

Abstract
Key points
The local arteriolar dilatation produced by contraction of skeletal muscle is dependent upon multiple signalling mechanisms.
In addition to the many metabolic signals that mediate this vasodilatation, we show here that the extracellular matrix protein fibronectin also contributes to the response.
This vasodilatory signal requires the heparin‐binding matricryptic RWRPK sequence in the first type III repeat of fibrillar fibronectin.
The fibronectin‐dependent component of the integrated muscle contraction‐dependent arteriolar vasodilatation is coupled through an endothelial cell‐dependent signalling pathway.
Abstract
Recent studies in contracting skeletal muscle have shown that functional vasodilatation in resistance arterioles has an endothelial cell (EC)‐dependent component, and, separately have shown that the extracellular matrix protein fibronectin (FN) contributes to functional dilatation in these arterioles. Here we test the hypotheses that (i) the matricryptic heparin‐binding region of the first type III repeat of fibrillar FN (FNIII1H) mediates vasodilatation, and (ii) this response is EC dependent. Engineered FN fragments with differing (defined) heparin‐ and integrin‐binding capacities were applied directly to resistance arterioles in cremaster muscles of anaesthetized (pentobarbital sodium, 65 mg kg−1) mice. Both FNIII1H,8‐10 and FNIII1H induced dilatations (12.2 ± 1.7 μm, n = 12 and 17.2 ± 2.4 μm, n = 14, respectively) whereas mutation of the active sequence (R613WRPK) of the heparin binding region significantly diminished the dilatation (3.2 ± 1.8 μm, n = 10). Contraction of skeletal muscle fibres via electrical field stimulation produced a vasodilatation (19.4 ± 1.2 μm, n = 12) that was significantly decreased (to 7.0 ± 2.7 μm, n = 7, P < 0.05) in the presence of FNIII1Peptide 6, which blocks extracellular matrix (ECM) FN and FNIII1H signalling. Furthermore, FNIII1H,8‐10 and FNIII1H applied to EC‐denuded arterioles failed to produce any dilatation indicating that endothelium was required for the response. Finally, FNIII1H significantly increased EC Ca2+ (relative fluorescence 0.98 ± 0.02 in controls versus 1.12 ± 0.05, n = 17, P < 0.05). Thus, we conclude that ECM FN‐dependent vasodilatation is mediated by the heparin‐binding (RWRPK) sequence of FNIII1 in an EC‐dependent manner. Importantly, blocking this signalling sequence decreased the dilatation to skeletal muscle contraction, indicating that there is a physiological role for this FN‐dependent mechanism.
Key points
The local arteriolar dilatation produced by contraction of skeletal muscle is dependent upon multiple signalling mechanisms.
In addition to the many metabolic signals that mediate this vasodilatation, we show here that the extracellular matrix protein fibronectin also contributes to the response.
This vasodilatory signal requires the heparin‐binding matricryptic RWRPK sequence in the first type III repeat of fibrillar fibronectin.
The fibronectin‐dependent component of the integrated muscle contraction‐dependent arteriolar vasodilatation is coupled through an endothelial cell‐dependent signalling pathway.
Abbreviations
- EC
endothelial cell
- ECM
extracellular matrix
- FN
fibronectin
- FNIII1
first type III repeat of fibrillar fibronectin
- GST
glutathione S‐transferase
- LNNA
N ω‐nitro‐l‐arginine
- VSM
vascular smooth muscle
Introduction
It is well established that blood flow in exercising muscle is tightly coupled to metabolic needs (Stainsby & Otis, 1964; Sarelius & Pohl, 2010; Murrant & Sarelius, 2015). Despite this, there is much that remains to be understood about metabolic coupling in skeletal muscle, particularly with respect to mechanisms that locally produce functional dilatation in arterioles. Recent studies in contracting skeletal muscle have served to re‐emphasize the integrated nature of this local vasodilatation. In particular, it is becoming clear that many vasodilatory responses in resistance arterioles have an endothelial cell (EC)‐dependent component. As examples, roles for nitric oxide (NO) and prostaglandins (both of which originate in ECs) in the arteriolar dilatation produced by skeletal muscle contraction have been identified (Cowley et al. 1985; Hester et al. 1993; Lau et al. 2000; Murrant & Sarelius, 2002; Schrage et al. 2004; Murrant et al. 2014). Importantly, we have shown that ECs are required for arteriolar dilatation in response to skeletal muscle contraction (Duza & Sarelius, 2004 a), and also in response to the metabolically derived purine, adenosine (Duza & Sarelius, 2004 a; Maimon et al. 2014). In further support of the role of ECs in metabolic vasodilatation, we have also shown that both muscle contraction (Duza & Sarelius, 2004 a) and adenosine (Duza & Sarelius, 2003; Maimon et al. 2014) cause an increase in EC calcium, which is a widely reported early signalling step for many EC‐dependent vasodilatations (Falcone et al. 1993; Cohen & Vanhoutte, 1995; Dora & Garland, 2013). Interestingly, in intact arterioles, each individual signalling pathway contributes variably to the full manifestation of the dilatation, with most contributing only about one‐quarter to one‐third of the full response (Poucher et al. 1990; Radegran & Calbet, 2001; Murrant & Sarelius, 2002; Maimon et al. 2014; Murrant & Sarelius, 2015); this clearly illustrates and underscores the integrated nature of endothelial‐ and smooth muscle‐derived signals in determining functional responses in intact tissue.
Recently, we identified an additional contributor to local dilatation in arterioles (Hocking et al. 2008). Using contracting hamster cremaster muscle as a model of functional hyperaemia, we showed that the extracellular matrix (ECM) protein fibronectin contributes to arteriolar dilatation via a matricryptic signalling region located in the first type III repeat of fibrillar fibronectin (FNIII1). Using the anti‐fibronectin antibody 9D2 (Chernousov et al. 1991), a portion of which maps to and blocks the matricryptic site within FNIII1 (Gui et al. 2006), we obtained data suggesting that skeletal muscle contraction transiently exposes the matricryptic site within interstitial fibronectin fibrils, and that exposure of this normally concealed site plays a role in functional dilatation (Hocking et al. 2008). Additionally, we showed that a recombinant fibronectin fragment engineered to mimic the fibrillar form of ECM fibronectin (FNIII1H,8‐10) could directly induce vasodilatation via an NO‐dependent pathway (Hocking et al. 2008). This engineered fragment (FNIII1H,8‐10) directly couples the matricryptic domain in an ‘open’ conformation (FNIII1H) to the α5β1 integrin‐binding domain (FNIII8–10). Interestingly, the vasodilatory response to this fibronectin fragment appeared to be independent of integrin receptor ligation.
