

Abstract
Spinal cord injury (SCI) promotes inflammation along the neuroaxis that jeopardizes plasticity, intrinsic repair and recovery. While inflammation at the injury site is well-established, less is known within remote spinal networks. The presence of bone marrow-derived immune (myeloid) cells in these areas may further impede functional recovery. Previously, high levels of the gelatinase, matrix metalloproteinase-9 (MMP-9) occurred within the lumbar enlargement after thoracic SCI and impeded activity-dependent recovery. Since SCI-induced MMP-9 potentially increases vascular permeability, myeloid cell infiltration may drive inflammatory toxicity in locomotor networks. Therefore, we examined neurovascular reactivity and myeloid cell infiltration in the lumbar cord after thoracic SCI. We show evidence of region-specific recruitment of myeloid cells into the lumbar but not cervical region. Myeloid infiltration occurred with concomitant increases in chemoattractants (CCL2) and cell adhesion molecules (ICAM-1) around lumbar vasculature 24 hours and 7 days post injury. Bone marrow GFP chimeric mice established robust infiltration of bone marrow-derived myeloid cells into the lumbar gray matter 24 hours after SCI. This cell infiltration occurred when the blood-spinal cord barrier was intact, suggesting active recruitment across the endothelium. Myeloid cells persisted as ramified macrophages at 7 days post injury in parallel with increased inhibitory GAD67 labeling. Importantly, macrophage infiltration required MMP-9.
Keywords: spinal cord injury, inflammation, MMP-9, macrophage, blood brain barrier
Introduction
Spinal cord injury (SCI) is a devastating condition that affects 12,000 patients each year in the United States (National Spinal Cord Injury Statistical Center, 2014). Despite decades of basic science and clinical efforts, there is no treatment that fully reverses paralysis. Increasing evidence identifies a central role of the immune system and prolonged opening of the blood-spinal cord barrier (BSCB) in limiting recovery after SCI (Noble et al., 2002; Whetstone et al., 2003). The BSCB is comprised of a complex network of junction-bound endothelia bordered by glial endfeet that maintain synaptic homeostasis of resident neurons (Bechmann et al., 2007). Injured and regenerating blood vessels display increased permeability allowing parenchymal infiltration of bone marrow (BM)-derived immune cells to the lesion epicenter (Noble et al., 2002; Popovich et al., 1996; Whetstone et al., 2003). Such permeability after SCI appears to approach the lower thoracic and first lumbar segments (Popovich et al., 1996). By contrast, vascular integrity and immune cell infiltration in regions well rostral and caudal to spinal cord pathology have not been examined. These remote regions are comprised of neural networks critical for functional recovery. Here, we postulate that vascular breakdown after thoracic SCI occurs well away from the epicenter and provokes an aggressive inflammatory response around putative central pattern generator (CPG) networks in the lumbar cord that control locomotion.
Following traumatic injury to the spinal cord, proximal and distal resident glia have altered phenotypes that may disrupt normal neuronal function. Within hours of thoracic SCI, glial activation occurs in the lumbar enlargement (Gwak et al., 2012; Hansen et al., 2013). Consequently, cytokine production in these segments induces mechanical hypersensitivity and renders neural networks refractive to locomotor training (Detloff et al., 2008; Hansen et al., 2013). Cervical and lumbar segments house dynamic networks of pattern generating interneurons that mediate reciprocal excitation and inhibition of motor neuron pools during reaching (Alilain et al., 2008; Alstermark and Isa, 2012; Alstermark et al., 2011; Lane et al., 2012) and walking (Grillner et al., 1998; Rossignol et al., 2006). Thus, strategies to attenuate inflammation within cervical and/or lumbar microenvironment are needed to potentiate the recovery of function.
Recently, our work and others suggests that resident inflammatory cells may not be the sole source of inflammation remote to the injury (Gwak et al., 2012; Hansen et al., 2013; Shin et al., 2014). For example, we identified barrier-degrading matrix metalloproteinase-9 (MMP-9) around lumbar vasculature early after SCI (Hansen et al., 2013). MMP-9 activity is critical for extracellular matrix degradation and migration of immune cells across the BSCB. Indeed, eliminating MMP-9 attenuated remote cytokine expression and enhanced activity-dependent recovery (Hansen et al., 2013). Therefore, it is possible that BM-derived immune cells gain access to these distal sites and drive an inflammatory response that inhibits functional recovery. Given the large distance from blunt tissue damage, we reasoned that neuroimmune signaling may be similar to non-trauma models such as stress. Specifically, psychological stress in mice leads to the recruitment of inflammatory myeloid cells into the brain. Moreover, this myeloid cell infiltration induced IL-1 signaling and caused prolonged behavioral dysfunction (Wohleb et al., 2014; Wohleb et al., 2013). Thus, infiltration of inflammatory immune cells can change brain physiology and behavior (Wohleb et al., 2015).
The aim of this study was to examine if thoracic SCI initiates remote myeloid infiltration into the cervical and lumbar parenchyma and jeopardizes the neural networks that control reaching and locomotion. Here, we provide the first evidence of monocytes within intact lumbar gray matter distal to thoracic SCI. Within 24 h, extravascular BM-derived cells were evident throughout lumbar gray matter. By 7 days post injury (dpi), these myeloid cells adopted a ramified phenotype. The presence of ramified macrophages coincided with MMP-9 dependent breakdown of the BSCB and enhanced GABAergic activity within locomotor networks.
Material and Methods
Mice
Experiments were conducted in accordance with The Ohio State University Institutional Laboratory Animal Care and Use Committee. A total of 85 mice were used in these studies. Adult (3-4 months of age) female C57BL/6J wild-type (WT) mice were obtained from Jackson Laboratories and n=4 were excluded as described below. WT mice were partitioned into sister groups for ELISA, histology, measures of vascular permeability, or chimerization. The chimeric mice were engineered by adoptively transferring bone marrow from a C57BL/6-Tg(CAG-EGFP) donor (3-4 months of age, Jackson) into a busulfan-treated C57BL/6J recipient mice (1-2 months of age, Jackson). MMP-9 null (MMP9KO) female mice (B6.FVB(Cg)-Mmp9tm1Tvu/J) were obtained from Jackson Laboratories. Mice were housed 3-5 per cage and provided food and water ad libitum.
Engineering of GFP+ BM-chimera
Recipient BM C57BL/6 female mice (6-8 weeks old) were injected intraperitoneally once daily for two consecutive days with busulfan in a 1:1 solution of DMSO and deionized H2O (30 mg/kg/100 μl). The selected dosage of busulfan results in partial BM ablation and limited morbidity (Wohleb et al., 2013). Donor mice were euthanized with carbon dioxide and the femur was extracted. Donor BM-derived cells were isolated from the femur and passed through a 70 μm cell strainer. Total number of cells was determined with a BD Coulter Particle Count and Size Analyzer (Beckman Coulter). BM-derived cells (1 × 106) were transferred to recipient mice by tail vein injection (100 μl) 48 h after the second dose of busulfan. Mice were left undisturbed for 4 weeks to allow engraftment. Engraftment was verified by determining the percentage of GFP+ cells in the BM and the blood. Mice that had <30% BM engraftment were excluded from the study (n=2).
Spinal cord injury
Contusion of the spinal cord was perfomed as previously described (Hansen et al., 2013). In brief, mice were anesthetized with a ketamine (138 mg/kg)-xylazine (20 mg/kg) cocktail and given prophylactic antibiotics (gentocin, 1mg/kg). Using aseptic techniques, removal of the spinous process and lamina of T9 exposed the dura. After stabilizing the vertebral column, the Infinite Horizon (IH) device delivered 75 kilodynes of force to induce a severe contusion injury. Biomechanics of the injury were screened on day 0, and outlier forces or displacements were excluded (n=2). Force and displacement trends were similar to those established by Ghasemlou and colleagues (2005) (mean force: 78.66 ± 0.57; mean displacement: 681.28 ±23.97). The incision was closed in layers and 2cc of sterile saline was given subcutaneously to prevent dehydration. Randomized group assignment occurred. During recovery, mice received antibiotics (1mg/kg gentocin, s.q.) and saline for 5 days and bladders were manually expressed twice per day (Hoschouer et al., 2010).
Flow cytometry on blood
Blood was collected from the heart at perfusion (naïve, 1, 7 dpi; n=16) and red blood cells were lysed. Blood leukocytes were washed and the Fc receptors were blocked with anti-CD16/CD32 antibody (eBioscience). Cells were incubated with the appropriate antibodies (CD11b, GR-1; eBioscience), and Ly6C (BD Biosciences) for 1 h at 4°C. Cells were washed and re-suspended in FACS buffer for analysis. Non-specific binding was assessed by using isotype-matched antibodies. Antigen expression was determined using a Becton-Dickinson FACSCaliber four-color cytometer (BD Biosciences). Data were analyzed using FlowJo software (Tree Star) and gating for each antibody was determined based on isotype stained controls.
Purification of CD11b+ cells from spinal cord
Enriched CD11b+ cells were isolated from the spinal cord (Naïve, 1dpi; n=7) as previously described (Donnelly et al., 2011). In brief, the spinal cord was removed and the cervical, epicenter, and lumbar regions were dissected. Each segment was homogenized using a potter homogenizer and centrifuged to collect a cell pellet. Cells were then suspended in a discontinuous Percoll density gradient (70%/35%/0%) and centrifuged. Microglia and BM-derived CD11b+ cells were collected at the 70%/35% Percoll interphase.
Flow cytometry on spinal cord tissue
Percoll enriched CD11b+ cells were incubated with an Fc receptors block (anti-CD16/CD32) antibody followed with rat anti-mouse CD45-PerCP-Cy5.5 and CD11b-APC antibodies (eBioscience, CA). Expression of these surface receptors was determined using a Becton-Dickinson FACSCaliber four color Cytometer (BD, NJ). Microglia and BM- derived macrophages were identified by CD11b+/CD45lo and CD11b+/CD45high expression, respectively (Wohleb et al., 2011). Flow data were analyzed using FlowJo software (Tree Star, CA).
Protein isolation and quantification for ELISA
Spinal cord segments were isolated from the lumbar enlargement (Naïve, 1, 2, 3, 7 dpi; n=17) of naïve and SCI mice. ELISA was run in replicate groups alongside naïve controls (n=3-5/SCI groups, n=2-3 naives for each replicate group). Mice were perfused with sterile saline, and spinal cords were quickly dissected, snap frozen, and stored at -80°C. Segments from L4-5 were homogenized in RIPA lysis buffer (Pierce) and protease inhibitor cocktail (Roche). After centrifugation at 10,000 rpm for 5 min, protein concentrations were determined by a BSA protein assay. Quantification of proteins was determined using a custom SearchLight Multiplex ELISA Array and performed by Aushon Biosystems (Billerica, MA, USA). Custom arrays were spotted with capture antibodies specific to Intracellular cell adhesion molecule-1 (ICAM-1), Monocyte chemoattractant protein (CCL2), and stromal cell-derived factor-1 (CXCL12). The bound proteins were then detected with a biotinylated detection antibody, followed by streptavidin-horseradish peroxidase (HRP) and visualized with SuperSignal ELISA Femto Chemiluminescent substrate. The luminescent signal produced from the HRP-catalyzed oxidation of the substrate was measured using the SearchLight Imaging System (Pierce) and protein concentrations extrapolated from a standard curve using ArrayVision (Pierce). All protein levels were analyzed relative to uninjured WT controls and are expressed throughout as percent naïve.
Assessment of vascular permeability
To measure vascular permeability, mice (n=25) were intraperitoneally injected with a 2% solution of evans blue dye (EBD) dissolved in normal saline (Manaenko et al., 2011). The injectate circulated for 30 minutes before perfusion and fixation. After flushing with 100ml cold saline, mice were perfused with 100ml of 4% PFA. Tissue was collected and protected from light. To localize EBD fluorescence, we did not generate tissue homogenates. Instead, we compared proportional area of fluorescence after confocal imaging. Upon sectioning (20μm), albumin-bound evans blue leakage was detected using confocal microscopy (Olympus FluoView FV1000 at 633nm wavelength). From confocal images, we used digital image analysis of EBD fluorescence on representative sections from L1-L2, L3-L4, and L5-6. Thresholds for positive staining were performed by a blinded investigator, and images were then processed using densitometric scanning of threshold targets using ImageJ software. Proportional area is reported as the average percent area in positive threshold for all images. Comparisons were made from averages of the entire lumbar cord.
Histology
The WT (n=17), GFP+ Chimera WT (n=16), EBD (n=25), and MMP9KO (n=6) mice were transcardially perfused with 0.1 M phosphate buffered saline (PBS; pH 7.4) followed by 4% paraformaldehyde (pH 7.2) at naive (n=3-4/group), 24 h post SCI (n=3-4/group), and 7 dpi (n=3-4/group) timepoints. Spinal cord segments from the cervical cord, T9 lesion site and from spinal levels L1-L6 were post-fixed for 1h in 4% paraformaldehyde, rinsed overnight in 0.2M phosphate buffer (PB, pH 7.4) then cryoprotected in 30% sucrose before being embedded in M-1 Embedding Matrix (Thermoscientific) and frozen on dry ice (Hansen et al., 2013; Ma et al., 2001; Ma et al., 2004). Cervical, epicenter and lumbar blocks were sectioned in their entirety at 20 μm on a Microm HM505E cryostat and collected in series of equally spaced sections.
Fluorescent immunohistochemistry was performed to identify microglia/macrophages and visualize vascular proteins in transverse spinal cord sections of lumbar segments. To examine microglia/macrophages, a 1:200 dilution was used for rabbit anti Iba-1 (Wako, 019-19741). The antibody was prepared in blocking solution containing 1% BSA, 0.1% FG, 3% NGS, 0.2% Tx-100 or 1% BSA, 0.1% FG, 10% NDS, 0.2% Tx-100 in PBS. Incubation of the primary antibody occurred overnight at 4°C. Two secondaries were used to visualize Iba-1 labeling: a goat anti-rabbit secondary antibody at a 1:400 dilution (AlexaFluor 488; Molecular Probes, A21103) and a donkey anti-rabbit secondary at a 1:200 dilution (AlexaFluor 550; Abcam, ab96892). To identify vascular anatomy, we used a 1:500 dilution for a rat anti-mouse monoclonal antibody to Ly6C for 48 h at 4°C (ER-MP20; Abcam, ab15627). The antibody was prepared in blocking solution containing 1% BSA, 2% NGS in PBS. A goat anti-rat secondary antibody was used to visualize Ly6C antibody at a 1:500 dilution, incubated overnight at 4°C (A594; Abcam, ab6565). To identify ICAM-1 labeling, we used a 1:250 dilution of goat anti ICAM-1 (R&D Systems, AF796) with overnight incubation 4°C. The antibody was prepared in a blocking solution containing 1% BSA in PBS. A donkey anti-goat secondary (AlexaFluor 550; Abcam, ab150129) was used to visualize the ICAM-1 antibody at a 1:250 dilution, incubated overnight at 4°C. To identify BM- derived cells in non-chimeric mice, we used a 1:500 concentration of rat anti-mouse CD45 antibody (Abcam, ab25603) diluted in a 1% BSA in PBS blocking solution. The primary antibody incubated overnight at 4°C. We then used a 1:1000 dilution of donkey anti-rat secondary (AlexaFlour488; Abcam, ab150153) to visualize the CD45 antibody. To identify GAD67 labeling, we used a 1:200 dilution of mouse anti GAD67 (Millipore, MAB5406) in 3% Triton-X and 10% NGS overnight at 4°C. A goat anti-mouse secondary (AlexaFlour 594; Life Technologies, a11005) at 1:100 was used to visualize GAD67. For all staining, control sections were processed by eliminating the primary antibody and replacing with blocking solution to ensure positive labeling.
Image Quantification
Sections were imaged using a Zeiss 510 Laser Confocal Microscope (The Ohio State University Confocal Microscopy Imaging Facility). Predefined anatomical maps were used to delineate dorsal (laminae 1, 2, 3, 4), intermediate (laminae 5, 6, 7), and ventral (laminae 8, 9) gray matter. Quantifications of GFP+ Iba-1+ co-labeling and cell morphology were performed in the same spinal cord sections by two raters blinded to group assignment. The blinded counts were averaged for comparison and had interrater r2 values greater than 0.93. To quantify GAD67 labeling, digital image analysis of staining was performed on lumbar sections (Donnelly and Popovich, 2008). Thresholds for positive staining were determined by a blinded investigator then processed by densitometric scanning of threshold targets using ImageJ software. Proportional area is reported as the average percent area in the positive threshold for all pictures. To quantify infiltrating immune cells in non-chimeric mice, the number of CD45 positive cells were counted live by a single blinded rater using a Nikon Eclipse fluorescence microscope. All gray matter CD45+ cells were quantified from four tissue sections per animal (n=5 to 7 per group).
Statistics
All data was analyzed using ANOVA with Tukey's post hoc testing or a t-test when appropriate. Means and standard error of the mean (SEM) are reported throughout. Protein levels are displayed as percent of naïve. Significance was set at p<0.05.
Results
Increased presence of monocytes and granulocytes in circulation 24 hours and 7 days after thoracic SCI
While inflammation at the lesion epicenter is well established (Greenhalgh and David, 2014; Noble et al., 2002; Popovich et al., 1996; Whetstone et al., 2003; Zhang et al., 2011), inflammation within remote lumbar locomotor networks is less understood. This is relevant because the presence of BM-derived immune cells within locomotor networks after injury may further impede functional recovery. Thus, the purpose of this study was to determine whether BM-derived immune cells infiltrate and persist in the lumbar cord after SCI.
First, the presence of monocytes and granulocytes were determined in the blood 24 h and 7 days after SCI. Fig. 1A shows representative dot plots of CD11b and Ly6C labeling of monocytes (CD11b+/Ly6C+). These data indicate an increase in the percentage of CD11b+/Ly6C+ monocytes 24 h after SCI (30%) compared to controls (20%, p < 0.05). This increase in circulating monocytes was also detected 7 days after SCI (28% compared to 20%, p < 0.05, Fig. 1B). While the total population of CD11b+/Ly6C cells increased after SCI, the number of highly inflammatory monocytes (Ly6Chigh) in circulation decreased 24 h after SCI and returned to baseline levels at 7 dpi (Fig. 1B).
Figure 1. Increased presence of monocytes and granulocytes in circulation 24 h and 7 days after thoracic SCI.
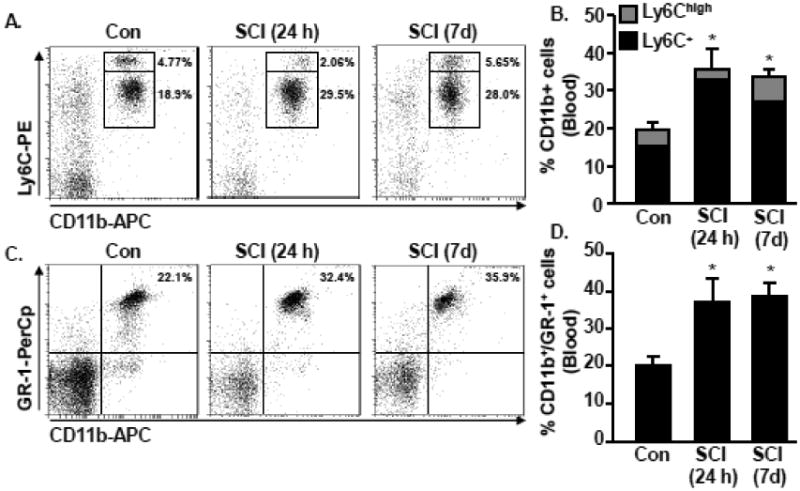
C57BL6 mice were naïve or subjected to a Mid-thoracic SCI. Blood was collected 24 h or 7 d later and the percentage of monocytes and granulocytes were assessed. A) Representative bivariate dot plots of CD11b and Ly6C labeling of monocytes. B) The percentage of CD11b+ cells that were Ly6C+ or Ly6Chigh in circulation 24 h and 7 d after SCI is shown. C) Representative bivariate dot plots of CD11b and GR-1 labeling of granulocytes. D) The percentage of granulocytes (CD11b+/GR-1+) in circulation 24 h and 7 d after SCI is shown. Bars represent the mean + SEM. Means with (*) are significantly different than naïve controls. Data were analyzed using one-way ANOVA and Tukey's HSD post hoc tests for significant main effects (n=4).
In the same samples, the percentage of granulocytes in circulation was determined. Fig. 1C shows representative dot plots of CD11b and GR-1 labeling. Similar to monocytes, there were increased granulocytes in circulation 24 h and 7 days after SCI (32% and 36%, p < 0.05 for each, Fig. 1D). Overall, increased granulocytes and monocytes occurred in circulation within 24 h after thoracic SCI that persisted to 7 dpi.
Trafficking of myeloid cells into the epicenter and lumbar regions after thoracic SCI
To determine whether circulating monocytes infiltrate the spinal cord in a localized or distributed manner, we examined myeloid cells above, at and below the thoracic contusion based on CD45 expression. In naïve mice, there was limited presence of CD45high expressing myeloid cells throughout the cord (2.3±0.5% of all CD11b+ cells, Fig. 2A). In contrast, robust infiltration of CD45high cells occurred in the lumbar cord (51±7.6% of all CD11b+ cells, p<0.05 vs. Naive) and at the lesion epicenter (79±8.2% of all CD11b+ cells, p<0.05 vs. Naive), although lumbar infiltration was lower than the epicenter region (p<0.05 from epicenter, Fig. 2B). However, there was little or no presence of CD11b+/CD45high expressing BM-derived myeloid cells above the lesion in the cervical region 24 h after SCI (6±1.3% of all CD11b+ cells, p>0.6 vs. Naive).
Figure 2. Trafficking of peripheral myeloid cells into the epicenter and lumbar regions after thoracic SCI.
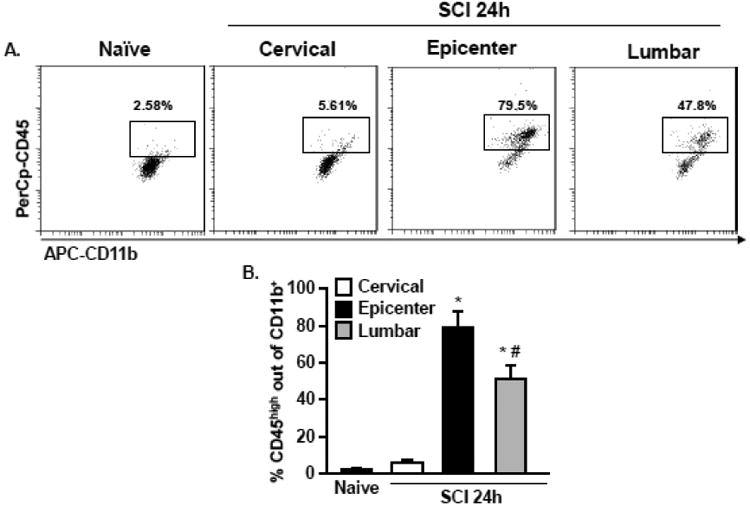
C57BL6 mice were naïve or subjected to a mid-thoracic SCI. The cervical region, epicenter region, and lumbar region was collected 24 h later and myeloid cells were isolated from the tissue. A) Representative bivariate dot plots of CD11b and CD45 labeling of myeloid cells. B) The percentage of peripherally derived CD11b+/CD45high cells was quantified. Bars represent the mean + SEM. Means with (*) are significantly different than naive. Mean with (#) is significantly different than epicenter. Data were analyzed using one-way ANOVA and Tukey's HSD post hoc tests for significant main effects (n=4).
Thoracic SCI results in increased in ICAM-1 and chemokine protein expression in remote lumbar segments
Evidence of region specific recruitment of BM derived cells into the lumbar cord exists in chemokine and adhesion molecule expression below the lesion (lumbar segments L4-5) 24 h – 7 days after a mid-thoracic SCI. Fig. 3A shows protein expression levels of ICAM-1, CCL2 and CXCL12 in the lumbar cord 24 h, 3 days, or 7 days after SCI. These results are shown as percent increase from the naïve control mice (100%). Increased ICAM-1 occurred by 24 h after SCI within the lumbar cord (150%, p < 0.05) and reached peak levels by 3 days after SCI (300%, p < 0.05). Additionally, CCL2 protein increased 24 h after SCI within the lumbar cord (145%, p < 0.05) and returned to baseline levels by 7 days. There was no induction of CXCL12 at any time after SCI in the lumbar cord. In fact, there was a reduction in CXCL12 protein at 24 h. This effect is contrary to the lesion epicenter, where increased CXCL12 works synergistically with MMP-9 to facilitate BM-cell infiltration (Zhang et al., 2011). This suggests that a distinct inflammatory response occurs in the remote lumbar cord that differs from the lesion epicenter, specifically through increases in CCL2 and ICAM-1 expression early after injury.
Figure 3. Thoracic SCI increased ICAM-1 and CCL2 protein expression within remote lumbar segments.
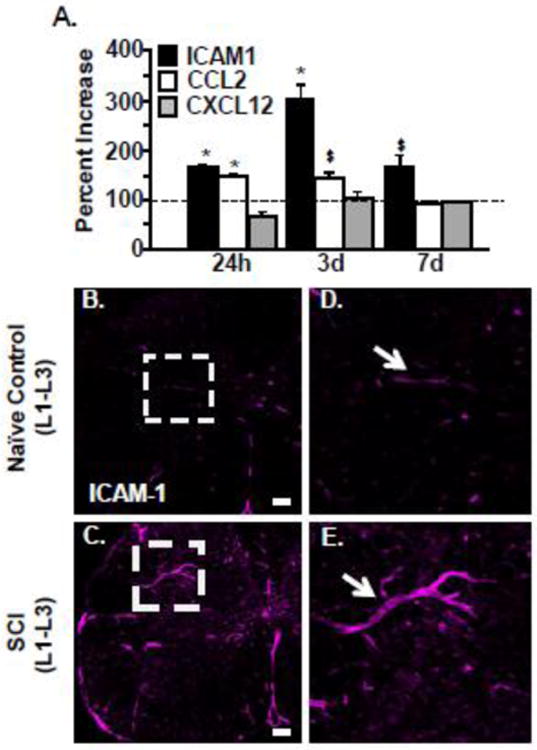
C57BL6 mice were naïve or subjected to a mid-thoracic SCI. A) The lumbar cord was collected 24 h, 3 d, 7 d after SCI and the protein levels of ICAM1, CCL2, and CXCL12 were determined. Data are presented as percent change from naïve controls (dotted line). Bars represent mean + SEM. Data were analyzed using two-way ANOVA and post hoc t-tests for significant main effects Means with (*) are significantly different than naive controls. Means with ($) have p-values ≤0.06. Data were analyzed using one-way ANOVA and Tukey's HSD post hoc tests for significant main effects (n=3-5). In a related experiment, C57BL6 mice were naïve or subjected to a mid-thoracic SCI. Mice were perfused, fixed and the spinal cord was collected 7 days after injury. Representative images of ICAM-1 labeling in the lumbar cord (L1-L3) of B) naïve and C) SCI mice. Scale bar is 100 μm. Dashed white boxes outline blood vessels that were positive for ICAM labeling. Representative enlarged images of ICAM-1+ blood vessels (white arrow) from D) naïve and E) SCI mice.
Based on the robust increase in ICAM protein within the lumbar cord after SCI, vascular ICAM expression was determined in lumbar segments. Representative images in Fig. 3 B&C show vascular endothelial of labeling of ICAM-1. Increased ICAM-1 protein localized to endothelia throughout dorsal and ventral gray matter 7 days (Fig. 3C) after injury compared to controls (Fig. 3B). High magnification of ICAM-1 positive vasculature is shown in Fig. 3D and Fig. 3E. Taken together, there is increased expression of key mediators associated with immune cell adhesion and recruitment in lumbar areas that are distal from the spinal cord injury site.
Region specific infiltration of GFP+ BM-derived cells 24 hours after thoracic SCI
To further characterize recruitment of myeloid cells into the injured spinal cord, we established BM GFP+ chimera mice as previously described (Wohleb et al., 2014; Wohleb et al., 2013). The BM ablation and reconstitution procedure is outlined in Fig. 4A. In these experiments, mice had SCI and the presence of GFP+ cells was determined 24 h later. Fig. 4 shows representative images of GFP+ cell in the thoracic region of a naïve mouse and in the cervical, epicenter, and lumbar region at 24 h after SCI. Consistent with our previous results using CD45 expression, there were few GFP+ cells found in the spinal cord of naïve mice (Fig. 4B). Furthermore, there was no evidence of GFP+ cell infiltration into the cervical region (Fig. 4C). Robust infiltration of GFP+ cells occurred in the epicenter (Fig. 4D) and lumbar cord (Fig. 4E). These data show that the GFP+ bone marrow chimeric mice can be used to track BM derived myeloid cells, and confirm our flow cytometry findings of region specific trafficking into the lumbar cord after thoracic SCI.
Figure 4. Region specific infiltration of GFP+ BM-derived cells into lumbar cord 24 hours after thoracic SCI.
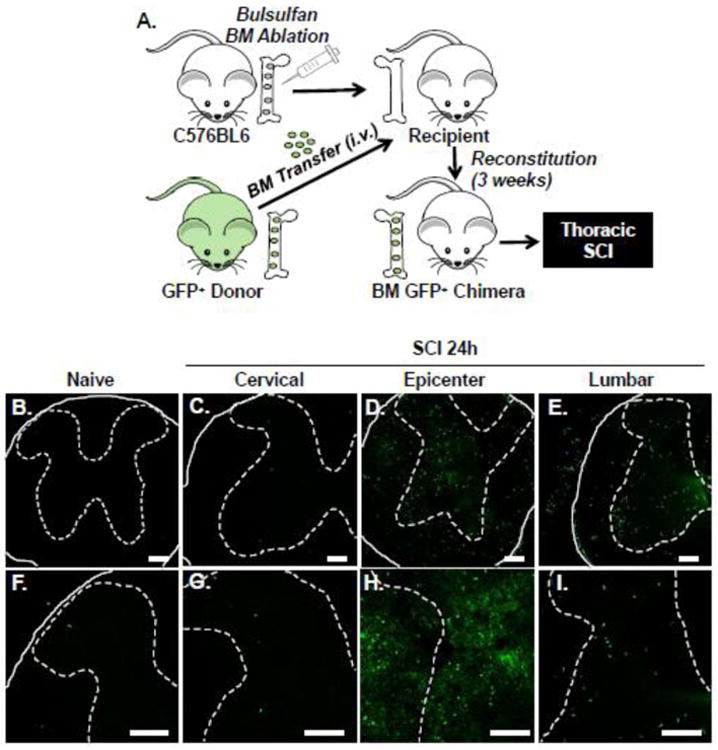
A) C57BL6 mice were subject to busulfan treatment to ablate the bone marrow. Next, busulfan treated mice received a BM transfer of cells from a GFP+ C57BL6 donor mouse. After BM reconstitution (3 weeks), GFP+ BM chimeric mice were assigned to naïve or thoracic SCI groups. Mice were perfused and fixed and the spinal cord was collected 24 h later. B-I) Representative images of GFP+ cells in the spinal cord of naïve mice (B, F) and 24 h after SCI in the cervical (C, G), epicenter (D, H), and lumbar (E, I) regions. High magnification images show (F-I) show increased detail of GFP+ cells. Solid lines indicate the outer border of the spinal cord, while dashed lines outline the gray matter. GFP+ cells were also noted in the peripheral nerve roots after SCI (not quantified; B, D, E, F). Data is further quantified via cell counts and flow cytometry in subsequent figures. Scale bar is 100 μm.
Increased infiltration of GFP+ BM-derived cells into locomotor networks 24 hours after thoracic SCI
To further quantify infiltration of GFP+ cells into the lumbar cord and determine their distribution, lumbar spinal cord sections were labeled with Ly6C (Wohleb et al., 2013) to identify the CNS vasculature. Ly6C is expressed in CNS vasculature but not in resident CNS microglia. As such, it has been used as a histological marker of the vasculature in the brain and spinal cord (Alliot et al., 1998; D'Mello et al., 2009). Fig. 5 shows representative images of GFP (green) and Ly6C (red) labeling in naïve and SCI mice. In naïve mice, there was no evidence of infiltration of GFP+ cells in any gray matter areas (dorsal horn, intermediate laminae, and ventral horn) of the lumbar segments. In fact, the rare BM-derived GFP+ cells were detected inside the Ly6C+ vasculature (Fig. 5C, arrow). In SCI mice, however, there was significant infiltration of BM derived GFP+ cells into the lumbar cord. Fig. 5D-F show an increased number of GFP+ cells in the lumbar gray matter (dorsal horn, intermediate laminae, and ventral horn) after SCI. There were GFP+ cells within the vasculature (yellow), but the majority of GFP+ cells in lumbar gray matter were extravascular by 24 h after SCI. Notably, GFP+ cells were also evident in the sacral segments of the cord after SCI (data not shown). Quantification of GFP+ myeloid cells at 24 h (Fig. 5G-I) confirmed a significant increase throughout dorsal (8.66 +/-2.12 vs. Naïve; p<0.05), intermediate (9.83 +/- 1.59 vs. Naïve; p<0.05) and ventral horn gray matter (18.5 +/- 2.31 vs. Naive; p<0.05). Overall, mid-thoracic SCI resulted in significant, rapid infiltration of GFP+ BM derived cells into the gray matter of lumbar segments of the spinal cord.
Figure 5. Increased infiltration of GFP+ BM derived cells into lumbar cord 24 hours after thoracic SCI.
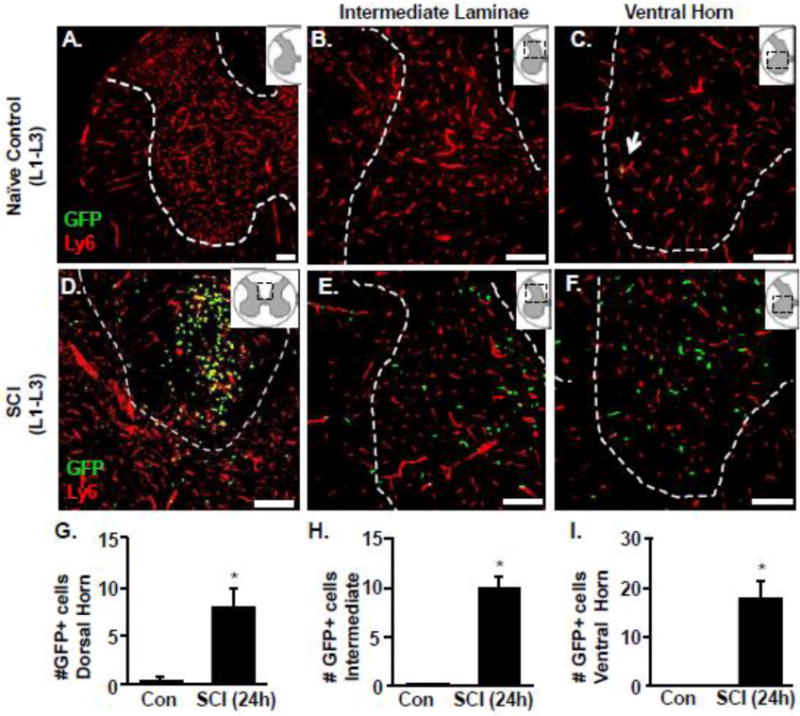
GFP+ BM chimeric mice were assigned to naïve or to a thoracic SCI. Mice were perfused and fixed and the spinal cord was collected 24 h later. The presence of GFP+ cells in and out of the vasculature (Ly6C+) of the lumbar cord (L1-L3) was determined. Representative images of the GFP+ (green) and Ly6C (red) labeling in naïve mice within the dorsal horn, intermediate laminae and ventral horn gray matter of the lumbar cord (A-C). Representative images of the GFP+ (green) and Ly6C (red) labeling in SCI mice with the dorsal column, intermediate laminae and ventral horn gray matter (D-F) of the lumbar cord. Inserts show the anatomical region within the spinal cord used. Dashed white outline shows the gray matter of the cord in labeled sections. Scale bar is 100μm. The number of GFP+ cells in the G) dorsal horn, H) intermediate laminae and I) ventral horn matter of the lumbar cord. Bars represent the mean + SEM. Means with (*) are significantly different than controls. Data were analyzed using one-way ANOVA (n=4).
BM-derived GFP+ macrophages persisted in the parenchyma of the lumbar cord through 7 days
Next, the presence of GFP+/Iba-1+ cells were determined in the lumbar cord 24 h and 7 days after thoracic SCI. Similar to the data in Fig. 5, GFP+ cells were detected in the parenchyma of the lumbar gray matter within 24 h after thoracic SCI. The representative image in Fig. 6A shows that these GFP+ cells were outside the vasculature and displayed either a rod or circular morphological profile. This phenotype is suggestive of a premature monocyte. At 24 h after SCI, these cells did not co-label with Iba-1 (Fig. 6B). GFP+ cells were also present in the lumbar cord 7 days after SCI and labeled with Iba-1 (Fig. 6B, right panel). Fig. 6A (right panel) shows that these GFP+ cells have a more ramified morphology with numerous processes by 7 days after injury. Quantification of the ramified GFP+ cells at 24 h and 7 days after SCI indicate that there was a time dependent increase in the number of ramified GFP+ cells in the intermediate laminae (Fig. 6D 42.3% vs. 24 h; p<0.05) and ventral regions (Fig. 6E, 36.5% vs. 24 h; p<0.05). In addition, by 7 days, the proportion of GFP+/Iba-1+ cells increased in the parenchyma of the ventral horn (Fig. 6H). The time dependent increase of GFP+ cells becoming Iba-1 positive was limited to the dorsal and ventral horn and was not readily detected in the intermediate laminae of the lumbar cord.
Figure 6. BM-derived GFP+ macrophages persisted in the parenchyma of the lumbar cord at 7 days.
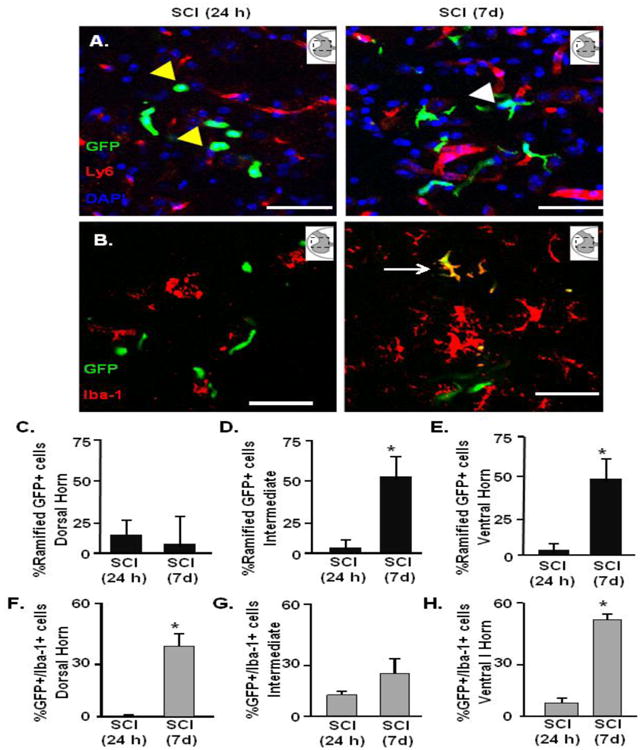
GFP+ chimeric C57BL6 mice were naïve or subjected to thoracic SCI. Mice were perfused and fixed and the spinal cord was collected at 24 h or 7 d. A) Representative images of the GFP+ (green), Ly6C (red), and DAPI (blue) labeling in the ventral horn 24 h (left) and 7 days (right) after SCI. Inserts show the anatomical region within the spinal cord used. Yellow arrow highlights rod or circular GFP+ cell and the white arrows highlight ramified GFP+ cells. Scale bar is 50um. The presence of GFP+ and Iba-1+ cells was determined alongside a quantification of cells that displayed a ramified morphology. B) Representative images of the GFP+ (green) and Iba-1 (red) in the ventral horn. White arrows highlight GFP+/Iba-1 cell in the lumbar cord 7 days after SCI. The number of ramified GFP+ cells in the parenchyma (GFP+/Ly6C-) 24 h and 7 d after SCI was determined in C) dorsal horn, D) intermediate laminae, and E) ventral horn of the lumbar cord. The number of GFP+/Iba+1 cells in the parenchyma 24 h and 7 d after SCI was determined in the F) dorsal horn, G) intermediate and H) ventral horn of the lumbar cord. Bars represent the mean + SEM. Means with (*) are significantly different between groups. Data were analyzed using one-way ANOVA (n=4).
MMP-9-dependent breakdown of lumbar vasculature 7 days after thoracic SCI
Our previous work shows that MMP-9 was detected within the lumbar enlargement, 9 segments caudal to a mid-thoracic SCI and impeded exercise-based recovery. Here, MMP9WT or MMP9KO mice were subjected to SCI and vascular permeability was determined in the lumbar cord 24 h and 7 days after SCI. Fig. 7 shows representative images of evans blue labeling. Vascular permeability was minimal throughout putative CPG regions (L1/L2 and L5/6) of the lumbar cord 24 h after SCI in WT mice (Fig. 7A). There was, however, evidence of increased vascular permeability throughout lumbar gray matter by 7 days after SCI (p<0.05, Fig. 7B). For example, patches of EBD fluorescence were evident in dorsal and intermediate gray matter regions of upper and lower lumbar segments of MMP9WT mice (Fig. 7B). In MMP9KO mice, there was no evidence of vascular permeability in lumbar segments 7 days after thoracic SCI (Fig. 7C). These data are quantified in Fig. 7D. Taken together, vascular permeability in the lumbar cord was evident 7 dpi and was dependent on MMP-9 (Fig. 7D).
Figure 7. MMP-9-dependent breakdown of the lumbosacral vasculature 7 days after thoracic SCI.

MMP9WT or MMP9KO mice were naïve or subjected to a thoracic SCI. Mice were injected with evans blue dye (EBD) as naive, and at 24 h or 7 d after SCI. Mice were perfused 30 minutes after injection, fixed, and the spinal cord was collected. Vascular permeability was described via fluorescence using confocal microscopy. Representative images of evans blue labeling in L5/L6 of the lumbar cord of A) MMP9WT 24 h, B) MMP9WT 7 day, and C) MMP9KO 7 day mice. D) Quantification of evans blue labeling through the lumbar cord. Bars represent the mean + SEM. Means with (*) are significantly different from 24 h. The same mice were labeled for CD45 expression in the lumbar cord. Representative images of CD45 labeling in lumbar cord of E) naïve, F) MMP9WT 7 days, and G) MMP9KO 7 day mice. The number of CD45 positive cells is quantified in H. Bars represent the mean + SEM. Means with (*) are significantly different from naive. Data were analyzed using one-way ANOVA and Tukey's HSD post hoc tests for significant main effects (n=5-7). Inserts show the anatomical region within the spinal cord used. Scale bar is 100um.
To determine whether MMP-9 is also required for trafficking of myeloid cells into the lumbar cord, the number of CD45+ cells was quantified in naïve, MMP9WT, and MMP9KO mice. Consistent with our findings of increased vascular permeability in the lumbar cord, we observed an increased number of BM-derived CD45+ cells in the lumbar gray matter 7 days after SCI in MMP9WT mice (Fig. 7E-H; p<0.01). Importantly, along with decreased vascular permeability, MMP9KO mice (Fig. 7G) had significantly fewer CD45+ cells than MMP9WT mice (Fig. 7F, H; p<0.05) and were not different from uninjured WT mice (Fig. 7E, H; p>.05). This further illustrates the essential role of MMP-9 for the infiltration of BM-derived immune cells into remote spinal cord regions after injury.
MMP-9 dependent enhancement of GAD67 labeling in lumbar cord 7 days after thoracic SCI
Based on our previous work (Hansen et al., 2013) and a paucity of data, we addressed the hypothesis that remote neuroinflammation influences inhibitory contacts within presumptive central pattern generator (CPG) regions after thoracic SCI by examining GAD67 expression. Central pattern generation depends on distinct periods of neuronal activation and inhibition. GABA is an inhibitory neurotransmitter that is synthesized by two isoforms of glutamic acid decarboxylase (GAD65 and GAD67). GAD67 is significantly increased in the lumbar cord after thoracic SCI and is expressed in a task-specific manner based on neuronal activation patterns (Tillakaratne et al., 2000; Wang et al., 2006). Thus, we used GAD67 labeling to identify localized inhibitory contacts that may be a surrogate measure of spinal shock early after SCI. Fig. 8 shows GAD67 labeling in the lumbar cord of MMP9WT or MMP9KO mice after SCI. The highest levels of GAD67 expression were in MMP9WT mice 7 days after SCI, suggesting broad distribution of inhibitory inputs. For example, marked upregulation of GAD67 expression was detected within the dorsal horn (Fig. 8G, p <0.05) and intermediate laminae (Fig. 8H, p<0.05) of lumbar segments compared to naive controls. Notably, there was no significant induction in the ventral horn of the lumbar cord 7 dpi (Fig 8I). These findings suggest that remote injury mechanisms cause broad inhibition in the lumbar cord which prevents early locomotor behavior and may induce long term deficits. In MMP9KO mice with SCI, GAD67 expression in the lumbar cord was attenuated compared to SCI-MMP9WT mice. In fact, GAD67 expression in SCI-MMP9KO was the same as naïve controls (Fig. 8G-I). Taken together, the SCI associated increase in vascular permeability and enhanced GABAergic activity likely associated with spinal shock within the lumbar cord were absent without functional MMP-9 activity.
Figure 8. MMP-9-dependent enhancement of GAD67 labeling in lumbar cord 7 days after thoracic SCI.
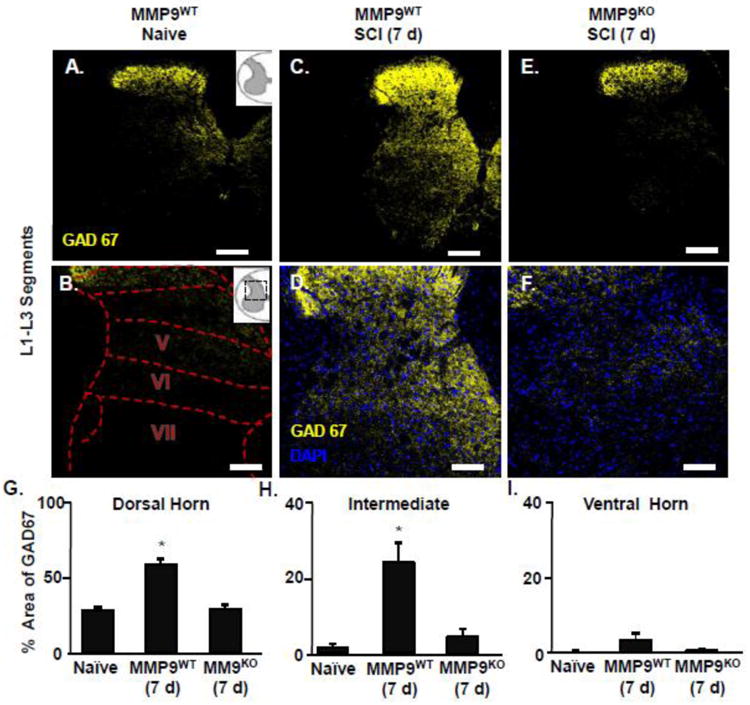
MMP9WT or MMP9KO mice were assigned to naïve or to thoracic SCI and survived for 7 d. Mice perfused, fixed, and the spinal cord was collected. A) Representative images of GAD67 (yellow) labeling in naïve mice or 7 days after SCI in L1-L3 segments of the lumbar cord. B) Inserts show the anatomical cord segments examined. C) Representative images of GAD67 (yellow) and DAPI (blue) labeling 7 days after SCI in L1-L3 of the lumbar cord. The proportional area of labeling for GAD67 in the D) dorsal horn, E) intermediate laminae, and F) ventral horn of the lumbar cord. Means with (*) are significantly different than naïve control. Data were analyzed using one-way ANOVA and Tukey's HSD post hoc tests for significant main effects (n=3-4). Inserts show the anatomical region within the spinal cord used. Scale bar is 100um.
Discussion
Recovery after SCI is largely dependent on segmental pattern generation within cervical and lumbar locomotor networks (Alilain et al., 2008; Alstermark and Isa, 2012; Alstermark et al., 2011; Edgerton et al., 2004; Grillner and Wallen, 1985; Lane et al., 2012; Rossignol and Frigon, 2011). Within hours, neuronal communication in the remote lumbar region is compromised by inflammatory signaling. The inflammatory signature of the lumbar cord has been associated with the development of neuropathic pain, spasticity, and the impairment of locomotor networks during activity-based training (Detloff et al., 2008; Gwak et al., 2012; Hains and Waxman, 2006; Hansen et al., 2013; Popovich et al., 1996). This study provides the first evidence of how neuroinflammation is initiated specifically in the lumbar but not cervical cord after midthoracic contusion. Here, we identify rapid neurovascular alterations localized to the lumbar cord that facilitate the infiltration of BM-derived myeloid cells. This is a critical finding given that blood brain barrier assessments have been characterized for over two decades. Our work identifies a novel injury response caudal but not rostral to SCI that likely undermines recovery and further compounds the functional consequence of axon loss at the lesion epicenter. In a companion manuscript, we have further described segmental functional impairments of neural networks in the lumbar cord at 7d after a moderate/severe thoracic SCI (Hansen et al., 2016). Behaviorally, we found that lumbar segments were incapable of generating adaptive plasticity and learning. This was accompanied by aberrant dendritic spine growth within lamina 7 interneurons that likely comprise part of the CPG. Here, we begin to identify some cell types and signaling mechanisms that impact remote lumbar dysfunction after thoracic SCI.
It is surprising that myeloid trafficking occurred so distal to the SCI lesion site. Until now, such long-distant inflammatory responses were postulated to be a central microglial response. However, early work showing greater uptake of a small molecule by vasculature in the thoracic cord and L1 after SCI provides strong rationale for the cellular-based studies here (Popovich et al., 1996). Immune cell diapedesis and BSCB breakdown has been extensively studied at the epicenter (Greenhalgh and David, 2014; Noble et al., 2002; Popovich et al., 1996; Soderblom et al., 2013; Whetstone et al., 2003) but not within the cervical and lumbar enlargement where high metabolic demand and vascular density exist (Mautes et al., 2000). However, our findings of caudal but not rostral myeloid infiltration has less to do with metabolic demand and points to specialized function of the lumbar vasculature. Indeed, our work supports evidence from multiple sclerosis models that vasculature in the lumbar enlargement is an important access point for BM derived cells (Arima et al., 2012). Here, we document robust active myeloid trafficking into the lumbar enlargement within 24 h and well-before vascular breakdown at 7 dpi. The amount of trafficking is likely underestimated given that only ∼65% BM-cells display a GFP tag in our chimera model. Extravascular GFP+ cells were evident throughout lumbar and sacral segments (sacral data not shown). Our work shows that thoracic SCI markedly alters the blood brain barrier throughout the vasculature in the lumbar enlargement. In the cervical cord, we found little or no myeloid cell infiltration. Clearly, parenchymal access is regulated differently in the lumbar cord, distal to SCI. While others suggest that lumbar immune access is activity-dependent in experimental autoimmune encephalomyelitis (EAE)) (Arima et al., 2012), we speculate that the changes we see with SCI result from a regional difference in blood spinal cord barrier composition. It is widely accepted that the blood brain barrier does not have uniform configuration throughout the body. Many locations in the CNS are described as having an absent or weakened blood brain barrier. Examples include the circumventricular organs like area postrema and subfornical organ, as well as secretory organs such as the pituitary and pineal glands. While the lumbar barrier is intact in the uninjured cord, we suggest that the remote neurovascular unit responds differently to degenerating axons and/or long distance glial communication than the vascular beds in the cervical cord. This may be due to the unique metabolic demands of respiratory and locomotor pattern generator networks and warrants additional investigation. It may also be due to the anatomy of a thoracic lesion. It is unclear whether, for instance, a cervical lesion would produce a differential long-distance inflammatory response. Based on our collected data, we hypothesize that embedding a lesion in a region where the blood-brain-barrier is more structurally sound, like the cervical cord, may restrict lesion pathology.
Another important finding in this study is that we identified phenotypic alterations of infiltrating myeloid cells over time. Upon infiltration, GFP+ cells display rod and circular shapes (24 h after SCI) that are consistent with monocytes. These cells were present in and outside the blood vasculature of the lumbar gray matter. This occurs alongside a bushy or activated phenotype in resident microglia which may contribute to increased production of monocyte chemoattractant CCL2 at 24 h (Hansen et al., 2013). A bushy phenotype describes an exaggerated activation profile in microglia, known to secrete CCL2 (Soltys et al., 2001). It is also possible that other cell types in the CNS such as neurons and astrocytes contribute to the increased CCL2 protein in the lumbar cord within 24 after SCI. Similar to events near the lesion epicenter, we speculate that activation of microglia in the lumbar cord initiates a chemokine gradient that recruits monocytes into the CNS parenchyma (Greenhalgh and David, 2014). However, early diapadesis of these cells did not disrupt BSCB integrity. Based on evans blue leakage, permeability was evident at 7 dpi by which time BM-cells display a macrophage phenotype and a toxic milieu of TNFα and MMP-9 developed (Hansen et al., 2013). By 7 dpi, infiltrating macrophages had increased Iba-1 expression and a more ramified morphology, similar to that of resident microglia. Additionally, ramified macrophages begin to downregulate CD45 while rod-shaped macrophages (based on GFP+ cell counts and morphological assessments) have high CD45 expression. These alterations in macrophage morphology and Iba-1/CD45 expression suggest that the 24 h - 7 d time window provides an opportunity to shape macrophage heterogeneity in the lumbar cord. Interventions that prevent infiltration or shift infiltrated macrophage phenotype based on environmental signals may prevent vascular leakage and attenuate the remote injury sequella. We demonstrate that mechanisms that initiate active myeloid recruitment to the lumbar cord are substantially different than the passive infiltration at the lesion epicenter (Greenhalgh and David, 2014; Kigerl et al., 2009; Kigerl et al., 2006). For example, we show that infiltration of CD45 cells into the lumbar cord was dependent on MMP-9. Our findings identify a novel target for SCI therapies that may improve locomotion and other below-level functional deficits.
There are several potential mechanisms of monocyte transvascular migration into the lumbar cord at a time when the blood spinal cord barrier appears intact (Abbott et al., 2010; Imhof and Aurrand-Lions, 2004; Muller, 2011, 2013, 2015). Our data suggests that early monocyte recruitment by CCL2 and endothelial attachment at regions of high ICAM expression initiate migration. We previously showed that the first week after SCI (24-7d) coincides with a progressive increase of MMP-9 production in the lumbar cord (Hansen et al. 2013). We postulate that these monocytes have elevated MMP-9 which is secreted and degrades claudin and occludin to allow targeted migration through endothelial tight junctions. By 7d, the accumulation of infiltrated macrophages may produce toxic levels of MMP-9 that further breaks down the endothelial junction from inside the CNS parenchyma. Perhaps opening of the blood spinal cord barrier is a consequence rather than a cause of macrophage entry within the first week of SCI. Our present work identifies an Iba-1+ macrophage phenotype that is likely responsible for the MMP-9 production and barrier breakdown. It may be possible that increased barrier permeability during subacute periods may play an important role in sustaining populations of monocytes in the lumbar cord.
There is significant interest in understanding macrophage heterogeneity after SCI. Macrophages play diverse roles in CNS trauma and elicit mechanisms associated with both injury and repair (Greenhalgh and David, 2014; Kigerl et al., 2009). An expanded continuum of heterogeneity in macrophage phenotypes has recently been described in injury and stress models (Fenn et al., 2014; Mosser and Edwards, 2008; Wohleb et al., 2014; Wohleb et al., 2013). It has been well documented that repeated social stressors promote the recruitment of BM derived immune cells into the brain, promote anxiety and impair cognitive tasks (Wohleb et al., 2011; Wohleb et al., 2014; Wohleb et al., 2013). We have previously shown that exercise-stressors produce functional declines early after SCI (Hansen et al., 2013). It may be possible that exercise interventions result in sympathetic outflow from the bone marrow that primes BM-derived cells to produce a toxic interaction. Stress responses initiate a release of inflammatory monocytes that infiltrate the brain and cause anxiety (Wohleb et al., 2014; Wohleb et al., 2013). Similarly, in our SCI model, we found that injury initiated the release of monocytes into circulation and that this was maintained through 7 dpi. Based on these findings, alongside work in our companion manuscript demonstrating segmental functional impairments in the lumbar cord (Hansen et al., 2016), we postulate that infiltrated macrophages around locomotor networks do not support adaptive function or repair. Instead, we suggest that lumbar macrophages take on a detrimental phenotype early after SCI that renders lumbar networks refractive to function and initiates an acute period of spinal shock.
Our results show that reducing the inflammatory signature and stabilizing the BSCB in the remote lumbar cord prevents aberrant inhibitory contacts within locomotor neuron networks. Similar to work done in rat and cat models, thoracic injury results in the upregulation of inhibitory GAD67 expression in dorsal and intermediate laminae by 7 days after SCI (Tillakaratne et al., 2002). This period of remote synaptic inhibition may reflect the early period of spinal shock after SCI (DiTunno et al., 2004). Spinal shock results from structural and biochemical indices of maladaptive neuroplasticity regulated by neuroinflammation. Findings of aberrant spine formations throughout dorsal, intermediate, and ventral neuron networks support this mechanism (Bandaru et al., 2015; Hains and Waxman, 2006; Hansen et al., 2016; Tan et al., 2008). We postulate that remote inflammatory signaling jeopardizes the homeostatic balance of excitatory and inhibitory synaptic contacts in locomotor networks after SCI. In support of this idea, TNFα signaling initiates AMPA receptor insertion that mediates aberrant excitability (Beattie et al., 2002; Huie et al., 2012). Other inflammatory actions modulate synaptic activity by altering GABAergic activity (Grau et al., 2014). Here, we show that MMP9KO mice display a frank reduction in vascular leakage alongside normalized expression of GAD67 within presumptive CPG laminae. By preventing remote inflammation, we create a more permissive balance of GAD67 expression to facilitate plasticity amenable to activity-dependent training. Moreover, a balance of inhibitory synaptic contacts within shared locomotor networks may ameliorate below-level deficits such as neuropathic pain and spasticity.
Early prevention of aberrant inhibitory synaptic contacts in lumbar neuron networks provides an opportunity for activity-dependent plasticity and recovery. Indeed, MMP9KO mice demonstrate improved recovery only in combination with lumbar-focused treadmill training (Hansen et al., 2013). Thus, optimal locomotor recovery likely depends on combinatorial therapies that unmask adaptive plasticity by reducing inflammation in the lumbar cord. Reducing the toxic milieu surrounding CPG networks may optimize function and more readily generate plasticity after SCI. From here, work is needed to selectively target lumbar inflammation and establish below level sensory and motor changes that are independent of axon sparing at the lesion site. Effective therapies that target remote neuroinflammation will likely depend on specific evaluations of microglia vs. macrophage phenotypes under conditions of neurorehabilitation.
Conclusions
The data in this manuscript uncover a far-reaching neuroimmune response after CNS injury. Until now, the role of the peripheral immune system in the remote lumbar cord has been only speculative. Identification of this response provides a novel opportunity to abate neuroinflammation and rescue below-level plasticity and function after SCI. Pharmacological therapies that target remote myeloid trafficking in the lumbar cord will be essential for unmasking a window of plasticity in the CNS that can be shaped with activity-based therapies. Future therapies must recognize the expansive nature of the SCI lesion, especially caudally, and address pathology at- and beyond the injury epicenter.
Highlights.
Myeloid infiltration occur in the lumbar cord within hours of thoracic SCI
Upregulation of signaling molecules around the vasculature suggests active trafficking
Myeloid progenitor cells in the lumbar cord develop a mature microglial phenotype 1 week post injury
Myeloid infiltration into locomotor networks corresponded with increases in inhibitory GAD67 expression
Acknowledgments
The authors would like to thank Lesley Fisher for technical support and assistance with manuscript preparation; and Samantha Kerr for her technical work, particularly on the evans blue study.
Funding Sources: This work is funded by 1F31NS080512-01 (CNH); 1RO1NS074882-01A1 (DMB); R21NS090265 (DMB, JG, JS); P30- NS04758 (CBSCR); Foundation for Physical Therapy Promotion of Doctoral Studies Level I Scholarship (TDF). None of the funding sources had input in the study design, data analysis or reporting of the results.
Footnotes
Conflict of Interest Statement: The authors have declared that no conflict of interest exists.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiol Dis. 2010;37:13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- Alilain WJ, Li X, Horn KP, Dhingra R, Dick TE, Herlitze S, Silver J. Light-induced rescue of breathing after spinal cord injury. J Neurosci. 2008;28:11862–11870. doi: 10.1523/JNEUROSCI.3378-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alliot F, Rutin J, Pessac B. Ly-6C is expressed in brain vessels endothelial cells but not in microglia of the mouse. Neurosci Lett. 1998;251:37–40. doi: 10.1016/s0304-3940(98)00473-x. [DOI] [PubMed] [Google Scholar]
- Alstermark B, Isa T. Circuits for skilled reaching and grasping. Annu Rev Neurosci. 2012;35:559–578. doi: 10.1146/annurev-neuro-062111-150527. [DOI] [PubMed] [Google Scholar]
- Alstermark B, Pettersson LG, Nishimura Y, Yoshino-Saito K, Tsuboi F, Takahashi M, Isa T. Motor command for precision grip in the macaque monkey can be mediated by spinal interneurons. J Neurophysiol. 2011;106:122–126. doi: 10.1152/jn.00089.2011. [DOI] [PubMed] [Google Scholar]
- Arima Y, Harada M, Kamimura D, Park JH, Kawano F, Yull FE, Kawamoto T, Iwakura Y, Betz UA, Marquez G, Blackwell TS, Ohira Y, Hirano T, Murakami M. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell. 2012;148:447–457. doi: 10.1016/j.cell.2012.01.022. [DOI] [PubMed] [Google Scholar]
- Bandaru SP, Liu S, Waxman SG, Tan AM. Dendritic spine dysgenesis contributes to hyperreflexia after spinal cord injury. J Neurophysiol. 2015;113:1598–1615. doi: 10.1152/jn.00566.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Galea I, Perry VH. What is the blood-brain barrier (not)? Trends Immunol. 2007;28:5–11. doi: 10.1016/j.it.2006.11.007. [DOI] [PubMed] [Google Scholar]
- D'Mello C, Le T, Swain MG. Cerebral microglia recruit monocytes into the brain in response to tumor necrosis factoralpha signaling during peripheral organ inflammation. J Neurosci. 2009;29:2089–2102. doi: 10.1523/JNEUROSCI.3567-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Detloff MR, Fisher LC, McGaughy V, Longbrake EE, Popovich PG, Basso DM. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol. 2008;212:337–347. doi: 10.1016/j.expneurol.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiTunno JF, Little JW, Tessler A, Burns AS. Spinal shock revisited: a four-phase model. Spinal Cord. 2004;42:383–395. doi: 10.1038/sj.sc.3101603. [DOI] [PubMed] [Google Scholar]
- Donnelly DJ, Longbrake EE, Shawler TM, Kigerl KA, Lai W, Tovar CA, Ransohoff RM, Popovich PG. Deficient CX3CR1 signaling promotes recovery after mouse spinal cord injury by limiting the recruitment and activation of Ly6Clo/iNOS+ macrophages. J Neurosci. 2011;31:9910–9922. doi: 10.1523/JNEUROSCI.2114-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgerton VR, Tillakaratne NJK, Bigbee AJ, de Leon RD, Roy RR. Plasticity of the spinal neural circuitry after injury. Annu Rev Neurosci. 2004;27:145–167. doi: 10.1146/annurev.neuro.27.070203.144308. [DOI] [PubMed] [Google Scholar]
