

Abstract
Key points
β‐adrenergic stimulation increases cardiac myosin binding protein C (MyBP‐C) and troponin I phosphorylation to accelerate pressure development and relaxation in vivo, although their relative contributions remain unknown.
Using a novel mouse model lacking protein kinase A‐phosphorylatable troponin I (TnI) and MyBP‐C, we examined in vivo haemodynamic function before and after infusion of the β‐agonist dobutamine.
Mice expressing phospho‐ablated MyBP‐C displayed cardiac hypertrophy and prevented full acceleration of pressure development and relaxation in response to dobutamine, whereas expression of phosphor‐ablated TnI alone had little effect on the acceleration of contractile function in response to dobutamine.
Our data demonstrate that MyBP‐C phosphorylation is the principal mediator of the contractile response to increased β‐agonist stimulation in vivo.
These results help us understand why MyBP‐C dephosphorylation in the failing heart contributes to contractile dysfunction and decreased adrenergic reserve in response to acute stress.
Abstract
β‐adrenergic stimulation plays a critical role in accelerating ventricular contraction and speeding relaxation to match cardiac output to changing circulatory demands. Two key myofilaments proteins, troponin I (TnI) and myosin binding protein‐C (MyBP‐C), are phosphorylated following β‐adrenergic stimulation; however, their relative contributions to the enhancement of in vivo cardiac contractility are unknown. To examine the roles of TnI and MyBP‐C phosphorylation in β‐adrenergic‐mediated enhancement of cardiac function, transgenic (TG) mice expressing non‐phosphorylatable TnI protein kinase A (PKA) residues (i.e. serine to alanine substitution at Ser23/24; TnIPKA−) were bred with mice expressing non‐phosphorylatable MyBP‐C PKA residues (i.e. serine to alanine substitution at Ser273, Ser282 and Ser302; MyBPCPKA−) to generate a novel mouse model expressing non‐phosphorylatable PKA residues in TnI and MyBP‐C (DBLPKA−). MyBP‐C dephosphorylation produced cardiac hypertrophy and increased wall thickness in MyBPCPKA− and DBLPKA− mice, and in vivo echocardiography and pressure–volume catheterization studies revealed impaired systolic function and prolonged diastolic relaxation compared to wild‐type and TnIPKA– mice. Infusion of the β‐agonist dobutamine resulted in accelerated rates of pressure development and relaxation in all mice; however, MyBPCPKA− and DBLPKA− mice displayed a blunted contractile response compared to wild‐type and TnIPKA– mice. Furthermore, unanaesthesized MyBPCPKA− and DBLPKA− mice displayed depressed maximum systolic pressure in response to dobutamine as measured using implantable telemetry devices. Taken together, our data show that MyBP‐C phosphorylation is a critical modulator of the in vivo acceleration of pressure development and relaxation as a result of enhanced β‐adrenergic stimulation, and reduced MyBP‐C phosphorylation may underlie depressed adrenergic reserve in heart failure.
Key points
β‐adrenergic stimulation increases cardiac myosin binding protein C (MyBP‐C) and troponin I phosphorylation to accelerate pressure development and relaxation in vivo, although their relative contributions remain unknown.
Using a novel mouse model lacking protein kinase A‐phosphorylatable troponin I (TnI) and MyBP‐C, we examined in vivo haemodynamic function before and after infusion of the β‐agonist dobutamine.
Mice expressing phospho‐ablated MyBP‐C displayed cardiac hypertrophy and prevented full acceleration of pressure development and relaxation in response to dobutamine, whereas expression of phosphor‐ablated TnI alone had little effect on the acceleration of contractile function in response to dobutamine.
Our data demonstrate that MyBP‐C phosphorylation is the principal mediator of the contractile response to increased β‐agonist stimulation in vivo.
These results help us understand why MyBP‐C dephosphorylation in the failing heart contributes to contractile dysfunction and decreased adrenergic reserve in response to acute stress.
Abbreviations
- dp/dt
rate of LV pressure development
- dp/dtmax
peak rate of left ventricular pressure development
- dp/dtmin
peak rate of left ventricular pressure relaxation
- EDP
end‐diastolic pressure
- EF
ejection fraction
- FS
fractional shortening
- HR
heart rate
- IVC
isovolumic contraction
- IVR
isovolumic relaxation
- IVRT
isovolumic relaxation time
- LV
left ventricle
- MyBP‐C
cardiac myosin binding protein C
- PKA
protein kinase A
- P–V
pressure‐volume
- Pmax
maximum systolic pressure
- PER
peak ejection rate normalized to end diastolic volume
- PFR
peak filling rate normalized to end diastolic volume
- PLB
phospholamban
- PVDF
poly(vinylidene difluoride)
- τ
rate constant of pressure relaxation
- TG
transgenic
- TnI
troponin I
- XB
cross‐bridge
- WT
wild‐type
Introduction
Increased cardiac output in response to changing circulatory demands depends, in part, on enhanced rates of pressure development and pressure relaxation mediated through increased β‐adrenergic signalling. Augmented in vivo cardiac contractility because of enhanced β‐adrenergic stimulation is a result of both accelerated Ca2+ release into the cytoplasm and reuptake into the sarcoplasmic reticulum (Bers, 2008; Fearnley et al. 2011) and, at the level of the myofilaments, decreased thin filament Ca2+ sensitivity and increased rates of cross‐bridge (XB) kinetics through protein kinase A (PKA)‐dependent phosphorylation of cardiac troponin I (TnI) (Sakthivel et al. 2005; Bilchick et al. 2007; Stelzer et al. 2007) and myosin binding protein C (MyBP‐C) (Stelzer et al. 2006, 2007; Tong et al. 2008), which together result in accelerated maximum shortening velocity and increased maximum power output (Herron et al. 2001). Although in vitro studies suggest that PKA‐mediated phosphorylation of TnI and MyBP‐C play a central role in enhancement cardiac contractility, the relative contributions of TnI and MyBP‐C phosphorylation to the in vivo acceleration of pressure development and relaxation remain largely unknown.
Transgenic (TG) mouse lines that mimic either constitutive phosphorylation (i.e. aspartic acid for serine substitutions) or dephosphorylation (i.e. alanine for serine substitutions) in PKA‐targeted troponin I (TnI) or MyBP‐C residues (Takimoto et al. 2004; Sakthivel et al. 2005; Nagayama et al. 2007; Tong et al. 2008) have been used to examine haemodynamic function and provided evidence indicating that phosphorylation of both sarcomeric proteins contributes to the maintenance of basal cardiac function. TG mice expressing non‐phosphorylatable MyBP‐C develop pathological cardiac hypertrophy (Sadayappan et al. 2005; Tong et al. 2008; Colson et al. 2012) and display a reduced ejection fraction (Nagayama et al. 2007) and impaired relaxation (Tong et al. 2008), along with a blunted systolic response to dobutamine administration (Nagayama et al. 2007). TG expression of TnI mimicking constitutive PKA phosphorylation enhanced basal systolic and diastolic function (Takimoto et al. 2004; Sakthivel et al. 2005), whereas TnI N‐terminal truncation to remove the PKA sites impairs relaxation and blunted the effects of β‐adrenergic activation (Fentzke et al. 1999; Layland et al. 2004). However, phospho‐ablation of the PKA sites on TnI did not produce a hypertrophic phenotype (Sakthivel et al. 2005; Stelzer et al. 2007). Although the evidence suggests that, individually, TnI and MyBP‐C phosphorylation play a role in maintaining basal cardiac function and in the response to increased β‐adrenergic signalling, their relative contributions are poorly understood.
A central feature of heart failure (HF) is contractile dysfunction (either systolic or diastolic) accompanied by ventricular wall remodelling (Hamdani et al. 2008; van der Velden, 2011; Wilson & Lucchesi, 2014) and impaired β‐adrenergic signalling that disrupts downstream protein phosphorylation (Post et al. 1999). In myocardial samples isolated from human HF patients, PKA activation and subunit expression were altered, resulting in decreased TnI and MyBP‐C phosphorylation (Han et al. 2013), which is a consistent finding among studies that have examined myofilament protein phosphorylation in human cardiovascular disease (Zakhary et al. 1999; Messer et al. 2007; Jacques et al. 2008; Kooij et al. 2010). These studies emphasize the importance of TnI and MyBP‐C dephosphorylation in contractile dysfunction and are supported by findings suggesting that experimental models of heart disease produce chronic TnI and MyBP‐C dephosphorylation (Decker et al. 2005, 2012; Sadayappan et al. 2005; El‐Armouche et al. 2007). Therefore, there is a clear need to develop a better understanding of the contributions of TnI and MyBP‐C dephosphorylation to contractile dysfunction and to the progression of cardiovascular disease.
The need to understand the relative contributions of TnI and MyBP‐C dephosphorylation to the deterioration of cardiac function in heart disease requires the development of models that closely mimic the conditions observed in HF patients. To examine the combined effects of chronic TnI and MyBP‐C dephosphorylation, we crossed two TG mouse lines that express either non‐phosphorylatable TnI (Ser23 and Ser24 mutated to alanines) or MyBP‐C (Ser273, Ser282 and Ser302 mutated to alanines) to produce a novel TG mouse line that lacks PKA‐phosphorylatable residues on both TnI and MyBP‐C. Using this model, we examined in vivo contractile and haemodynamic function before and after β‐agonist administration aiming to determine the relative contributions of TnI and MyBP‐C phosphorylation to the β‐adrenergic‐mediated enhancement of systolic and diastolic function.
Methods
Ethical approval and generation of mouse models
All procedures for anaesthesia, surgery and general care of the animals used in the present study were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University, and the study was conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85‐23, revised 1996). Adult mice (aged 3–6 months) of both sexes were used, and all mouse lines used were of the SV/129 strain. Transgenic (TG) mice expressing non‐phosphorylatable MyBP‐C with serine (Ser) to alanine (Ala) mutations at Ser273, Ser282 and Ser302 (MyBPCPKA−) on a MyBP‐C null background were generated previously (Tong et al. 2008). TG mice expressing non‐phosphorylatable TnI with Ser to Ala mutations at Ser23 and Ser24 (TnIPKA−) on a TnI null background were generated as described previously (Pi et al. 2003). MyBPCPKA− and TnIPKA− mice were bred to generate mice with TG expression of the non‐phosphorylatable MyBP‐C and TnI constructs (DBLPKA−), in the complete absence of endogenous MyBP‐C and TnI expression. Non‐transgenic wild‐type (WT) mice were used as controls.
