

Abstract
Type 2 diabetes, fuelled by the obesity epidemic, is an escalating worldwide cause of personal hardship and public cost. Diabetes incidence increases with age, and many studies link the classic senescence and ageing protein p16INK4A to diabetes pathophysiology via pancreatic islet biology. Genome-wide association studies (GWASs) have unequivocally linked the CDKN2A/B locus, which encodes p16 inhibitor of cyclin-dependent kinase (p16INK4A) and three other gene products, p14 alternate reading frame (p14ARF), p15INK4B and antisense non-coding RNA in the INK4 locus (ANRIL), with human diabetes risk. However, the mechanism by which the CDKN2A/B locus influences diabetes risk remains uncertain. Here, we weigh the evidence that CDKN2A/B polymorphisms impact metabolic health via islet biology vs effects in other tissues. Structured in a bedside-to-bench-to-bedside approach, we begin with a summary of the evidence that the CDKN2A/B locus impacts diabetes risk and a brief review of the basic biology of CDKN2A/B gene products. The main emphasis of this work is an in-depth look at the nuanced roles that CDKN2A/B gene products and related proteins play in the regulation of beta cell mass, proliferation and insulin secretory function, as well as roles in other metabolic tissues. We finish with a synthesis of basic biology and clinical observations, incorporating human physiology data. We conclude that it is likely that the CDKN2A/B locus influences diabetes risk through both islet and non-islet mechanisms.
Keywords: Ageing, ANRIL, Beta cell mass, Cdkn2A, Cdkn2B, CDKN2B-AS, Insulin secretion, Oncogene, p14, p14ARF, p15, p15INK4B, p16, p16INK4A, Pancreatic beta cell, Proliferation, Review, Senescence, Tumour suppressor
The CDKN2A/B locus impacts human diabetes risk
The CDKN2A/B locus, at chromosome 9p21, influences diabetes risk across varied ethnicities and geography [1–3], including people of European [4–9], Asian [10–24], Indian [25], Pakistani [26], Mexican [27–29] and Arab [30–32] descent. CDKN2A/B polymorphisms impact risk of gestational diabetes [33–35], early progression to type 2 diabetes after gestational diabetes [36], post-transplant diabetes [37, 38] and cystic fibrosis-related diabetes [39]. CDKN2A/B is not associated with type 1 diabetes risk [40–42], but is associated with rapid decline in beta cell function [43], and progression to diabetic nephropathy, in individuals with type 1 diabetes [44]. CDKN2A/B polymorphisms contribute only a fraction of observed heritable type 2 diabetes risk [2]. How CDKN2A/B influences diabetes risk remains uncertain [1–3].
CDKN2A/B is a hotspot influencing genetic risk for many diseases
The CDKN2A/B locus also influences risk for vascular conditions [1], including coronary artery disease [45–49], aneurysm and ischaemic stroke [50, 51], as well as glaucoma [52, 53], Alzheimer's disease [54], endometriosis [55], periodontitis [56], ageing-related diseases [3, 57] and numerous cancers [58]. Intriguingly, the region of CDKN2A/B influencing type 2 diabetes risk is physically separated from regions contributing risk for other diseases, even for type 2 diabetes-related disorders such as cancer and cardiovascular disease [59–61].
General biology of CDKN2A/B locus genes
In order to consider how single nucleotide polymorphisms (SNPs) in the CDKN2A/B region may impact type 2 diabetes risk, an important starting point is the known biology of local genes.
The CDKN2A/B locus encodes three coding transcripts and a long non-coding RNA
The human CDKN2A/B locus (Fig. 1) encodes three proteins: p14 alternate reading frame (p14ARF) (p19ARF in mice), p15 and p16 inhibitors of cyclin dependent kinase 4 (p15INK4B and p16INK4A), and a long non-coding RNA (lncRNA) called ANRIL (also known as CDKN2B-AS) [1, 62, 63]. p16INK4A and p14ARF, encoded by the CDKN2A gene, share common second and third exons but have different first exons and promoters about 20 kb apart [64]. p16INK4A and p14ARF are in alternate reading frames, resulting in unrelated peptide sequences despite the common mRNA sequence. CDKN2B, about 30 kb from CDKN2A, encodes p15INK4B [65, 66]. Smaller splice variants of both p16INK4A (p12) and p15INK4B (p10) have been described [64, 67, 68]. ANRIL, which overlaps the p14ARF promoter and two exons of p15INK4B [69], transcribed by RNA polymerase II [70] in the antisense direction to CDKN2B, is spliced into linear or circular isoforms [69, 71, 72]. The mouse Cdkn2a/b locus, on chromosome 4, encodes p16INK4A, p19ARF and p15INK4B in a similar arrangement to the human locus, but with a different lncRNA called AK148321 in a position similar to ANRIL [73].
Fig. 1.

The human CDKN2A/B locus at 9p21 contains genes encoding p16INK4A, p14ARF, p15INK4B and the lncRNA ANRIL. CDKN2A encodes both p16INK4A and p14ARF, which share exons 2 and 3 but in different reading frames, producing unrelated peptides. Polymorphisms influencing type 2 diabetes risk are physically separate from the coronary artery disease risk interval. Diagram not to scale. CAD, coronary artery disease; T2D, type 2 diabetes
p14ARF, p15INK4B and p16INK4A are cell cycle inhibitors
CDKN2A/B proteins block cell cycle progression and influence tumorigenesis, senescence and ageing [62, 63]. p16INK4A and p15INK4B are cyclin-dependent kinase (CDK) inhibitors that prevent activation of CDK4/6 by D-cyclins (Fig. 2). CDK4/6 phosphorylates retinoblastoma (Rb); hypophosphorylated Rb represses early region 2 transcription factor (E2F) to prevent cell cycle entry [74]. p14ARF, also antiproliferative, acts by stabilising the tumour suppressor p53 by sequestering its negative regulator, mouse double minute 2 homologue (MDM2) [75]. p53 reduces cell cycle entry through the CDK-interacting protein/kinase inhibitory protein (CIP/KIP) family inhibitor p21. The p10 and p12 splice variants of p16INK4A and p15INK4B also inhibit the cell cycle, but through slightly different mechanisms [64, 67]. ANRIL has widespread influences on gene expression, and impacts the cell cycle by regulating the expression of p14ARF, p15INK4B and p16INK4A [73].
Fig. 2.
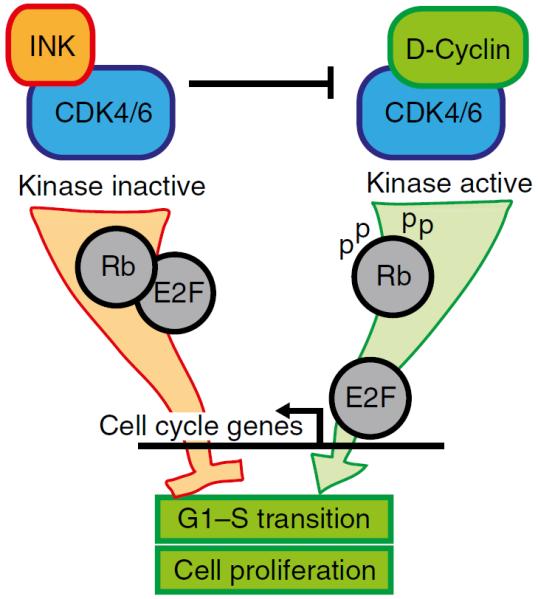
INK family inhibitors (p15INK4B, p16INK4A, p18INK4C and p19INK4D) bind CDK4/6 and prevent CDK binding and activation by D-cyclins. Hypophosphorylated Rb sequesters E2Fs; active (cyclin-bound) CDK phosphorylates Rb, de-repressing transcriptional activity of activating E2Fs to promote cell cycle entry
CDKN2A/B and tumorigenesis, senescence and ageing
The CDKN2A/B locus is inactivated by deletion, methylation or mutation in many cancers; CDKN2A/B aberrations correlate with advanced tumour stage and reduced overall and disease-free survival [58, 76]. Conversely, restoring p16INK4A suppresses tumour growth [77]. CDKN2A was the first familial melanoma gene identified [78]; germline loss-of-function CDKN2A mutations are the most frequent genetic events underlying familial melanoma susceptibility [79]. Activating CDK4 mutations (R24C and R24H) that prevent inhibition by p16INK4A increase melanoma risk [80]. Intriguingly, loss of p16INK4A also contributes to pancreatic neuroendocrine tumours, which include islet tumours such as insulinomas [81–85]. Paradoxically, when not deleted, p16INK4A may be overexpressed in tumours and transformed cells [86, 87]. p14ARF loss is oncogenic as well, via MDM2/p53 and other mechanisms [62]. p15INK4B may be co-deleted with p16INK4A in haematological malignancies [62], and plays a role in TGF-β-induced pancreatic cancer [88, 89]. ANRIL is also implicated in many cancer pathways, downstream of oncogenic Ras, phospholipase D and specificity protein 1 (SP1), and upstream of Kruppel-like factor 2, p21 and CDKN2A/2B [90–93].
p16INK4A is an effector of senescence, an irreversible growth arrest that occurs when a cell reaches the end of its replicative lifespan [63, 94]. p16INK4A expression increases with age [62], triggered by ageing-dependent gene demethylation, telomere shortening and other senescence inducers such as oncogenic activation, oxidative stress, nutrient deprivation and DNA damage [95, 96]. Mice and humans with the R24C activating mutation of CDK4 are resistant to senescence [97]. A recent study has shown that ablating senescent cells, as defined by p16INK4A expression, extends longevity and forestalls ageing-related tissue functional decline [98]. p14ARF is also linked to senescence, independent of p16INK4A [99].
