

Abstract
The emergence of homeostatic mechanisms that enabled maintenance of an intracellular steady-state during growth was critical to the advent of cellular life. Here, we show that concentration-dependent reversible binding of short oligonucleotides, of both specific and random sequence, can modulate ribozyme activity. In both cases, catalysis is inhibited at high concentrations, and dilution activates the ribozyme via inhibitor dissociation, thus maintaining near-constant ribozyme specific activity throughout protocell growth. To mimic the result of RNA synthesis within non-growing protocells, we co-encapsulated high concentrations of ribozyme and oligonucleotides within fatty acid vesicles; ribozyme activity was inhibited. Following vesicle growth, the resulting internal dilution produced ribozyme activation. This simple physical system enables a primitive homeostatic behavior: the maintenance of constant ribozyme activity per unit volume during protocell volume changes. We suggest such systems, wherein short oligonucleotides reversibly inhibit functional RNAs, could have preceded sophisticated modern RNA regulatory mechanisms, such as those involving miRNAs.
Main Text
Modern organisms employ a wide range of sophisticated homeostatic mechanisms in order to regulate their internal state in response to both internal and external fluctuations in conditions. The fact that modern homeostatic mechanisms use complex biochemical machinery raises the question of whether the earliest cells entirely lacked homeostatic processes, or whether intrinsic physical processes that conferred a degree of homeostasis were present from the beginning. Fatty acid vesicles – models of primitive cells – are dynamic systems that can grow in response to osmotic stress, the addition of fatty acid micelles, and the activity of encapsulated catalysts1, 2, 3. One example of such a dynamic behavior occurs in vesicles containing phospholipids or hydrophobic peptides, which can grow at the expense of surrounding vesicles that lack phospholipids or hydrophobic peptides3, 4, 5, 6, 7. Such abilities were likely critical elements of protocell fitness7, 8, but the volume increase resulting from growth dilutes encapsulated solutes, including catalysts such as ribozymes. The resulting decreased specific activity of cellular ribozymes would, in turn, have slowed RNA-catalyzed RNA replication, metabolism, and other cellular activities. While the vesicle membrane itself has been used as a reaction promoter, suggesting a partial solution to increased requirements for catalysts resulting from cellular (and, thus, membrane) growth3, 9, it remains unclear how membrane growth could have been coupled to the regulation of catalytic function within the vesicle lumen. Here, we demonstrate a simple physical process that could have increased the activity of ribozyme catalysts within growing primordial cells, thus compensating for the volume changes resulting from cell growth. Our demonstration of enhanced enzyme activity as a result of growth-driven dilution of cellular contents, which results in ca. constant enzyme specific activity (on a volume basis) before and after vesicle growth-induced dilution, shows that simple physical homeostatic mechanisms could have been operative in the earliest cells.
We examined whether the growth of a vesicle membrane and concomitant dilution of its contents could result in the activation of a ribozyme catalyst by dissociation of short oligonucleotide inhibitors, such as those expected to be generated by random RNA synthesis, partial RNA copying reactions, or nonspecific degradation of RNA. As a model functional RNA, we employed the well-characterized hammerhead ribozyme. This enzyme can be assembled from two oligonucleotides, HH-A and HH-B. The resulting HH-A/HH-B complex catalyzes the self-cleavage of HH-A10. We first screened in solution a series of ten 5-7 nt oligonucleotide inhibitors (HH-I-1 through HH-I-10) that were complementary to sequences within HH-B, by measuring cleavage yield after overnight incubation at saturating Mg2+. These oligonucleotides had predicted dissociation constants for HH-B of between ca. 250 nM and 800 μM (Supplementary Information Table 1). We carried out reactions at three different concentrations, with the inhibitor oligonucleotide present in 100-fold excess relative to HH-A and HH-B, which were present at 0.1-10 μM each. We reasoned that, at high concentration, these relatively weakly-binding oligonucleotides would inhibit formation of the active HH-A/HH-B ribozyme complex (Figure 1); following dilution, they would dissociate, allowing reconstitution of the functional ribozyme. Only the tightest-binding inhibitor, HH–I–10, exhibited significant (>50%) inhibition at high concentration (Supplementary Information Figure 1); HH-I-1 through 9 exhibited no significant inhibition of hammerhead activity. The addition of two more weakly-binding inhibitors, HH-I-3 and HH-I-9, enhanced the inhibition caused by HH–I–10, resulting in 98% suppression of ribozyme activity (as measured by overnight cleavage yield) at 10 μM enzyme concentration and 1 mM inhibitor concentration. However, 68% cleavage was observed upon dilution to 0.1 μM enzyme and 10 μM inhibitor concentration (Supplementary Information Figure 2). Interestingly, the weakly-binding oligonucleotide HH-I-3, with a predicted dissociation constant of ca. 160 μM, is a significant contributor to the suppression of enzyme activity when used in systems containing HH-I-10 as well (Supplementary Information Figures 2 and 3), demonstrating the importance of cooperative interactions in networks of interacting RNAs.