In the present work, our goals were to provide further evidence supporting a vasodilatory role in arterioles for the matricryptic, heparin‐binding region of FNIII1; to determine the involvement of integrin ligation in this vasodilatory pathway; and to test the hypothesis that the vasodilatory response to the matricryptic FNIII1 site is EC dependent. To do this, we used a series of engineered fibronectin fragments that have different heparin‐ and integrin‐binding capacities to explore and define the vasodilatory activity of ECM fibronectin. We studied both intact and EC‐denuded blood perfused arterioles in situ in cremaster muscles of anaesthetized mice.
Methods
Animals and tissue preparation
All experimental animal procedures were approved by the University Committee on Animal Resources at the University of Rochester. Male C57Bl/6 mice (The Jackson Laboratory, Bar Harbor, ME, USA; aged 12–15 weeks) were anaesthetized (pentobarbital sodium, 65 mg kg−1, i.p.), tracheotomised to ensure a patent airway, and had a right jugular catheter placed for administration of supplemental anaesthesia as needed. Body temperature was maintained via convective heating throughout the tissue preparation and protocols. The right cremaster muscle was prepared for intravital microscopy as described previously (Kim & Sarelius, 2004; Duza & Sarelius, 2004 a; Maimon et al. 2014). To summarize, the muscle was exteriorized via a longitudinal skin incision, separated from testis and epididymis and gently spread over a quartz pillar suitable for microscopy. During all procedures the tissue was continuously superfused at 5 ml min−1 with 36ºC physiological solution composed of (mm): 131.9 NaCl, 4.7 KCl, 2.0 CaCl2, 1.2 MgSO4 and 18 NaHCO3, and equilibrated with gas containing 5% CO2–95% N2 to maintain pH at 7.4. Arterioles were visualized using an Olympus BX50WI microscope, ×25 objective (NA 0.35) and 1.6× magnification changer. A video camera (Dage‐MTI, Michigan City, IN, USA, CCD72) was used to capture images, which were displayed on a monitor and recorded to DVD (Sony DVO‐1000MD) for off‐line analysis. Prior to data acquisition, the tissue preparation was allowed to stabilize for approximately 30 min. Arterioles (maximal diameter 30–45 μm) were chosen based on vessel size, presence of vascular tone and vessel clarity. On completion of protocols, animals were killed by anaesthetic overdose followed by pneumothorax.
Endothelial denudation
For studies examining the role of ECs, the endothelium was disrupted in selected regions (up to 200 μm in length) using a locally introduced air bubble, as described previously (Duza & Sarelius 2004 b). Briefly, a sharp, triple‐bevelled micropipette was used to introduce an air bubble into the arteriole feeding a targeted microvascular region. Brief rapid pressurization of the pipette was used to drive the air bubble into the associated microvessel branches. The pipette was immediately depressurized and withdrawn, which allowed blood flow to resume. The air bubble dispersed into the microcirculatory network in approximately 5 min. Lack of dilatation to a brief local application of acetylcholine (ACh; 10−4 m) and continued ability to dilate to locally applied sodium nitroprusside (SNP; 10−4 m) were used to verify EC disruption with continuing vascular smooth muscle function in the targeted arteriole.
Fibronectin fusion proteins and peptides
A schematic diagram of the fibronectin proteins and peptides used in this study is shown in Fig. 1; receptor‐binding capacities are summarized in Table 1.
Figure 1. Fibronectin fusion proteins and peptides .
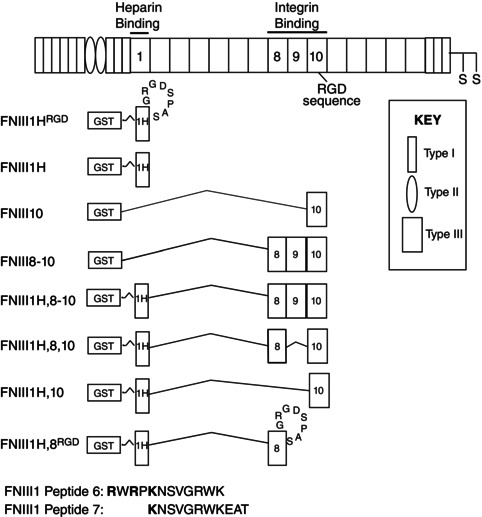
Schematic representation of a fibronectin subunit and recombinant fibronectin fusion proteins. Relevant type III repeats are numbered. FNIII1H is the heparin‐binding C‐terminal fragment of FNIII1. The heparin‐binding amino acids that form the matricryptic FNIII1 site are shown in bold.
Table 1.
Ligand binding properties of recombinant fibronectin fusion proteins and peptides
| Ligand | ||||
|---|---|---|---|---|
| FN fusion proteins and peptides | heparin | α5β1 integrins | αvβ3 integrins | Reference |
| FNIII1H,8‐10 | + | + | − | Hocking & Kowalski, 2002; Roy & Hocking, 2013 |
| FNIII1H | + | − | − | Hocking & Kowalski, 2002; Roy et al. 2011 |
| FNIII1HΔRRK | − | − | − | Roy et al. 2011 |
| FNIII8–10 | − | + | − | Hocking & Kowalski, 2002; Roy & Hocking, 2013 |
| FNIII1H,8,10 | + | + | + | Roy et al. 2011, Roy & Hocking, 2013 |
| FNIII1H,10 | + | − | + | Roy et al. 2011, Roy & Hocking, 2013 |
| FNIII1H,8RGD | + | − | + | Roy et al. 2011, Roy & Hocking, 2013 |
| FNIII1HRGD | + | − | + | Brennan & Hocking, 2015 |
| FNIII1HRGDΔRRK | − | − | + | Brennan & Hocking, 2015 |
| FN Peptide 6 | + | − | − | Gui et al. 2006, Hocking et al, 2008 |
| FN Peptide 7 | − | − | − | Gui et al. 2006, Hocking et al, 2008 |
The ability (+) or inability (−) of the various fibronectin fusion proteins and peptides to bind heparin and/or integrin receptors is noted.