Myofibril isolation and PKA treatment
Cardiac myofibrils were isolated as described previously (Cheng et al. 2013; Gresham et al. 2014). Frozen mouse cardiac tissue was thawed and homogenized in fresh relaxing solution on the day of the experiment. Myofibrils were skinned for 15 min using 1% Triton X‐100 on a mechanical rocker plate, centrifuged, and resuspended in fresh relax solution containing protease and phosphatase inhibitors (PhosSTOP and cOmplete ULTRA Tablets; Roche Applied Science, Indianapolis, IN, USA) and then kept on ice until use. Cardiac myofibrils were phosphorylated by PKA as described previously (Tong et al. 2008; Gresham et al. 2014). All solutions were brought to room temperature (22°C) for 10 min before initiating the reaction. Next, 100 μg of myofibrils were incubated with the catalytic subunit of bovine PKA (Sigma, St Louis, MO, USA) to a final concentration of 0.15 U PKA μg–1 myofibrils for 1 h at 30°C. Control myofibrils were incubated under the same conditions without PKA. Laemli buffer was added to stop the reaction and samples were heated at 90°C for 5 min and stored at −20°C.
Cardiac morphology and histological analysis of cardiac tissue
Examination of heart morphology was carried out as described previously (Cheng et al. 2013; Gresham et al. 2014). Hearts were excised and submerged in formalin for 4 h prior to sectioning at their mid‐left ventricles (LVs). Dissected hearts were kept in formalin overnight, paraffin embedded and sectioned at 5 μm thickness before staining with Masson's trichrome. Analysis of trichrome stained sections was performed on each cardiac section using ImageJ (National Institute of Health, Bethesda, MD, USA) and quantified using an ImageJ macro (Kennedy et al. 2006) that measured the number pixels in which the blue intensity exceeded the red intensity by >120%. For each section, two areas of interest were selected from the LV free wall, the right ventricular free wall, the interventricular septum and the apex. Fibrosis was quantified in four or five hearts for each mouse line and the results are presented as the percentage fibrotic area out of the total area examined (total area examined was not different between the groups).
Quantification of myofilament protein expression and phosphorylation levels
Determination of protein phosphorylation was carried out by western blotting and using the Pro‐Q Diamond Phosphoprotein stain (Life Technologies, Grand Island, NY, USA) as described previously (Mamidi et al. 2014; Gresham et al. 2014). Myofibril samples were thawed immediately before gel electrophoresis. For western blotting, 5 μg of solubilized myofibrils were loaded onto a 4–20% Tris‐glycine gel (Lonza, Allendale, NJ, USA) and electrophoretically separated at 180 V for 70 min. Proteins were transferred to poly(vinylidene difluoride) (PVDF) membranes and incubated overnight with one of the following primary antibodies: total TnI (Cell Signaling Technology, Beverly, MA, USA), TnI phospho‐serine 23 and 24 (detects phosphorylation of Ser23 and Ser24 of TnI; Cell Signaling), total MyBP‐C (Santa Cruz Biotechnology, Santa Cruz, CA, USA), MyBP‐C phospho‐serine 273, 282 or 302 (detects phosphorylation of Ser273, S282 or Ser302 of MyBP‐C; 21st Century Biochemicals, Marlborough, MA, USA) or HSC70 (Santa Cruz Biotechnology). Membranes were subsequently exposed to the appropriate secondary antibodies and imaged. For total protein phosphorylation analysis, 2.5 μg of myofibrils were separated at 180 V for 85 min, fixed and stained with Pro‐Q phosphostain before being imaged using a Typhoon gel scanner. Gels were counterstained with Coomassie blue to determine total protein load.
Measurement of phospholamban (PLB) phosphorylation in response to β‐adrenergic stimulation was carried out by western blotting of ventricular tissue isolated from mice following dobutamine administration. Mice were anaesthetized, given an i.p. injection of dobutamine (10 μg g−1), and the heart was removed and flash‐frozen in liquid nitrogen and stored at −80°C until use. Ventricular tissue was homogenized as reported previously (Cheng et al. 2013) in a buffer containing 20 mm Tris‐base, 137 mm NaCl, 2.7 mm KCl, 1 mm MgCl2‐6H2O, 1 mm (K2)EDTA, 10% glycerol and 1% Triton X‐100 (pH 7.8), with protease and phosphatase inhibitors. Non‐boiled ventricular samples were loaded onto 15% Tris‐glycine gels, transferred to a PVDF membrane, and probed using antibodies to detect either total phospholamban (Thermo Scientific, Waltham, MA, USA) or PLB phospho‐serine 16 (detects phosphorylation of Ser16 of PLB; Santa Cruz Biotechnology). Non‐treated hearts for each line were used as the baseline control for protein phosphorylation analysis.
Cardiac morphology and in vivo contractile and haemodynamic function
Overall cardiac morphology and in vivo function, including left‐ventricular wall thickness, ejection fraction (EF), fractional shortening (FS) and isovolumic relaxation time (IVRT), were evaluated by echocardiography as described previously (Cheng et al. 2013; Gresham et al. 2014). Mice were anaesthetized using isoflurane (1.5–2.0%) and scanned using a Sequoia C256 system (Siemens Medical, Erlangen, Germany). In vivo haemodynamic function was evaluated using pressure–volume (P–V) loop analysis as described previously (Cheng et al. 2013; Gresham et al. 2014). Mice were anaesthetized with isoflurane, ventilated and a pressure‐conductance catheter was inserted through the carotid artery into the left ventricle. Isovolumic contraction (IVC) was defined as the time from the start of contraction to dp/dt max, ejection was defined as the time from dp/dt max to dp/dt min, isovolumic relaxation (IVR) was defined as the time from dp/dtmin until pressure returned to the preceding end‐diastolic pressure (EDP) and diastolic filling was defined as the time from the end of IVR to the beginning of the next contraction. The time constant of pressure relaxation (τ) was calculated using a logistic function as previously described (Matsubara et al. 1995). Systemic blood pressure in conscious mice was monitored using implantable HD‐X11 transmitters (Data Sciences International, New Brighton, MN, USA). Mice were anaesthetized with isoflurane and a pressure catheter was inserted through the carotid artery and advanced to the aortic arch. Mice were allowed to recover from surgery and blood pressure measurements were made 3 days post‐surgery. For echocardiography and P–V loop analysis, a stable baseline was achieved and baseline measurements were made prior to i.p. injection of dobutamine (10 μg g−1) to measure the response to increased β‐adrenergic stimulation. For blood pressure analysis, a stable baseline segment was analysed immediately before dobutamine injection (10 μg g−1) and the post‐dobutamine data were analysed at the peak of the dobutamine response. P–V loops and blood pressure were analysed offline using LabChart7 software (AD Instruments, Sydney, Australia).
Statistical analysis
One‐way ANOVA with the Tukey–Kramer post hoc test was used to compare results between groups. Comparisons between baseline and post‐dobutamine treatment within groups were performed using a Student's t test as appropriate. All data are presented as the mean ± SEM. P < 0.05 was considered statistically significant.
Results
Quantification of myofilament protein expression and phosphorylation levels
To confirm the ablation of PKA‐phosphorylatable residues in TnI and MyBP‐C in the DBLPKA− mice, myocardial samples were probed with primary antibodies to detect phosphorylation of Ser23/24 in TnI or Ser273, Ser282 or Ser302 in MyBP‐C (Fig. 1 A) and stained with Pro‐Q diamond phospho‐stain to detect sarcomeric protein phosphorylation (Fig. 1 B). TnI phosphorylation was abundant in samples with endogenous TnI protein (i.e. WT and MyBPCPKA− samples) but was absent from samples with TG expression of phospho‐ablated TnI (i.e. TnIPKA− and DBLPKA− samples). Similarly, MyBP‐C phosphorylation was observed in samples expressing endogenous MyBP‐C (i.e. WT and TnIPKA− samples) but was not detected in samples expressing TG phospho‐ablated MyBP‐C (i.e. MyBPCPKA− and DBLPKA− tissue). MyBP‐C or TnI phospho‐ablation did not result in compensatory changes in phosphorylation levels of other myofilament proteins (Fig. 1) (Stelzer et al. 2007; Tong et al. 2008). PKA treatment increased TnI phosphorylation in WT and MyBPCPKA− samples and MyBP‐C phosphorylation in WT and TnIPKA− samples but produced no observable TnI phosphorylation in TnIPKA− and DBLPKA− samples or MyBP‐C phosphorylation in MyBPCPKA− and DBLPKA− samples (Fig. 1 A and B). Phosphorylation of troponin T, a thin filament regulatory protein, was increased by PKA incubation in every mouse line, and no differences were observed between groups before or after PKA incubation. Expression of TG MyBP‐C lacking PKA‐phosphorylatable residues in DBLPKA− samples was 73 ± 5% of WT MyBP‐C levels, which was similar to TG MyBP‐C expression levels in MyBPCPKA− (73 ± 8%) observed in the present study and in a previous study (Tong et al. 2008). These results confirm phospho‐ablation of PKA‐targeted MyBP‐C residues in MyBPCPKA− and TnI residues in TnIPKA− mice, and also validate our new model of combined PKA‐targeted phospho‐ablation of MyBP‐C and TnI in DBLPKA− mice.
Figure 1. MyBP‐C and TnI phosphorylation in TG lines .