Evidence that CDKN2A/B influences type 2 diabetes risk via beta cell mass and proliferation
The best-known role of CDKN2A/B gene products in a metabolic tissue is the ageing-related negative impact of p16INK4A on beta cell proliferation and regeneration [100, 101]. The critical dependence of mouse beta cell mass on CDK4 further supports the importance of this pathway in islet biology. Here, we will review the known roles of CDKN2A/B genes in islets.
|
| ||
| Effects of CDKN2A/B-related proteins on metabolic tissues | ||
| Tissue | Protein | Effects |
|
| ||
| Islets | p16INK4A | Restricts beta cell proliferation in ageing, restricts beta cell regeneration, mediates overnutrition-related senescence, reduces insulin secretory function |
| CDK4 | Required for postnatal beta cell mass expansion | |
| Cyclin D1, D2 | Required for postnatal beta cell mass expansion | |
| Adipose | p15INK4B | Inhibits adipocyte differentiation |
| p16INK4A | Modulates adipose macrophage activation and polarisation | |
| CDK4 | Promotes adipocyte differentiation | |
| Liver | p16INK4A | Restrains hepatic gluconeogenesis |
| Cyclin D1/CDK4 | Regulates fasted–fed transition | |
| Muscle | CDK4 | Impacts mitochondrial oxidative metabolism via E2F1 |
|
| ||
Beta cell proliferation decreases with age
Type 2 diabetes incidence increases with age, related to declining beta cell proliferation, beta cell function and/or insulin sensitivity [102–105]. Human autopsy studies report an age-dependent reduction in beta cell proliferation [102, 106, 107]. The importance of this has been contested on the basis that proliferation is rare even in young humans, such that a loss with ageing is not meaningful [108]. The demonstration that some measures of proliferation on autopsy specimens do not accurately reflect in vivo proliferation raises questions about the validity of postmortem analyses [109]. Beta cell mass is reported to be unchanged, or only modestly reduced, in ageing Japanese [108, 110]. As human beta cells may be extremely long lived, maintenance of beta cell mass may not require much ongoing proliferation [103]. Adult beta cell mass is thought to depend on early postnatal beta cell proliferation [106, 107]. In rodents, an age-associated decline in proliferation is clear [105]. Intriguingly, old islets exposed to young circulation via parabiosis or transplantation recovered a youthful proliferation frequency, suggesting the loss of proliferation is due to a circulating factor [111]. However, the rate of human beta cell proliferation is lower than that of rodents, and engrafting human islets into mice does not increase human beta cell proliferation to the rodent frequency [112, 113].
p16INK4A expression in rodent and human islets increases with age
Given the low beta cell replication frequency, cell cycle inhibitors have been a focus in this field. p16INK4A is expressed in mouse islets [101, 114–126]. Telomere shortening impacts islet p16INK4A expression as it does in other tissues [121]. The abundance of p16INK4A in young healthy islets is low, however, at or below the limit of detection in our hands by quantitative PCR using primers that do not amplify p14ARF (data not shown). Mouse islet p16INK4A abundance increases with ageing [101, 116, 117, 119, 121, 124–126]. p16INK4A is expressed in human islets [110, 127–132] in an age-dependent fashion [128], possibly related to progressive CDKN2A locus demethylation with age [96]. In human pancreas, nuclear p16INK4A staining was occasionally present in fetal beta cells, but was evident in 63% of adult beta cells [127]; p16INK4A staining was significantly lower in younger (age 0–9 years) than older (age 10–59 and 60–79 years) samples [110].
Regulation of islet p16INK4A expression and subcellular localisation
Islet p16INK4A expression is regulated at the RNA level via epigenetic histone modification by the polycomb repressors enhancer of zeste 2 (EZH2) and B cell-specific Moloney murine leukaemia virus insertion site 1 (BMI1). BMI1 and EZH2 bind the p16INK4A promoter to increase H3K27 trimethylation and reduce histone 2A (H2A) ubiquitination and H3K4 trimethylation via trithorax group (TrxG) mixed lineage leukaemia protein 1 (MLL1) [116, 119]. BMI1 and EZH2 occupancy of the p16INK4A promoter is reduced with ageing, resulting in de-repression of gene expression. Knockdown of EZH2 reduced BMI1 presence at the p16INK4A promoter, suggesting cooperative recruitment [116]. Overexpression of EZH2 suppressed p16INK4A expression in young but not old mice; in aged islets, MLL1 prevented EZH2 recruitment to the p16INK4A promoter [133]. EZH2 expression in human islets is inversely related to age [119]. These observations may apply to p16INK4A and p14ARF, but not p15INK4B [119].
Insulin signalling pathways impact islet p16INK4A expression. BMI1 is regulated by p38 mitogen-activated protein kinase (p38MAPK). Mice expressing an activation-resistant allele of p38Mapk (also known as Mapk14) had reduced abundance of p15INK4B, p16INK4A and p14ARF in aged islets; age-dependent BMI1 repressor loss at the p16INK4A promoter was reduced, possibly through p38MAPK target MAPK-activated protein kinase 3 (MK3) phosphorylation of BMI1 [134]. The age dependence of p38 activation was due to reduction of the wild-type p53-induced phosphatase 1 (WIP1) p38 phosphatase with age. Phosphoinositide 3-kinase (PI3K) signalling also influences p16INK4A expression; mice with phosphatase and tensin homologue (PTEN) deficiency had increased p16INK4A expression via a pathway involving cyclin D1, E2Fs and EZH2 [124, 125].
The protein subcellular localisation of cell cycle regulators likely impacts activity. In human beta cells, directly driving the cell cycle by overexpressing cyclin D3 and CDK6 caused p16INK4A, but not p15INK4B, to shift from the cytoplasm to the nucleus [130]. Nuclear relocalisation of p16INK4A depended on culture duration; at shorter time points after transduction (24–48 h) p16INK4A was less likely to be nuclear than in control cells, but after 72 h p16INK4A was more likely to be nuclear. The proportion of beta cells with nuclear p16INK4A expression increased with culture duration in control islets as well. Human islet cells undergo growth arrest after 10–15 divisions, with shortened telomeres and increased p16INK4A expression [135]. Taken together, this suggests that driving the cell cycle by activating CDK6 in human beta cells may hasten culture-related senescence.
p16INK4A mediates ageing-related loss in beta cell proliferation
Overexpression of p16INK4A reduced beta cell proliferation in young mice, and deletion of p16INK4A rescued the age-related loss of proliferation [101]. Mice lacking BMI1 or EZH2, such that p16INK4A is prematurely induced, have reduced beta cell proliferation; in the case of EZH2, dependence of proliferation loss on p16INK4A was confirmed [117, 119]. Overexpression of EZH2 repressed p16INK4A and increased proliferation in young mice; in older mice the same effects were seen but only if MLL1 was also reduced [133]. Ex vivo, knockdown of p16INK4A using small interfering (si)RNA rescued the loss of proliferation induced by NEFA exposure [123]. Alterations in the WIP1/p38MAPK/BMI1 or PTEN/E2F/EZH2 pathways that reduced p16INK4A expression increased beta cell proliferation in ageing mice [124, 134]. Loss of p16INK4A and a related INK family inhibitor, p18INK4c, synergistically increased beta cell proliferation in a CDK4-dependent manner [136]. p15INK4B also impacts beta cell proliferation; transgenic overexpression of TGF-β in alpha cells resulted in pancreas and islet hypoplasia associated with increased islet expression of p15INK4B [137].
p16INK4A restricts beta cell regeneration
The impact of ageing on beta cell regeneration is an area of controversy. Beta cell proliferation after partial pancreatectomy, exendin-4 treatment, high-fat feeding or streptozotocin ablation is lower in ageing mice than in young controls [101, 104, 117]. On the other hand, after diphtheria toxin ablation or glucokinase activation mice even beyond 2 years of age retained capacity for regenerative proliferation [138]. p16INK4A mediates age-related loss of islet regenerative capacity [101]. Loss of BMI1 and EZH2 impair beta cell proliferation in response to regenerative stimuli [117, 119]. BMI1 may also promote exocrine pancreas regeneration [139, 140]. A synthetic antagonist of hepatocyte nuclear factor (HNF)4α increased beta cell proliferation by suppressing the expression of CDK inhibitors including p16INK4A; inhibiting HNF4α induced alpha, beta and delta cell proliferation after beta cell ablation [141]. Beta cell proliferation in response to parathyroid hormone-related peptide (PTHrP) was associated with decreased expression of p16INK4A [122]. On the other hand, increased islet expression of p16INK4A may have prevented the expected increase in beta cell proliferation with overexpression of activated S6 kinase [120].
CDK4: critical for postnatal mouse beta cell mass
INK family inhibitors, which include p15INK4B and p16INK4A as well as p18INK4c and p19INK4d, bind to and inhibit CDK4/6 (Fig. 2) [74]. The essential role for CDK4 in mouse islet biology supports the importance of INK family inhibitors in islets. Mice lacking CDK4 develop severe insulin-deficient diabetes because of hypoplastic islets [142]. Intriguingly, CDK4 deletion also impacts other endocrine systems: male and female infertility, and poor growth, are possibly related to pituitary defects. Islet morphology is normal at birth, suggesting CDK4 is not required for pancreatic development, but islets fail to expand during postnatal growth. Conversely, mice expressing the INK-resistant mutant CDK4 `R24C' have hyperplastic islets due to increased postnatal beta cell proliferation [142, 143]. Mutation of the arginine at CDK4 position 24 is oncogenic in many tissues, despite also having impaired binding to D-cyclins and reduced efficacy as an Rb kinase [144]. Rodent insulinoma cell lines have increased expression of CDKs including 4 and 6; overexpressing CDK4/6 in rat islets increased proliferation [86].
d-cyclins and mouse beta cell proliferation
Cyclins D1, D2 and D3 activate CDK4/6 kinases, linking external growth signals with cell cycle regulation. INK family inhibitors bind CDKs near the D-cyclin binding site, preventing D-cyclin binding and kinase activation. Consistent with the importance of CDK4 in islets, D-cyclins also regulate beta cell proliferation in mice. Cyclin D2 and, to a lesser extent, cyclin D1, are required for postnatal beta cell proliferation [145, 146]. Like CDK4, D-cyclins are not required for prenatal pancreatic development. Upstream signalling events that promote beta cell proliferation, such as nutrient excess, insulin signalling and Wnt signalling, increase D-cyclin abundance in islets [122, 123, 147–155]. Islet overexpression of a stabilised cyclin D2 increased islet mass, although proliferation was not increased at the time points tested [151].
