

Update to: European Journal of Human Genetics (2012) 20; doi:10.1038/ejhg.2011.227; published online 14 December 2011
1. Disease characteristics
1.1 Name of the disease (synonyms)
Acrodermatitis enteropathica (AEZ).
1.2 OMIM# of the disease
201100.
1.3 Name of the analyzed genes or DNA/chromosome segments
SLC39A4.1
1.4 OMIM# of the gene(s)
607059.
1.5 Variant spectrum
AEZ is a rare autosomal recessive form of zinc deficiency. Individuals with a genuine AEZ have either homozygous or compound heterozygous variants of SLC39A4. Thirty-four variants clearly affecting SLC39A4 function have been reported so far: 16 missense, four nonsense, three splice-site, and 11 frameshifting indels.2, 3, 4 Seven additional variants of unknown significance were also reported,3 among which three can be considered likely deleterious—two missense and a splice-site variant. No hotspot-variant region is observed, as alterations are evenly distributed all along the 12 exons. Except for a few cases where a founder effect is suspected, most of the variants affecting SLC39A4 function are private. All published variants that affect SLC39A4 function and variants of unknown significance will be available soon in the public LOVD database under construction that is dedicated to SLC39A4 (www.LOVD.nl/SLC39A4).
1.6 Analytical methods
Single-nucleotide variants and indels are detected by bidirectional sequencing of the 12 exons of SLC39A4 and their flanking intronic sequences, using either Sanger sequencing of PCR products or high-throughput sequencing (HTS) targeting the regions of interest with a recommended minimal read depth of 30 × and preferably of at least 100 ×. Whole exome5 or even whole-genome sequencing may also be used for variant detection, provided the sequence quality meets diagnostic requirements. Larger deletions or duplications involving at least one exon can be found by analysis of HTS data or they can be screened by a quantitative method such as quantitative multiplex PCR of short fluorescent fragments or multiplex ligation-dependent probe amplification.
1.7 Analytical validation
The first level of validation is the sequencing of both strands. Any suspected new variant present on both strands is then submitted to an internal validation through analysis of variants found in previous patients; it is also compared with public variant databases (eg, dbSNP (http://www.ncbi.nlm.nih.gov/SNP/), Exome Variant Server (http://evs.gs.washington.edu/niehsExome/), 1000 genomes (http://www.1000genomes.org/), ExAC Browser (http://exac.broadinstitute.org/), or LOVD (www.LOVD.nl/SLC39A4)) and its potential deleterious effect on SLC39A4 function is searched through prediction by bioinformatic tools. Single-nucleotide variants or copy-number variations identified through HTS have to be confirmed by Sanger sequencing or by a quantitative method, respectively. Whenever possible, segregation between the variant and the phenotype should be assessed by testing affected/unaffected relatives and obligatory carriers in the family, to exclude possible variants that would not affect SLC39A4 function.
1.8 Estimated frequency of the disease (Incidence at birth (birth prevalence) or population prevalence)
The only data available in literature indicates an incidence of 1:500 000 births in Denmark6 (http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf). This is consistent with the frequency of rare variants known to affect or potentially affecting SLC39A4 function, which are released in public variant database, considering an autosomal recessive mode of inheritance. An unofficial study conducted in France in the late 90s identified 15 families. The frequency seems artificially much higher in countries of the Mediterranean basin, probably because of a founder effect due to a higher incidence of consanguinity.
1.9 If applicable, prevalence in the ethnic group of investigated person
Not applicable.
1.10 Diagnostic setting

Comment: AEZ is an autosomal recessive syndrome of severe permanent zinc deficiency due to a malabsorption of zinc by a defective zinc transporter, SLC39A4 (or ZIP4), expressed mainly in the brush border of jejunum and duodenum enterocytes.1 (Differential) diagnostics—Testing of SLC39A4 is indicated in patients who developed clinical symptoms and biological signs of a severe zinc deficiency, at birth or after weaning (clinical and biological features are described in section 3.1.2). The identification of a molecular anomaly in SLC39A4 gives definitive proof of AEZ and enables its distinction from transient forms of zinc deficiency that can: (1) have a genetic component, such as the transient neonatal zinc deficiency (TNZD) caused by variant affecting SLC30A2 function, which reduces milk zinc concentration in nursing mothers of exclusively breastfed infants,4, 7 or (2) be acquired by an unbalanced diet too rich in phytates—found notably in cereals—by total parenteral nutrition or prematurity. In a few cases, genetic testing may also differentiate AEZ from biotin deficiency, atopic dermatitis, or rare genodermatoses mimicking AEZ (Küry et al. manuscript under preparation), especially when biochemical results are ambiguous (see 3.1.3). Predictive testing—it is not applicable in most cases, because AEZ symptoms and signs occurs in the perinatal period. The value of the genetic test would therefore rather be considered diagnostic than predictive. Prenatal—it is technically feasible, but it is not recommended. Zinc supplementation can indeed be applied immediately in a newborn suspected of AEZ on the basis of a zinc dosage (see 3.1.2). Besides, the risk for the mother inherent to prenatal testing cannot be neglected. Genetic testing comes in a second time, in order to confirm clinical diagnosis.