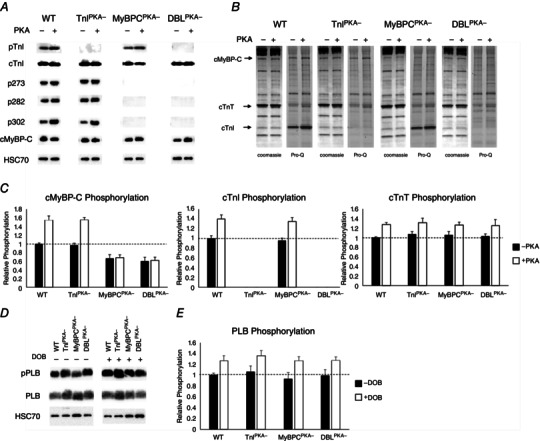
A, representative western blots of WT, TnIPKA−, MyBPCPKA− and DBLPKA− cardiac samples demonstrating TnI and/or MyBP‐C phospho‐ablation. PVDF membranes were probed with primary antibodies specific for TnI phosphorylation (Ser23/24), total TnI, MyBP‐C phosphorylation (Ser273, Ser282 or Ser302), total MyBP‐C or HSC70. B, representative Coomasie and Pro‐Q stained cardiac myofibrils from TG and WT lines that were incubated in the presence or absence of the catalytic subunit of PKA (see Methods). C, quantification of MyBP‐C, TnI and troponin T phosphorylation from cardiac myofibrils for each line with or without PKA incubation. The intensity of the Pro‐Q band was normalized to the Coomassie band intensity and non‐PKA treated WT myofibril protein phosphorylation was set as 1. Myofibrils were isolated from six hearts for each mouse line. D, representative western blots of WT, TnIPKA−, MyBPCPKA− and DBLPKA− cardiac ventricular samples demonstrating PLB phosphorylation. PVDF membranes were probed with primary antibodies specific for PLB phosphorylation (Ser16) in the absence (–) or presence (+) of dobutamine administration. E, quantification of PLB phosphorylation from cardiac samples for each line with or without dobutamine administration. PLB phosphorylation was normalized to total PLB protein expression and non‐dobutamine treated WT PLB phosphorylation was set as 1. Ventricular samples were isolated from three or four hearts for each mouse line.
To determine whether the β‐adrenergic signalling pathway has been altered in any of the mouse lines used in the present study, we performed western blots to determine the phosphorylation levels of PLB in response to dobutamine administration (Fig. 1 D). Western blotting demonstrated that dobutamine administration increased PLB phosphorylation to a similar extent in all of the mouse lines, and that there were no differences in PLB phosphorylation levels at baseline (Fig. 1 E). These results suggest that β‐adrenergic signalling is not altered in any of the TG lines and that the impaired adrenergic reserve observed in DBLPKA− and MyBPCPKA‐ mice is not a result of alterations in the phosphorylation of Ca2+‐handling proteins targeted β‐adrenergic pathway.
Cardiac morphology
We examined the morphology of excised mouse hearts to determine whether combined MyBP‐C and TnI phospho‐ablation would produce pathological cardiac remodelling. Representative formalin fixed hearts from each TG line are shown in Fig. 2 demonstrating overall cardiac morphology (Fig. 2 A) and cross‐sectional wall thickness (Fig. 2 B). DBLPKA− hearts displayed an enlarged overall heart size with a corresponding increase in LV wall thickness compared to WT control hearts. The hypertrophic appearance of DBLPKA− hearts closely resembled the increase in heart size observed in MyBPCPKA− mice (Fig. 2 B). TnIPKA− hearts did not display a noticeable increase in heart size or wall thickness and were similar in appearance to WT hearts. None of the mouse lines examined in the present study displayed an observable increase in ventricular fibrosis when examined using Masson's trichrome staining (WT, 0.068 ± 0.013%; TnIPKA−, 0.077 ± 0.012%; MyBPCPKA−, 0.093 ± 0.007%; DBLPKA−, 0.085 ± 0.010%; n = 4–5 hearts per group) (Figs. 2 C and E).
Figure 2. Analysis of cardiac morphology .
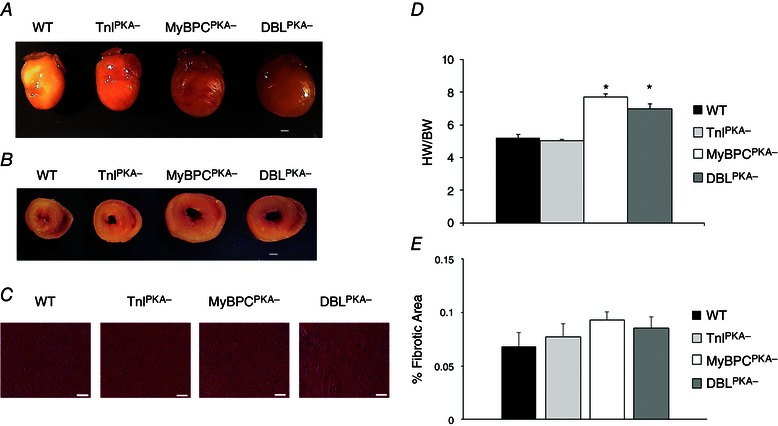
Representative formalin fixed hearts from WT, TnIPKA−, MyBPCPKA− and DBLPKA− mice. After formalin fixation, representative hearts were imaged (A) (scale bar = 1 mm) before being sectioned at the mid LV and imaged again (B) (scale bar = 1 mm). C, cross‐sections of the mid LV were stained with Masson's trichrome and imaged at 100× magnification (scale bar = 50 μm). D, summary data showing heart weight normalized to body weight for each line. Values are expressed as the mean ± SEM from four or five mice per line. *Significantly different from WT (P < 0.05). E, quantification of fibrosis from Masson's trichrome cardiac sections in (C). Values are expressed as the mean ± SEM and cardiac sections were analysed from four or five mice per line.
Echocardiography was performed on each mouse line to corroborate the changes in morphology observed in DBLPKA− and MyBPCPKA− mice (Table 1). An overall increase in LV mass with a parallel increase in posterior wall thickness was observed in DBLPKA− mice, similar to MyBPCPKA− hearts. By contrast, TnIPKA− hearts did not display an increase in cardiac mass or wall dimensions, suggesting the absence of cardiac hypertrophy. Our results indicate that the absence of MyBP‐C phosphorylation leads to cardiac hypertrophy, whereas the absence of TnI phosphorylation does not induce pathological remodelling.
Table 1.
LV morphology and in vivo cardiac performance measured by echocardiography
| WT | TnIPKA− | MyBPCPKA− | DBLPKA− | |
|---|---|---|---|---|
| BW (g) | 28.04 ± 0.65 | 27.50 ± 0.94 | 26.47 ± 1.43 | 25.40 ± 2.15 |
| LV mass/BW | 3.67 ± 0.11 | 3.80 ± 0.19 | 5.60 ± 0.39† | 5.11 ± 0.21† |
| PWd (mm) | 0.85 ± 0.02 | 0.88 ± 0.01 | 1.05 ± 0.05† | 0.98 ± 0.04† |
| PWs (mm) | 1.19 ± 0.03 | 1.25 ± 0.02 | 1.35 ± 0.05† | 1.29 ± 0.03 |
| EDV (μL) | 50.6 ± 2.3 | 55.6 ± 2.7 | 74.4 ± 4.7† | 70.7 ± 3.3† |
| ESV (μL) | 11.1 ± 1.1 | 12.3 ± 1.6 | 29.8 ± 3.3† | 27.2 ± 2.2† |
| IVRT (ms) | 19.44 ± 1.14 | 18.78 ± 1.42 | 27.31 ± 1.43† | 28.80 ± 1.02† |
| HR (beats min–1) | 416 ± 11 | 434 ± 12 | 439 ± 10 | 415 ± 10 |
| HR (beats/min) + dobutamine | 496 ± 9* | 514 ± 9* | 502 ± 6* | 489 ± 7* |
| FS (%) | 37.14 ± 1.31 | 37.50 ± 1.34 | 30.12 ± 1.73† | 29.17 ± 1.69† |
| FS (%) + dobutamine | 58.90 ± 2.04* | 54.23 ± 2.23* | 36.90 ± 1.45*, † | 34.46 ± 2.03*, † |
| EF (%) | 74.15 ± 1.54 | 73.51 ± 1.76 | 64.84 ± 2.68† | 62.39 ± 2.35† |
| EF (%) + dobutamine | 93.00 ± 0.99* | 88.81 ± 1.62* | 74.16 ± 1.77*, † | 70.71 ± 2.65*, † |
LV mass/BW, ratio of LV and body weight; PWd, posterior wall thickness in diastole; PWs, posterior wall thickness in systole; EDV, end diastolic volume; ESV, end systolic volume. Values are expressed as the mean ± SEM from eight mice per group. *Significantly different from the corresponding baseline group (without dobutamine treatment) (P < 0.05). †Significantly different from WT (P < 0.05).
In vivo basal cardiac function
To determine the effects of combined MyBP‐C and TnI phospho‐ablation on cardiac contractile function, we performed echocardiography to examine in vivo cardiac performance. DBLPKA− mice displayed reduced EF and FS compared to WT mice (EF: 62.39 ± 2.35% and FS: 29.17 ± 1.69% for DBLPKA− compared to 74.15 ± 1.54% and 37.14 ± 1.31% for WT; respectively, P < 0.05) (Table 1). DBLPKA− mice also displayed impaired cardiac relaxation as evidenced by significantly longer IVRT (28.80 ± 1.02 ms) compared to WT hearts (19.44 ± 1.14; P < 0.05). The impaired cardiac function in DBLPKA− mice was similar to MyBPCPKA− mice, which also displayed reduced EF and FS with a prolonged IVRT (EF: 64.84 ± 2.68%; FS: 30.12 ± 1.73%; IVRT: 27.31 ± 1.43 ms; P < 0.005). By contrast, the EF and FS in TnIPKA− mice was not different compared to WT controls and these mice also displayed normal LV relaxation (EF: 73.51 ± 1.76%; FS: 37.50 ± 1.34%; IVRT: 18.78 ± 1.42 ms), indicating normal systolic and diastolic cardiac function.