Metabolic insults may cause premature islet ageing
Metabolic stress influences ageing-related markers in general, and p16INK4A expression specifically. Failed human islet grafts in diabetic nude mice showed widespread p16INK4A immunostaining [129]. Energy restriction, an intervention that reduces metabolic load and delays ageing, decreased ageing-related p16INK4A expression in many tissues [126]. On the other hand, intrauterine protein-energy malnutrition increases type 2 diabetes risk. In rats, in utero nutrient deprivation reduced postnatal beta cell proliferation and led to premature ageing, with telomere shortening and induction of senescence markers including p16INK4A [156]. Maternal islets are also sensitive; postpartum, but not never-pregnant, female mice treated with the endocrine disruptor bisphenol-A developed glucose intolerance related to lost beta cell mass and insulin secretory function, with increased p16INK4A and p53 and reduced cyclin D2 and CDK4 [118].
The impact of lipid excess on beta cell proliferation remains controversial [123, 157, 158]. In vivo or ex vivo exposure to glucolipotoxicity reduced proliferation and induced islet p16INK4A expression in young mice [123]. Senescence markers in islets heralded metabolic decompensation after long-term high-fat feeding [159]. In contrast, short-term intravenous infusion with glucose and lipid in rats did not induce p16INK4A expression under conditions where beta cell proliferation was high [158]. Nutrition deprivation followed by nutrient excess induced islet senescence markers in rats [156]. The impact of nutrients on islet p16INK4A expression and senescence may depend on other factors related to development, genetics, duration of insult, overall stress load and stress tolerance.
Differences between cell cycle regulation in mouse and human beta cells: do D-cyclins, CDK4/6 or p16INK4A influence human beta cell proliferation?
Surveys of cell cycle regulator expression report similar patterns for rodent and human islets; of 34 proteins tested, only CDK6, E2F3, E2F7 and E2F2 show differences, with CDK6, E2F3 and E2F7 present in human islets but absent in mouse islets, and the converse for E2F2 [132, 160, 161]. Some reports suggest cyclin D2 may be less abundant than D1 and D3 in human islets [128, 132, 162]; however, recent reports link the CCND2 genomic locus, which encodes cyclin D2, with human type 2 diabetes risk [163–165]. A low-frequency non-coding variant at CCND2 reduced type 2 diabetes risk by about half in European populations; risk reduction was due to increased insulin secretion [164]. The impact of CCND2 was sex dependent, with a larger effect in men than women [166]. In ex vivo human islet cultures D-cyclins and CDK4/6 were detected in the cytoplasm rather than the nuclear compartment [131]; the same was true in rat islet cells for those cell cycle activators tested (cyclins D1/D2 and CDK2) [130]. Overexpression of D-cyclins was not as effective as CDKs in increasing human beta cell proliferation, this was perhaps related to the cytoplasmic location of CDKs, the abundance of INK family inhibitors [131, 162] or synergy with nutrient signals [167]. Data linking p16INK4A to reduced proliferation in human beta cells are somewhat limited. In autopsy material, p16INK4A was never detected in actively proliferating beta cells [127], and p16INK4A staining inversely correlated with beta cell mass [110]. Ex vivo human beta cells with nuclear p16INK4A after overexpression of cyclin D3 and CDK6 did not stain for the proliferation marker Ki67, despite 10–20% of beta cells in these cultures staining for Ki67 [130]. Knockdown of p16INK4A in the transformed EndoC-bH1 human beta cell line did not impact proliferation; however, transformation bypasses normal cell cycle regulation [168].
CDKN2A/B gene products may impact type 2 diabetes risk via other mechanisms
Evidence suggesting p16INK4A does not increase type 2 diabetes risk by reducing islet mass
Several observations contradict the concept that p16INK4A impacts diabetes incidence via effects on beta cell mass. The combined loss of p16INK4A and p14ARF was not sufficient to restore the glucose induction of mitotic genes lost in aged islets [169]. Old islets that recovered proliferation when exposed to young mouse circulation did not have reduced p16INK4A expression [111]. Perhaps most puzzling from an islet-centric viewpoint are observations from the Super-Ink4/ARF mouse, which contains an extra copy of the entire Cdkn2a/b locus [170]. Despite modestly increased expression of p15INK4B, p16INK4A and p14ARF in several tissues, these mice have improved glucose tolerance with ageing from enhanced insulin sensitivity in liver and muscle. Beta cell proliferation and islet number are unaffected. Reduced insulin secretion is observed, but is likely to be secondary to the improved insulin sensitivity. Also curious, a survey of islet gene expression at type 2 diabetes-linked loci in diabetes-prone New Zealand Obese (NZO) mice vs diabetes-resistant ob/ob mice revealed increased p15INK4B and p16INK4A expression in the diabetes-resistant mice [115].
p16INK4A may influence insulin secretory function, insulin clearance and insulin sensitivity
Some observations suggest an impact on insulin secretory function independent of the effects on beta cell mass (see the text box `Effects of CDKN2A/B-related proteins on metabolic tissues'). Knockdown of p16INK4A in the EndoC-bH1 human beta cell line increased insulin secretion [168]. In mice, haploinsufficiency for telomerase increased p16INK4A expression and impaired insulin secretion via altered regulation of exocytosis. These mice were glucose intolerant even though beta cell mass was normal, suggesting a primary effect on beta cell function rather than proliferation [121]. CDK4 also regulates insulin secretion, through Rb-dependent transcriptional regulation of potassium inward rectifying channel 6.2 (Kir6.2) [171]. In human studies, individuals from familial melanoma kindreds with heterozygous loss of function of CDKN2A had increased insulin secretion, impaired insulin sensitivity and reduced hepatic insulin clearance [168].
CDKN2A/B locus genes impact adipose, liver and muscle
CDKN2A/B locus genes and their CDK targets influence adipose, liver and muscle biology. Deletion of a region of mouse chromosome 4, orthologous to the human 9p21 cardiovascular disease risk interval, reduced local gene expression and increased body weight, linking the region with obesity and metabolic risk [172]. ANRIL regulates genes involved in glucose and fatty acid metabolism [173]. p15INK4B is highly expressed in subcutaneous adipose tissue and may inhibit expandability of the subcutaneous fat depot, a critical protection against overnutrition toxicity [174, 175]. Mice lacking CDK4 in all tissues are smaller, with less fat mass, and have insulin resistance in addition to insulin deficiency [176]. CDK4 regulates adipogenesis, via both Rb-dependent and Rb-independent mechanisms, including direct phosphorylation of insulin signalling intermediates [177–179]. CDKN2A/B locus genes also impact adipose tissue inflammation; p16INK4A modulates the activation and polarisation of adipose-associated macrophages [180], although deleting p16INK4A in bone marrow-derived cells did not impact glucose metabolism, even under obese conditions [181]. In liver, p16INK4A regulates hepatic gluconeogenesis independently of the cell cycle [182]. p16INK4A deficiency increased liver glucose production via a protein kinase A (PKA)-mediated induction of gluconeogenic gene expression. CDK4 participates in the hepatocyte fasting–fed transition by phosphorylating and activating the general control of amino acid synthesis protein 5-like 2 (GCN5) histone acetyltransferase, which regulates PPARG coactivator 1 α (PGC-1A) and hepatic glucose metabolism [183]. CDK4 also impacts muscle mitochondrial oxidative metabolism [184]. These observations, collectively, greatly broaden the potential mechanism(s) by which CDKN2A/B polymorphisms might impact diabetes risk.
How might CDKN2A/B polymorphisms influence type 2 diabetes risk?
Type 2 diabetes SNPs at CDKN2A/B are non-coding, and high-resolution mapping of the CDKN2A/B locus did not reveal SNPs with greater disease association than known SNPs [185]. Attention has thus turned to the regulation of local genes.
Do CDKN2A/B polymorphisms influence local gene expression?
Testing whether genomic polymorphisms alter mRNA abundance is performed by expression quantitative trait locus (eQTL) analysis. Thus far, however, eQTL analysis for the CDKN2A/B locus has failed to produce major mechanistic breakthroughs. Type 2 diabetes SNPs at CDKN2A/B were not associated with expression of CDKN2A or CDKN2B in pancreas, liver or colon [186], nor in pancreatic islets themselves [187]. CDKN2A/B SNPs impacting cardiovascular risk, outside the type 2 diabetes region, influence ANRIL expression [71, 188]. Both type 2 diabetes and non-type 2 diabetes SNPs at this locus appear to have a stronger effect on ANRIL expression than CDKN2A or CDKN2B expression [189]. Expression of ANRIL is reported to correlate with expression of CDKN2A and CDKN2B in many tissues, suggesting coordinated local regulation [69, 71, 76, 189], although independent regulation is also reported [76, 126]. Deletion of a large region including the type 2 diabetes-risk locus plus part of ANRIL reduced CDKN2A and CDKN2B expression in several vascular-relevant cell types in mice [172]. ANRIL regulates gene expression at the CDKN2A/B locus by recruiting polycomb proteins chromobox homologue 7 (CBX7 [PRC1]) and suppressor of zeste 12 (SUZ12 [PRC2]) to modulate epigenetic repression by H3K27 methylation [70, 72, 73, 90, 190]. ANRIL can also regulate distant genes with retrotransposon Alu repeats in their promoters [191]. In sum, existing eQTL analyses do not identify a mechanism by which polymorphisms at CDKN2A/B influence type 2 diabetes risk through local gene expression, but studies may have been performed in the wrong cell type, developmental, environmental or nutritional state to identify the point of activity. The requirement to perform these studies in human tissues, the difficulty in obtaining human samples, and caveats introduced through postmortem state, tissue collection and ex vivo culture are barriers to this type of study.