Figure 1. Regulation of enzyme activity in model protocells by dissociation of short complementary oligonucleotides.
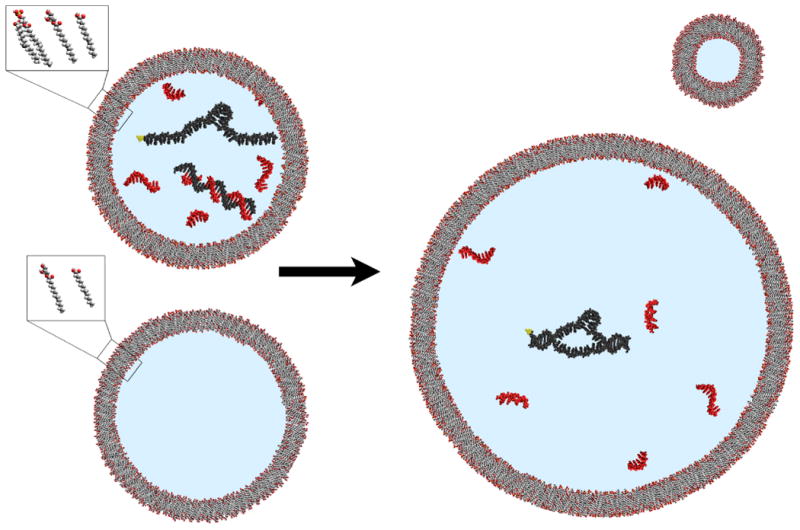
Mixed fatty acid-glycerol ester-phospholipid vesicles that contain split ribozymes (blue) and high concentrations of short oligonucleotides (red) exhibit no ribozyme activity, due to inhibition by duplex formation between the ribozyme fragments and complementary oligonucleotides (top left). When mixed with vesicles lacking phospholipid (bottom left), the phospholipid-containing vesicles grow at the expense of the phospholipid-lacking vesicles. This growth results in dilution of vesicle contents, inhibitor dissociation, and ribozyme reconstitution (right), increasing catalyst activity in the enlarged vesicles.
Having screened these inhibitors by endpoint analysis, we examined the reaction kinetics of ribozyme-inhibitor mixtures in more detail. The three-inhibitor HH-I-3/9/10 system, as well as those containing each single inhibitor alone, exhibit kinetics consistent with the results above; at 10 μM HH-A/HH-B concentration, 1 mM each of HH–I-3/9/10 results in a >100-fold reduction in apparent rate constant relative to the uninhibited HH-A/HH-B enzyme – a significantly greater reduction in rate than resulted from any single inhibitor (Figure 2, Supplementary Information Table 2). When this system is diluted 100-fold (to 100 nM ribozyme concentration), the weakly binding inhibitors dissociate, with the HH–I–3/9/10 system exhibiting 59% of the apparent rate constant of the uninhibited HH-A/HH-B system at this concentration. The order of addition (inhibitors added to HH-B followed by HH-A, or inhibitors added to a pre-formed HH-A/HH-B complex) did not affect concentration-dependent ribozyme inhibition (Supplementary Information Figure 4), suggesting that the active and inhibited complexes were in fast exchange.