To further investigate the effects of combined MyBP‐C and TnI phospho‐ablation on cardiac function, we assessed LV haemodynamics by analysis of P–V loops generated from invasive LV catheterization. Baseline steady‐state haemodynamics were recorded once a stable heart rate (HR) was achieved under constant isoflurane administration (Table 2). Representative P–V loops from each line are shown in Fig. 3. The maximum systolic pressure (P max), the maximum rate of pressure development (dp/dt max) and the time constant for relaxation (τ) were not significantly different between DBLPKA− and WT mice (88.3 ± 3.3 mmHg P max, 7599 ± 798 mmHg s−1 dp/dt max and 6.6 ± 0.2 ms τ for DBLPKA− compared to 95.3 ± 5.2 mmHg P max, 7134 ± 570 mmHg s−1 dp/dt max and 6.4 ± 0.1 ms τ for WT). Additionally, peak ejection rate (PER) and peak filling rate (PFR) were unaltered by combined MyBP‐C and TnI phospho‐ablation (11.2 ± 1.6 μl s−1 PER and 13.0 ± 0.8 μl s−1 PFR for DBLPKA− compared to 12.8 ± 1.2 μl s−1 PER and 13.0 ± 1.5 μl s−1 PFR for WT). EDP, however, was elevated in DBLPKA− mice compared to WT mice (10.7 ± 2.1 mmHg in DBLPKA− vs. 5.0 ± 0.7 mmHg in WT; P < 0.05) and TnIPKA− mice displayed a similar trend (9.6 ± 1.0 mmHg in TnIPKA− vs. 5.0 ± 0.7 mmHg in WT; P = 0.11), whereas EDP in MyBPCPKA− mice was not different than WT mice (Table 2). These results suggest that TnI dephosphorylation may contribute to increased EDP, thereby resulting in incomplete cardiac relaxation in diastole.
Table 2.
Left ventricular haemodynamic function measured by P–V loop analysis
| Group | P max (mmHg) | EDP (mmHg) | PFR (μl s−1) | PER (μl s−1) | HR (beats min–1) | dp/dtmax (mmHg s−1) | τ (ms) |
|---|---|---|---|---|---|---|---|
| Baseline | |||||||
| WT | 95.3 ± 5.2 | 5.0 ± 0.7 | 13.0 ± 1.5 | 12.8 ± 1.2 | 452 ± 12 | 7,134 ± 570 | 6.4 ± 0.1 |
| TnIPKA− | 102.5 ± 3.4 | 9.6 ± 1.0 | 10.6 ± 0.6 | 8.7 ± 1.0 | 430 ± 13 | 7,082 ± 627 | 6.8 ± 0.3 |
| MyBPCPKA− | 94.0 ± 5.8 | 6.6 ± 1.0 | 11.7 ± 1.0 | 11.9 ± 1.9 | 450 ± 9 | 7,618 ± 585 | 6.8 ± 0.3 |
| DBLPKA− | 88.3 ± 3.3 | 10.7 ± 2.1† | 13.0 ± 0.8 | 11.2 ± 1.6 | 427 ± 7 | 7,599 ± 798 | 6.6 ± 0.2 |
| + Dobutamine | |||||||
| WT | 98.1 ± 4.7 | 4.4 ± 0.6 | 24.9 ± 1.2* | 32.8 ± 1.8* | 541 ± 9* | 13,756 ± 967* | 4.9 ± 0.2* |
| TnIPKA− | 108.4 ± 4.7 | 6.4 ± 0.7* | 21.1 ± 1.2* | 27.9 ± 1.6* | 579 ± 14* | 14,879 ± 947* | 4.7 ± 0.2* |
| MyBPCPKA− | 88.21 ± 5.7 | 4.3 ± 0.5* | 14.9 ± 1.5† | 14.4 ± 1.6† | 529 ± 9* | 8,437 ± 791† | 6.2 ± 0.3† |
| DBLPKA− | 81.6 ± 4.2 | 4.6 ± 0.6* | 18.1 ± 1.5* † | 17.1 ± 1.9* † | 528 ± 15* | 9,130 ± 942† | 6.1 ± 0.3† |
Values are the mean ± SEM from seven to 10 mice per group. *Significantly different from the corresponding baseline group (without dobutamine treatment) (P < 0.05). †Significantly different from WT (P < 0.05).
Figure 3. Representative P–V loops from TG lines .
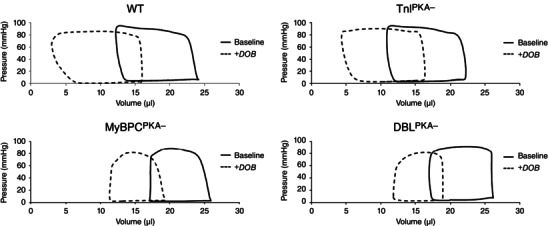
Representative P–V loops from WT, TnIPKA−, MyBPCPKA− and DBLPKA‐ mice. Representative loops were averaged from at least 10 P–V loops before (solid trace) and after (dashed trace) dobutamine administration for each animal.
Phospho‐ablation of MyBP‐C appeared to prolong the early phase of pressure relaxation from peak systolic pressure to dp/dt min (Fig. 4). In DBLPKA− mice, the time from peak pressure to dp/dt min was significantly longer and the slope of the pressure trace from these two points was significantly shallower compared to WT controls (25 ± 1 ms and 1.2 ± 0.1 mmHg ms−1 for DBLPKA− compared to 14 ± 1 ms and 2.4 ± 0.3 mmHg ms−1 for WT mice; P < 0.05). MyBPCPKA− mice also displayed a prolonged early pressure relaxation with a significantly shallower slope of pressure decline (29 ± 2 ms and 1.4 ± 0.1 mmHg ms−1), both of which were unaltered in TnIPKA− mice (14 ± 2 ms and 2.4 ± 0.3 mmHg ms−1). These results suggest that the absence of MyBP‐C phosphorylation contributes to significantly slow the rate of early pressure decline.
Figure 4. MyBP‐C phospho‐ablation slows early pressure decline .
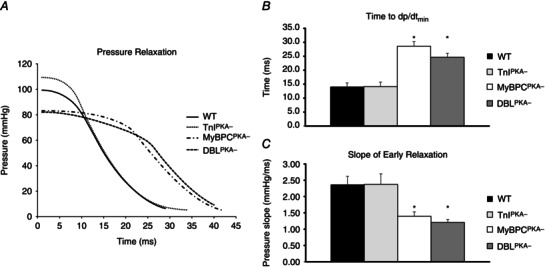
A, representative pressure traces showing peak pressure decline from peak pressure. MyBPCPKA‐ and DBLPKA− mice show a prolonged early pressure decline from peak pressure to dp/dtmin compared to WT and TnIPKA− mice. B, MyBPCPKA− and DBLPKA− mice display a significantly prolonged time from peak pressure to dp/dtmin compared to WT and TnIPKA− mice. C, the slope of the pressure trace from peak pressure to dp/dtmin was significantly shallower in MyBPCPKA− and DBLPKA− mice compared to WT and TnIPKA− mice, suggesting slower pressure relaxation. Values are expressed as the mean ± SEM from seven to 10 mice per line. *Significantly different from WT (P < 0.05).
Adrenergic reserve
To examine how phosphorylation of TnI and MyBP‐C contributes to adrenergic reserve, we administered the β‐agonist dobutamine and measured haemodynamic parameters at the point of peak dobutamine response. The systolic response to dobutamine was severely blunted in DBLPKA− and closely matched the impaired adrenergic response observed in MyBPCPKA− mice (Fig. 5 and Table 2). Early pressure development during the isovolumic period of the cardiac cycle was accelerated by dobutamine in WT mice and could be measured as an increase in dp/dtmax (Fig. 5 B). This acceleration in systolic pressure development was accompanied by enhanced maximal ventricular power generation (Powermax) (Fig. 5 C) and an increased rate of ventricular ejection (PER) (Fig. 5 D). The enhancement in pressure development and systolic function in response to dobutamine was significantly blunted in mice with MyBP‐C phospho‐ablation (DBLPKA− mice had an increase of 1434 ± 591 mmHg s−1 in dp/dtmax, 606 ± 205 mmHg*μl s−1 in Powermax and 6.0 ± 1.6 μl s−1 PER compared to an increase of 6621 ± 817 mmHg s−1 in dp/dtmax, 1823 ± 290 mmHg*μl s−1 in Powermax and 20.0 ± 1.9 μl s−1 PER in WT mice; P < 0.05). Figure 5 A depicts representative traces showing early pressure development before (black traces) and after (grey traces) dobutamine injection and demonstrates the impaired acceleration in pressure development by MyBP‐C phospho‐ablation. MyBPCPKA− mice also demonstrated an impaired systolic dobutamine response that closely matched the impaired response observed in DBLPKA− mice (increase of 819 ± 491 mmHg s−1 in dp/dtmax, 180 ± 65 mmHg*μl s−1 in Powermax and 2.5 ± 1.0 μl s−1 PER; P < 0.05). By contrast, the dobutamine‐mediated enhancement of systolic function was preserved in TnIPKA− mice compared to WT controls (increase of 7796 ± 836 mmHg s−1 in dp/dtmax, 2107 ± 182 mmHg*μl s−1 in Powermax and 19.1 ± 1.4 μl s−1 PER), suggesting the rapid acceleration of systolic pressure development after dobutamine administration is largely dependent on MyBP‐C phosphorylation.
Figure 5. MyBP‐C phospho‐ablation blunts the adrenergic mediated enhancement of systolic function .
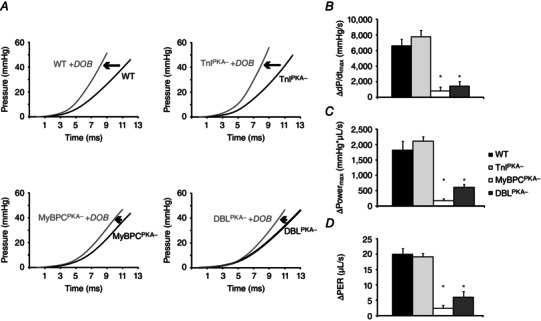
A, early pressure development in WT and TnIPKA− mice is accelerated by β‐adrenergic activation, although this acceleration is blunted in MyBPCPKA‐ and DBLPKA− mice. Black traces represent baseline LV developed pressure, whereas grey traces represent post‐dobutamine pressure development. MyBP‐C phospho‐ablation also blunts the β‐adrenergic mediated enhancement of the maximum rate of pressure development ( dp/dtmax) (B, maximum power development (C) and the peak ejection rate (PER, normalized to end diastolic volume) (D). All values in (B) to (D) are represented as the values at the peak dobutamine response minus baseline values. Values are expressed as the mean ± SEM from seven to 10 mice per line. *Significantly different from WT (P < 0.05).