CDKN2A/B polymorphisms are located in regulatory enhancers
Cis-regulatory elements are found near the type 2 diabetes-risk interval [192–194]. Disease-associated SNPs are predicted to disrupt transcription factor binding sites, including some factors with known roles in beta cell development or survival, including, v-maf avian musculoaponeurotic fibrosarcoma oncogene homologue B (MAFB), NK homeobox protein 6.1 (NKX6.1), nuclear factor of activated T lymphocytes (NFAT), FOXA2, forkhead box A2 (FOXA2), nuclear factor κβ (NFκB), hepatocyte nuclear factor 1 (HNF1) and CCAAT-enhancer-binding protein homologous protein (CHOP) [90, 192, 194]. Polymorphisms may also impact microRNA regulation of transcription or translation [195], although most SNPs at the type 2 diabetes locus are non-coding. An enhancer identified in tumours, RDINK4/ARF, interacts with oncoproteins to silence the CDKN2A/B locus [196, 197]. Deletions of RDINK4/ARF have been detected in pancreatic neuroendocrine tumours, implying activity in islet cells [198].
Do polymorphisms influence epigenetic regulation at the CDKN2A/B locus?
Epigenetic modification regulates gene expression at this locus in islets [116, 119]. The type 2 diabetes-associated rs564398 removes a DNA methylation CpG site, reducing methylation of other local CpG sites and decreasing insulin content in human islets, although without impacting local gene expression [199]. Histone modifications can be influenced by polymorphisms as well; many epigenetic modifiers act at the CDKN2A/B locus [200]. Metabolic inputs, such as overfeeding or energy restriction, influence CpG methylation and histone modification at CDKN2A/B in humans [201, 202]. Whether this is influenced by type 2 diabetes-risk polymorphisms remains unknown. Taken together, although many mechanisms exist by which polymorphisms at CDKN2A/B might influence local gene expression, none has yet been proved.
Evidence that human CDKN2A/B-related type 2 diabetes risk involves beta cells
CDKN2A/B genotype influences insulin secretory capacity
Many type 2 diabetes risk polymorphisms, including at CDKN2A/B, may impact human diabetes risk by altering insulin secretory capacity [203]. Age influences the association between CDKN2A/B polymorphism rs10811661 and type 2 diabetes, consistent with the known interaction between age and p16INK4A activity in islets [204]. The rs10811661 `T' (risk) allele is associated with reduced insulin secretion after both oral and intravenous glucose challenge [205, 206]. In Europeans undergoing hyperglycaemic clamp, although CDKN2A/B was not independently associated with beta cell function, a composite including CDKN2A/B among eight loci predicted reduced first-phase insulin secretion [207]. In this cohort, each additional risk allele lowered glucose-stimulated insulin secretion (GSIS) by 5%, but no effect was seen on insulin sensitivity. The detrimental effect on insulin secretion was of similar magnitude in individuals with normal glucose tolerance and impaired glucose tolerance, suggesting CDNK2A/B impact pre-dates metabolic decompensation. In a large meta-analysis including more than 58,000 individuals, CDKN2A/B was associated with insulinogenic index, acute insulin response, fasting glucose or HOMA-B, but not with insulin sensitivity index, fasting insulin, fasting proinsulin, or HOMA-IR [208]. A parallel study of whether CDKN2A/B genotype impacts islet function in vivo and ex vivo revealed an in vivo association with reduced insulin secretion and disposition index, but not insulin sensitivity or glucagon secretion, but no striking impact on ex vivo function [209]. Individuals with reduced p16INK4A or p16INK4A/p14ARF activity have increased basal and stimulated insulin secretion [168].
Little evidence for or against a role in CDKN2A/B in human beta cell mass accrual
The observed reduction in insulin secretory capacity with CDKN2A/B polymorphisms could be due to reduced beta cell function (glucose sensing, insulin production, stimulus-secretion coupling) or reduced beta cell mass. Despite the data linking p16INK4A to beta cell mass in mice, few studies have assessed whether CDKN2A/B genotype impacts human beta cell proliferation or mass. A limitation is that human beta cell mass and proliferation cannot easily be measured in living people. In a single study, CDKN2A/B polymorphisms were not associated with AIRmax, a surrogate for beta cell mass [207]. On the other hand, the contribution of p16INK4A loss to pancreatic neuro endocrine tumour risk may imply a role in human islet cell proliferation [81–85].
Evidence that CDKN2A/B influences type 2 diabetes risk via non-islet mechanisms
Evidence that CDKN2A/B increases diabetes risk without impacting islet function
CDKN2A/B was not associated with proinsulin conversion to insulin [210] or the effect of ambient glycaemia on insulin secretion [211]. Although CDKN2A/B significantly impacted risk of diabetes in a Han Chinese population, this was not related to reduced HOMA-B, although other loci (CDKAL1, IGF2BP2 and SLC30A8) did impact beta cell function in this population [212]. In a relatively young cohort of European individuals tested by OGTT, CDKN2A/B was not associated with insulin secretion or glucose sensitivity, although CDKAL1 and HHEX were [212]; intriguingly, in this cohort two of three CDKN2A/B risk alleles tested (rs10757283 and rs564398 but not rs10811661) showed a trend towards reduced insulin sensitivity. In a hyperglycaemic clamp study, CDKAL1 and IGF2BP2, but not CDKN2A/B, were associated with reduced first-phase insulin secretion [213]. Complicating matters, the influence of CDKN2A/B type 2 diabetes polymorphisms on insulin secretion may depend on ambient insulin sensitivity [214].
CDKN2A/B impacts beta cell response to particular diabetes therapies
In a post-hoc analysis of the Diabetes Prevention Program (DPP), CDKN2A/B identity was not related to baseline insulin secretory capacity; however, unique among the eight loci tested, CDKN2A/B predicted response to therapy. Insulin secretion, but not insulin sensitivity, was improved in protective-allele carriers, but only in the thiazolidinedione treatment arm [215]. In another pharmacogenetics study, of 27 diabetes loci tested, only CDKN2A/B rs10811661 predicted response to therapy: the protective allele was associated with greater response to sulfonylurea therapy [216]. The CDKN2A/B locus may [217] or may not [218] influence the metabolic response to exercise.
Evidence that CDKN2A/B impacts diabetes risk through other metabolic tissues
The intriguing relationship between CDKN2A/B genotype and response to thiazolidinedione in the DPP cohort suggests activity in adipose tissue; as lipids negatively impact pancreatic beta cell proliferation and function this may represent an adipocyte–beta cell axis [123, 157, 215]. In a Japanese population, CDKN2A/B was not related to visceral fat accumulation [219], but in an Indian sibling-pair study, CDKN2A/B polymorphisms impacted fasting insulin and HOMA-IR but not HOMA-B, suggesting a primary effect on insulin sensitivity [220]. The melanoma kindred study also found that individuals haploinsufficient for p16INK4A had impaired insulin sensitivity [168]. In the Helsinki Birth Cohort, CDKN2A/B type 2 diabetes-risk polymorphisms were associated with reduced birthweight, suggesting the possibility that CDKN2A/B diabetes risk might be related to developmental impact of this locus, bringing an additional temporal variable to play [221].
Type 2 diabetes CDKN2A/B polymorphisms are not yet clinically useful
Initial attempts to use type 2 diabetes genome-wide association study (GWAS) information to predict disease risk, optimise therapeutic impact and estimate prognosis have been disappointing. Even combining risk alleles only marginally improves type 2 diabetes prediction over clinical factors [222–224]. As described above, some progress has been made using the CDKN2A/B genotype to predict response to therapy [215, 216]. Although CDKN2A/B polymorphisms are not generally associated with longevity [225], cardiovascular mortality was paradoxically reduced in individuals homozygous for the type 2 diabetes-risk allele at rs10811661 [226]. In sum, despite progress, genotype information at the CDKN2A/B locus does not yet have meaningful clinical implications for individual patients.
Future directions and unresolved questions
Great progress has been made in identifying roles played by CDKN2A/B gene products in islets and other metabolic tissues, mostly in rodents but also in humans. p16INK4A and related proteins are critical regulators of rodent beta cell mass, but whether human CDKN2A/B polymorphisms influence type 2 diabetes risk via islet biology remains uncertain. Gene regulation analyses have not yet proved a relationship between type 2 diabetes SNPs and local gene expression, but the analyses to date may not have been performed in the relevant tissue, developmental stage, metabolic milieu and/or human subpopulation. CDKN2A/B genes impact islet, adipose, muscle, liver and immune cell function, at stages ranging from in utero development to ageing. Human biological variation likely influences CDKN2A/B effects. The availability of human samples across tissues and stages is a serious limitation in this field. We are hopeful that, in the future, CDKN2A/B polymorphisms will improve diabetes understanding and inform clinical decisions.
Summary points.
Non-coding polymorphisms near the CDKN2A/B locus influence risk of several type 2 diabetes-related forms of diabetes
The CDKN2A/B locus encodes p16INK4A, p15INK4B, p14ARF and ANRIL, which regulate proliferation, oncogenesis, senescence and ageing
p16INK4A mediates an ageing-related decline in beta cell proliferation in mice, and may regulate insulin secretion independently of beta cell mass
CDKN2A/B and related genes also regulate adipocyte differentiation, inflammation, hepatic insulin clearance, fasted–fed transition and muscle metabolism
Polymorphisms at CDKN2A/B have not yet been clearly linked to local gene expression in any metabolic tissue
The mechanisms linking the CDKN2A/B locus to type 2 diabetes risk in human populations remain unknown
Acknowledgements
We thank members of the Beta Cell Biology Group at the University of Massachusetts Medical School, including R. Bortell, A. Jurczyk, C. Yang, L. Covassin, A. Rittenhouse, S. Wolfe, J. Wang, R. Zhuge, and D. Harlan of the Department of Medicine, University of Massachusetts, for helpful discussions.
Funding This work was supported by American Diabetes Association 7-14-BS-003 in collaboration with Order of the Amaranth (LCA), by NIH/NIDDK R01-DK095140 (LCA), and by the George F. and Sybil H. Fuller Foundation (LCA).