2. Test characteristics
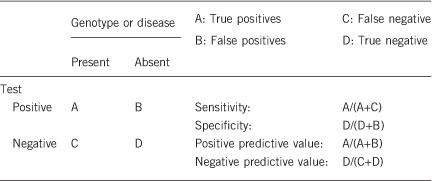
2.1 Analytical sensitivity
(proportion of positive tests if the genotype is present)
Theoretically 100% if variants affecting SLC39A4 function are localized within exons or flanking introns sequences, which encompasses at least 80% of this type of variants.
2.2 Analytical specificity
(proportion of negative tests if the genotype is not present)
100%.
2.3 Clinical sensitivity
(proportion of positive tests if the disease is present)
The clinical sensitivity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if quantification can only be made case by case.
Overall, variants affecting SLC39A4 function are observed in about 48% of the index cases tested: ~40% of the patients are homozygotes or compound heterozygotes, whereas 8% are heterozygotes only.8 In these last ones, we cannot exclude the presence of a second anomaly in unexplored regions of the SLC39A4 gene (intronic, promoting, untranslated or even regulatory regions) or in another gene directly involved in zinc homeostasis.9 Besides, the participation of epigenetic events such as methylation or miRNA regulation cannot be ruled out either. The same hypotheses are also valuable to explain the failure in variant detection in about 52% of the patients.9 In addition, the difficulty—or even sometimes the impossibility—to distinguish congenital zinc deficiency AEZ from acquired zinc deficiency largely accounts for these negative tests; except for the age at onset, which can be much later in acquired zinc deficiency, both entities share indeed the same clinical features. A few other negative tests are attributable to the partial clinical overlap between AEZ and biotin deficiency, atopic dermatitis or exceedingly rare genodermatoses (Küry et al. manuscript under preparation).
2.4 Clinical specificity
(proportion of negative tests if the disease is not present)
The clinical specificity can be dependent on variable factors such as age or family history. In such cases a general statement should be given, even if a quantification can only be made case by case.
100%.
2.5 Positive clinical predictive value
(life time risk to develop the disease if the test is positive)
If homozygous or compound heterozygous variants affecting SLC39A4 function are identified in still asymptomatic breastfed children—their zinc deficiency is masked by zinc from maternal milk—the risk to develop AEZ after weaning is 100%.
2.6 Negative clinical predictive value (probability not to develop the disease if the test is negative)
Assume an increased risk based on family history for a non-affected person. Allelic and locus heterogeneity may need to be considered.
Index case in that family had been tested:
When a variant affecting SLC39A4 function is identified in the index case, the negative predictive value is 100%.
Index case in that family had not been tested:
Not applicable.
3. Clinical utility
3.1 (Differential) diagnostics: The tested person is clinically affected
(To be answered if in 1.10 ‘A' was marked)
3.1.1 Can a diagnosis be made other than through a genetic test?

3.1.2. Describe the burden of alternative diagnostic methods to the patient
Diagnosis can be established by the concomitant presence of characteristic clinical features and biological signs. Historically, AEZ was defined by a triad of symptoms, including a periacral and periorificial dermatitis, an alopecia and a diarrhea.10 In practical terms, this pathognomonic triad is seen in only 25% of the cases.11 The most constant clinical symptoms are cutaneous lesions (first eczematous, then quickly erosive, psoriasiform or vesiculopustulous) always located at least in periorificial areas, and very frequently associated with acral lesions. Secondary fungal and bacterial infections, erosions of the buccal mucosa, integument disorders (alopecia and nail dystrophy), diarrhea, neuropsychiatric symptoms (irritability, apathy or psychomotor delay), and failure to thrive are also frequently reported.11 The characteristic biological sign of AEZ is a decrease in body zinc levels, usually assessed by a dosage of zinc plasma or serum levels; low levels of zinc-dependent enzymes, such as alkaline phosphatase, are also observed. Consequently, a key element in the diagnosis of AEZ is the excellent responsiveness of zinc deficiency symptoms and signs to oral zinc supplementation; all of them should indeed resolve within days/weeks. It is worth noting, however, that such a responsiveness to zinc therapy is also observed in transient neonatal zinc deficiency (TNZD), in which maternal milk does not provide enough zinc to meet the infant's needs, despite a normal intestinal absorption.7 Eventually, interruption of zinc therapy may help to distinguish between inherited (AEZ) and acquired zinc deficiency, as, on the one hand, a relapse is always observed in AEZ, whereas, on the other hand, no further supplementation is required after weaning in TNZD.
3.1.3 How is the cost effectiveness of alternative diagnostic methods to be judged?
Establishing a diagnosis of AEZ is not very easy, because of the rarity of the disease, which often leads to a long diagnostic delay. Moreover, clinical and biochemical diagnostic methods are rarely sufficient to establish a definitive diagnosis of AEZ, notably because acquired zinc deficiency causes are varied and not always obvious to determine.12, 13 More rarely, biotinidase deficiency and atopic dermatitis are sometimes misdiagnosed as AEZ, especially when zinc levels are found subnormal and symptoms seem zinc responsive. Such situations can be considered as exceptions, but they nevertheless illustrate the difficulty to rely upon zinc dosage and clinical features only, all the more that true AEZ patients also happen to exhibit very marginal decreases in plasma zinc levels. As a matter of fact, zinc homeostasis is so tightly regulated that even the slightest biochemical abnormality can stress a more profound zinc deficiency. Methods for zinc dosage and definition of standard values for zinc levels are not harmonized either between laboratories/countries, which can induce misinterpretation of biochemical results. In any case, the ultimate diagnostic proof of AEZ is therefore brought by the identification of a molecular anomaly in SLC39A4.
3.1.4 Will disease management be influenced by the result of a genetic test?

3.2 Predictive setting: The tested person is clinically unaffected but carries an increased risk based on family history
(To be answered if in 1.10 ‘B' was marked)
3.2.1 Will the result of a genetic test influence lifestyle and prevention?
If the test result is positive (please describe). Not applicable.
If the test result is negative (please describe). Not applicable.
3.2.2 Which options in view of lifestyle and prevention does a person at-risk have if no genetic test has been done (please describe)?
Not applicable.
3.3 Genetic risk assessment in family members of a diseased person
(To be answered if in 1.10 ‘C' was marked)
3.3.1 Does the result of a genetic test resolve the genetic situation in that family?
Yes; given the autosomal recessive mode of inheritance, newborns can be evaluated immediately after birth for the disorder.
3.3.2 Can a genetic test in the index patient save genetic or other tests in family members?
No.
3.3.3 Does a positive genetic test result in the index patient enable a predictive test in a family member?
Yes.
3.4 Prenatal diagnosis
(To be answered if in 1.10 ‘D' was marked)
3.4.1 Does a positive genetic test result in the index patient enable a prenatal diagnosis?
Not applicable.
4. If applicable, further consequences of testing
Please assume that the result of a genetic test has no immediate medical consequences. Is there any evidence that a genetic test is nevertheless useful for the patient or his/her relatives? (Please describe)
Results of SLC39A4 genetic testing may have no immediate medical consequence, because zinc supplementation has almost always been proposed to patients prior to the request for genetic analysis. Yet, in addition to its influence on disease management in homozygotes or compound heterozygotes (see section 3.1.4), the results of SLC39A4 genetic testing may also have consequences for their relatives. Variant carriers are worth detecting, as heterozygous carriers of a variant affecting function would be hypersensitive to zinc deficiency.14 Their zinc status should therefore be regularly followed up to prevent possible symptoms of zinc deficiency. Besides, genetic testing of SLC39A4 may avoid zinc oversupplementation and secondary copper or iron deficiency in transiently zinc-deficient neonates, who do not need any further zinc therapy after weaning. Of note, in TNZD, a biallelic or heterozygous variant affecting SLC30A2 function is present in the lactating mother, which induces AEZ-like zinc deficiency in the breastfed infant.4, 7 Beyond SLC39A4 screening, additional genetic testings are therefore required for this other form of zinc deficiency, and they may be considered for possible alternative inherited forms of AEZ-like entities suspected to be due to variants in zinc transporters or metallothioneins.9 This suggests a genetic heterogeneity and the need for further investigations to unravel the whole pathogenic mechanism, leading to transient TNZD and alternative inherited forms of transient zinc deficiency.
Acknowledgments
This work was supported by EuroGentest2 (Unit 2: ‘Genetic testing as part of health care'), a Coordination Action under FP7 (Grant Agreement Number 261469) and the European Society of Human Genetics. SK, SS and SB are partially supported by a donation from the laboratory LABCATAL for their fundamental research on AEZ.
The authors declare no conflict of interest.
References
- Küry S, Dréno B, Bézieau S et al: Identification of SLC39A4, a gene involved in acrodermatitis enteropathica. Nat Genet 2002; 31: 239–240. [DOI] [PubMed] [Google Scholar]
- Küry S, Kharfi M, Kamoun R et al: Mutation spectrum of human SLC39A4 in a panel of patients with acrodermatitis enteropathica. Hum Mutat 2003; 22: 337–338. [DOI] [PubMed] [Google Scholar]
- Schmitt S, Küry S, Giraud M, Dréno B, Kharfi M, Bézieau S: An update on mutations of the SLC39A4 gene in acrodermatitis enteropathica. Hum Mutat 2009; 30: 926–933. [DOI] [PubMed] [Google Scholar]
- Kasana S, Din J, Maret W: Genetic causes and gene-nutrient interactions in mammalian zinc deficiencies: acrodermatitis enteropathica and transient neonatal zinc deficiency as examples. J Trace Elem Med Biol 2014; 29C: 47–62. [DOI] [PubMed] [Google Scholar]
- Takeichi T, Nanda A, Liu L et al: Impact of next generation sequencing on diagnostics in a genetic skin disease clinic. Exp Dermatol 2013; 22: 825–831. [DOI] [PubMed] [Google Scholar]
- Van Wouwe JP: Clinical and laboratory diagnosis of acrodermatitis enteropathica. Eur J Pediatr 1989; 149: 2–8. [DOI] [PubMed] [Google Scholar]
- Chowanadisai W, Lonnerdal B, Kelleher SL: Identification of a mutation in SLC30A2 (ZnT-2) in women with low milk zinc concentration that results in transient neonatal zinc deficiency. J Biol Chem 2006; 281: 39699–39707. [DOI] [PubMed] [Google Scholar]
- Küry S, Schmitt S, Giraud M et al: A nine-year experience with the genetic testing of the rare disease acrodermatitis enteropathica; (Abstract #1062 T). Presented at the 12th International Congress of Human Genetics/61st Annual Meeting of The American Society of Human Genetics; 13 October 2011; Montreal, Québec, Canada.
- Küry S, Schmitt S, Blouin E, Bézieau S: Deciphering the genetics of inherited zinc deficiencies; Presented at the European Human Genetic Conference 2013, 8–11 June 2013; Paris, France: Eur J Hum Genet 2013; 21 (Supplement 2): 84. [Google Scholar]
- Danbolt N, Closs K: Acrodermatitis enteropathica. Acta Dermatol Venereol 1942; 23: 127–169. [Google Scholar]
- Kharfi M, El Fekih N, Aounallah-Skhiri H et al: Acrodermatitis enteropathica: a review of 29 Tunisian cases. Int J Dermatol 2010; 49: 1038–1044. [DOI] [PubMed] [Google Scholar]
- Perafan-Riveros C, Franca LF, Alves AC, Sanches JA Jr: Acrodermatitis enteropathica: case report and review of the literature. Pediatr Dermatol 2002; 19: 426–431. [DOI] [PubMed] [Google Scholar]
- Maverakis E, Fung MA, Lynch PJ et al: Acrodermatitis enteropathica and an overview of zinc metabolism. J Am Acad Dermatol 2007; 56: 116–124. [DOI] [PubMed] [Google Scholar]
- Dufner-Beattie J, Weaver BP, Geiser J et al: The mouse acrodermatitis enteropathica gene Slc39a4 (Zip4) is essential for early development and heterozygosity causes hypersensitivity to zinc deficiency. Hum Mol Genet 2007; 16: 1391–1399. [DOI] [PubMed] [Google Scholar]