In addition to impaired acceleration of pressure development after dobutamine administration, MyBP‐C phospho‐ablation significantly blunted the acceleration in relaxation after dobutamine treatment (Fig. 6 and Table 2). In WT mice, pressure relaxation during diastole was accelerated by dobutamine and could be measured as a decrease in τ (Fig. 6 B) and the slope of the pressure trace (from 50% to 25% of peak pressure) (Fig. 6 C). The enhancement of diastolic function also corresponded with an accelerated peak rate of left ventricular filling (PFR) (Fig. 6 D and Table 2). Compared to WT mice, however, DBLPKA‐ mice displayed a significantly blunted enhancement of diastolic relaxation and ventricular filling (a decrease of 0.5 ± 0.3 ms in τ, 130 ± 132 mmHg s−1 in pressure slope and an increase of 5.6 ± 1.1 μl s−1 in PFR compared to a decrease of 1.5 ± 0.1 ms in τ, 1086 ± 71 mmHg s−1 in pressure slope and an increase of 12.0 ± 1.7 μl s−1 in PFR; P < 0.05). The blunted diastolic dobutamine response in DBLPKA− mice closely paralleled the impaired enhancement of relaxation observed in MyBPCPKA− mice (a decrease of 0.6 ± 0.1 ms in τ, 134 ± 106 mmHg s−1 in pressure slope and an increase of 3.2 ± 0.8 μl s−1 in PFR; P < 0.05). By contrast, τ and PFR were similar in TnIPKA− and WT mice after dobutamine (Table 2); however, the absolute change in pressure relaxation was significantly larger in TnIPKA− mice (a decrease of 2.1 ± 0.1 ms in τ and 2341 ± 371 mmHg s−1 in pressure slope; P < 0.05), whereas the increase in PFR was unaltered (10.5 ± 1.5 μl s−1) compared to WT controls (Fig. 6). Our results suggest that the absence of MyBP‐C phosphorylation significantly limits the ability of the heart to accelerate pressure relaxation in response to increased β‐adrenergic stimulation.
Figure 6. MyBP‐C phospho‐ablation blunts the adrenergic mediated enhancement of diastolic relaxation .
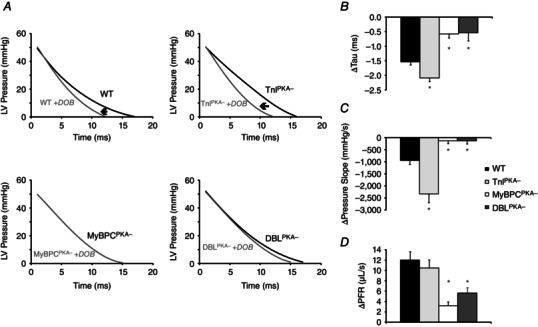
A, ventricular pressure relaxation in WT and TnIPKA− mice is accelerated by β‐adrenergic activation, although this acceleration is blunted in MyBPCPKA‐ and DBLPKA− mice. Black traces represent baseline LV developed pressure during diastolic relaxation, whereas grey traces represent post‐dobutamine pressure relaxation. MyBP‐C phospho‐ablation also blunts the β‐adrenergic mediated acceleration of the relaxation time constant (Tau, B), the decrease in the slope of the pressure trace from 50% to 25% of maximal pressure (C) and the peak filling rate (PFR, normalized to end diastolic volume) (D). All values in (B) to (D) are represented as the values at the peak dobutamine response minus baseline values. Values are expressed as the mean ± SEM from seven to 10 mice per line. *Significantly different from WT (P < 0.05).
To determine how phospho‐ablation impacts the relative durations of the phases of the cardiac cycle, we examined the durations of IVC, ejection, IVR and diastolic filling. At baseline, the duration of each portion of the cardiac cycle was similar across the mouse lines, although relative changes in duration after dobutamine administration were observed in mice with non‐phosphorylatable MyBP‐C (Fig. 7 A). In WT mice, dobutamine infusion shortened the relative durations of IVC, ejection and IVR at the same time as increasing the relative duration of diastolic filling. Mice with MyBP‐C phospho‐ablation, however, displayed a relatively prolonged ejection and IVR phase, a decreased filling time and a relatively unchanged IVC phase. TnIPKA− mice displayed no differences in the relative duration of the phases of the cardiac cycle after dobutamine administration. The data demonstrate that, in WT mice, dobutamine infusion significantly accelerates systolic pressure development and shortens the relative duration of systole compared to the relative duration of diastole (Fig. 7 B); however, in mice with MyBP‐C phospho‐ablation, the opposite effect is observed because an inability to accelerate systolic pressure development results in a relative lengthening of systole compared to the relative duration of diastole (Fig. 7 B). This disruption in the normal timing of the cardiac cycle following dobutamine infusion may underlie the decreased contractile performance and adrenergic reserve in conditions of increased workload in hearts with non‐phosphorylatable MyBP‐C.
Figure 7. MyBP‐C phospho‐ablation alters the timing of the cardiac cycle after β‐adrenergic stimulation .
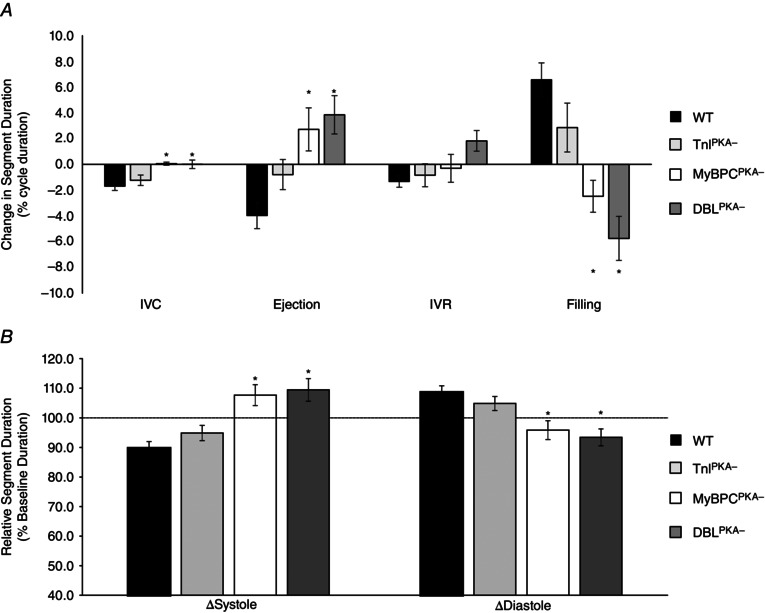
A, in WT and TnIPKA− mice, dobutamine administration shortens the relative duration of IVC, ejection and IVR, and lengthens the relative duration of diastolic filling, whereas MyBPCPKA‐ and DBLPKA− mice displayed an increased relative duration of ejection time, a decreased relative duration of filling time and an unchanged IVC duration after dobutamine. The relative duration of IVR was increased by dobutamine in DBLPKA− mice but unchanged in MyBPCPKA− mice. All durations were normalized to the total length of the cardiac cycle and are presented as the percentage change from baseline values. B, dobutamine administration significantly shortened the relative duration of systole and prolonged the relative duration of diastole in WT and TnIPKA− mice compared to baseline, whereas systole was relatively prolonged and diastole relatively shortened in MyBPCPKA‐ and DBLPKA− mice after dobutamine. All durations were normalized to the total length of the cardiac cycle and all values are presented as a percentage of baseline. *Significantly different from WT values (P < 0.05).
Assessment of in vivo systemic blood pressure in unanesthesized mice
To determine the effects of MyBP‐C and TnI phospho‐ablation on haemodynamic function in conscious, unanesthetized mice, we employed implantable telemetry transmitters to measure systolic and diastolic blood pressure before and after dobutamine administration. At baseline, there were no differences in either systolic or diastolic blood pressure between any of the groups (Fig. 8 and Table 3). In WT mice, dobutamine administration increased HR and significantly reduced systolic and diastolic blood pressure (Table 3). In DBLPKA− mice, dobutamine administration produced a significant increase in HR compared to baseline and also reduced systolic and diastolic blood pressure compared to baseline; however, systolic blood pressure was significantly lower in DBLPKA− mice after dobutamine infusion compared to WT mice (81.9 ± 3.6 mmHg for DBLPKA− mice compared to 99.6 ± 3.3 mmHg for WT; P < 0.05). Additionally, although there were no differences in the time to peak systolic pressure at baseline between DBLPKA− and WT mice (28 ± 4 ms in DBLPKA− compared to 27 ± 2 ms in WT), peak pressure was reached significantly later in DBLPKA− mice after dobutamine compared to WT mice (24 ± 1 ms in DBLPKA− compared to 19 ± 1 ms in WT; P < 0.05) (Fig. 8 and Table 3). Systolic blood pressure was also significantly lower in MyBPCPKA‐ mice after dobutamine administration (78.1 ± 4.1 mmHg; P < 0.05) and peak pressure took significantly longer to develop after dobutamine infusion (26 ± 2 ms; P < 0.05) compared to WT controls. Diastolic blood pressure was significantly reduced in both DBLPKA− and MyBPCPKA− after dobutamine to a similar extent as that in WT mice (67.3 ± 2.6 mmHg for DBLPKA−, 63.1 ± 6.6 for MyBPCPKA− and 74.1 ± 3.6 for WT). Both systolic blood pressure and diastolic blood pressure in TnIPKA− mice were similar to WT mice both at baseline and after dobutamine infusion (Fig. 8 and Table 3). Our data suggest that MyBP‐C phospho‐ablation results in delayed achievement of peak systolic pressure and an inability to maintain peak systolic pressure following dobutamine infusion, thereby contributing to reduced contractility and cardiac output in conditions of increased workload.
Figure 8. MyBP‐C phospho‐ablation disrupts in vivo systolic pressure development after β‐adrenergic stimulation .
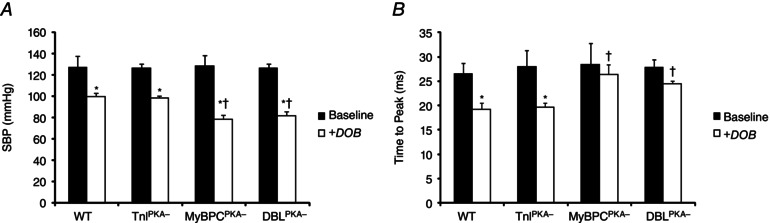
A, β‐adrenergic stimulation by dobutamine administration decreases maximum systolic pressure development to accommodate an increased HR (see WT and TnIPKA−). MyBP‐C phospho‐ablation, however, results in significantly reduced systolic blood pressure after β‐adrenergic stimulation because of an inability to fully accelerate pressure development in the absence of MyBP‐C phosphorylation. SBP, systolic blood pressure. B, time to peak systolic blood pressure was significantly decreased by dobutamine administration in WT and TnIPKA− mice but was not significantly shortened from baseline in MyBPCPKA‐ and DBLPKA− mice and was significantly longer (after dobutamine) compared to WT. *Significantly different from the corresponding baseline group (without dobutamine treatment) (P < 0.05). †Significantly different from WT under the same treatment (i.e. post‐dobutamine) (P < 0.05).
Table 3.
In vivo assessment of blood pressure using implantable telemetry
| Group | Systolic pressure (mmHg) | Diastolic pressure (mmHg) | Time to peak (ms) | HR (beats min–1) |
|---|---|---|---|---|
| Baseline | ||||
| WT | 127.0 ± 11.0 | 110.6 ± 12.3 | 27 ± 2 | 675 ± 19 |
| TnIPKA− | 126.6 ± 3.8 | 96.0 ± 5.7 | 28 ± 3 | 658 ± 15 |
| MyBPCPKA− | 128.7 ± 9.7 | 106.7 ± 11.5 | 28 ± 4 | 624 ± 42 |
| DBLPKA− | 126.9 ± 6.8 | 106.1 ± 6.1 | 28 ± 2 | 604 ± 27 |
| + Dobutamine | ||||
| WT | 99.6 ± 3.3* | 74.1 ± 3.6* | 19 ± 1* | 757 ± 8* |
| TnIPKA− | 98.4 ± 2.0* | 71.9 ± 3.6* | 20 ± 1* | 823 ± 34* |
| MyBPCPKA− | 78.1 ± 4.1*, † | 63.1 ± 6.6* | 26 ± 2† | 690 ± 38* |
| DBLPKA− | 81.9 ± 3.6*, † | 67.3 ± 2.6* | 24 ± 1† | 737 ± 22* |
Values are expressed as the mean ± SEM from four or five mice per group. *Significantly different from the corresponding baseline group (without dobutamine treatment) (P < 0.05). †Significantly different from WT (P < 0.05).
Discussion
The β‐adrenergic signalling pathway is a critical regulator of cardiac output, in part through the phosphorylation of two key sarcomeric proteins: TnI and MyBP‐C. However, the relative contributions of TnI and MyBP‐C phosphorylation to the β‐adrenergic mediated acceleration of pressure development and relaxation remains an area of intense investigation. The present study aimed to determine the respective roles of TnI and MyBP‐C phosphorylation in contributing to in vivo haemodynamic function both at baseline and after increased β‐adrenergic stimulation. To address these questions, we generated a novel TG mouse line expressing non‐phosphorylatable TnI and MyBP‐C to examine in vivo cardiac contractile function at baseline and after dobutamine administration. Abolishing MyBP‐C phosphorylation produced a hypertrophic phenotype characterized by increased wall thickness and enlarged cardiac mass, in agreement with previous studies (Sadayappan et al. 2005; Tong et al. 2008; Colson et al. 2012). Baseline systolic and diastolic function was impaired in mice expressing phospho‐ablated MyBP‐C when studied by echocardiography and the time course of pressure relaxation was prolonged when studied by P–V loop analysis. Mice expressing non‐phosphorylatable MyBP‐C also displayed a blunted acceleration of pressure development during systole and an impaired enhancement of pressure relaxation during diastole in response to β‐adrenergic stimulation. By contrast, mice expressing only non‐phosphorylatable TnI displayed no detectable pathological phenotype and displayed relatively normal basal cardiac haemodynamic function and enhancement of cardiac function in response to dobutamine. Taken together our data support a critical role for MyBP‐C phosphorylation in mediating the cardiac haemodynamic response to increased β‐adrenergic stress.
Effects of MyBP‐C and TnI phospho‐ablation on systolic function
The rate of pressure development during early systole depends on the availability of myosin binding sites on the thin filament and the rate of XB attachment and cycling (Hinken & Solaro, 2007; Stehle & Iorga, 2010). Ca2+ binding to troponin C (TnC) and activation of the thin filament allows initial binding of XBs to actin, and the transition of XBs to strongly‐bound force‐generating states activates neighbouring thin filament regulatory units, thereby enabling further XB recruitment (Moss & Fitzsimons, 2010), suggesting that the rate at which force develops in the myocardium is dependent on the rate at which XBs are co‐operatively recruited. At submaximal Ca2+ concentrations when relatively few regulatory units are directly activated by Ca2+ binding to TnC, the time course of force development is highly dependent on co‐operative XB recruitment and their transitions from weakly‐bound to the strongly bound force generating states, which slows the overall rate of rate of force development (Campbell, 1997). MyBP‐C phosphorylation speeds the recruitment of XBs and the spread of thin filament activation, and thereby serves to accelerate the rate of force development at the myofilament level and the rate of pressure development in vivo. By contrast, MyBP‐C dephosphorylation impedes co‐operative XB recruitment and slows the spread of thin filament activation to limit the acceleration in the rate of force generation and pressure development in vivo.
Changes in the contractile state of the ventricle can be measured in vivo as the maximum rate of pressure development (dp/dtmax) and the time required to reach maximum pressure development (time to dp/dtmax) (Adler et al. 1996 b, 1996 a); an enhancement in the rate of thin filament activation and recruitment of strongly‐bound force‐generating XBs would be reflected as an acceleration in dp/dtmax and a decrease in the amount of time required to reach dp/dtmax. In the present study, MyBP‐C phospho‐ablated MyBPCPKA− and DBLPKA− mice displayed a blunted acceleration in pressure development after dobutamine administration as indicated by a reduced dp/dtmax (Fig. 5 B and Table 2) and a blunted shortening in the time required to reach maximum pressure development (i.e. a longer time to dp/dtmax after dobutamine; data not shown) compared to WT mice. These results are in agreement with a previous study reporting that MyBP‐C phosphorylation is important for the acceleration of pressure development during IVC (Nagayama et al. 2007). Previous studies have demonstrated that MyBP‐C phospho‐ablation does not result in alterations in Ca2+ transient properties (Rosas et al. 2015; Tong et al. 2015); therefore, the inability to accelerate pressure development following dobutamine infusion in mice with phospho‐ablated MyBP‐C in the present study is probably a result of impaired co‐operative XB recruitment and activation of the thin filament.
The rapid rise in ventricular pressure during IVC continues until ventricular pressure exceeds aortic pressure, at which point the aortic valve opens as blood is ejected into the periphery; during this period of ejection, ventricular pressure continues to develop but at a much slower rate than during IVC. The maximum ventricular pressure achieved during the cardiac cycle is most probably controlled by a number of interdependent factors, including the level of thin filament activation, the extent and rate of sarcomere shortening, and the level of preload and afterload (Hinken & Solaro, 2007). However, peak pressure is ultimately proportional to the number of force‐generating XBs attached to the thin filament (Hinken & Solaro, 2007), and an increase in the number of force‐generating XBs would be expected to increase the amount of pressure developed during the cardiac cycle, whereas a reduction in force‐generating XBs would be expected to have the opposite effect. In the present study, we observed a decrease in peak maximal pressure in unanesthetized MyBPCPKA− and DBLPKA− mice in response to dobutamine administration compared to WT mice (Fig. 8). This impaired development of peak pressure following acute stress parallels an impaired enhancement of force development observed in isolated intact papillary muscles from MyBP‐C phospho‐ablated mice in response to increased pacing frequency or acute dobutamine administration (Tong et al. 2015), suggesting that MyBPC dephosphorylation reduces the number of force generating XBs recruited to the thin filament at high workloads and thereby contributes to diminished enhancement of maximum power generation and blood ejection from the ventricle (Fig. 5 C and D) during the cardiac cycle. A reduction in cardiac reserve as a result of MyBP‐C dephosphorylation would severely limit the ability to enhance cardiac output in conditions of increased circulatory demands such as in exercise, and could potentially limit the ability to adapt to chronic pathological stress such as hypertension.
Effects of MyBP‐C and TnI phospho‐ablation on diastolic function
Ventricular relaxation is initiated by the removal of Ca2+ from the cytosol and reuptake into the sarcoplasmic reticulum or extrusion across the sarcoplasmic membrane; however, the intracellular Ca2+ transient decline precedes the decline in twitch force, suggesting that other processes regulate the rate of ventricular pressure relaxation (Hinken & Solaro, 2007; Biesiadecki et al. 2014). The decline in force mediating myocardial relaxation is most probably controlled by two interdependent processes: the rate of thin filament deactivation and the rate of XB detachment from the thin filament. Ca2+ dissociation from TnC promotes a rearrangement of the troponin complex, shifting tropomyosin to a position that blocks the interaction between myosin heads and the actin filament; however, strongly‐bound XBs can maintain the level of thin filament activation (Metzger, 1995; Davis et al. 2007) and slowed XB detachment from the thin filament slows the rate of thin filament deactivation. The combined effects of Ca2+ dissociation from TnC and the declining co‐operative activation of the thin filament reduces the availability of actin binding sites for the myosin XBs and, as the number of force producing XBs decreases, the ventricular pressure declines towards diastolic pressure levels. Ventricular relaxation, then, is enhanced by the combined acceleration of Ca2+ removal from the cytosol, thin filament deactivation and XB detachment rates after β‐adrenergic stimulation (Biesiadecki et al. 2014; Hanft et al. 2008). Extensive in vitro studies have demonstrated that PKA mediated phosphorylation of TnI and MyBP‐C decreases TnC‐Ca2+ affinity and enhances XB kinetics to speed relaxation (Sakthivel et al. 2005; Stelzer et al. 2006, 2007; Bilchick et al. 2007; Tong et al. 2008); however, the relative roles of PKA‐mediated TnI and MyBP‐C phosphorylation in accelerating in vivo relaxation following β‐adrenergic stimulation are unclear.