Abbreviations
- ANRIL
Antisense non-coding RNA in the INK4 locus
- ARF
Alternate reading frame
- BMI1
B cell-specific Moloney murine leukaemia virus insertion site 1
- CBX7
Chromobox homologue 7
- CDK
Cyclin-dependent kinase
- CDKN2
Cyclin-dependent kinase inhibitor 2
- DPP
Diabetes Prevention Program
- E2F
Early region 2 transcription factor
- eQTL
Expression quantitative trait locus
- EZH2
Enhancer of zeste 2
- GWAS
Genome-wide association study
- HNF
Hepatic nuclear factor
- INK
Inhibitor of cyclin-dependent kinase
- lncRNA
Long non-coding RNA
- MDM2
Mouse double minute 2 homologue
- MLL1
Mixed lineage leukaemia protein 1
- P38MAPK
p38 mitogen-activated protein kinase
- PTEN
Phosphatase and tensin homologue
- Rb
Retinoblastoma
- SNP
Single nucleotide polymorphism
- WIP1
Wild-type p53-induced phosphatase 1
Footnotes
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Contribution statement All authors reviewed the literature, drafted and revised the manuscript and approved the final submitted manuscript.
References
- 1.Hannou SA, Wouters K, Paumelle R, Staels B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: what have we learned from GWASs? Trends Endocrinol Metab. 2015;26:176–184. doi: 10.1016/j.tem.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Rutter GA. Dorothy Hodgkin Lecture 2014. Understanding genes identified by genome-wide association studies for type 2 diabetes. Diabet Med. 2014;31:1480–1487. doi: 10.1111/dme.12579. [DOI] [PubMed] [Google Scholar]
- 3.Jeck WR, Siebold AP, Sharpless NE. Review: A meta-analysis of GWAS and age-associated diseases. Aging Cell. 2012;11:727–731. doi: 10.1111/j.1474-9726.2012.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Diabetes Genetics Initiative of Broad Institute of Harvard and MIT, Lund University, and Novartis Institutes of BioMedical Research. Saxena R, Voight BF, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- 5.Scott LJ, Mohlke KL, Bonnycastle LL, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeggini E, Weedon MN, Lindgren CM, et al. Replication of genome-wide association signals in UK samples reveals risk loci for type 2 diabetes. Science. 2007;316:1336–1341. doi: 10.1126/science.1142364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duesing K, Fatemifar G, Charpentier G, et al. Strong association of common variants in the CDKN2A/CDKN2B region with type 2 diabetes in French Europids. Diabetologia. 2008;51:821–826. doi: 10.1007/s00125-008-0973-4. [DOI] [PubMed] [Google Scholar]
- 8.Gori F, Specchia C, Pietri S, et al. Common genetic variants on chromosome 9p21 are associated with myocardial infarction and type 2 diabetes in an Italian population. BMC Med Genet. 2010;11:60. doi: 10.1186/1471-2350-11-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42:579–589. doi: 10.1038/ng.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng MCY, Park KS, Oh B, et al. Implication of genetic variants near TCF7L2, SLC30A8, HHEX, CDKAL1, CDKN2A/B, IGF2BP2, and FTO in type 2 diabetes and obesity in 6,719 Asians. Diabetes. 2008;57:2226–2233. doi: 10.2337/db07-1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omori S, Tanaka Y, Takahashi A, et al. Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes. 2008;57:791–795. doi: 10.2337/db07-0979. [DOI] [PubMed] [Google Scholar]
- 12.Lee Y-H, Kang ES, Kim SH, et al. Association between polymorphisms in SLC30A8, HHEX, CDKN2A/B, IGF2BP2, FTO, WFS1, CDKAL1, KCNQ1 and type 2 diabetes in the Korean population. J Hum Genet. 2008;53:991–998. doi: 10.1007/s10038-008-0341-8. [DOI] [PubMed] [Google Scholar]
- 13.Tabara Y, Osawa H, Kawamoto R, et al. Replication study of candidate genes associated with type 2 diabetes based on genome-wide screening. Diabetes. 2009;58:493–498. doi: 10.2337/db07-1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Takeuchi F, Serizawa M, Yamamoto K, et al. Confirmation of multiple risk Loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes. 2009;58:1690–1699. doi: 10.2337/db08-1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu C, Zhang R, Wang C, et al. PPARG, KCNJ11, CDKAL1, CDKN2A-CDKN2B, IDE-KIF11-HHEX, IGF2BP2 and SLC30A8 are associated with type 2 diabetes in a Chinese population. PLoS One. 2009;4:e7643. doi: 10.1371/journal.pone.0007643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan JT, Ng DPK, Nurbaya S, et al. Polymorphisms identified through genome-wide association studies and their associations with type 2 diabetes in Chinese, Malays, and Asian-Indians in Singapore. J Clin Endocrinol Metab. 2010;95:390–397. doi: 10.1210/jc.2009-0688. [DOI] [PubMed] [Google Scholar]
- 17.Wen J, Rönn T, Olsson A, et al. Investigation of type 2 diabetes risk alleles support CDKN2A/B, CDKAL1, and TCF7L2 as susceptibility genes in a Han Chinese cohort. PLoS One. 2010;5:e9153. doi: 10.1371/journal.pone.0009153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han X, Luo Y, Ren Q, et al. Implication of genetic variants near SLC30A8, HHEX, CDKAL1, CDKN2A/B, IGF2BP2, FTO, TCF2, KCNQ1, and WFS1 in type 2 diabetes in a Chinese population. BMC Med Genet. 2010;11:81. doi: 10.1186/1471-2350-11-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chidambaram M, Radha V, Mohan V. Replication of recently described type 2 diabetes gene variants in a South Indian population. Metabolism. 2010;59:1760–1766. doi: 10.1016/j.metabol.2010.04.024. [DOI] [PubMed] [Google Scholar]
- 20.Xu M, Bi Y, Xu Y, et al. Combined effects of 19 common variations on type 2 diabetes in Chinese: results from two community-based studies. PLoS One. 2010;5:e14022. doi: 10.1371/journal.pone.0014022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Gan W, Lu L, et al. A genome-wide association study identifies GRK5 and RASGRP1 as type 2 diabetes loci in Chinese Hans. Diabetes. 2013;62:291–298. doi: 10.2337/db12-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuo JZ, Sheu WH-H, Assimes TL, et al. Trans-ethnic fine mapping identifies a novel independent locus at the 3′ end of CDKAL1 and novel variants of several susceptibility loci for type 2 diabetes in a Han Chinese population. Diabetologia. 2013;56:2619–2628. doi: 10.1007/s00125-013-3047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wei F, Cai C, Feng S, et al. TOX and CDKN2A/B gene polymorphisms are associated with type 2 diabetes in Han Chinese. Sci Rep. 2015;5:11900. doi: 10.1038/srep11900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Qian Y, Lu F, Dong M, et al. Cumulative effect and predictive value of genetic variants associated with type 2 diabetes in Han Chinese: a case-control study. PLoS One. 2015;10:e0116537. doi: 10.1371/journal.pone.0116537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chauhan G, Spurgeon CJ, Tabassum R, et al. Impact of common variants of PPARG, KCNJ11, TCF7L2, SLC30A8, HHEX, CDKN2A, IGF2BP2, and CDKAL1 on the risk of type 2 diabetes in 5,164 Indians. Diabetes. 2010;59:2068–2074. doi: 10.2337/db09-1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rees SD, Hydrie MZI, Shera AS, et al. Replication of 13 genome-wide association (GWA)-validated risk variants for type 2 diabetes in Pakistani populations. Diabetologia. 2011;54:1368–1374. doi: 10.1007/s00125-011-2063-2. [DOI] [PubMed] [Google Scholar]
- 27.Parra EJ, Below JE, Krithika S, et al. Genome-wide association study of type 2 diabetes in a sample from Mexico City and a meta-analysis of a Mexican-American sample from Starr County, Texas. Diabetologia. 2011;54:2038–2046. doi: 10.1007/s00125-011-2172-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gamboa-Meléndez MA, Huerta-Chagoya A, Moreno-Macías H, et al. Contribution of common genetic variation to the risk of type 2 diabetes in the Mexican Mestizo population. Diabetes. 2012;61:3314–3321. doi: 10.2337/db11-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lara-Riegos JC, Ortiz-López MG, Peña-Espinoza BI, et al. Diabetes susceptibility in Mayas: evidence for the involvement of polymorphisms in HHEX, HNF4α, KCNJ11, PPARγ, CDKN2A/2B, SLC30A8, CDC123/CAMK1D, TCF7L2, ABCA1 and SLC16A11 genes. Gene. 2015;565:68–75. doi: 10.1016/j.gene.2015.03.065. [DOI] [PubMed] [Google Scholar]
- 30.Cauchi S, Ezzidi I, El Achhab Y, et al. European genetic variants associated with type 2 diabetes in North African Arabs. Diabetes Metab. 2012;38:316–323. doi: 10.1016/j.diabet.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 31.Al-Sinani S, Woodhouse N, Al-Mamari A, et al. Association of gene variants with susceptibility to type 2 diabetes among Omanis. World J Diabetes. 2015;6:358–366. doi: 10.4239/wjd.v6.i2.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Turki A, Al-Zaben GS, Khirallah M, Marmouch H, Mahjoub T, Almawi WY. Gender-dependent associations of CDKN2A/2B, KCNJ11, POLI, SLC30A8, and TCF7L2 variants with type 2 diabetes in (North African) Tunisian Arabs. Diabetes Res Clin Pract. 2014;103:e40–e43. doi: 10.1016/j.diabres.2013.12.040. [DOI] [PubMed] [Google Scholar]
- 33.Lauenborg J, Grarup N, Damm P, et al. Common type 2 diabetes risk gene variants associate with gestational diabetes. J Clin Endocrinol Metab. 2009;94:145–150. doi: 10.1210/jc.2008-1336. [DOI] [PubMed] [Google Scholar]
- 34.Cho YM, Kim TH, Lim S, et al. Type 2 diabetes-associated genetic variants discovered in the recent genome-wide association studies are related to gestational diabetes mellitus in the Korean population. Diabetologia. 2009;52:253–261. doi: 10.1007/s00125-008-1196-4. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Nie M, Li W, et al. Association of six single nucleotide polymorphisms with gestational diabetes mellitus in a Chinese population. PLoS One. 2011;6:e26953. doi: 10.1371/journal.pone.0026953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwak SH, Choi SH, Jung HS, et al. Clinical and genetic risk factors for type 2 diabetes at early or late post partum after gestational diabetes mellitus. J Clin Endocrinol Metab. 2013;98:E744–E752. doi: 10.1210/jc.2012-3324. [DOI] [PubMed] [Google Scholar]