Glutathione S‐transferase (GST)‐tagged fusion proteins FNIII1H; FNIII10; FNIII8–10; FNIII1H,8‐10; FNIII1H,8,10; FNIII1H,10; and FNIII1H,8RGD were produced in Escherichia coli and isolated by glutathione‐Sepharose (GE Healthcare, Piscataway, NJ, USA) affinity chromatography, as described previously (Hocking & Kowalski, 2002; Gui et al. 2006; Roy et al. 2011; Roy & Hocking, 2013). FNIII1H is a carboxyl‐terminal fragment of the first type III repeat of fibronectin (FNIII1) and is comprised of amino acids I597–T673 (bases 1802–2032). FNIII1HRGD was engineered by inserting the integrin‐binding RGD loop between the F and G β strands of FNIII1H, analogous to its native location in FNIII10. FNIII1HRGD was produced using the mutant sense primer: 5′‐GGTATACGAGGGCCAGCTCATATCGATTCAGGGCCGTGGAGACTCGCCGGC AAGCCAAGAAGTGACTCGCTTTGAC‐3′. Bases 1976 to 1987 (Q655 to H658) of FNIII1H were replaced with the underlined bases, which encode the amino acids G1491RGDSPAS from FNIII10, leading to a net addition of four amino acids to FNIII1. The FNIII10 insert contains an engineered NgoMIV site as a marker (shown in italics). The BstZ17I site is shown in bold. The antisense primer for FNIII1HRGD (5′‐CCCGAATTCCTATGTGCTGGTGCTGGTGGTG‐3′) contains an EcoRI site, shown in bold. The PCR product of this reaction was ligated into pGEX2T/FNIII1H after removal of the corresponding BstZ17I and EcoRI fragment and cloned into DH5α bacteria.
FNIII1HRGDΔRRK was produced from pGEX2TK/FNIII1H,8–10ΔRRK (Roy et al. 2011). The sense primer 5′‐CCCGGATCCATCCAGTGGAATGCACCACAG‐3′ (BamHI site shown in bold) and antisense primer 5′‐CCCGAATTCCTATGTGCTGGTGCTGGTGGTG‐3′ (EcoRI site in bold) were used to generate a PCR fragment from this vector comprising bases 1802–2032, and containing the mutations R613T, R615T and K617A. This fragment was ligated into pGEX2T to generate pGEX2T/FNIII1H∆RRK. The wild‐type BstZ17I–EcoRI fragment from this construct was then replaced with the corresponding fragment from pGEX2T/FNIII1HRGD to generate pGEX2T/FNIII1HRGDΔRRK. DNA was sequenced to confirm the presence of the mutations. GST‐tagged fusion proteins were isolated on glutathione‐Sepharose and dialysed extensively against phosphate‐buffered saline (PBS). Proteins were filter‐sterilized and purity was assessed by SDS‐polyacrylamide gel electrophoresis. Purified proteins were stored in aliquots at −80°C. We confirmed that the integrin binding capacity of both FNIII1HRGD and FNIII1HRGDΔRRK was not altered by mutation of the heparin binding sequence. To do this, tissue culture plates were coated with increasing concentrations of the various fibronectin fusion proteins, washed 3 times with PBS, and then blocked with 1% BSA. Fibronectin‐null mouse embryonic fibroblasts (Sottile et al. 1998) were seeded onto protein‐coated wells (1.5 × 105 cells cm−2) and allowed to attach for 30 min. Wells were then washed with PBS and fixed with 1% paraformaldehyde. Cell number was quantified by staining with crystal violet, as described previously (Roy et al. 2011). Figure 2 shows that the adhesive properties of these constructs are not altered compared to that previously established for the fusion protein FNIII10, which contains no heparin binding region (Roy et al. 2011).
Figure 2. Integrin binding capacity of FNIII1HRGD is not altered by mutation of the heparin binding sequence .
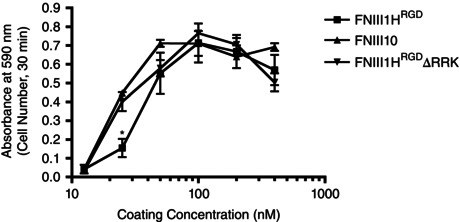
Fibronectin‐null mouse embryonic fibroblasts were grown on selected peptide constructs and cell number at different coating concentrations was measured. *Significantly different from FNIII10 and FNIII1HRGDΔRRK at corresponding coating concentrations, P < 0.05 (ANOVA).
Fibronectin fusion proteins were applied to tissues locally via a micropipette at a concentration of 15 μm (Hocking et al. 2008); preliminary protocols were used to verify effective concentrations for each protein (data not shown). In the skeletal muscle contraction experiments (see General protocols), a FNIII1 blocking peptide (Peptide 6: RWRPKNSVGRWK) or its inactive control (Peptide 7: KNSVGRWKEAT) (Gui et al. 2006) were added to the superfusate at a concentration of 5 mm for 10 min before, and during skeletal muscle contraction.
Endothelial cell calcium measurements
EC calcium levels in confocally imaged blood perfused arterioles in situ were measured using Fluo‐4, as described previously (Duza & Sarelius 2004 a,b). In brief summary, an arteriole upstream of the targeted arteriole was cannulated using a triple bevelled micropipette containing Fluo‐4 AM (5 μm; Duza & Sarelius 2004 a,b); dye was perfused for approximately 15 min, after which the pipette was withdrawn and blood flow allowed to resume. A period of 10–15 min was allowed for the intracellular dye to de‐esterify, and for arteriolar tone to be re‐established. EC loading was confirmed in the chosen arteriolar region by monitoring the EC Ca2+ response to locally applied 10−4 m ACh. EC Ca2+ changes were measured off‐line in individual ECs by defining a region of interest that encompassed individual ECs selected because they were in focus in the confocal plane, as described previously (Duza & Sarelius 2004 a,b); changes in total fluorescence signal were measured for each sampled EC during the observation period. Fluorescence intensity was background subtracted and normalized to baseline as described elsewhere (Duza & Sarelius 2004 a,b).