Figure 2. A small ribozyme exhibits diminished activity upon dilution; the same ribozyme, in the presence of short oligonucleotide inhibitors, exhibits enhanced activity upon dilution.

a-c) Time courses of hammerhead ribozyme cleavage, expressed as natural log of remaining fraction of uncleaved HH-A vs. time. Error bars represent S.E.M. of ribozyme cleavage product yields, N=3. a) No inhibitors. White squares: 100 nM each HH-A/HH-B, grey circles: 10 μM each HH–A/HH‐B. b) HH-I-3/9/10 inhibitor oligonucleotides present. Red squares: 100 nM each HH-A/HH-B, 10 μM each HH-I-3/9/10 (not inhibited); dark red circles: 10 μM each HH‐A/HH-B, 1 mM each HH-I-3/9/10 (inhibited). c) Random-sequence inhibitor oligonucleotides r(N6) present. Blue squares: 100 nM each HH-A/HH-B, 30 μM r(N6) (not inhibited); dark blue circles: 10 μM each HH-A/HH-B, 3 mM r(N6) (inhibited). d) Rates of ribozyme cleavage in the conditions specified in panels a-c; error bars represent S.E.M., N=3. e) Representative polyacrylamide gel electrophoresis (PAGE) analyses of hammerhead ribozyme reactions. Substrate (HH-A) is denoted as S, cleavage product is denoted as P.
We next sought to examine whether such interactions could be relevant to a system of model protocell vesicles comprised of fatty acids and encapsulated RNAs. We employed a fatty acid vesicle system consisting of a 2:1 mixture of myristoleic acid and glycerol monomyristoleate (MA/GMO vesicles). This system was previously reported to be robust to at least 4 mM magnesium (Mg2+) and is capable of supporting hammerhead ribozyme activity11. We have previously shown that oleate vesicles containing a small fraction of phospholipid grow when surrounded by pure oleate vesicles; this competitive growth is a result of the slower off-rate of fatty acid molecules from membranes that contain some phospholipid7. We therefore examined the 2:1 myristoleic acid:glycerol monomyristoleate vesicle system, to see if it, too, would undergo competition-based growth when doped with a small amount (10 mol%) of diacylphospholipid (here, DOPA, or dioleylphosphatidic acid). When 2:1 myristoleic acid:glycerol monomyristoleate vesicles containing 10 mol% DOPA (MA/GMO/DOPA vesicles) were mixed with 20 eq. of pure MA/GMO vesicles, they grew in surface area ca. 3-fold (corresponding to a 5-fold volume increase, given spherical vesicles, Supplementary Information Figure 5); full volume relaxation occurred over ca. 3 h (Supplementary Information Figure 6).
We examined hammerhead function in this vesicle system at 1 μM ribozyme, reasoning that an intermediate concentration of the HH-A/HH-B/HH-I-3/9/10 system would be most sensitive to concentration changes. The HH-A/HH-B complex without inhibitors, encapsulated in MA/GMO/DOPA vesicles at 1 μM ribozyme concentration, exhibited an apparent rate constant for self-cleavage of 0.23 h-1 (Figure 3, Supplementary Information Table 2), similar to the unencapsulated control. Upon mixture with 20 eq. of MA/GMO vesicles and vesicle swelling (with concomitant dilution of contents), a rate depression to 0.11 h-1 was observed, somewhat lower than that observed with unencapsulated ribozyme. This rate decrease is presumably a result of dissociation of the HH-A/HH-B complex, owing, in both cases, to the HH-A/HH-B concentration approaching the Kd of hammerhead ribozymes of this stem length10. The overall lower rate in liposomes is likely a result of chelation of Mg2+ by fatty acids.
Figure 3. Competition-driven growth relieves inhibition of ribozyme activity in vesicles.