- 37.Kang ES, Kim MS, Kim CH, et al. Association of common type 2 diabetes risk gene variants and posttransplantation diabetes mellitus in renal allograft recipients in Korea. Transplantation. 2009;88:693–698. doi: 10.1097/TP.0b013e3181b29c41. [DOI] [PubMed] [Google Scholar]
- 38.Kurzawski M, Dziewanowski K, Łapczuk J, Wajda A, Droździk M. Analysis of common type 2 diabetes mellitus genetic risk factors in new-onset diabetes after transplantation in kidney transplant patients medicated with tacrolimus. Eur J Clin Pharmacol. 2012;68:1587–1594. doi: 10.1007/s00228-012-1292-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blackman SM, Commander CW, Watson C, et al. Genetic modifiers of cystic fibrosis-related diabetes. Diabetes. 2013;62:3627–3635. doi: 10.2337/db13-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Qu H-Q, Grant SFA, Bradfield JP, et al. Association analysis of type 2 diabetes loci in type 1 diabetes. Diabetes. 2008;57:1983–1986. doi: 10.2337/db08-0270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raj SM, Howson JMM, Walker NM, et al. No association of multiple type 2 diabetes loci with type 1 diabetes. Diabetologia. 2009;52:2109–2116. doi: 10.1007/s00125-009-1391-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Winkler C, Raab J, Grallert H, Ziegler A-G. Lack of association of type 2 diabetes susceptibility genotypes and body weight on the development of islet autoimmunity and type 1 diabetes. PLoS One. 2012;7:e35410. doi: 10.1371/journal.pone.0035410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Andersen MLM, Rasmussen MA, Pörksen S, et al. Complex multi-block analysis identifies new immunologic and genetic disease progression patterns associated with the residual β-cell function 1 year after diagnosis of type 1 diabetes. PLoS One. 2013;8:e64632. doi: 10.1371/journal.pone.0064632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fagerholm E, Ahlqvist E, Forsblom C, et al. SNP in the genome-wide association study hotspot on chromosome 9p21 confers susceptibility to diabetic nephropathy in type 1 diabetes. Diabetologia. 2012;55:2386–2393. doi: 10.1007/s00125-012-2587-0. [DOI] [PubMed] [Google Scholar]
- 45.Wellcome Trust Case Control Consortium Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Samani NJ, Erdmann J, Hall AS, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. 2007;357:443–453. doi: 10.1056/NEJMoa072366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broadbent HM, Peden JF, Lorkowski S, et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum Mol Genet. 2008;17:806–814. doi: 10.1093/hmg/ddm352. [DOI] [PubMed] [Google Scholar]
- 48.Helgadottir A, Thorleifsson G, Manolescu A, et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science. 2007;316:1491–1493. doi: 10.1126/science.1142842. [DOI] [PubMed] [Google Scholar]
- 49.Pasmant E, Sabbagh A, Vidaud M, Bièche I. ANRIL, a long, noncoding RNA, is an unexpected major hotspot in GWAS. FASEB J. 2011;25:444–448. doi: 10.1096/fj.10-172452. [DOI] [PubMed] [Google Scholar]
- 50.Matarin M, Brown WM, Singleton A, Hardy JA, Meschia JF, ISGS investigators Whole genome analyses suggest ischemic stroke and heart disease share an association with polymorphisms on chromosome 9p21. Stroke J Cereb Circ. 2008;39:1586–1589. doi: 10.1161/STROKEAHA.107.502963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gschwendtner A, Bevan S, Cole JW, et al. Sequence variants on chromosome 9p21.3 confer risk for atherosclerotic stroke. Ann Neurol. 2009;65:531–539. doi: 10.1002/ana.21590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burdon KP, Macgregor S, Hewitt AW, et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat Genet. 2011;43:574–578. doi: 10.1038/ng.824. [DOI] [PubMed] [Google Scholar]
- 53.Ramdas WD, van Koolwijk LME, Lemij HG, et al. Common genetic variants associated with open-angle glaucoma. Hum Mol Genet. 2011;20:2464–2471. doi: 10.1093/hmg/ddr120. [DOI] [PubMed] [Google Scholar]
- 54.Emanuele E, Lista S, Ghidoni R, et al. Chromosome 9p21.3 genotype is associated with vascular dementia and Alzheimer's disease. Neurobiol Aging. 2011;32:1231–1235. doi: 10.1016/j.neurobiolaging.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 55.Uno S, Zembutsu H, Hirasawa A, et al. A genome-wide association study identifies genetic variants in the CDKN2BAS locus associated with endometriosis in Japanese. Nat Genet. 2010;42:707–710. doi: 10.1038/ng.612. [DOI] [PubMed] [Google Scholar]
- 56.Schaefer AS, Richter GM, Groessner-Schreiber B, et al. Identification of a shared genetic susceptibility locus for coronary heart disease and periodontitis. PLoS Genet. 2009;5:e1000378. doi: 10.1371/journal.pgen.1000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Melzer D, Frayling TM, Murray A, et al. A common variant of the p16(INK4A) genetic region is associated with physical function in older people. Mech Ageing Dev. 2007;128:370–377. doi: 10.1016/j.mad.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li W-Q, Pfeiffer RM, Hyland PL, et al. Genetic polymorphisms in the 9p21 region associated with risk of multiple cancers. Carcinogenesis. 2014;35:2698–2705. doi: 10.1093/carcin/bgu203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang L, Li J, Duan F, et al. Interaction of type 2 diabetes mellitus with chromosome 9p21 rs10757274 polymorphism on the risk of myocardial infarction: a case-control study in Chinese population. BMC Cardiovasc Disord. 2014;14:170. doi: 10.1186/1471-2261-14-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ma RCW, So WY, Tam CHT, et al. Genetic variants for type 2 diabetes and new-onset cancer in Chinese with type 2 diabetes. Diabetes Res Clin Pract. 2014;103:328–337. doi: 10.1016/j.diabres.2013.12.016. [DOI] [PubMed] [Google Scholar]
- 61.Shea J, Agarwala V, Philippakis AA, et al. Comparing strategies to fine-map the association of common SNPs at chromosome 9p21 with type 2 diabetes and myocardial infarction. Nat Genet. 2011;43:801–805. doi: 10.1038/ng.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim WY, Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006;127:265–275. doi: 10.1016/j.cell.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 63.Sharpless NE, Sherr CJ. Forging a signature of in vivo senescence. Nat Rev Cancer. 2015;15:397–408. doi: 10.1038/nrc3960. [DOI] [PubMed] [Google Scholar]
- 64.Robertson KD, Jones PA. Tissue-specific alternative splicing in the human INK4A/ARF cell cycle regulatory locus. Oncogene. 1999;18:3810–3820. doi: 10.1038/sj.onc.1202737. [DOI] [PubMed] [Google Scholar]
- 65.Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature. 1994;371:257–261. doi: 10.1038/371257a0. [DOI] [PubMed] [Google Scholar]
- 66.Quelle DE, Ashmun RA, Hannon GJ, et al. Cloning and characterization of murine p16INK4A and p15INK4B genes. Oncogene. 1995;11:635–645. [PubMed] [Google Scholar]
- 67.Poi MJ, Knobloch TJ, Yuan C, Tsai M-D, Weghorst CM, Li J. Evidence that P12, a specific variant of P16(INK4A), plays a suppressive role in human pancreatic carcinogenesis. Biochem Biophys Res Commun. 2013;436:217–222. doi: 10.1016/j.bbrc.2013.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pérez de Castro I, Benet M, Jiménez M, Alzabin S, Malumbres M, Pellicer A. Mouse p10, an alternative spliced form of p15INK4B, inhibits cell cycle progression and malignant transformation. Cancer Res. 2005;65:3249–3256. doi: 10.1158/0008-5472.CAN-03-3445. [DOI] [PubMed] [Google Scholar]
- 69.Pasmant E, Laurendeau I, Héron D, Vidaud M, Vidaud D, Bièche I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007;67:3963–3969. doi: 10.1158/0008-5472.CAN-06-2004. [DOI] [PubMed] [Google Scholar]
- 70.Yap KL, Li S, Muñoz-Cabello AM, et al. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4A. Mol Cell. 2010;38:662–674. doi: 10.1016/j.molcel.2010.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Folkersen L, Kyriakou T, Goel A, et al. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS One. 2009;4:e7677. doi: 10.1371/journal.pone.0007677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burd CE, Jeck WR, Liu Y, Sanoff HK, Wang Z, Sharpless NE. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010;6:e1001233. doi: 10.1371/journal.pgen.1001233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Congrains A, Kamide K, Ohishi M, Rakugi H. ANRIL: molecular mechanisms and implications in human health. Int J Mol Sci. 2013;14:1278–1292. doi: 10.3390/ijms14011278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993;366:704–707. doi: 10.1038/366704a0. [DOI] [PubMed] [Google Scholar]
- 75.Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature. 1991;351:453–456. doi: 10.1038/351453a0. [DOI] [PubMed] [Google Scholar]
- 76.Gil J, Peters G. Regulation of the INK4B-ARF-INK4A tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 77.Singh SK, Ellenrieder V. Senescence in pancreatic carcinogenesis: from signalling to chromatin remodelling and epigenetics. Gut. 2013;62:1364–1372. doi: 10.1136/gutjnl-2012-302793. [DOI] [PubMed] [Google Scholar]
- 78.Hussussian CJ, Struewing JP, Goldstein AM, et al. Germline p16 mutations in familial melanoma. Nat Genet. 1994;8:15–21. doi: 10.1038/ng0994-15. [DOI] [PubMed] [Google Scholar]
- 79.Goldstein AM, Chan M, Harland M, et al. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J Med Genet. 2007;44:99–106. doi: 10.1136/jmg.2006.043802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Walker GJ, Hayward NK. p16INK4A and p14ARF tumour suppressors in melanoma: lessons from the mouse. Lancet. 2002;359:7–8. doi: 10.1016/S0140-6736(02)07271-9. [DOI] [PubMed] [Google Scholar]