- Fenn AM, Hall JCE, Gensel JC, Popovich PG, Godbout JP. IL-4 signaling drives a unique arginase+/IL-1β+ microglia phenotype and recruits macrophages to the inflammatory CNS: consequences of age-related deficits in IL-4Rα after traumatic spinal cord injury. J Neurosci. 2014;34:8904–8917. doi: 10.1523/JNEUROSCI.1146-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghasemlou N, Kerr BJ, David S. Tissue displacement and impact force are important contributors to outcome after spinal cord contusion injury. Exp Neurol. 2005;196:9–17. doi: 10.1016/j.expneurol.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Grau JW, Huie JR, Lee KH, Hoy KC, Huang YJ, Turtle JD, Strain MM, Baumbauer KM, Miranda RM, Hook MA, Ferguson AR, Garraway SM. Metaplasticity and behavior: how training and inflammation affect plastic potential within the spinal cord and recovery after injury. Front Neural Circuits. 2014;8:100–100. doi: 10.3389/fncir.2014.00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh AD, David S. Differences in the phagocytic response of microglia and peripheral macrophages after spinal cord injury and its effects on cell death. J Neurosci. 2014;34:6316–6322. doi: 10.1523/JNEUROSCI.4912-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grillner S, Buchanan JT, Wallén P, Brodin L. Neural control of locomotion in lower vertebrates: From behavior to ionic mechanisms. In: Cohen AH, Rossignol S, Grillner S, editors. Neural Control of Rhythmic Movement in Vertebrates. John Wiley and Sons; New York: 1998. pp. 1–40. [Google Scholar]
- Grillner S, Wallen P. Central pattern generators for locomotion, with special reference to vertebrates. Annu Rev Neurosci. 1985;8:233–261. doi: 10.1146/annurev.ne.08.030185.001313. [DOI] [PubMed] [Google Scholar]
- Gwak YS, Kang J, Unabia GC, Hulsebosch CE. Spatial and temporal activation of spinal glial cells: role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp Neurol. 2012;234:362–372. doi: 10.1016/j.expneurol.2011.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hains BC, Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci. 2006;26:4308–4317. doi: 10.1523/JNEUROSCI.0003-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CN, Faw TD, Buford JA, White S, Grau JW, Basso DM. Sparing of descending axons rescues interneuron plasticity in the lumbar cord to allow adaptive learning after thoracic spinal cord injury. Front Neural Circuits. 2016 doi: 10.3389/fncir.2016.00011. 10.3389/fncir.2016.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen CN, Fisher LC, Deibert RJ, Jakeman LB, Zhang H, Noble-Haeusslein L, White S, Basso DM. Elevated MMP-9 in the lumbar cord early after thoracic spinal cord injury impedes motor relearning in mice. J Neurosci. 2013;33:13101–13111. doi: 10.1523/JNEUROSCI.1576-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoschouer EL, Basso DM, Jakeman LB. Aberrant sensory responses are dependent on lesion severity after spinal cord contusion injury in mice. Pain. 2010;148:328–342. doi: 10.1016/j.pain.2009.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huie JR, Baumbauer KM, Lee KH, Bresnahan JC, Beattie MS, Ferguson AR, Grau JW. Glial tumor necrosis factor alpha (TNFa) generates metaplastic inhibition of spinal learning. Plos One. 2012;7:e39751–e39751. doi: 10.1371/journal.pone.0039751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nature reviews. Immunology. 2004;4:432–444. doi: 10.1038/nri1375. [DOI] [PubMed] [Google Scholar]
- Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kigerl KA, McGaughy VM, Popovich PG. Comparative analysis of lesion development and intraspinal inflammation in four strains of mice following spinal contusion injury. J Comp Neurol. 2006;494:578–594. doi: 10.1002/cne.20827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane MA, Lee KZ, Salazar K, O'Steen BE, Bloom DC, Fuller DD, Reier PJ. Respiratory function following bilateral mid-cervical contusion injury in the adult rat. Exp Neurol. 2012;235:197–210. doi: 10.1016/j.expneurol.2011.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma M, Basso DM, Walters P, Stokes BT, Jakeman LB. Behavioral and histological outcomes following graded spinal cord contusion injury in the C57Bl/6 mouse. Exp Neurol. 2001;169:239–254. doi: 10.1006/exnr.2001.7679. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB. Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol. 2004;474:469–486. doi: 10.1002/cne.20149. [DOI] [PubMed] [Google Scholar]
- Manaenko A, Chen H, Kammer J, Zhang JH, Tang J. Comparison Evans Blue injection routes: Intravenous versus intraperitoneal, for measurement of blood-brain barrier in a mice hemorrhage model. J Neurosci Methods. 2011;195:206–210. doi: 10.1016/j.jneumeth.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mautes AE, Weinzierl MR, Donovan F, Noble LJ. Vascular events after spinal cord injury: contribution to secondary pathogenesis. Phys Ther. 2000;80:673–687. [PubMed] [Google Scholar]
- Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. 2011;6:323–344. doi: 10.1146/annurev-pathol-011110-130224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. 2013;50:7–22. doi: 10.1177/0300985812469883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller WA. The regulation of transendothelial migration: new knowledge and new questions. Cardiovasc Res. 2015;107:310–320. doi: 10.1093/cvr/cvv145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Spinal Cord Injury Statistical Center. Spinal Cord Injury Facts and Figures at a Glance. University of Alabama; Birmingham: 2014. [Google Scholar]
- Noble LJ, Donovan F, Igarashi T, Goussev S, Werb Z. Matrix metalloproteinases limit functional recovery after spinal cord injury by modulation of early vascular events. J Neurosci. 2002;22:7526–7535. doi: 10.1523/JNEUROSCI.22-17-07526.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popovich PG, Horner PJ, Mullin BB, Stokes BT. A quantitative spatial analysis of the blood-spinal cord barrier. I. Permeability changes after experimental spinal contusion injury. Exp Neurol. 1996;142:258–275. doi: 10.1006/exnr.1996.0196. [DOI] [PubMed] [Google Scholar]
- Rossignol S, Dubuc R, Gossard JP. Dynamic sensorimotor interactions in locomotion. Physiol Rev. 2006;86:89–154. doi: 10.1152/physrev.00028.2005. [DOI] [PubMed] [Google Scholar]
- Rossignol S, Frigon A. Recovery of locomotion after spinal cord injury: some facts and mechanisms. Annu Rev Neurosci. 2011;34:413–440. doi: 10.1146/annurev-neuro-061010-113746. [DOI] [PubMed] [Google Scholar]
- Shin HY, Kim H, Kwon MJ, Hwang DH, Lee K, Kim BG. Molecular and cellular changes in the lumbar spinal cord following thoracic injury: regulation by treadmill locomotor training. Plos One. 2014;9:e88215–e88215. doi: 10.1371/journal.pone.0088215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soderblom C, Luo X, Blumenthal E, Bray E, Lyapichev K, Ramos J, Krishnan V, Lai-Hsu C, Park KK, Tsoulfas P, Lee JK. Perivascular fibroblasts form the fibrotic scar after contusive spinal cord injury. J Neurosci. 2013;33:13882–13887. doi: 10.1523/JNEUROSCI.2524-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AM, Stamboulian S, Chang YW, Zhao P, Hains AB, Waxman SG, Hains BC. Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J Neurosci. 2008;28:13173–13183. doi: 10.1523/JNEUROSCI.3142-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillakaratne NJ, Mouria M, Ziv NB, Roy RR, Edgerton VR, Tobin AJ. Increased expression of glutamate decarboxylase (GAD(67)) in feline lumbar spinal cord after complete thoracic spinal cord transection. J Neurosci Res. 2000;60:219–230. doi: 10.1002/(SICI)1097-4547(20000415)60:2<219::AID-JNR11>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Tillakaratne NJK, de Leon RD, Hoang TX, Roy RR, Edgerton VR, Tobin AJ. Use-dependent modulation of inhibitory capacity in the feline lumbar spinal cord. J Neurosci. 2002;22:3130–3143. doi: 10.1523/JNEUROSCI.22-08-03130.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Pillai S, Wolpaw JR, Chen XY. Motor learning changes GABAergic terminals on spinal motoneurons in normal rats. Eur J Neurosci. 2006;23:141–150. doi: 10.1111/j.1460-9568.2005.04547.x. [DOI] [PubMed] [Google Scholar]
- Whetstone WD, Hsu JY, Eisenberg M, Werb Z, Noble-Haeusslein LJ. Blood-spinal cord barrier after spinal cord injury: relation to revascularization and wound healing. J Neurosci Res. 2003;74:227–239. doi: 10.1002/jnr.10759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LTM, Bailey MT, Nelson RJ, Godbout JP, Sheridan JF. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J Neurosci. 2011;31:6277–6288. doi: 10.1523/JNEUROSCI.0450-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, McKim DB, Sheridan JF, Godbout JP. Monocyte trafficking to the brain with stress and inflammation: a novel axis of immune-to-brain communication that influences mood and behavior. Front Neurosci. 2015;8:447. doi: 10.3389/fnins.2014.00447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Patterson JM, Sharma V, Quan N, Godbout JP, Sheridan JF. Knockdown of interleukin-1 receptor type-1 on endothelial cells attenuated stress-induced neuroinflammation and prevented anxiety-like behavior. J Neurosci. 2014;34:2583–2591. doi: 10.1523/JNEUROSCI.3723-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohleb ES, Powell ND, Godbout JP, Sheridan JF. Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci. 2013;33:13820–13833. doi: 10.1523/JNEUROSCI.1671-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Chang M, Hansen CN, Basso DM, Noble-Haeusslein LJ. Role of Matrix Metalloproteinases and Therapeutic Benefits of Their Inhibition in Spinal Cord Injury. Neurotherapeutics. 2011;8:206–220. doi: 10.1007/s13311-011-0038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]