a-c) Time courses of hammerhead ribozyme cleavage, expressed as natural log of remaining fraction of uncleaved HH-A vs. time. All reactions were performed in MA/GMO/DOPA vesicles containing an initial concentration of 1 μM each HH-A and HH-B and 100 μM each HH-I-3/9/10 or 300 μM r(N6) (if inhibitors were present). Error bars represent S.E.M. of ribozyme cleavage product yields, N=4. a) No inhibitors. Grey circles: no vesicle growth; white squares: vesicle growth induced by mixing with 20 eq. (relative to lipid) MA/GMO vesicles. b) HH-I-3/9/10 inhibitor oligonucleotides present. Dark red circles: no vesicle growth; Red squares, vesicle growth induced as above. c) Random-sequence inhibitor oligonucleotides r(N6) present. Dark blue circles: no vesicle growth; blue squares, vesicle growth induced as above. d) Rates of ribozyme cleavage in the conditions specified in panels a-c; error bars represent S.E.M., N=4. e) Representative polyacrylamide gel electrophoresis (PAGE) analyses of hammerhead ribozyme reactions. Substrate (HH-A) is denoted as S, cleavage product is denoted as P. Oligonucleotides of lengths spanning a range of those used were retained within swollen, Mg2+-treated vesicles (Supplementary Information Figure 10), demonstrating that the observed reaction occurs within vesicles.
Having validated hammerhead function in this system, we examined the kinetics of this enzyme in the presence of inhibitors. The presence of 100 μM each of HH-I-3/9/10 in unswollen vesicles resulted in almost total abolition of 1 μM HH-A/HH-B ribozyme activity, with an intra-vesicle apparent rate constant of 11% of the uninhibited reaction (Figure 3). The addition of 20 eq. of MA/GMO vesicles to these vesicles and subsequent swelling and dilution of their contents resulted in reconstitution of ribozyme activity to an apparent rate constant of 0.12 h-1 – essentially identical to the uninhibited rate constant in vesicles after volume growth (Figure 3, Supplementary Information Table 2). Thus, the increased volume of the vesicle lumen following membrane growth resulted in ribozyme activation.
Encouraged by these results, we asked whether random oligonucleotides, such as those generated by untemplated synthesis, could exhibit a similar effect. Remarkably, in an unencapsulated system, the random oligonucleotide r(N6), when present at 300 eq. relative to HH-A/HH-B, suppressed catalytic activity to near-undetectable levels (kapp<0.005 h-1) at 10 μM HH-A/HH-B concentration (3 mM r(N6)), with recovery to an apparent rate constant of 0.10 h-1 upon dilution to 0.1 μM HH-A/HH-B (30 μM r(N6), Supplementary Information Table 2). A mixture of 2 mM r(N6) and 1 mM r(N5) also exhibited concentration-dependent inhibition, with an apparent rate constant of 0.0057 h-1 observed for 10 μM HH-A/HH-B (Supplementary Information Table 2); 100-fold dilution (to 0.1 μM HH-A/HH-B, 20 μM r(N6), 10 μM r(N5)) gave an apparent rate constant of 0.17 h-1. We examined each of these systems in MA/GMO/DOPA vesicles as before. In encapsulated systems containing 1 μM HH-A/HH-B, 300 eq. (300 μM) r(N6) gave an apparent rate constant of 0.027 h-1, corresponding to 88% inhibition; a mixture of 200 eq. r(N6) and 100 eq. r(N5) gave an apparent rate constant of 0.043 h-1, corresponding to 81% inhibition (Supplementary Information Table 2, Figure 3). Vesicle growth induced by the addition of 20 eq. of MA/GMO vesicles resulted in ribozyme reconstitution in both systems, with an apparent rate constant of 0.099 h-1 for r(N6) alone and 0.11 h-1 for the mixed random-inhibitor system. These changes represent 3.7- and 2.6-fold increases in rate constant, in contrast to the ca 2-fold decrease in the rate constant of the uninhibited HH–A/HH-B system upon vesicle growth. These results show that even a random-sequence pool of short oligonucleotides could have contributed to homeostatic behavior in primitive cells. Given that the short RNA pool in an early cell would have exhibited at least partial sequence complementarity to longer RNAs, owing to templating effects and enhanced chemical stability of base-paired RNAs, we expect that the magnitude of these phenomena in primordial cells would be intermediate to those observed with the HH-I-3/9/10 and r(N6) inhibitor systems.