- 81.Muscarella P, Melvin WS, Fisher WE, et al. Genetic alterations in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors: an analysis of p16/MTS1 tumor suppressor gene inactivation. Cancer Res. 1998;58:237–240. [PubMed] [Google Scholar]
- 82.Serrano J, Goebel SU, Peghini PL, Lubensky IA, Gibril F, Jensen RT. Alterations in the p16INK4A/CDKN2A tumor suppressor gene in gastrinomas. J Clin Endocrinol Metab. 2000;85:4146–4156. doi: 10.1210/jcem.85.11.6970. [DOI] [PubMed] [Google Scholar]
- 83.House MG, Herman JG, Guo MZ, et al. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann Surg. 2003;238:423–431. doi: 10.1097/01.sla.0000086659.49569.9e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lopez JR, Claessen SMH, Macville MVE, Albrechts JCM, Skogseid B, Speel E-JM. Spectral karyotypic and comparative genomic analysis of the endocrine pancreatic tumor cell line BON-1. Neuroendocrinology. 2010;91:131–141. doi: 10.1159/000254483. [DOI] [PubMed] [Google Scholar]
- 85.Speisky D, Duces A, Bièche I, et al. Molecular profiling of pancreatic neuroendocrine tumors in sporadic and Von Hippel-Lindau patients. Clin Cancer Res. 2012;18:2838–2849. doi: 10.1158/1078-0432.CCR-11-2759. [DOI] [PubMed] [Google Scholar]
- 86.Cozar-Castellano I, Harb G, Selk K, et al. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes. 2008;57:3056–3068. doi: 10.2337/db08-0393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Romagosa C, Simonetti S, López-Vicente L, et al. p16Ink4a overexpression in cancer: a tumor suppressor gene associated with senescence and high-grade tumors. Oncogene. 2011;30:2087–2097. doi: 10.1038/onc.2010.614. [DOI] [PubMed] [Google Scholar]
- 88.Villanueva A, García C, Paules AB, et al. Disruption of the antiproliferative TGF-beta signaling pathways in human pancreatic cancer cells. Oncogene. 1998;17:1969–1978. doi: 10.1038/sj.onc.1202118. [DOI] [PubMed] [Google Scholar]
- 89.Morisset J, Aliaga JC, Calvo EL, Bourassa J, Rivard N. Expression and modulation of p42/p44 MAPKs and cell cycle regulatory proteins in rat pancreas regeneration. Am J Physiol. 1999;277:G953–G959. doi: 10.1152/ajpgi.1999.277.5.G953. [DOI] [PubMed] [Google Scholar]
- 90.Kotake Y, Nakagawa T, Kitagawa K, et al. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene. 2011;30:1956–1962. doi: 10.1038/onc.2010.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang M, Chen W, Qi F, et al. Long non-coding RNA ANRIL is upregulated in hepatocellular carcinoma and regulates cell apoptosis by epigenetic silencing of KLF2. J Hematol Oncol. 2015;8:50. doi: 10.1186/s13045-015-0146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kang Y-H, Kim D, Jin E-J. Down-regulation of phospholipase D stimulates death of lung cancer cells involving up-regulation of the long ncRNA ANRIL. Anticancer Res. 2015;35:2795–2803. [PubMed] [Google Scholar]
- 93.Nie F, Sun M, Yang J, et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol Cancer Ther. 2015;14:268–277. doi: 10.1158/1535-7163.MCT-14-0492. [DOI] [PubMed] [Google Scholar]
- 94.Hayflick L, Moorhead PS. The serial cultivation of human diploid cell strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 95.Kong Y, Cui H, Ramkumar C, Zhang H. Regulation of senescence in cancer and ageing. J Aging Res. 2011;2011:963172. doi: 10.4061/2011/963172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Avrahami D, Li C, Zhang J, et al. Aging-dependent demethylation of regulatory elements correlates with chromatin state and improved β cell function. Cell Metab. 2015;22:619–632. doi: 10.1016/j.cmet.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rane SG, Cosenza SC, Mettus RV, Reddy EP. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol. 2002;22:644–656. doi: 10.1128/MCB.22.2.644-656.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Baker DJ, Childs BG, Durik M, et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016;530:184–189. doi: 10.1038/nature16932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kamijo T, Zindy F, Roussel MF, et al. Tumor suppression at the mouse INK4A locus mediated by the alternative reading frame product p19ARF. Cell. 1997;91:649–659. doi: 10.1016/s0092-8674(00)80452-3. [DOI] [PubMed] [Google Scholar]
- 100.Salas E, Rabhi N, Froguel P, et al. Role of Ink4a/Arf locus in beta cell mass expansion under physiological and pathological conditions. J Diabetes Res. 2014;2014:e873679. doi: 10.1155/2014/873679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krishnamurthy J, Ramsey MR, Ligon KL, et al. p16INK4A induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 102.Kushner JA. The role of ageing upon β cell turnover. J Clin Invest. 2013;123:990–995. doi: 10.1172/JCI64095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Perl S, Kushner JA, Buchholz BA, et al. Significant human beta-cell turnover is limited to the first three decades of life as determined by in vivo thymidine analog incorporation and radiocarbon dating. J Clin Endocrinol Metab. 2010;95:E234–E239. doi: 10.1210/jc.2010-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rankin MM, Kushner JA. Adaptive beta-cell proliferation is severely restricted with advanced age. Diabetes. 2009;58:1365–1372. doi: 10.2337/db08-1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- 106.Gregg BE, Moore PC, Demozay D, et al. Formation of a human β-cell population within pancreatic islets is set early in life. J Clin Endocrinol Metab. 2012;97:3197–3206. doi: 10.1210/jc.2012-1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Meier JJ, Butler AE, Saisho Y, et al. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saisho Y, Butler AE, Manesso E, Elashoff D, Rizza RA, Butler PC. β-cell mass and turnover in humans: effects of obesity and ageing. Diabetes Care. 2013;36:111–117. doi: 10.2337/dc12-0421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sullivan BA, Hollister-Lock J, Bonner-Weir S, Weir GC. Reduced Ki67 staining in the postmortem state calls into question past conclusions about the lack of turnover of adult human β-cells. Diabetes. 2015;64:1698–1702. doi: 10.2337/db14-1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mizukami H, Takahashi K, Inaba W, et al. Age-associated changes of islet endocrine cells and the effects of body mass index in Japanese. J Diabetes Investig. 2014;5:38–47. doi: 10.1111/jdi.12118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Salpeter SJ, Khalaileh A, Weinberg-Corem N, Ziv O, Glaser B, Dor Y. Systemic regulation of the age-related decline of pancreatic β-cell replication. Diabetes. 2013;62:2843–2848. doi: 10.2337/db13-0160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Levitt HE, Cyphert TJ, Pascoe JL, et al. Glucose stimulates human beta cell replication in vivo in islets transplanted into NOD-severe combined immunodeficiency (SCID) mice. Diabetologia. 2011;54:572–582. doi: 10.1007/s00125-010-1919-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Diiorio P, Jurczyk A, Yang C, et al. Hyperglycemia-induced proliferation of adult human beta cells engrafted into spontaneously diabetic immunodeficient NOD-Rag1null IL2rγnull Ins2Akita mice. Pancreas. 2011;40:1147–1149. doi: 10.1097/MPA.0b013e31821ffabe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cozar-Castellano I, Weinstock M, Haught M, Velázquez-Garcia S, Sipula D, Stewart AF. Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes. 2006;55:70–77. [PubMed] [Google Scholar]
- 115.Kluth O, Matzke D, Schulze G, Schwenk RW, Joost H-G, Schürmann A. Differential transcriptome analysis of diabetes-resistant and - sensitive mouse islets reveals significant overlap with human diabetes susceptibility genes. Diabetes. 2014;63:4230–4238. doi: 10.2337/db14-0425. [DOI] [PubMed] [Google Scholar]
- 116.Dhawan S, Tschen S-I, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic β-cell proliferation. Genes Dev. 2009;23:906–911. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tschen S-I, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in β-cell proliferation restricts the capacity of β-cell regeneration in mice. Diabetes. 2009;58:1312–1320. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Alonso-Magdalena P, García-Arévalo M, Quesada I, Nadal Á . Bisphenol-A treatment during pregnancy in mice: a new window of susceptibility for the development of diabetes in mothers later in life. Endocrinology. 2015;156:1659–1670. doi: 10.1210/en.2014-1952. [DOI] [PubMed] [Google Scholar]
- 119.Chen H, Gu X, Su IH, et al. Polycomb protein Ezh2 regulates pancreatic β-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23:975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Elghazi L, Balcazar N, Blandino-Rosano M, et al. Decreased IRS signaling impairs β-cell cycle progression and survival in transgenic mice overexpressing S6K in β-Cells. Diabetes. 2010;59:2390–2399. doi: 10.2337/db09-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guo N, Parry EM, Li L-S, et al. Short telomeres compromise β-cell signaling and survival. PLoS One. 2011;6:e17858. doi: 10.1371/journal.pone.0017858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Williams K, Abanquah D, Joshi-Gokhale S, et al. Systemic and acute administration of parathyroid hormone-related peptide(1–36) stimulates endogenous beta cell proliferation while preserving function in adult mice. Diabetologia. 2011;54:2867–2877. doi: 10.1007/s00125-011-2260-z. [DOI] [PubMed] [Google Scholar]
- 123.Pascoe J, Hollern D, Stamateris R, et al. Free fatty acids block glucose-induced β-cell proliferation in mice by inducing cell cycle inhibitors p16 and p18. Diabetes. 2012;61:632–641. doi: 10.2337/db11-0991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Zeng N, Yang K-T, Bayan J-A, et al. PTEN controls β-cell regeneration in aged mice by regulating cell cycle inhibitor p16ink4a. Aging Cell. 2013;12:1000–1011. doi: 10.1111/acel.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Yang K-T, Bayan J-A, Zeng N, et al. Adult-onset deletion of Pten increases islet mass and beta cell proliferation in mice. Diabetologia. 2014;57:352–361. doi: 10.1007/s00125-013-3085-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Krishnamurthy J, Torrice C, Ramsey MR, et al. Ink4a/Arf expression is a biomarker of ageing. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Köhler CU, Olewinski M, Tannapfel A, Schmidt WE, Fritsch H, Meier JJ. Cell cycle control of β-cell replication in the prenatal and postnatal human pancreas. Am J Physiol Endocrinol Metab. 2011;300:E221–E230. doi: 10.1152/ajpendo.00496.2010. [DOI] [PubMed] [Google Scholar]
- 128.Taneera J, Fadista J, Ahlqvist E, et al. Expression profiling of cell cycle genes in human pancreatic islets with and without type 2 diabetes. Mol Cell Endocrinol. 2013;375:35–42. doi: 10.1016/j.mce.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 129.Davalli AM, Perego L, Bertuzzi F, et al. Disproportionate hyperproinsulinemia, beta-cell restricted prohormone convertase 2 deficiency, and cell cycle inhibitors expression by human islets transplanted into athymic nude mice: insights into nonimmune-mediated mechanisms of delayed islet graft failure. Cell Transplant. 2008;17:1323–1336. doi: 10.3727/096368908787648137. [DOI] [PubMed] [Google Scholar]
- 130.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Cytoplasmic-nuclear trafficking of G1/s cell cycle molecules and adult human β-cell replication: a revised model of human β-cell G1/S control. Diabetes. 2013;62:2460–2470. doi: 10.2337/db12-0778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fiaschi-Taesch NM, Kleinberger JW, Salim FG, et al. Human pancreatic β-cell g1/s molecule cell cycle atlas. Diabetes. 2013;62:2450–2459. doi: 10.2337/db12-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Fiaschi-Taesch N, Bigatel TA, Sicari B, et al. Survey of the human pancreatic β-cell g1/s proteome reveals a potential therapeutic role for CDK-6 and cyclin D1 in enhancing human β-cell replication and function in vivo. Diabetes. 2009;58:882–893. doi: 10.2337/db08-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zhou JX, Dhawan S, Fu H, et al. Combined modulation of polycomb and trithorax genes rejuvenates β cell replication. J Clin Invest. 2013;123:4849–4858. doi: 10.1172/JCI69468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wong ESM, Le Guezennec X, Demidov ON, et al. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell. 2009;17:142–149. doi: 10.1016/j.devcel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 135.Halvorsen TL, Beattie GM, Lopez AD, Hayek A, Levine F. Accelerated telomere shortening and senescence in human pancreatic islet cells stimulated to divide in vitro. J Endocrinol. 2000;166:103–109. doi: 10.1677/joe.0.1660103. [DOI] [PubMed] [Google Scholar]
- 136.Ramsey MR, Krishnamurthy J, Pei X-H, et al. Expression of p16Ink4a compensates for p18Ink4c loss in cyclin-dependent kinase 4/6–dependent tumors and tissues. Cancer Res. 2007;67:4732–4741. doi: 10.1158/0008-5472.CAN-06-3437. [DOI] [PubMed] [Google Scholar]
- 137.Moritani M, Yamasaki S, Kagami M, et al. Hypoplasia of endocrine and exocrine pancreas in homozygous transgenic TGF-β1. Mol Cell Endocrinol. 2005;229:175–184. doi: 10.1016/j.mce.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 138.Stolovich-Rain M, Hija A, Grimsby J, Glaser B, Dor Y. Pancreatic beta cells in very old mice retain capacity for compensatory proliferation. J Biol Chem. 2012;287:27407–27414. doi: 10.1074/jbc.M112.350736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Sangiorgi E, Capecchi MR. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc Natl Acad Sci U S A. 2009;106:7101–7106. doi: 10.1073/pnas.0902508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fukuda A, Morris JP, Hebrok M. Bmi1 is required for regeneration of the exocrine pancreas in mice. Gastroenterology. 2012;143:821–831. e1–2. doi: 10.1053/j.gastro.2012.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Lee S-H, Piran R, Keinan E, Pinkerton A, Levine F. Induction of β-cell replication by a synthetic HNF4α antagonist. Stem Cells. 2013;31:2396–2407. doi: 10.1002/stem.1496. [DOI] [PubMed] [Google Scholar]
- 142.Rane SG, Dubus P, Mettus RV, et al. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in β-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- 143.Marzo N, Mora C, Fabregat ME, et al. Pancreatic islets from cyclin-dependent kinase 4/R24C (Cdk4) knockin mice have significantly increased beta cell mass and are physiologically functional, indicating that Cdk4 is a potential target for pancreatic beta cell mass regeneration in type 1 diabetes. Diabetologia. 2004;47:686–694. doi: 10.1007/s00125-004-1372-0. [DOI] [PubMed] [Google Scholar]
- 144.Coleman KG, Wautlet BS, Morrissey D, et al. Identification of CDK4 sequences involved in cyclin D1 and p16 binding. J Biol Chem. 1997;272:18869–18874. doi: 10.1074/jbc.272.30.18869. [DOI] [PubMed] [Google Scholar]
- 145.Kushner JA, Ciemerych MA, Sicinska E, et al. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Georgia S, Hinault C, Kawamori D, et al. Cyclin D2 Is essential for the compensatory β-cell hyperplastic response to insulin resistance in rodents. Diabetes. 2010;59:987–996. doi: 10.2337/db09-0838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Alonso LC, Yokoe T, Zhang P, et al. Glucose infusion in mice: a new model to induce beta-cell replication. Diabetes. 2007;56:1792–1801. doi: 10.2337/db06-1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Stamateris RE, Sharma RB, Hollern DA, Alonso LC. Adaptive β-cell proliferation increases early in high-fat feeding in mice, concurrent with metabolic changes, with induction of islet cyclin D2 expression. Am J Physiol Endocrinol Metab. 2013;305:E149–E159. doi: 10.1152/ajpendo.00040.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Balcazar N, Sathyamurthy A, Elghazi L, et al. mTORC1 activation regulates β-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem. 2009;284:7832–7842. doi: 10.1074/jbc.M807458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fatrai S, Elghazi L, Balcazar N, et al. Akt induces β-cell proliferation by regulating cyclin D1, cyclin D2, and p21 levels and cyclin-dependent kinase-4 activity. Diabetes. 2006;55:318–325. doi: 10.2337/diabetes.55.02.06.db05-0757. [DOI] [PubMed] [Google Scholar]
- 151.He LM, Sartori DJ, Teta M, et al. Cyclin D2 protein stability is regulated in pancreatic β-cells. Mol Endocrinol. 2009;23:1865–1875. doi: 10.1210/me.2009-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Salpeter SJ, Klochendler A, Weinberg-Corem N, et al. Glucose regulates cyclin d2 expression in quiescent and replicating pancreatic β-cells through glycolysis and calcium channels. Endocrinology. 2011;152:2589–2598. doi: 10.1210/en.2010-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Metukuri MR, Zhang P, Basantani MK, et al. ChREBP mediates glucose-stimulated pancreatic β-cell proliferation. Diabetes. 2012;61:2004–2015. doi: 10.2337/db11-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Velazquez-Garcia S, Valle S, Rosa TC, et al. Activation of protein kinase C-ζ in pancreatic β-cells in vivo improves glucose tolerance and induces β-cell expansion via mTOR activation. Diabetes. 2011;60:2546–2559. doi: 10.2337/db10-1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Takamoto I, Kubota N, Nakaya K, et al. TCF7L2 in mouse pancreatic beta cells plays a crucial role in glucose homeostasis by regulating beta cell mass. Diabetologia. 2014;57:542–553. doi: 10.1007/s00125-013-3131-6. [DOI] [PubMed] [Google Scholar]
- 156.Tarry-Adkins JL, Chen JH, Smith NS, Jones RH, Cherif H, Ozanne SE. Poor maternal nutrition followed by accelerated postnatal growth leads to telomere shortening and increased markers of cell senescence in rat islets. FASEB J. 2009;23:1521–1528. doi: 10.1096/fj.08-122796. [DOI] [PubMed] [Google Scholar]
- 157.Sharma RB, Alonso LC. Lipotoxicity in the pancreatic beta cell: not just survival and function, but proliferation as well? Curr Diab Rep. 2014;14:492. doi: 10.1007/s11892-014-0492-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fontés G, Zarrouki B, Hagman DK, et al. Glucolipotoxicity age-dependently impairs beta cell function in rats despite a marked increase in beta cell mass. Diabetologia. 2010;53:2369–2379. doi: 10.1007/s00125-010-1850-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48:58–67. doi: 10.1007/s00125-004-1605-2. [DOI] [PubMed] [Google Scholar]
- 160.Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, et al. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006;27:356–370. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- 161.Martín J, Hunt SL, Dubus P, et al. Genetic rescue of Cdk4 null mice restores pancreatic beta-cell proliferation but not homeostatic cell number. Oncogene. 2003;22:5261–5269. doi: 10.1038/sj.onc.1206506. [DOI] [PubMed] [Google Scholar]
- 162.Fiaschi-Taesch NM, Salim F, Kleinberger J, et al. Induction of human β-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes. 2010;59:1926–1936. doi: 10.2337/db09-1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Perry JRB, McCarthy MI, Hattersley AT, et al. Interrogating type 2 diabetes genome-wide association data using a biological pathway-based approach. