs within 200 kb on either flank of the reported index variant. Multiple regions defined within the same gene are combined into one single locus if the distance between the index SNPs is within 100 kb. MANTRA was used to perform the meta-analysis of the three GWAS cohorts. It takes a Bayesian approach to calculate a Baye's factor (BF) in favor of association at each SNP by allowing for allelic effect heterogeneity between diverse populations.10 Based on the number of SNPs considered (n=1204), we adopted a significance threshold of 4 × 10−5 as the criterion for discovery in the meta-analysis of the three GWAS cohorts, which was equivalent to a log10 BF of 3.38 in MANTRA, according to the conversion formula −log10(P-value)=0.85+1.05log10BF between P-values and BFs, established by Wang and colleagues38 between P-values and BFs.10 The proportion of phenotypic variance explained by the identified protein-altering variants at each locus is estimated within a regression framework by considering the difference in the regression R2 values obtained by a set of SNPs with and without including the SNP of interest. To assess the improvement in fine-mapping resolution because of transethnic meta-analysis, we defined ‘99% credible set' of SNPs that harbors the ‘causal' variant. At the jth SNP, the posterior probability that the SNP is ‘causal' is calculated by 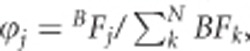 where N is the total number of SNPs. The ‘99% credible set' is then derived by agglomerating ϕj with the largest value until the cumulative posterior probability exceeded 0.99.39 In this manuscript, protein-altering variants are restricted to the nonsynonymous variants only (missense, nonsense, frameshift and so on). VarLD9 is used to perform interpopulation comparison of regional patterns of LD.
where N is the total number of SNPs. The ‘99% credible set' is then derived by agglomerating ϕj with the largest value until the cumulative posterior probability exceeded 0.99.39 In this manuscript, protein-altering variants are restricted to the nonsynonymous variants only (missense, nonsense, frameshift and so on). VarLD9 is used to perform interpopulation comparison of regional patterns of LD.
Results
Rank of the association signals at the causal variant
Regardless of whether fine mapping was attempted within a single-population setting or in a meta-analysis of all three case–control collections, the simulated causal variants did not always emerge as the SNP with the strongest statistical evidence (Figure 1). In fact, this happened only in 23% and 26% of the 2000 collections for the European and East Asian ancestries, respectively. The causal variants exhibited the most significant evidence in 43% of the simulated African case–control collections and in 59% of the meta-analyses, where the higher proportions were likely due to gains from shorter LD blocks inherently present in African populations. We extended the search of the causal variants in the ‘99% credible set', a statistically rigorous approach used in MANTRA, which identifies that a list of SNPs cumulatively confer a 99% chance of harboring the causal variant. As expected, the causal variant was identified in the ‘99% credible set' in 99.2% of the transethnic fine-mapping analyses.
Figure 1.

Histograms (gray vertical bars) and cumulative frequencies (red lines) on the ranks of the simulated causal variants out of 2000 rounds of simulations. The horizontal axes indicate the rank of the association evidence at the rediscovered causal variant in comparison with all neighboring variants. Three of the display panels indicate the causal variant ranking when the analysis is performed with the data from a single ancestry panel, whereas the fourth display panel indicates the rank when data from three ancestry groups are combined in a transethnic fine-mapping.
Transethnic fine-mapping GWAS loci for eye traits and blood lipids
Using a log10 BF of 3.38 as the Bonferroni-corrected threshold for the mean of 1204 SNPs in a region of 400 kb length across the 176 loci from GWAS of eye traits and blood lipids, our meta-analysis reproduced the associations seen at 26 loci with the respective phenotypes (Table 1), thus qualifying these loci to the next stage of causal variant fine mapping. The index SNPs with the strongest evidence at these loci were all common (MAF >5%) in the three cohorts, although only the index SNP (rs7412—hg19 chr19: g.44908822C>T, a missense SNP) at TOMM40-APOE for LDL-C association was a protein-altering variant, except that this was already reported previously.40 When we expanded the search to the ‘99% credible set' at each locus, four other protein-altering variants were identified at the ABCA1 locus for HDL-C (rs2230808), CARD10 locus for ODA (rs9610775), LPL locus for TG (rs328) and PDGFRA locus for CA (rs35597368).