We have previously suggested that short oligonucleotides generated in primordial cells as a consequence of partial replication reactions could act as replication intermediates in a hierarchical template-copying process12. As it is likely that each copy of a functional RNA synthesized during replication is produced along with many partial copies13, 14, 15, 16, such short oligonucleotides were likely abundant in primitive cells. Here, we have demonstrated that such short oligonucleotides can also act as concentration-dependent ribozyme inhibitors. These inhibitors afford model protocell vesicles the ability to activate ribozymes following growth via competitive processes, as shown here, or by other, non-competitive mechanisms, such as the growth of vesicles following sporadic exposure to fatty acid micelles. Such concentration-dependent inhibition could, in turn, have led to simple homeostatic mechanisms that may have been functional in the earliest cells. For example, if a protocell contained ribozymes that contributed to RNA replication or metabolic reactions, concentration-dependent inhibition would tend to slow down RNA replication or metabolism when internal RNA concentrations were high, but RNA-based catalysts of replication or other metabolic processes would activate following cell growth, thus contributing to the maintenance of a constant internal environment. In our experiments, the specific activity of an encapsulated ribozyme following growth is only ∼10% of that prior to growth (2-fold loss of total activity due to strand dissociation, together with five-fold dilution; Supplementary Information Table 2). In contrast, for encapsulated ribozyme-inhibitor systems, the initial rate of product formation per unit volume after growth is ∼50-100% of that prior to growth. These results suggest that the mechanisms we have described could have helped maintain a constant internal environment by regulating the activity of ribozymes required for metabolic processes in growing cells (such as polymerases, proofreading enzymes, etc.). Thus, the activation of catalysts in response to the dilution associated with cellular growth could have afforded one of the earliest means of maintaining intracellular homeostasis – a critical step in early cellular evolution. We speculate that the mechanism described here could have been elaborated through the evolution of increasingly finely tuned ribozyme inhibitors optimized for affinity and specificity, as well as increasingly sophisticated ribozymes enabling more complex cellular functions. The hammerhead ribozyme employed here produces fragments that, themselves, could act as product inhibitors in multiple-turnover catalysis, with predicted dissociation constants on the order of 10-9 and 10-7 M (Supplementary Information Table 1). We note that robust multiple turnover is possible with this hammerhead construct10, and any such product inhibition would be highly dependent on stem sequences. For example, the two stems found in the enzyme employed in this work are both hexanucleotides, but they have predicted KDs differing by ca. two orders of magnitude. This suggests a simple means by which primitive nucleases could have evolved a means to regulate their capacity for multiple turnover catalysis.
The predicted yields of full-length functional RNAs and short inhibitors, based on stepwise coupling efficiencies (Supplementary Information Figure 7, Supplementary Information Table 3), suggest that inhibitor:functional RNA ratios similar to those employed in this work would arise naturally as a result of RNA synthesis processes with ca. 85% stepwise coupling efficiency. This lies within the range of coupling efficiencies observed for the most effective known ribozyme RNA polymerases13,14. Additionally, it is likely that transesterification-mediated degradation of unstructured RNAs helped contribute to the short RNA pool in primitive cells15.
Such primitive RNA regulatory mechanisms could have contributed to the development of complex RNA-based cells and the ability of such cells to adapt to divergent and variable environments. Indeed, given the rich array of regulatory behaviors modulated by short non-coding RNAs in contemporary life17, it is not surprising that an early form of life with primarily RNA-based catalysts might have exploited such phenomena. The mechanisms we have described here represent a primitive form of ncRNA-based regulation of cellular behavior that may have been operative in the earliest forms of life.
Methods
Oligonucleotides
All oligonucleotides were obtained from IDT (Coralville, IA). HH-A (r(Fluorescein-CG CGC CGA AAC ACC GUG UCU CGA GC)) and HH-B (r(GGC UCG ACU GAU GAG GCG CG)) were obtained with HPLC purification. Random-sequence oligonucleotides r(N6) (r(NNN NNN)) and r(N5) (r(NNN NN)), HH-I-1 through HH-I-10, and FAM-DNA-HH-I-3 (d(Fluorescein-TC GAG), the fluorescently labeled DNA equivalent of HH-I-3) were obtained desalted but not HPLC purified. r(N6) and r(N5) were prepared by mix-on-machine delivery of all four RNA phosphoramidites in equimolar amounts. Affinities of HH-I-1 through HH-I-10 and the sequences comprising the 5′ and 3′ stems formed in the HH-A/HH-B complex, HH–A–5pStem and HH-A-3pStem, for HH-B, were calculated using MELTING 5.1.0 (Supplementary Information Table 1)18,19.
Lipids
Myristoleic acid and glycerol monomyristoleate were obtained from Nu-Chek (Elysian, MN), Dioleylphosphatidic acid (DOPA) was obtained from Avanti Polar Lipids (Alabaster, AL), and rhodamine DHPE (lissamine rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine) and NBD DHPE (NBD 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine) were from Life Technologies (Woburn, MA).
Other Materials
Tris-HCl (1 M solution), MgCl2 (1 M solution), and RNAse-free water (non-DEPC-treated) were from Life Technologies (Woburn, MA). Hand-poured denaturing polyacrylamide gels (20%) were prepared using the UreaGel concentrate/diluent system from National Diagnostics (Atlanta, GA). Pre-cast denaturing polyacrylamide gels (15%) were Novex TBE-Urea gels from Life Technologies (Woburn, MA). TBE (in hand-poured gels and running buffer) was 1 ×, prepared from a 10 × stock solution (0.89 M tris, 0.89 M boric acid, 0.01 M EDTA). Track-etched membranes for vesicle extrusion were Nucleopore brand from Whatman/GE (Little Chalfont, Buckinghamshire, UK). Other reagents and solvents were from Sigma (St. Louis, MO), Fisher (Waltham, MA), or VWR (Radnor, PA).
Vesicles
Thin films of lipids were prepared by drying a chloroform or dichloromethane solution of the desired final lipid composition in a glass vial under a stream of argon. The resulting film was resuspended in a solution of 250 mM tris-HCl pH 8 with 0.5 eq NaOH relative to unesterified carboxylic acid (i.e., myristoleic acid) and tumbled for 12–18 h, after which vesicles were sized and made unilamellar by nine extrusions through a 100 nm track-etched membrane.
Vesicles to encapsulate RNA were prepared using a resuspension solution containing the species to be encapsulated; extruded vesicles were purified over a Sepharose 4B column in running buffer containing vesicles of identical composition to the mixture to be purified. Vesicle-containing fractions were identified by the emission of the fluorescein tag on encapsulated oligonucleotides (λex=495 nm and λem=520 nm).
The composition of the MA/GMO vesicles was 66.7 mM/33.3 mM, and the composition of the MA/GMO/DOPA vesicles was 60 mM/30 mM/10 mM. All vesicles were always incubated with tumbling.
Vesicle Growth
Vesicle growth was performed by mixing MA/GMO/DOPA vesicles with MA/GMO vesicles (20 eq., 4 volumes at 5 × lipid concentration), several inversions of the vial containing the vesicles, and tumbling for 3 h prior to subsequent manipulations. This incubation was sufficient to allow 90‐95% volume equilibration (Supplementary Information Figure 6).
Where vesicle growth was monitored by FRET, FRET is reported as Fd/Fa, or the ratio of the donor (NBD) fluorescence (λem=530 nm) to acceptor (rhodamine) fluorescence (λem=586 nm) with λex=430 nm. Lipids labeled with these dyes were present at an initial concentration of 0.2 mol% total dye (i.e., 0.1% each lipid), and changes in surface area due to membrane growth were calculated from a standard curve of lipid dye concentration in the membrane, relative to Fd/Fa (Supplementary Information Figure 5). Fluorescence measurements of vesicles were read at the bottom of the plate to minimize scattering.
Hammerhead Reactions
Except where specified, hammerhead reactions were performed in 250 mM tris-HCl, pH 8 with 4 mM MgCl2. Reaction mixtures were generated by mixing all reaction components except HH–A and MgCl2 to generate HH-B-inhibitor complexes; HH-A was then added, giving a reaction mixture containing the highest used HH-A/HH-B concentration (i.e., 10 μM, or 1 μM for encapsulation mixtures). An inhibited ribozyme that could be activated to the same extent by dilution could also be generated from preformed HH-A/HH-B, suggesting an equilibrium mixture of HH-A/HH-B/inhibitors is present in these reactions (Supplementary Information Figure 4). Reaction mixtures were then diluted or encapsulated in vesicles (as described above) as desired, and the cleavage reaction was initiated by the addition of a 10 × (i.e., 40 mM) solution of MgCl2.
Reaction volumes were 10 μL for unencapsulated 10 μM and 1 μM reactions and 100 μL for unencapsulated 0.1 μM reactions. Encapsulated reactions were prepared in 100 μL volumes. Following column purification, this volume increased to ca. 750-900 μL. Vesicles were purified in running buffer of identical lipid concentration, ensuring the internal volume of the vesicles (and, therefore, concentrations of encapsulated RNAs) remained constant during initial purification. A typical sample was 120 μL, mixed with four volumes of vesicles at 5× (i.e., 500 mM total lipid) concentration. Vesicles undergoing growth were mixed with vesicles lacking phospholipid; those not undergoing growth were subjected to a dummy treatment of mixing with vesicles of identical composition. Encapsulated reactions were stopped by vesicle lysis with 1% Triton X-100, followed by ethanol precipitation overnight at −20 °C (300-350 μL of vesicle sample was typically precipitated), three additional washes with 100 μL 70% ethanol and resuspension in 10 μL 8M urea, 1 × TBE, which was used to load the sample on gels for analysis. The precipitation and wash procedure gave identical recovery efficiency for both HH-A and its cleavage product (Supplementary Information Figure 8).
Non-encapsulated reactions were stopped at the desired time points by addition of an aliquot (1–10 μL) of the reaction mixture to gel loading buffer containing 8 M urea, 1 × TBE, and 20 mM additional EDTA (10-99 μL), yielding 50-100 nM HH-A/HH-B in loading buffer. The resulting samples were loaded directly (i.e., without ethanol precipitation) in 5-10 μL volumes containing 250-500 fmol end-labeled oligonucleotide.
Reactions were analyzed by 20% or 15% PAGE and imaged on a Typhoon phosphorimager by monitoring the fluorescence of the 5′–fluorescein tag on HH–A; all other strands employed were not end-labeled. The green (532 nm) laser was employed as the excitation source, and the 526-nm short-pass filter was used as the emission filter. Gel integrations were performed with GelQuant.NET. An electropherogram showing HH-A (25 nt) and its cleavage product (19 nt) alongside a size marker ladder (10, 15, 20, 25, 30 ssRNA nt) is presented in Supplementary Information Figure 9, demonstrating the cleavage product is of the expected length.
Both the longest strand employed (HH-A) and a fluorescently-labeled DNA analogue of the shortest strand employed (FAM-DNA-HH-I-3), were retained within vesicles, even after growth and magnesium addition, over the experimental time course (Supplementary Information Figure 10), demonstrating that the reaction takes place within the fatty acid vesicles.
Supplementary Material
Acknowledgments
We thank K.A. Björkbom, T. Walton, N. Kamat, C. Hentrich, L. Jin, and other Szostak lab members for discussions. This work was supported in part by NASA Exobiology grant NNX11AD56G to J.W.S. and a grant (290363) from the Simons Collaboration on the Origin of Life to J.W.S. A.E.E. was supported by an appointment to the NASA Postdoctoral Program, administered by Oak Ridge Associated Universities through a contract with NASA, and by a Tosteson Fellowship from the Massachusetts General Hospital Executive Committee on Research. J.W.S. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions: A.E.E., K.A., and J.W.S. designed experiments, analyzed data, and wrote the manuscript. A.E.E. and K.A. performed experiments. A.E.E. and K.A. contributed equally to this work.
Reprints and permissions information is available at www.nature.com/reprints.
The authors declare no competing financial interests.
References
- 1.Chen IA, Szostak JW. A kinetic study of the growth of fatty acid vesicles. Biophysical Journal. 2004;87:988–998. doi: 10.1529/biophysj.104.039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen IA, Roberts RW, Szostak JW. The emergence of competition between model protocells. Science. 2004;305(5689):1474–1476. doi: 10.1126/science.1100757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Adamala K, Szostak JW. Competition between model protocells driven by an encapsulated catalyst. Nat Chem. 2013;5(6):495–501. doi: 10.1038/nchem.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peterlin P, Arrigler V, Kogej K, Svetina S, Walde P. Growth and shape transformations of giant phospholipid vesicles upon interaction with an aqueous oleic acid suspension. Chem Phys Lipids. 2009;159(2):67–76. doi: 10.1016/j.chemphyslip.2009.03.005. [DOI] [PubMed] [Google Scholar]
- 5.Rogerson ML, Robinson BH, Bucak S, Walde P. Kinetic studies of the interaction of fatty acids with phosphatidylcholine vesicles (liposomes) Colloids and surfaces B, Biointerfaces. 2006;48(1):24–34. doi: 10.1016/j.colsurfb.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 6.Cheng Z, Luisi PL. Coexistence and Mutual Competition of Vesicles with Different Size Distributions. J Phys Chem B. 2003;107(39):10940–10945. [Google Scholar]
- 7.Budin I, Szostak JW. Physical effects underlying the transition from primitive to modern cell membranes. Proc Natl Acad Sci U S A. 2011;108(13):5249–5254. doi: 10.1073/pnas.1100498108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stano P, Luisi PL. Achievements and open questions in the self-reproduction of vesicles and synthetic minimal cells. Chem Commun (Camb) 2010;46(21):3639–3653. doi: 10.1039/b913997d. [DOI] [PubMed] [Google Scholar]
- 9.Walde P, Umakoshi H, Stano P, Mavelli F. Emergent properties arising from the assembly of amphiphiles. Artificial vesicle membranes as reaction promoters and regulators. Chem Commun (Camb) 2014;50(71):10177–10197. doi: 10.1039/c4cc02812k. [DOI] [PubMed] [Google Scholar]
- 10.Uhlenbeck OC. A small catalytic oligoribonucleotide. Nature. 1987;328(6131):596–600. doi: 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- 11.Chen IA, Salehi-Ashtiani K, Szostak JW. RNA catalysis in model protocell vesicles. J Am Chem Soc. 2005;127(38):13213–13219. doi: 10.1021/ja051784p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Szostak JW. An optimal degree of physical and chemical heterogeneity for the origin of life? Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2011;366(1580):2894–2901. doi: 10.1098/rstb.2011.0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011;332(6026):209–212. doi: 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- 14.Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science. 2001;292(5520):1319–1325. doi: 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- 15.Soukup GA, Breaker RR. Relationship between internucleotide linkage geometry and the stability of RNA. RNA. 1999;5(10):1308–1325. doi: 10.1017/s1355838299990891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Adamala K, Engelhart AE, Szostak JW. Generation of Functional RNAs from Inactive Oligonucleotide Complexes by Non-enzymatic Primer Extension. J Am Chem Soc. 2015;137(1):483–489. doi: 10.1021/ja511564d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mattick JS, Makunin IV. Non-coding RNA. Human molecular genetics. 2006;15 Spec 1:R17–29. doi: 10.1093/hmg/ddl046. [DOI] [PubMed] [Google Scholar]
- 18.Le Novère N. MELTING, computing the melting temperature of nucleic acid duplex. Bioinformatics. 2001;17:1226–1227. doi: 10.1093/bioinformatics/17.12.1226. [DOI] [PubMed] [Google Scholar]
- 19.Dumousseau M, Rodriguez N, Juty N, Le Novère N. MELTING, a flexible platform to predict the melting temperatures of nucleic acids BMC. Bioinformatics. 2012;13:101. doi: 10.1186/1471-2105-13-101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.