General protocols
Fibronectin fusion proteins (15 μm, in superfusion solution) were delivered locally to the target arteriole via a pressurized (approx. 30 cmH2O ejection pressure to generate flow out of the pipette) glass micropipette placed in close proximity to the arteriolar wall, as described previously (Hocking et al. 2008). To confirm flow out of the pipette, and to verify that the flow was across the arteriole, trace fluorescein isothiocyanate (FITC)‐dextran (molecular mass, 4000 Da) was added to the micropipette contents and brief epifluorescence was used to visualize the flow. Arteriolar responses are not affected by this tracer (Frame & Sarelius, 1995). For all arteriolar diameter measurements, vessels were recorded during a 1 min baseline period and during the 10 min exposure to pipette contents. Recovery of the vessel to its baseline diameter was monitored after the exposure but was not used in the analyses. Unless stated otherwise, the mean response at 10 min to exposure to the test protein was used to characterize the response. For the muscle contraction protocol, electrical field stimulation from silver foil electrodes placed at the muscle origin and perimeter was used as described previously (Duza & Sarelius, 2004 a; Maimon et al. 2014) to produce a maximal tetanic contraction (30 Hz, 0.2 ms duration, 5–10 V) for 15 s. The diameter at 0.5 s post‐stimulation was used to characterize the arteriolar response to muscle contraction. At the conclusion of each protocol, maximal diameter was obtained for each arteriole following at least 10 min superfusion of the tissue with either adenosine (10−4 m) or SNP (10−4 m), or a combination of both.
Diameters were measured off‐line using video calipers (Colorado Video, Boulder, CO, USA, model 308A) and expressed as change in diameter from baseline, or as a fraction of the maximal capacity to dilate, which was calculated as (D test − D baseline)/(D max − D baseline). Group means ± SEM are reported. Numbers of observations (n) are given in the figure legends or text: n refers to the number of arteriolar sites, with not more than three sites per tissue and at least three animals per group, unless stated otherwise. Data were compared using paired and unpaired Student's t test, or ANOVA, as appropriate; when an ANOVA showed significant differences, Bonferroni's post hoc analysis was used to compare means between conditions. Differences were considered significant if P ≤ 0.05.
Results
The arterioles used in these protocols had a mean maximal diameter of 33.4 ± 0.7 μm and resting diameter of 5.8 ± 0.4 μm: there were no significant differences among experimental groups except for the protocols undertaken in EC‐denuded arterioles. For the EC‐denuded vessels, mean baseline diameter was 9.3 ± 0.8 μm and maximal diameter was 27.9 ± 1.8 μm: again, there were no significant differences among the different protocols undertaken in EC‐denuded vessels.
FNIII1H induces arteriolar vasodilatation
To functionally mimic insoluble fibronectin fibrils, a series of soluble, recombinant fibronectin fragments was developed wherein the ‘open’ heparin‐binding fragment of FNIII1 (FNIII1H) was directly coupled to various portions of the integrin‐binding domain (Hocking & Kowalski, 2002; Roy et al. 2011). In previous studies (Hocking et al. 2008), we demonstrated that direct application of the full‐length fibronectin matrix mimetic FNIII1H,8‐10 to hamster skeletal muscle in situ produces vasodilatation in small resistance arterioles via the matricryptic, heparin‐binding site in FNIII1 (Hocking et al. 2008). Thus, our first goal in the present study was to establish that the matricryptic site of FNIII1 mediates a similar vasodilatory response in the present murine model. Figure 3 shows that vasodilatation was produced by local application of either the full‐length construct, FNIII1H,8‐10, (12.2 ± 1.7 μm, n = 12), or a smaller construct that contains only the heparin‐binding fragment of FNIII1 and lacks any known integrin‐binding sequences (FNIII1H; 17.2 ± 2.4 μm, n = 14). Moreover, vasodilatation in response to FNIII1H was mediated by the functional, heparin‐binding amino acid sequence RWRPK (Gui et al. 2006), as local application of a construct in which the active sequence (R613WRPK) was mutated to non‐charged amino acids (TWTPA) exhibited a significantly attenuated arteriolar dilatation (FNIII1HΔRRK; 3.2 ± 1.8 μm, n = 10). Thus, these data extend our previous findings (Hocking et al. 2008) and indicate that the ability of the matricryptic site in FNIII1 to induce vasodilatation is characteristic of skeletal muscle resistance arterioles.
Figure 3. FNIII1H induces vasodilatation in mouse skeletal muscle arterioles .
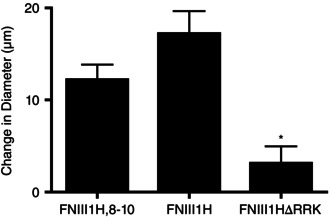
Arteriolar diameter was monitored before and during exposure to 15 μm FNIII1H,8‐10 (n = 12), FNIII1H (n = 14), or FNIII1HΔRRK (n = 10). Data are presented as mean change in diameter after 10 min protein exposure ± SEM. *Significantly different from other treatments; ANOVA, P < 0.05.