Table 1. Twenty-six loci with significant association evidence in the meta-analysis of the three Asian cohorts.
| Traits | Locus | Top SNP | Chr | Position (bp) GRCh37 (hg19) | Effect allele | Other allele | Effect allele frequency in SiMES/SINDI/SCES | Annotation | Log10 BFa | Potential functional variantb |
|---|---|---|---|---|---|---|---|---|---|---|
| CA | SUCLG2 | rs4856867 | 3 | 67 549 438 | A | G | 0.181/0.276/0.214 | Intron | 3.719 | NA |
| CA | PDGFRA | rs7660560 | 4 | 55 134 394 | A | G | 0.172/0.253/0.263 | Intron | 3.816 | NA |
| CC | MTOR | rs113124929 | 1 | 11 240 111 | G | A | 0.134/0.199/0.239 | Intron | 12.573 | NA |
| CC | RPL22P13 | rs4864863 | 4 | 55 100 831 | G | A | 0.186/0.256/0.258 | Intron | 7.682 | NA |
| CCT | COL8A2 | rs96067 | 1 | 36 571 920 | G | A | 0.476/0.407/0.364 | Unknown | 9.656 | NA |
| CCT | COL5A1 | rs3132307 | 9 | 137 436 214 | C | G | 0.389/0.404/0.450 | Intron | 14.017 | NA |
| CCT | PDE8A | rs7165242 | 15 | 85 845 848 | T | G | 0.527/0.449/0.447 | Unknown | 6.255 | NA |
| CCT | ZNF469 | rs34715091 | 16 | 88 326 782 | G | A | 0.187/0.320/0.399 | Unknown | 14.626 | NA |
| ODA | RPL39P13 | rs1192419 | 1 | 92 080 059 | A | G | 0.887/0.149/0.691 | Unknown | 19.904 | NA |
| ODA | ATOH7 | rs3858144 | 10 | 70 011 354 | C | T | 0.672/0.174/0.690 | Unknown | 19.554 | NA |
| ODA | UNGP1 | rs1121635 | 16 | 51 647 562 | A | T | 0.573/0.534/0.614 | Unknown | 3.415 | NA |
| ODA | CARD10 | rs9610778 | 22 | 37 914 526 | A | G | 0.180/0.258/0.260 | Intron | 5.622 | rs9610775 |
| HDL | LPL | rs2119690 | 8 | 19 859 539 | A | G | 0.211/0.157/0.248 | Unknown | 6.449 | NA |
| HDL | ABCA1 | rs2777802 | 9 | 107 569 337 | T | C | 0.410/0.475/0.408 | Intron | 3.958 | rs2230808 |
| HDL | ZNF259 | rs651821 | 11 | 116 662 579 | C | T | 0.256/0.290/0.204 | Untranslated 5 | 15.873 | NA |
| HDL | LIPC | rs2043085 | 15 | 586 80 954 | C | T | 0.432/0.465/0.430 | Unknown | 11.162 | NA |
| HDL | CETP | rs247616 | 16 | 569 89 590 | T | C | 0.157/0.149/0.294 | Unknown | 25.748 | NA |
| HDL | LIPG | rs9958734 | 18 | 47 118 398 | C | T | 0.406/0.301/0.086 | Untranslated 3 | 6.069 | NA |
| LDL | CELSR2 | rs611917 | 1 | 109 815 252 | G | A | 0.066/0.075/0.291 | Intron | 5.641 | NA |
| LDL | HMGCR | rs6453131 | 5 | 74 644 706 | G | T | 0.502/0.466/0.524 | Intron | 6.831 | NA |
| LDL | TOMM40 | rs7412 | 19 | 45 412 079 | T | C | 0.091/0.116/0.087 | Missense | 42.436 | rs7412 |
| TG | DOCK7 | rs1168036 | 1 | 62 962 734 | A | G | 0.247/0.288/0.474 | Intron | 4.045 | NA |
| TG | LPL | rs78404258 | 8 | 19 881 058 | G | A | 0.103/0.072/0.106 | Unknown | 5.53 | NA |
| TG | BUD13 | rs651821 | 11 | 116 662 579 | C | T | 0.256/0.294/0.204 | Untranslated 5 | 30.035 | NA |
| TG | CILP2 | rs73004951 | 19 | 19 695 228 | T | C | 0.095/0.224/0.159 | Intron | 6.015 | NA |
| TG | APOE | rs483082 | 19 | 45 416 178 | T | G | 0.194/0.266/0.135 | Near-gene 5 | 10.83676 | NA |
Log10 BF (Baye's factor) of 3.38 is used as the Bonferroni-corrected threshold.
Highest-ranking function-altering SNP in the top 10 associated SNPs. An ‘NA' entry indicates that there were no function-altering SNPs in the top 10 SNPs.
Transethnic fine-mapping narrows associated regions
To assess how transethnic fine-mapping narrows the associated regions, we compared the number of SNPs and the size of the genomic region covered by the ‘99% credible set' in each of the single-population fine mapping, as well as in the transethnic fine mapping (Table 2). As this assumes there is a single causal variant shared across multiple populations, six loci with multiple independent signals were excluded from this analysis (see Supplementary Material and Supplementary Table S3). In all but one region (APOE), transethnic analyses reduced the number of variants in the ‘99% credible set'. The genomic intervals of the region spanned by the credible set also shortened considerably in most of the loci considered. The greatest reduction was observed at CELSR2 for LDL-C, where the interval was reduced from 400 kb within individual populations to only 6 kb after transethnic analysis, and the number of SNPs in the credible set was reduced from several hundreds to only nine, although none of the nine SNPs alter function (Figure 2).
Table 2. Properties of the 99% credible sets of SNPs at significant loci.
| 99% Credible set for transethnic fine mapping | 99% Credible set for Singapore Chinese (SCES) | 99% Credible set for Singapore Malay (SiMES) | 99% Credible set for Singapore Indian (SINDI) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Traits | Chr | Locus | Top SNPs | SNPs | Interval (bp) | SNPs | Interval (bp) | SNPs | Interval (bp) | SNPs | Interval (bp) |
| CA | 3 | SUCLG2 | rs4856867 | 189 | 381 274 | 855 | 392 558 | 205 | 390 394 | 846 | 392 558 |
| CA | 4 | PDGFRA | rs7660560 | 109 | 104 064 | 633 | 397 663 | 459 | 397 663 | 534 | 397 663 |
| CC | 1 | MTOR | rs113124929 | 31 | 107 541 | 646 | 395 738 | 94 | 222 826 | 239 | 216 842 |
| CC | 4 | RPL22P13 | rs4864863 | 108 | 77 138 | 572 | 398 741 | 115 | 81 761 | 325 | 398 225 |
| CCT | 1 | COL8A2 | rs96067 | 8 | 14 473 | 347 | 412 589 | 313 | 412 589 | 23 | 81 170 |
| CCT | 15 | PDE8A | rs7165242 | 82 | 123 807 | 503 | 398 043 | 279 | 398 063 | 463 | 395 868 |
| CCT | 16 | ZNF469 | rs34715091 | 14 | 9602 | 304 | 341 010 | 30 | 38 889 | 24 | 22 977 |
| ODA | 1 | RPL39P13 | rs1192419 | 5 | 7162 | 32 | 13 058 | 83 | 195 445 | 6 | 7162 |
| ODA | 10 | ATOH7 | rs3858144 | 28 | 46 645 | 158 | 229 792 | 147 | 103 646 | 24 | 53 748 |
| ODA | 16 | UNGP1 | rs1121635 | 573 | 398 754 | 631 | 399 438 | 624 | 399 438 | 601 | 399 438 |
| ODA | 22 | CARD10 | rs9610778 | 39 | 27 300 | 700 | 396 912 | 547 | 396 912 | 46 | 209 430 |
| HDL | 8 | LPL | rs2119690 | 69 | 58 010 | 341 | 304 655 | 1316 | 424 571 | 86 | 58 019 |
| HDL | 18 | LIPG | rs9958734 | 11 | 63 270 | 112 | 174 921 | 1109 | 421 106 | 1191 | 421 807 |
| LDL | 1 | CELSR2 | rs611917 | 9 | 6756 | 534 | 403 529 | 249 | 397 578 | 434 | 384 041 |
| LDL | 5 | HMGCR | rs6453131 | 34 | 251 791 | 1020 | 430 814 | 568 | 430 521 | 569 | 430 531 |
| TG | 1 | DOCK7 | rs1168036 | 193 | 233 119 | 1068 | 658 589 | 839 | 659 423 | 1073 | 659 663 |
| TG | 8 | LPL | rs78404258 | 155 | 104 099 | 786 | 428 023 | 1301 | 424 571 | 166 | 269 809 |
| TG | 11 | BUD13 | rs651821 | 2 | 1128 | 2 | 1128 | 23 | 103 260 | 19 | 56 941 |
| TG | 19 | CILP2 | rs73004951 | 5 | 21 330 | 760 | 402 517 | 724 | 402 517 | 768 | 402 517 |
| TG | 19 | APOE | rs483082 | 5 | 19 959 | 818 | 405 467 | 18 | 29 144 | 2 | 2483 |
Figure 2.

Regional plots of SNPs at the LDL-C locus, CELSR2, for the Chinese (SCES), Malays (SiMES), Indians (SINDI) and the transethnic fine mapping of all three cohorts. The vertical axes measure the statistical evidence of association with the log10 BF, and each SNP is indicated by a colored circle, diamond or triangle. In each panel, the index SNP rs611917 is indicated by the purple diamond, whereas all remaining SNPs are assigned colors according to the extent of LD, with the lead SNP in six categories: (i) r2≥0.8 (red); (ii) 0.6≤r2<0.8 (gold); (iii) 0.4≤r2<0.6 (green); (iv) 0.2≤r2<0.4 (cyan); (v) (ii) r2<0.2 (blue); and (vi) unknown r2 (gray). Diamonds represent variants that are found within the ‘99% credible set' and function-altering variants are represented with triangles. Recombination rates estimated from the International HapMAp Project are superimposed with blue lines, and all gene annotations are obtained from the University of California Santa Cruz genome browser.
Population-specific versus 1KGP cosmopolitan reference panel
The two sets of imputation performed on each of the three GWAS cohorts provided the opportunity to assess whether the use of a larger cosmopolitan reference panel will produce different index SNPs at the 26 loci, compared to the use of population-specific panels that are significantly smaller in sample sizes. We observed that in 20 out of the 26 loci, the top index SNPs were different between the two sets of analyses, although most of these index SNPs were within the ‘99% credible set ‘on either lists (Table 3). There is no significant difference in the log10 BF of the top SNPs.
Table 3. Comparison between population-specific and 1KGP cosmopolitan reference panels.
| Population Reference panel discovery | Replication in 1000G ref | Rank in 1KG ref | 1000G reference panel discovery | Rank in Pop ref | Concordance check | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Traits | Chr | Locus | Top SNP | Log10 BFa | Log10 BFa | Rank | Top SNP | Log10 BFa | Rank | |
| CA | 3 | SUCLG2 | rs4856867 | 3.719 | 3.857 | 1 | rs4856867 | 3.857 | 1 | Yes |
| CA | 4 | PDGFRA | rs7660560 | 3.816 | 3.725 | 6 | rs1565670 | 3.852 | 24 | No |
| CC | 1 | MTOR | rs113124929 | 12.573 | 12.456 | 5 | rs74225573 | 12.553 | 7 | No |
| CC | 4 | RPL22P13 | rs4864863 | 7.682 | 7.101 | 47 | rs1800813 | 7.615 | 4 | No |
| CCT | 1 | COL8A2 | rs96067 | 9.656 | 8.569 | 1 | rs96067 | 8.569 | 1 | Yes |
| CCT | 9 | COL5A1 | rs3132307 | 14.017 | 14.103 | 3 | rs3132309 | 14.218 | 2 | No |
| CCT | 15 | PDE8A | rs7165242 | 6.255 | 5.925 | 8 | rs7172789 | 6.123 | 11 | No |
| CCT | 16 | ZNF469 | rs34715091 | 14.626 | 13.833 | 7 | rs28411862 | 14.086 | 2 | No |
| ODA | 1 | RPL39P13 | rs1192419 | 19.904 | 19.758 | 2 | rs1192415 | 19.799 | 10 | No |
| ODA | 10 | ATOH7 | rs3858144 | 19.554 | 17.76 | 22 | rs9783176 | 19.7 | 4 | No |
| ODA | 16 | UNGP1 | rs1121635 | 3.415 | 1.041 | 16 | rs2647987 | 2.307 | 2 | No |
| ODA | 22 | CARD10 | rs9610778 | 5.622 | 4.96 | 7 | rs2092171 | 5.114 | 5 | No |
| HDL | 8 | LPL | rs2119690 | 6.449 | 5.946 | 44 | rs3208305 | 6.749 | 41 | No |
| HDL | 9 | ABCA1 | rs2777802 | 3.958 | 3.704 | 5 | rs2740480 | 4.107 | 5 | No |
| HDL | 11 | ZNF259 | rs651821 | 15.873 | 14.678 | 2 | rs662799 | 15.484 | 2 | No |
| HDL | 15 | LIPC | rs2043085 | 11.162 | 10.708 | 2 | rs1532085 | 11.25 | 2 | No |
| HDL | 16 | CETP | rs247616 | 25.748 | 25.226 | 5 | rs3764261 | 25.565 | 4 | No |
| HDL | 18 | LIPG | rs9958734 | 6.069 | 6.184 | 1 | rs9958734 | 6.184 | 1 | Yes |
| LDL | 1 | CELSR2 | rs611917 | 5.641 | 5.395 | 1 | rs611917 | 5.395 | 1 | Yes |
| LDL | 5 | HMGCR | rs6453131 | 6.831 | 7.257 | 3 | rs10045497 | 7.41 | 0b | No |
| LDL | 19 | TOMM40 | rs7412 | 42.436 | 22.931 | 9 | rs72654473 | 44.675 | 4 | No |
| TRI | 1 | DOCK7 | rs1168036 | 4.045 | 3.78 | 52 | rs6693353 | 4.158 | 187 | No |
| TRI | 8 | LPL | rs78404258 | 5.53 | 4.875 | 53 | rs287 | 6.099 | 5 | No |
| TRI | 11 | BUD13 | rs651821 | 30.035 | 31.437 | 1 | rs651821 | 31.437 | 1 | Yes |
| TRI | 19 | CILP2 | rs73004951 | 6.015 | 5.368 | 1 | rs73004951 | 5.368 | 1 | Yes |
| TRI | 19 | APOE | rs483082 | 10.837 | 9.859 | 3 | rs438811 | 10.75 | 21 | No |
Log10 BF (Baye's factor) of 3.38 is used as the Bonferroni-corrected threshold.
Rank 0 means the variant is not found in the population-specific MANTRA result.
The only exception was the identification of rs7412 in TOMM40-APOE in the meta-analysis of the population-specific imputed data, which yielded a log10 BF of 42.4 compared with the 1KGP equivalent of 22.9 (Figure 3). Instead, the meta-analysis of the 1KGP-imputed data identified a proxy SNP (rs72654473) with the strongest evidence (log10 BF=44.7). The reduction in the BF by 20 orders of magnitude was the consequence of excluding the Malay cohort in the transethnic meta-analysis at rs7412, as the quality of the imputation of this SNP was below the quality check threshold in the SiMES cohort using the 1KGP reference panel (Table 4). To explain the difference in the imputation quality, we assessed the variation in LD patterns between the study populations and the reference panels with varLD.9 We observed significant variations in LD structures at the TOMM40-APOE region between the study population and the cosmopolitan reference panel, but not the population-specific reference panels.
Figure 3.

Regional plots of SNPs at the LDL-C locus TOMM40-APOE from two transethnic meta-analyses using either the population-specific reference panels or the cosmopolitan reference panel from the 1000 Genomes Project. In each panel, the index SNP is represented in purple with either a circle or a diamond, with the latter used if the variant is annotated to alter function by the University of California Santa Cruz genome browser. All remaining SNPs are assigned colors according to the extent of LD with the lead SNP in six categories: (i) r2≥0.8 (red); (ii) 0.6≤r2<0.8 (gold); (iii) 0.4≤r2<0.6 (green); (iv) 0.2≤r2<0.4 (cyan); (v) (ii) r2<0.2 (blue); and (vi) unknown r2 (gray).
Table 4. Comparison of regional varLD between study populations and different reference panels at the TOMM40-APOE region.
| Study populations | Reference panels | Regional varLD | Imput-Info at rs7412 |
|---|---|---|---|
| SCES | 1 kg-ASN | 0.432 | 0.82098 |
| SCES | 1 kg-CEU | 0.0001 | 0.53886 |
| SiMES | 1 kg-ASN | 0.0009 | 0.44695 |
| SiMES | 1 kg-CEU | 0.0001 | |
| SiMES | SSMP | 0.1365 | 0.57477 |
| SINDI | 1 kg-ASN | 0.0001 | 0.50768 |
| SINDI | 1 kg-CEU | 0.0001 | |
| SINDI | SSIP | 0.1037 | 0.70117 |
Discussion
There have been several efforts to locate the causal variants driving GWAS signals with and without the use of data from diverse genetic ancestries.9, 10 These studies have relied on the causal variants emerging with the strongest evidence of association, as was seen in fine mapping the sickle cell variant in the malaria study conducted in The Gambia.5 However, we have shown, in our simulations, that even in the scenario where the GWAS was imputed against a perfectly matched reference panel, the causal variant did not emerge as the top-ranking SNP in about 57% of the simulations in single-population fine mapping. Combining data from diverse ancestries to perform transethnic fine mapping can increase the chance of accurately identifying the causal variant by leveraging the LD difference between the causal variant and the marker SNP across populations as well as increasing sample size, although it is difficult to qualify how much of the improvement is attributed to each of the two factors. Presently, varLD serves to quantify the interpopulation differences in LD patterns, but the varLD metric does not yield sufficient resolution on whether the observed difference in LD is expected to benefit transethnic analyses. For example, it is not possible to claim that a genomic region with a varLD score greater than a pre-set threshold will definitely benefit from a combined analysis between the different populations.
The current stage of GWAS fine-mapping studies are highly dependent on imputation with the 1KGP cosmopolitan reference panel. There are many examples of success in studies from European and East Asian ancestries, where the cosmopolitan panel is adequately representative of the study populations. However, our analyses have shown that for populations with ancestries that may not be similar to those in the reference panel, such as the Southeast Asian Malays, the evidence from fine mapping can decrease by almost 20 orders of magnitude because of the inadvertent exclusion of a possible missense SNP as a result of imperfect imputation in one cohort using the cosmopolitan panel. This single example highlights the possibility that, for a process as sensitive as fine-mapping, the use of population-specific panels can be important as it is almost impossible to predict or detect when a cosmopolitan panel will fail to identify an unknown causal variant.
There were several reasons why the fine-mapping success at HbS was exceptional: (i) the variant confers almost 10-fold protection against severe malaria, which is a condition often fatal for young children in impoverished health systems;5 (ii) the ongoing balancing selection between sickle cell anemia and severe malaria resulted in the presence of a single long haplotype in The Gambia, which carried the protective allele (measured by a high D′ of 1.00); (iii) the shorter LD blocks inherently present in African populations (measured by a low r2);41 and (iv) the convergent evolution of the HbS locus resulted in the protective allele residing on fundamentally different haplotype backgrounds in different populations.42 None of these four conditions were likely to be present for causal variants driving non-communicable diseases and common traits.
Many studies have reported that population-specific panels, even if they are considerably smaller in sample size, can yield higher imputation accuracy compared with the 1KGP panel.5, 43, 44 For example, the SSMP showed that for common variants (MAF >5%), a population-specific panel of 96 subjects can produce more accurate imputation compared with the 1KGP cosmopolitan panel, which is more than 10-fold larger in sample size, although the opposite was true when it comes to low-frequency and rare variants. Contrary to the perception that this is only true in populations not represented in the 1KGP, a recent study by the Genome of the Netherlands Consortium reported significantly better imputation over 1KGP of low-frequency and rare variants with a population-specific panel built by sequencing 769 Dutch individuals.43 It is thus important to contextualize previous reports that a well-defined cosmopolitan panel can provide accurate imputation of unobserved variants, even for those with low allele frequencies:12, 45, 46 these studies usually rely on statistics on imputation performance that are averaged across the whole genome, and knowledge of these statistics may only be peripherally useful when deciding whether a specific genomic region has been accurately imputed. Do we even know how common the scenario is where a causal variant is excluded from a transethnic meta-analysis, simply because the imputation accuracy failed to meet some predetermined threshold and was thus filtered out in one of the contributing GWAS?