At baseline, MyBP‐C dephosphorylation negatively impacted early relaxation by prolonging the time required to reach the maximum rate of relaxation (i.e. time to dp/dtmin) and reducing the slope of the pressure trace during early relaxation (i.e. a reduced rate of pressure decline) (Fig. 4). During this part of early relaxation, intracellular Ca2+ levels have already declined from their peak as Ca2+ is rapidly being sequestered into the SR and ventricular pressure is maintained by the level of thin filament activation and the number of strongly‐bound XBs in force‐generating states (Hinken & Solaro, 2007; Solaro, 2010). MyBP‐C dephosphorylation may act to prolong this early phase of ventricular pressure relaxation by slowing the rate of XB detachment and maintaining the population of strongly‐bound XBs attached to the thin filament, which would slow thin filament deactivation. MyBP‐C phospho‐ablation also prevents the rapid acceleration of XB detachment mediated by PKA (Tong et al. 2008), which would be expected to blunt the acceleration in ventricular pressure relaxation in response to increased β‐adrenergic stimulation in vivo. In the present study, infusion of dobutamine in mice lacking PKA‐phosphorylatable MyBP‐C residues resulted in a severely blunted acceleration of pressure relaxation when compared to mice expressing phosphorylatable MyBP‐C (Fig. 6 and Table 2), suggesting that MyBP‐C phosphorylation is necessary for full acceleration of ventricular relaxation, most probably because MyBP‐C phosphorylation results in an acceleration of XB detachment, which allows for faster thin filament deactivation, and thereby pressure relaxation (Stelzer et al. 2006; Rosas et al. 2015).
Preventing MyBP‐C phosphorylation in response to β‐adrenergic signalling could also limit the acceleration in relaxation by slowing the rate of thin filament deactivation as a result of reduced sarcomere shortening. After the ventricular pressure generated by the myocardium exceeds aortic pressure, the aortic valve opens and the ventricle loses volume as blood is ejected into the periphery. During this ejection phase, as the ventricular muscle transitions from isometric to concentric contraction, the tension generated by XBs actively shortens the sarcomeres and promotes thin filament deactivation (Hanft et al. 2008). MyBP‐C phosphorylation releases the restraint on the myosin head (Gruen et al. 1999) and increases the rate of transition through the XB cycle (Stelzer et al. 2007; Tong et al. 2008), contributing to the acceleration in sarcomere shortening after PKA‐mediated phosphorylation (McDonald et al. 2012). By preventing MyBP‐C phosphorylation and limiting the acceleration in sarcomere shortening and the acceleration in thin filament deactivation, the subsequent enhancement of ventricular relaxation would be blunted and diastolic relaxation prolonged, as observed in the present study.
Physiological significance of MyBP‐C dephosphorylation
Normal ventricular function requires that cardiac output and venous return be coupled to systemic demands (Hanft et al. 2008) to maintain adequate delivery of blood to the periphery. During acute stress, enhanced β‐adrenergic signalling activates PKA, which increases phosphorylation of myocardial proteins (Fu et al. 2013), resulting in a decrease in the duration of the cardiac cycle at the same time as increasing the rate of ventricular blood ejection and filling. At baseline, WT mice displayed a close match between the rates of ventricular ejection and ventricular filling (1.04 ± 0.10 ratio between PER and PFR) (Table 2) and the relative durations of the ejection and filling periods (1.03 ± 0.04 ratio between ejection and filling duration; data not shown). Infusion of dobutamine increased the rate of ventricular filling and ejection, although the maximum rate of ejection was enhanced to a great extent relative to filling (1.33 ± 0.09 ratio between PER and PFR) at the same time as shortening the duration of ejection relative to filling (0.85 ± 0.06 ratio between ejection and filling duration; data not shown). These data suggest that, in the present study, β‐adrenergic stimulation enhances systolic function to a greater extent than diastolic function and that the systolic portion of the cardiac cycle is relatively shortened (Fig. 7) to maintain adequate ventricular filling. The enhancement in the rate of ventricular ejection and the shortening of the duration of ejection in relation to filling after dobutamine administration are disrupted by MyBP‐C phospho‐ablation, creating a mismatch between ejection and filling. In MyBPCPKA− and DBLPKA− mice, the ratio between peak ejection rate and peak filling rate, which was maintained at baseline, was unaltered by dobutamine administration (1.01 ± 0.10 in MyBPCPKA− mice; 0.95 ± 0.07 in DBLPKA− DBLPKA− mice after dobutamine) (Table 2), whereas the duration of ejection was relatively prolonged compared to filling (data not shown), lengthening the relative duration of systole (Fig. 7). This apparent mismatch between the enhancement of ejection and filling and the disruption of the duration of the different segments of the cardiac cycle (Fig. 7) when MyBP‐C phosphorylation is prevented, may underlie the reduction in cardiac reserve and disrupt the enhancement in cardiac output in response to acute stress.
Limitations
Although the results of the present study strongly support the role of MyBP‐C Ser273, Ser282 and Ser302 phosphorylation as a critical regulator of adrenergic reserve, several potentially confounding factors should be considered. Although we have examined phosphorylation of several cardiac proteins before and after dobutamine or PKA treatment to eliminate the potential influence of protein phosphorylation (outside of MyBP‐C and TnI) in the present study, it is possible that post‐translation modification of proteins not examined here could impact the results obtained. For example, titin isoform expression and phosphorylation can directly impact myocardial stiffness, which could impact ventricular relaxation (Fukuda et al. 2005), although this was not examined in the present study. Additionally, ventricular hypertrophy is often accompanied by impaired ventricular relaxation and diastolic filling (Lorell & Carabello, 2000), which could impact our interpretation of the data following dobutamine administration in MyBPCPKA− and DBLPKA− mice. However, in a previous study, we demonstrated that mice with partial phospho‐ablation of MyBP‐C (Ser282 was mutated to Ala but Ser273 and Ser302 were not affected) displayed an impaired adrenergic reserve in the absence of significant hypertrophy (Gresham et al. 2014), suggesting that there is strong link between MyBP‐C phosphorylation and adrenergic reserve, and that complete MyBP‐C phospho‐ablation of all three PKA sites is most probably the main cause for the observed impaired adrenergic reserve in MyBPCPKA− and DBLPKA− mice in the present study. Additionally, because diastolic function is impaired by increased stiffness as a result of increased cardiac fibrosis, and because MyBPCPKA− and DBLPKA− mice did not display an increase in ventricular fibrosis, the impaired acceleration of pressure relaxation in MyBPCPKA− and DBLPKA− mice is most probably a result of MyBP‐C phospho‐ablation.
To adapt to the impaired ventricular adrenergic reserve observed with MyBP‐C phospho‐ablation, it is possible that TG mice may modulate arterial tone to maintain adequate systemic blood delivery. A decrease in vascular resistance in MyBPCPKA− and DBLPKA− mice could accommodate increased blood flow even at lower ventricular developed pressures, which could potentially increase systemic blood perfusion after β‐adrenergic stimulation despite impaired ventricular pressure development. However, a decrease in afterload because of a drop in vascular resistance would be expected to accelerate the time course of ventricular relaxation, which is the opposite of what we observed in mice with TG MyBP‐C phospho‐ablation. Therefore, the data reported in the present study support a role for MyBP‐C phosphorylation as a critical mediator of β‐adrenergic mediated acceleration in pressure development and relaxation in vivo.
Conclusions
Our data demonstrate a central role for MyBP‐C phosphorylation in the regulation of cardiac contractile and haemodynamic response to increased β‐adrenergic stimulation in vivo. Here, we show that a mouse model expressing PKA non‐phosphorylatable residues in MyBP‐C and TnI displayed impairments in the ability to accelerate pressure generation and relaxation following dobutamine infusion in vivo similar to mice expressing only PKA non‐phosphorylatable MyBP‐C. Phospho‐ablation of MyBP‐C resulted in a significant decrease in the rate of rise of systolic pressure generation and also the magnitude of systolic peak pressure following acute β‐agonist infusion. Furthermore, MyBP‐C phospho‐ablation significantly disrupted diastolic pressure relaxation and reduced diastolic filling following β‐agonist infusion, demonstrating that MyBP‐C phosphorylation is also an important regulator of in vivo cardiac diastolic function. The inability to phosphorylate MyBP‐C with increased cardiac workload reduces cardiac reserve and creates a mismatch between systolic ejection and diastolic filling that results in decreased cardiac output. Our results suggest that reduced MyBP‐C phosphorylation may be a significant contributor to depressed contractile function in heart failure.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contribution
All experiments were performed at the Department of Physiology and Biophysics, Case Western Reserve University, Cleveland, Ohio, USA. KSG and JES participated in performing the experiments and data collection, the conception and design of the experiments, the analysis and interpretation of data, and in writing and revising the article. Both authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This work was supported by the National Heart, Lung, and Blood Institute Grant (HL‐114770).
Acknowledgements
This research was supported by the Tissue Resources Core Facility of the Case Comprehensive Cancer Centre (P30CA043703). We would like to thank Scott Howell, PhD, Department of Ophthalmology and The Visual Sciences Research Centre at Case Western Reserve University, for assistance with histology and image acquisition; Xiaoqin Chen, MD, Department of Physiology and Biophysics at Case Western Reserve University for assistance with echocardiography; and Tracy McElfresh, BS, Department of Physiology and Biophysics at Case Western Reserve University for assistance with P–V catheterization.
References
- Adler D, Monrad ES, Hess OM, Krayenbuehl HP & Sonnenblick EH (1996. a). Time to dP/dt(max), a useful index for evaluation of contractility in the catheterization laboratory. Clin Cardiol 19, 397–403. [DOI] [PubMed] [Google Scholar]
- Adler D, Nikolic SD, Sonnenblick EH & Yellin EL (1996. b). Time to dP/dtmax, a preload‐independent index of contractility: open‐chest dog study. Basic Res Cardiol 91, 94–100. [DOI] [PubMed] [Google Scholar]
- Bers DM (2008). Calcium cycling and signaling in cardiac myocytes. Annu Rev Physiol 70, 23–49. [DOI] [PubMed] [Google Scholar]
- Biesiadecki BJ, Davis JP, Ziolo MT & Janssen PML (2014). Tri‐modal regulation of cardiac muscle relaxation; intracellular calcium decline, thin filament deactivation, and cross‐bridge cycling kinetics. Biophys Rev 6, 273–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilchick KC, Duncan JG, Ravi R, Takimoto E, Champion HC, Gao WD, Stull LB, Kass DA & Murphy AM (2007). Heart failure‐associated alterations in troponin I phosphorylation impair ventricular relaxation‐afterload and force‐frequency responses and systolic function. Am J Physiol Heart Circ Physiol 292, H318–H325. [DOI] [PubMed] [Google Scholar]
- Campbell K (1997). Rate constant of muscle force redevelopment reflects cooperative activation as well as cross‐bridge kinetics. Biophys J 72, 254–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wan X, McElfresh TA, Chen X, Gresham KS, Rosenbaum DS, Chandler MP & Stelzer JE (2013). Impaired contractile function due to decreased cardiac myosin binding protein C content in the sarcomere. Am J Physiol Heart Circ Physiol 305, H52–H65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colson BA, Patel JR, Chen PP, Bekyarova T, Abdalla MI, Tong CW, Fitzsimons DP, Irving TC & Moss RL (2012). Myosin binding protein‐C phosphorylation is the principal mediator of protein kinase A effects on thick filament structure in myocardium. J Mol Cell Cardiol 53, 609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis JP, Norman C, Kobayashi T, Solaro RJ, Swartz DR & Tikunova SB (2007). Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys J 92, 3195–3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker RS, Decker ML, Kulikovskaya I, Nakamura S, Lee DC, Harris K, Klocke FJ & Winegrad S (2005). Myosin‐binding protein C phosphorylation, myofibril structure, and contractile function during low‐flow ischemia. Circulation 111, 906–912. [DOI] [PubMed] [Google Scholar]
- Decker RS, Nakamura S, Decker ML, Sausamuta M, Sinno S, Harris K, Klocke FJ, Kulikovskaya I & Winegrad S (2012). The dynamic role of cardiac myosin binding protein‐C during ischemia. J Mol Cell Cardiol 52, 1145–1154. [DOI] [PubMed] [Google Scholar]
- El‐Armouche A, Pohlmann L, Schlossarek S, Starbatty J, Y‐H Yeh, Nattel S, Dobrev D, Eschenhagen T & Carrier L (2007). Decreased phosphorylation levels of cardiac myosin‐binding protein‐C in human and experimental heart failure. J Mol Cell Cardiol 43, 223–229. [DOI] [PubMed] [Google Scholar]
- Fearnley CJ, Roderick HL & Bootman MD (2011). Calcium signaling in cardiac myocytes. Cold Spring Harb Perspect Biol 3, a004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL & Leiden JM (1999). Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol 517, 143–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda N, Wu Y, Nair P & Granzier HL (2005). Phosphorylation of titin modulates passive stiffness of cardiac muscle in a titin isoform‐dependent manner. J Gen Physiol 125, 257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Q, Chen X & Xiang YK (2013). Compartmentalization of β‐adrenergic signals in cardiomyocytes. Trends Cardiovasc Med 23, 250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham KS, Mamidi R & Stelzer JE (2014). The contribution of cardiac myosin binding protein‐c Ser282 phosphorylation to the rate of force generation and in vivo cardiac contractility. J Physiol 592, 3747–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruen M, Prinz H & Gautel M (1999). cAPK‐phosphorylation controls the interaction of the regulatory domain of cardiac myosin binding protein C with myosin‐S2 in an on‐off fashion. FEBS Lett 453, 254–259. [DOI] [PubMed] [Google Scholar]
- Hamdani N, de Waard M, Messer AE, Boontje NM, Kooij V, van Dijk S, Versteilen A, Lamberts R, Merkus D, Dos Remedios C, Duncker DJ, Borbely A, Papp Z, Paulus W, Stienen GJM, Marston SB & van der Velden J (2008). Myofilament dysfunction in cardiac disease from mice to men. J Muscle Res Cell Motil 29, 189–201. [DOI] [PubMed] [Google Scholar]
- Hanft LM, Korte FS & McDonald KS (2008). Cardiac function and modulation of sarcomeric function by length. Cardiovasc Res 77, 627–636. [DOI] [PubMed] [Google Scholar]
- Han YS, Arroyo J & Ogut O (2013). Human heart failure is accompanied by altered protein kinase A subunit expression and post‐translational state. Arch Biochem Biophys 538, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herron TJ, Korte FS & McDonald KS (2001). Power output is increased after phosphorylation of myofibrillar proteins in rat skinned cardiac myocytes. Circ Res 89, 1184–1190. [DOI] [PubMed] [Google Scholar]
- Hinken AC & Solaro RJ (2007). A dominant role of cardiac molecular motors in the intrinsic regulation of ventricular ejection and relaxation. Physiol Bethesda Md 22, 73–80. [DOI] [PubMed] [Google Scholar]
- Jacques AM, Copeland O, Messer AE, Gallon CE, King K, McKenna WJ, Tsang VT & Marston SB (2008). Myosin binding protein C phosphorylation in normal, hypertrophic and failing human heart muscle. J Mol Cell Cardiol 45, 209–216. [DOI] [PubMed] [Google Scholar]
- Kennedy DJ, Vetteth S, Periyasamy SM, Kanj M, Fedorova L, Khouri S, Kahaleh MB, Xie Z, Malhotra D, Kolodkin NI, Lakatta EG, Fedorova OV, Bagrov AY & Shapiro JI (2006). Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 47, 488–495. [DOI] [PubMed] [Google Scholar]
- Kooij V, Saes M, Jaquet K, Zaremba R, Foster DB, Murphy AM, Dos Remedios C, van der Velden J & Stienen GJM (2010). Effect of troponin I Ser23/24 phosphorylation on Ca2+‐sensitivity in human myocardium depends on the phosphorylation background. J Mol Cell Cardiol 48, 954–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ & Shah AM (2004). Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol 556, 835–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorell BH & Carabello BA (2000). Left ventricular hypertrophy pathogenesis, detection, and prognosis. Circulation 102, 470–479. [DOI] [PubMed] [Google Scholar]
- Mamidi R, Gresham KS & Stelzer JE (2014). Length‐dependent changes in contractile dynamics are blunted due to cardiac myosin binding protein‐C ablation. Front Physiol 5, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara H, Takaki M, Yasuhara S, Araki J & Suga H (1995). Logistic time constant of isovolumic relaxation pressure – time curve in the canine left ventricle better alternative to exponential time constant. Circulation 92, 2318–2326. [DOI] [PubMed] [Google Scholar]
- McDonald KS, Hanft LM, Domeier TL & Emter CA (2012). Length and PKA dependence of force generation and loaded shortening in porcine cardiac myocytes. Biochem Res Int 2012, 371415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer AE, Jacques AM & Marston SB (2007). Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end‐stage heart failure. J Mol Cell Cardiol 42, 247–259. [DOI] [PubMed] [Google Scholar]
- Metzger JM (1995). Myosin binding‐induced cooperative activation of the thin filament in cardiac myocytes and skeletal muscle fibers. Biophys J 68, 1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss RL & Fitzsimons DP (2010). Regulation of contraction in mammalian striated muscles – the plot thickens. J Gen Physiol 136, 21–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagayama T, Takimoto E, Sadayappan S, Mudd JO, Seidman JG, Robbins J & Kass DA (2007). Control of in vivo left ventricular contraction/relaxation kinetics by myosin binding protein C: protein kinase A phosphorylation dependent and independent regulation. Circulation 116, 2399–2408. [DOI] [PubMed] [Google Scholar]
- Pi Y, Zhang D, Kemnitz KR, Wang H & Walker JW (2003). Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J Physiol 552, 845–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Post SR, Hammond HK & Insel PA (1999). β‐Adrenergic receptors and receptor signaling in heart failure. Annu Rev Pharmacol Toxicol 39, 343–360. [DOI] [PubMed] [Google Scholar]
- Rosas PC, Liu Y, Abdalla MI, Thomas CM, Kidwell DT, Dusio GF, Mukhopadhyay D, Kumar R, Baker KM, Mitchell BM, Powers PA, Fitzsimons DP, Patel BG, Warren CM, Solaro RJ, Moss RL & Tong CW (2015). Phosphorylation of cardiac myosin binding protein‐C is a critical mediator of diastolic function. Circ Heart Fail 8, 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW, Klevitsky R, Seidman CE, Seidman JG & Robbins J (2005). Cardiac myosin‐binding protein‐C phosphorylation and cardiac function. Circ Res 97, 1156–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakthivel S, Finley NL, Rosevear PR, Lorenz JN, Gulick J, Kim S, VanBuren P, Martin LA & Robbins J (2005). In vivo and in vitro analysis of cardiac troponin I phosphorylation. J Biol Chem 280, 703–714. [DOI] [PubMed] [Google Scholar]
- Solaro RJ (2010). Sarcomere control mechanisms and the dynamics of the cardiac cycle. J Biomed Biotechnol 2010, 105648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehle R & Iorga B (2010). Kinetics of cardiac sarcomeric processes and rate‐limiting steps in contraction and relaxation. J Mol Cell Cardiol 48, 843–850. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR & Moss RL (2006). Protein kinase A‐mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP‐C. Circ Res 99, 884–890. [DOI] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Walker JW & Moss RL (2007). Differential roles of cardiac myosin‐binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res 101, 503–511. [DOI] [PubMed] [Google Scholar]
- Takimoto E, Soergel DG, Janssen PML, Stull LB, Kass DA & Murphy AM (2004). Frequency‐ and afterload‐dependent cardiac modulation in vivo by troponin i with constitutively active protein kinase A phosphorylation sites. Circ Res 94, 496–504. [DOI] [PubMed] [Google Scholar]
- Tong CW, Stelzer JE, Greaser ML, Powers PA & Moss RL (2008). Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res 103, 974–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong CW, Wu X, Liu Y, Rosas PC, Sadayappan S, Hudmon A, Muthuchamy M, Powers PA, Valdivia HH & Moss RL (2015). Phosphoregulation of cardiac inotropy via myosin binding protein‐C during increased pacing frequency or β1‐adrenergic stimulation. Circ Heart Fail 8, 595–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Velden J (2011). Diastolic myofilament dysfunction in the failing human heart. Pflügers Arch 462, 155–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K & Lucchesi PA (2014). Myofilament dysfunction as an emerging mechanism of volume overload heart failure. Pflügers Arch Eur J Physiol 466, 1065–1077. [DOI] [PubMed] [Google Scholar]
- Zakhary DR, Moravec CS, Stewart RW & Bond M (1999). Protein kinase A (PKA)‐dependent troponin‐I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation 99, 505–510. [DOI] [PubMed] [Google Scholar]