FNIII1H‐dependent vasodilatation does not require integrin signalling
Given that vasoactive roles for integrin receptor ligation have been established (Martinez‐Lemus et al. 2003; Martinez‐Lemus et al. 2005; Wu et al. 2008), we next asked whether the vasodilatory action of FNIII1H could be mimicked or modified by co‐delivery of either the α5β1 or the αvβ3 integrin‐binding domains of fibronectin. To do this, FNIII1H‐based fusion proteins that selectively ligate α5β1 integrins (FNIII1H,8‐10), αvβ3 integrins (FNIII1H,10; FNIII1H,8RGD; FNIII1HRGD), or both α5β1 and αvβ3 integrins (FNIII1H,8,10) (Roy & Hocking, 2013) were tested for their ability to induce vasodilatation and/or modify the response to FNIII1H. Figure 4 shows that while the original construct containing both the matricryptic site and the full integrin binding region (FNIII1H,8‐10) was able to produce vasodilatation as expected (Fig. 3 and Hocking et al. (2008)), modifications to the integrin binding domain that altered integrin‐binding specificity did not significantly alter the ability to dilate. Thus, FNIII1H8,10, FNIII1H,10, FNIII1H,8RGD, or FNIII1HRGD, all of which have been shown to have biological activity as adhesive substrates (Roy & Hocking (2013) and Fig. 2), all produced vasodilatation (FNIII1H,8,10: 14.3 ± 2.9 μm, n = 11; FNIII1H,10: 10.5 ± 3.8 μm, n = 8; FNIII1H,8RGD: 13.5 ± 3.5 μm, n = 8; and FNIII1HRGD: 17.1 ± 2.3 μm, n = 11). These dilatations were not different from that produced by FNIII1H,8‐10 (12.2 ± 1.7 μm, n = 12). Importantly, Fig. 4 also shows that even in the presence of the integrin binding RGD sequence, mutation of the heparin‐binding sequence of FNIII1H to non‐charged amino acids (FNIII1HRGDΔRRK) significantly decreased the vasodilatory response (to 4.8 ± 1.8 μm, n = 10), providing strong support for the conclusion that the matricryptic, heparin‐binding region of FNIII1 alone is required for vasodilatation.
Figure 4. FNIII1H‐induced vasodilatation does not require co‐ligation of integrin receptors .
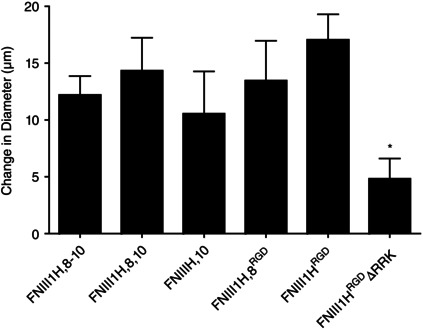
Arteriolar diameter was monitored before and during 10 min exposure to 15 μm FNIII1H,8‐10 (n = 12), FNIII1H,8,10 (n = 11), FNIII1H,10 (n = 8), FNIII1H,8RGD (n = 8), FNIII1HRGD (n = 11), or FNIII1HRGDΔRRK (n = 10). Data are presented as mean change in diameter after 10 min protein exposure. *Significantly different from all other treatments; ANOVA, P < 0.05.
The matricryptic RWRPK sequence contributes to functional dilatation of arterioles
Thus far, our data show that the matricryptic, heparin‐binding sequence of FNIII1H is capable of producing arteriolar vasodilatation when applied exogenously to intact tissue. An important question is therefore, is there a physiological role for this vasodilatory response? That is, does the connective tissue matrix in skeletal muscle contribute to the local functional dilatation produced by skeletal muscle contraction? To address this, we stimulated cremaster muscle to contract, using electrical field stimulation (see Methods) as a model of functional hyperaemia. Using this approach, we measured the local, muscle contraction‐induced dilatation in the presence or absence of FNIII1 Peptide 6, which contains the amino acid sequence RWRPK and has been shown to block signalling in response to both ECM fibronectin and FNIII1H (Gui et al. 2006). Figure 5 shows that application of FNIII1 Peptide 6 significantly decreased the vasodilatation produced by skeletal muscle contraction from 19.4 ± 1.2 μm (n = 12) in control conditions to 7.0 ± 2.7 μm (n = 7). In contrast, dilatation in the presence of the control peptide, FNIII1 Peptide 7 (20.0 ± 2.5 μm, n = 5), which does not contain the complete RWRPK sequence, was not different from controls (Fig. 5). Thus, these data provide direct evidence that the RWRPK sequence of FNIII1H in ECM fibronectin fibrils of intact tissues contributes to functional hyperaemia.
Figure 5. Vasodilatation induced by skeletal muscle contraction is blocked by a FNIII1H blocking peptide .
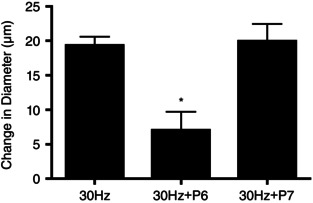
Arteriolar diameter measured immediately after 15 s of 30 Hz muscle contraction in the absence (30 Hz, n = 12) and presence of either FNIII1 Peptide 6 (P6, n = 7) or FNIII1 Peptide 7 (P7, n = 5). Shown is mean peak diameter change ± SEM. *Significantly different from 30 Hz control; ANOVA, P < 0.05.
Endothelium is required for the vasodilatation produced by FNIII1H
A critical question concerning the dilatation produced by FNIII1H is whether, in these small resistance arterioles, the signal from ECM fibronectin is coupled via endothelial cells (ECs) or vascular smooth muscle (VSM). In recent studies, we have shown that metabolic (functional) dilatation in these small arterioles is EC dependent (Duza & Sarelius, 2004 a; Maimon et al. 2014), and hence we hypothesized that FNIII1H‐dependent signalling is also EC dependent. To address this hypothesis, we measured the vasodilatory response to FNIII1H in arterioles denuded of their endothelium as described in Methods. Observations were made in vessel regions that demonstrated a significant decrease in the fractional capacity to dilate produced by ACh (Fig. 6: 0.19 ± 0.08, n = 8, grey bar versus 0.87 ± 0.08, n = 3, black bar) with ongoing vasodilatation in response to SNP (Fig. 6: 0.81 ± 0.07, n = 8, grey bar versus 0.88 ± 0.07, n = 3, black bar), thus verifying EC disruption with continuing ability of VSM to dilate. Figure 6 shows that dilatation in response to either FNIII1H,8‐10 (−0.06 ± 0.05, n = 8) or FNIII1H (−0.11 ± 0.14, n = 5) was effectively abolished with removal of ECs (Fig. 6, white bars), indicating that indeed, vasodilatation in response to FNIII1H requires intact endothelium.
Figure 6. Vasodilatation to fibronectin fusion proteins and peptides requires an intact endothelium .
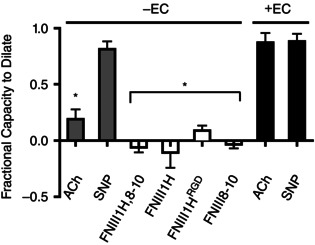
Endothelium (EC)‐denuded vessels (confirmed by the significant decrease in dilatation to ACh, grey bar) do not dilate to local application of FNIII1H,8‐10 or FNIII1H. The ability of EC‐denuded vessels to dilate is confirmed by the unchanged dilatation to SNP (grey bar) compared to EC intact vessels (black bar). Integrin‐binding peptides do not directly activate vascular smooth muscle in these small resistance arterioles as shown by the absence of significant response in EC‐denuded vessels (FNIII1HRGD and FNIII8–10). *Significantly different from ACh and SNP in intact vessels (black bars); ANOVA, P < 0.05.
Other published work (Martinez‐Lemus et al. 2003; Martinez‐Lemus et al. 2005; Wu et al. 2008) has shown that vascular smooth muscle (VSM) can respond to integrin‐mediated signalling, albeit more usually with vasoconstriction. As such, we asked whether either αvβ3 or α5β1 integrin binding fragments were vasoactive in these EC‐denuded vessel regions. Figure 6 shows that neither FNIII1HRGD (0.09 ± 0.04, n = 6), which is αvβ3 integrin‐binding, nor FNIII8–10 (−0.03 ± 0.04, n = 5), which is α5β1 integrin‐binding, were vasoactive in EC‐denuded regions, indicating that VSM‐mediated signalling via αvβ3 or α5β1 integrins is not a primary response pathway in these small resistance arterioles. The ability of these vessels to dilate via VSM‐dependent mechanisms is confirmed by the dilatation to SNP observed in EC‐denuded vessels; this dilatation (grey bar, Fig. 6) is not significantly different from the dilatation to SNP produced in arterioles with endothelium intact (black bar, Fig. 6).
Further support for the importance of EC‐dependent mechanisms in the dilatation to FNIII1H is provided by our observation (Fig. 7) that direct local application of FNIII1H to the arteriolar wall produced a significant increase in EC Ca2+ (relative fluorescence 1.12 ± 0.05, n = 17 cells versus 0.98 ± 0.02, n = 17 in controls, P < 0.05). We showed in earlier work (Duza & Sarelius, 2004 a) that increased EC Ca2+ is required for metabolic dilatation, and thus this finding adds to the evidence that supports a role for FN signalling in this important physiological response. Furthermore, FNIII1H‐dependent signalling was significantly decreased in the presence of 10−4 m N ω‐nitro‐l‐arginine (LNNA) (from 6.9 ± 0.4, n = 15 in controls to 2.5 ± 0.4, n = 14 with LNNA, P < 0.05), indicating that FNIII1H‐induced vasodilatation has an NO‐dependent signalling component. These data are consistent with the known contributions of both EC Ca2+ and NO in metabolic vasodilatation (Lau et al. 2000; Murrant & Sarelius 2002; Duza & Sarelius 2004 a).
Figure 7. FNIII1H increases intracellular calcium in endothelium .
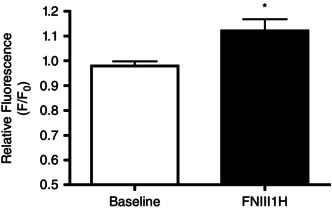
Relative fluorescence of Fluo‐4 loaded endothelial cells in in situ blood perfused arterioles in the absence (white bar) and presence (black bar) of FNIII1H when locally applied to the arteriolar wall. *Significantly different; t test, P < 0.05.
Discussion
In this study, we show that the heparin‐binding, RWRPK sequence in FNIII1 can stimulate vasodilatation in vivo, and that this matricryptic signalling pathway of connective tissue fibronectin fibrils contributes to skeletal muscle contraction‐induced dilatation. Importantly, we show that in the presence of a FNIII1H inhibiting peptide, the dilatation produced by muscle contraction is significantly reduced. Thus, ECM fibronectin must be added to the many previously identified initiators of signalling mechanisms that mediate functional dilatation in small resistance arterioles. Furthermore, we show that the FNIII1H signalling pathway is coupled through endothelial cells, is associated with increased EC Ca2+, and has an NO‐dependent component, thus providing further strong support for the conclusion that functional dilatation in skeletal muscle arterioles is EC dependent.
Although the resistance vessel dilatation produced locally by contraction of skeletal muscle fibres is often referred to as a ‘metabolic’ response, it has become very clear that many of the signalling pathways that contribute to this dilatation are not solely derived from metabolic products of working skeletal muscle, but also derive from other physiologically relevant aspects of muscle fibre contraction. Thus, for example, release of K+ from contracting fibres has long been identified as a key contributor to the integrated vasodilatation of arterioles (Kjellmer, 1965; Armstrong et al. 2007). In general, products related more directly to metabolic changes, of which adenosine and related purines are a classic and still prominent example (Radegran & Calbet, 2001; Murrant & Sarelius, 2002; Marshall, 2007; Maimon et al. 2014), are thought to contribute to the integrated dilatory response at later time points than dilators such as K+. How a mechanically coupled, fibronectin‐mediated dilatory component fits into the fully integrated functional response is an important question that will need to be addressed in future work.
Given that ECM fibronectin is a component of the interstitial tissue surrounding blood vessels (Hocking et al. 2008), we initially postulated that ECM fibronectin would contribute to the integrated functional dilatation via coupling between fibronectin fibrils and the vascular smooth muscle cells of the arteriolar wall. However, our study shows that FNIII1H‐dependent dilatation acts via endothelium. We note that an alternative explanation for the data shown in Fig. 6 is that the FNIII1H‐dependent signal could activate vascular smooth muscle, which in turn, via signal transfer through myoendothelial junctions, could cause activation of ECs which then send vasodilatory signals back to the smooth muscle. However, our data are consistent with previous findings that both functional (Duza & Sarelius 2004 a) and purinergic (Maimon et al. 2014) dilatations require endothelium. Furthermore, we confirmed in the present study that stimulation of the FNIII1H signalling site increases EC Ca2+, and that vasodilatation in response to FNIII1H is NO dependent; both responses are consistent with earlier observations on the critical role of EC Ca2+ increases (Duza & Sarelius, 2004 a) and the contribution of NO release (Lau et al. 2000; Murrant & Sarelius, 2002) in functional dilatation. Thus, the simplest interpretation of our findings is that the response is EC dependent. How might this work? It is now well established that endothelial and vascular smooth muscle cells in resistance arterioles are tightly coupled not only by locally released paracrine agents such as NO, but also via endothelial processes that penetrate the internal elastic lamina to directly interdigitate with smooth muscle cells and form a compartmentalized myoendothelial signalling region (Mather et al. 2005; Sandow et al. 2009; Garland et al. 2011). This not only facilitates communication between ECs and VSM, but importantly, allows signals arising in the ablumenal region of arterioles to impinge directly on ECs. In this scenario, fibrillar ECM structures (such as fibronectin fibrils) that surround individual cells of the arteriolar wall are able to communicate directly with the EC projections that are external to the internal elastic lamina. Thus, we can postulate that mechanically induced deformations (or other structural rearrangements) of this fibronectin fibrillar matrix by contraction of skeletal muscle myocytes would expose the matricryptic signalling site; the ensuing signals will be transferred directly to the endothelium. In turn, this would initiate EC‐dependent dilatory signalling in the arteriolar wall that would terminate once the fibronectin fibrils returned to their resting conformation. Clearly, an important question arising from our current study is: how might such mechanically induced deformations in the fibronectin matrix be coupled to EC signalling mechanisms? This will require new studies to identify the binding partner(s) for the RWRPK region of FNIII1H.
There is evidence that the integrin α5β1 can directly mediate responses in vascular smooth muscle via potentiation of large conductance Ca2+‐activated K+ channels, leading to vasodilatation (Wu et al. 2008). There is also evidence that both α5β1 and αvβ3 integrins can activate smooth muscle to produce either dilatation or constriction of blood vessels (Martinez‐Lemus et al. 2005). We were therefore interested to note that in EC‐denuded arterioles (where the vascular response reflects stimulation only of smooth muscle cells), the integrin binding motifs contained within either the full‐length fibronectin mimetic FNIII1H,8‐10 or FNIII8–10 alone were not able to induce any significant vascular responses (either constriction or dilatation), arguing that integrin‐mediated activation of vascular smooth muscle signalling is not a significant response mechanism in these small resistance vessels. The idea that the expression of key receptors and signalling mechanisms differs between large and small arterioles is not new (Anderson & Faber, 1991; Sarelius & Pohl, 2010), but whether this applies to integrin expression remains to be established. From our present data, we conclude that integrin‐mediated vascular responses are more likely to be a feature of larger distributing arterioles, where the ability to variously constrict or dilate enables redistribution of blood among tissues or tissue regions. In contrast, the ability to locally increase blood flow to working muscle fibres (via a non‐integrin‐dependent, but FNIII1‐dependent mechanism) is a feature of small resistance arterioles: this speculation will need to be addressed directly in future studies.
In conclusion, our study identifies a novel, mechanically coupled, component of the integrated functional dilatation that occurs in resistance arterioles in response to skeletal muscle contraction. This ECM‐mediated dilatation is dependent on an intact endothelium, and one of the implicated signalling pathways downstream of fibronectin activation involves increased EC Ca2+ and release of NO. Activation of this signalling pathway is dependent on the heparin‐binding RWRPK sequence of the first type III repeat of fibrillar fibronectin, a sequence that is normally buried in the tertiary structure of fibronectin. Thus, we speculate that this heparin‐binding matricryptic site is exposed by mechanical alterations that are induced in the fibronectin fibrils by skeletal muscle contraction: when the matricryptic site is exposed, this enables the transient ligation of an as‐yet‐unidentified receptor on endothelial cells.
Additional information
Competing interests
No conflicts of interests, financial or otherwise, are declared by the authors.
Author contributions
I.H.S. and D.C.H.: conception and design of research; P.A.T., N.M., W.O., S.W.M. and J.R.B. performed experiments; I.H.S., D.C.H., P.A.T., N.M., W.O. and J.R.B. analysed data; I.H.S., D.C.H., P.A.T. and J.B.R. prepared figures; I.H.S. drafted manuscript; I.H.S. and D.C.H. edited and revised the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported in part by grant R01HL105909 from the National Institutes of Health. J.R.B received support from the Harold C. Hodge Memorial Fund through the Department of Pharmacology and Physiology.
References
- Anderson KM & Faber JE (1991). Differential sensitivity of arteriolar alpha 1‐ and alpha 2‐adrenoceptor constriction to metabolic inhibition during rat skeletal muscle contraction. Circ Res 69, 174–184. [DOI] [PubMed] [Google Scholar]
- Armstrong ML, Dua AK & Murrant CL (2007). Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581, 841–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JR & Hocking DC (2015). Cooperative effects of fibronectin matrix assembly and initial cell‐substrate adhesion strength in cellular self‐assembly. Acta Biomater DOI:10.1016/j.act.bio.2015.12.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernousov MA, Fogerty FJ, Koteliansky VE & Mosher DF (1991). Role of the I‐9 and III‐1 modules of fibronectin in formation of an extracellular fibronectin matrix. J Biol Chem 266, 10851–10858. [PubMed] [Google Scholar]