The cost of whole-genome sequencing is dropping rapidly, and it is entirely plausible that future association studies will rely on whole-genome sequencing instead of genotyping surrogates. However, the challenge provided by long stretches of high LD will almost certainly remain to confound the search for the causal variants. Transethnic strategies can continue to provide a viable solution by leveraging on diverse LD patterns. Perhaps what is promising about this then is that imputation will no longer be required to fill in the blanks, which comes with the real risk that we exclude the very variant that we are looking for.
Acknowledgments
This project acknowledges the support of the Saw Swee Hock School of Public Health from the National University of Singapore. The Singapore Malay Eye Study (SiMES) was funded by the National Medical Research Council (NMRC 0796/2003 and NMRC/STaR/0003/2008) and Biomedical Research Council (BMRC, 09/1/3/19/616). The Singapore Indian Eye Study (SINDI) was funded by grants from Biomedical Research Council of Singapore (BMRC 09/1/35/19/616 and BMRC 08/1/35/19/550) and National Medical Research Council of Singapore (NMRC/STaR/0003/2008). Y-YT and XW acknowledge the support from the Singapore National Research Foundation, NRF-RF-2010-05.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Supplementary Material
References
- McCarthy MI, Abecasis GR, Cardon LR et al: Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 2008; 9: 356–369. [DOI] [PubMed] [Google Scholar]
- Manolio TA, Collins FS, Cox NJ et al: Finding the missing heritability of complex diseases. Nature 2009; 461: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abecasis GR, Auton A, Brooks LD et al: An integrated map of genetic variation from 1,092 human genomes. Nature 2012; 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL et al: A map of human genome variation from population-scale sequencing. Nature 2010; 467: 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jallow M, Teo YY, Small KS et al: Genome-wide and fine-resolution association analysis of malaria in West Africa. Nat Genet 2009; 41: 657–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo YY, Small KS, Kwiatkowski DP: Methodological challenges of genome-wide association analysis in Africa. Nat Rev Genet 2010; 11: 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Huang L, Jewett EM, Szpiech ZA, Jankovic I, Boehnke M: Genome-wide association studies in diverse populations. Nat Rev Genet 2010; 11: 356–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twee-Hee Ong R, Wang X, Liu X, Teo YY: Efficiency of trans-ethnic genome-wide meta-analysis and fine-mapping. Eur J Hum Genet 2012; 20: 1300–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo YY, Ong RT, Sim X, Tai ES, Chia KS: Identifying candidate causal variants via trans-population fine-mapping. Genet Epidemiol 2010; 34: 653–664. [DOI] [PubMed] [Google Scholar]
- Morris AP: Transethnic meta-analysis of genomewide association studies. Genet Epidemiol 2011; 35: 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan A, Go MJ, Zhang W et al: Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet 2014; 46: 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie B, Marchini J, Stephens M: Genotype imputation with thousands of genomes. G3 (Bethesda, MD) 2011; 1: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, Howie B: Genotype imputation for genome-wide association studies. Nat Rev Genet 2010; 11: 499–511. [DOI] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J: A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 2009; 5: e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J, Howie B, Myers S, McVean G, Donnelly P: A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007; 39: 906–913. [DOI] [PubMed] [Google Scholar]
- Wong LP, Ong RT, Poh WT et al: Deep whole-genome sequencing of 100 southeast Asian Malays. Am J Hum Genet 2013; 92: 52–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong LP, Lai JK, Saw WY et al: Insights into the genetic structure and diversity of 38 South Asian Indians from deep whole-genome sequencing. PLoS Genet 2014; 10: e1004377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindorff LA, Mehta J, Wise A et alEuropean Bioinformatics Institute.: A catalog of Published Genome-Wide Association Studies; Available at: www.genome.gov/gwastudies (last access date September 2014).
- Lopes MC, Hysi PG, Verhoeven VJ et al: Identification of a candidate gene for astigmatism. Invest Ophthalmol Vis Sci 2013; 54: 1260–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Q, Zhou X, Khor CC et al: Genome-wide meta-analysis of five Asian cohorts identifies PDGFRA as a susceptibility locus for corneal astigmatism. PLoS Genet 2011; 7: e1002402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khor CC, Ramdas WD, Vithana EN et al: Genome-wide association studies in Asians confirm the involvement of ATOH7 and TGFBR3, and further identify CARD10 as a novel locus influencing optic disc area. Hum Mol Genet 2011; 20: 1864–1872. [DOI] [PubMed] [Google Scholar]
- Ramdas WD, van Koolwijk LM, Ikram MK et al: A genome-wide association study of optic disc parameters. PLoS Genet 2010; 6: e1000978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggenheim JA, McMahon G, Kemp JP et al: A genome-wide association study for corneal curvature identifies the platelet-derived growth factor receptor alpha gene as a quantitative trait locus for eye size in white Europeans. Mol Vis 2013; 19: 243–253. [PMC free article] [PubMed] [Google Scholar]
- Mishra A, Yazar S, Hewitt AW et al: Genetic variants near PDGFRA are associated with corneal curvature in Australians. Invest Ophthalmol Vis Sci 2012; 53: 7131–7136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S, Chen P, Fan Q et al: Association of variants in FRAP1 and PDGFRA with corneal curvature in Asian populations from Singapore. Hum Mol Genet 2011; 20: 3693–3698. [DOI] [PubMed] [Google Scholar]
- Kathiresan S, Melander O, Guiducci C et al: Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet 2008; 40: 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Sanna S, Jackson AU et al: Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 2008; 40: 161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko YS, Ripatti S, Lindqvist I et al: Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet 2009; 41: 47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathiresan S, Willer CJ, Peloso GM et al: Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet 2009; 41: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teslovich TM, Musunuru K, Smith AV et al: Biological, clinical and population relevance of 95 loci for blood lipids. Nature 2010; 466: 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer CC, Su Z, Donnelly P, Marchini J: Designing genome-wide association studies: sample size, power, imputation, and the choice of genotyping chip. PLoS Genet 2009; 5: e1000477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frazer KA, Ballinger DG, Cox DR et al: A second generation human haplotype map of over 3.1 million SNPs. Nature 2007; 449: 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Chen CH, Hu C et al: Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet 2012; 44: 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim X, Ong RT, Suo C et al: Transferability of type 2 diabetes implicated loci in multi-ethnic cohorts from Southeast Asia. PLoS Genet 2011; 7: e1001363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bakker PI, Ferreira MA, Jia X, Neale BM, Raychaudhuri S, Voight BF: Practical aspects of imputation-driven meta-analysis of genome-wide association studies. Hum Mol Genet 2008; 17: R122–R128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeggini E, Scott LJ, Saxena R et al: Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet 2008; 40: 638–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithana EN, Aung T, Khor CC et al: Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum Mol Genet 2011; 20: 649–658. [DOI] [PubMed] [Google Scholar]
- Wang X, Chua HX, Chen P et al: Comparing methods for performing trans-ethnic meta-analysis of genome-wide association studies. Hum Mol Genet 2013; 22: 2303–2311. [DOI] [PubMed] [Google Scholar]
- Franceschini N, van Rooij FJ, Prins BP et al: Discovery and fine mapping of serum protein loci through transethnic meta-analysis. Am J Hum Genet 2012; 91: 744–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen-Torvik LJ, Pacheco JA, Wilke RA et al: High density GWAS for LDL cholesterol in African Americans using electronic medical records reveals a strong protective variant in APOE. Clin Transl Sci 2012; 5: 394–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Jakobsson M, Coop G et al: A worldwide survey of haplotype variation and linkage disequilibrium in the human genome. Nat Genet 2006; 38: 1251–1260. [DOI] [PubMed] [Google Scholar]
- Hanchard N, Elzein A, Trafford C et al: Classical sickle beta-globin haplotypes exhibit a high degree of long-range haplotype similarity in African and Afro-Caribbean populations. BMC Genet 2007; 8: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelen P, Menelaou A, van Leeuwen EM et al: Improved imputation quality of low-frequency and rare variants in European samples using the 'Genome of The Netherlands'. Eur J Hum Genet 2014; 22: 1321–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Haritunians T, Marjoram P et al: Genotype Imputation for Latinos Using the HapMap and 1000 Genomes Project Reference Panels. Front Genet 2012; 3: 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Li Y, Singleton AB et al: Genotype-imputation accuracy across worldwide human populations. Am J Hum Genet 2009; 84: 235–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Willer CJ, Ding J, Scheet P, Abecasis GR: MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet Epidemiol 2010; 34: 816–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.