- Cohen RA & Vanhoutte PM (1995). Endothelium‐dependent hyperpolarization. Beyond nitric oxide and cyclic GMP. Circulation 92, 3337–3349. [DOI] [PubMed] [Google Scholar]
- Cowley AJ, Stainer K, Rowley JM & Wilcox RG (1985). Effect of aspirin and indomethacin on exercise‐induced changes in blood pressure and limb blood flow in normal volunteers. Cardiovasc Res 19, 177–180. [DOI] [PubMed] [Google Scholar]
- Dora KA & Garland CJ (2013). Linking hyperpolarization to endothelial cell calcium events in arterioles. Microcirculation 20, 248–256. [DOI] [PubMed] [Google Scholar]
- Duza T & Sarelius IH (2003). Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+ . Am J Physiol Heart Circ Physiol 285, H26–H37. [DOI] [PubMed] [Google Scholar]
- Duza T & Sarelius IH (2004. a). Increase in endothelial cell Ca2+ in response to mouse cremaster muscle contraction. J Physiol 555, 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duza T & Sarelius IH (2004. b). Localized transient increases in endothelial cell Ca2+ in arterioles in situ: implications for coordination of vascular function. Am J Physiol Heart Circ Physiol 286, H2322–H2331. [DOI] [PubMed] [Google Scholar]
- Falcone JC, Kuo L & Meininger GA (1993). Endothelial cell calcium increases during flow‐induced dilation in isolated arterioles. Am J Physiol Heart Circ Physiol 264, H653–659. [DOI] [PubMed] [Google Scholar]
- Frame MD & Sarelius IH (1995). L‐arginine‐induced conducted signals alter upstream arteriolar responsivity to L‐arginine. Circ Res 77, 695–701. [DOI] [PubMed] [Google Scholar]
- Garland CJ, Hiley CR & Dora KA (2011). EDHF: spreading the influence of the endothelium. Br J Pharmacol 164, 839–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui L, Wojciechowski K, Gildner CD, Nedelkovska H & Hocking DC (2006). Identification of the heparin‐binding determinants within fibronectin repeat III1: role in cell spreading and growth. J Biol Chem 281, 34816–34825. [DOI] [PubMed] [Google Scholar]
- Hester RL, Eraslan A & Saito Y (1993). Differences in EDNO contribution to arteriolar diameters at rest and during functional dilation in striated muscle. Am J Physiol Heart Circ Physiol 265, H146–H151. [DOI] [PubMed] [Google Scholar]
- Hocking DC & Kowalski K (2002). A cryptic fragment from fibronectin's III1 module localizes to lipid rafts and stimulates cell growth and contractility. J Cell Biol 158, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocking DC, Titus PA, Sumagin R & Sarelius IH (2008). Extracellular matrix fibronectin mechanically couples skeletal muscle contraction with local vasodilation. Circ Res 102, 372–379. [DOI] [PubMed] [Google Scholar]
- Kim MB & Sarelius IH (2004). Role of shear forces and adhesion molecule distribution on P‐selectin‐mediated leukocyte rolling in postcapillary venules. Am J Physiol Heart Circ Physiol 287, H2705–H2711. [DOI] [PubMed] [Google Scholar]
- Kjellmer I (1965). The potassium ion as a vasodilator during muscular exercise. Acta Physiol Scand 63, 460–468. [DOI] [PubMed] [Google Scholar]
- Lau KS, Grange RW, Isotani E, Sarelius IH, Kamm KE, Huang PL & Stull JT (2000). nNOS and eNOS modulate cGMP formation and vascular response in contracting fast‐twitch skeletal muscle. Physiol Genomics 2, 21–27. [DOI] [PubMed] [Google Scholar]
- Maimon N, Titus PA & Sarelius IH (2014). Pre‐exposure to adenosine, acting via A2A receptors on endothelial cells, alters the protein kinase A dependence of adenosine‐induced dilation in skeletal muscle resistance arterioles. J Physiol 592, 2575–2590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall JM (2007). The roles of adenosine and related substances in exercise hyperaemia. J Physiol 583, 835–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez‐Lemus LA, Crow T, Davis MJ & Meininger GA (2005). αvβ3‐ and α5β1‐integrin blockade inhibits myogenic constriction of skeletal muscle resistance arterioles. Am J Physiol Heart Circ Physiol 289, H322–H329. [DOI] [PubMed] [Google Scholar]
- Martinez‐Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ & Meininger GA (2003). Integrins as unique receptors for vascular control. J Vasc Res 40, 211–233. [DOI] [PubMed] [Google Scholar]
- Mather S, Dora KA, Sandow SL, Winter P & Garland CJ (2005). Rapid endothelial cell‐selective loading of connexin 40 antibody blocks endothelium‐derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res 97, 399–407. [DOI] [PubMed] [Google Scholar]
- Murrant CL, Dodd JD, Foster AJ, Inch KA, Muckle FR, Ruiz DA, Simpson JA & Scholl JH (2014). Prostaglandins induce vasodilatation of the microvasculature during muscle contraction and induce vasodilatation independent of adenosine. J Physiol 592, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrant CL & Sarelius IH (2002). Multiple dilator pathways in skeletal muscle contraction‐induced arteriolar dilations. Am J Physiol Regul Integr Comp Physiol 282, R969–R978. [DOI] [PubMed] [Google Scholar]
- Murrant CL & Sarelius IH (2015). Local control of blood flow during active hyperemia: what kinds of integration are important? J Physiol 593, 4699–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poucher SM, Nowell CG & Collis MG (1990). The role of adenosine in exercise hyperaemia of the gracilis muscle in anaesthetized cats. J Physiol 427, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radegran G & Calbet JA (2001). Role of adenosine in exercise‐induced human skeletal muscle vasodilatation. Acta Physiol Scand 171, 177–185. [DOI] [PubMed] [Google Scholar]
- Roy DC & Hocking DC (2013). Recombinant fibronectin matrix mimetics specify integrin adhesion and extracellular matrix assembly. Tissue Eng Part A 19, 558–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy DC, Wilke‐Mounts SJ & Hocking DC (2011). Chimeric fibronectin matrix mimetic as a functional growth‐ and migration‐promoting adhesive substrate. Biomaterials 32, 2077–2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandow SL, Gzik DJ & Lee RM (2009). Arterial internal elastic lamina holes: relationship to function? J Anat 214, 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarelius I & Pohl U (2010). Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiol (Oxf) 199, 349–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrage WG, Joyner MJ & Dinenno FA (2004). Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557, 599–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile J, Hocking DC & Swiatek PJ (1998). Fibronectin matrix assembly enhances adhesion‐dependent cell growth. J Cell Sci 111, 2933–2943. [DOI] [PubMed] [Google Scholar]
- Stainsby WN & Otis AB (1964). Blood flow, blood oxygen tension, oxygen uptake, and oxygen transport in skeletal muscle. Am J Physiol 206, 858–866. [DOI] [PubMed] [Google Scholar]
- Wu X, Yang Y, Gui P, Sohma Y, Meininger GA, Davis GE, Braun AP & Davis MJ (2008). Potentiation of large conductance, Ca2+‐activated K+ (BK) channels by α5β1 integrin activation in arteriolar smooth muscle. J Physiol 586, 1699–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]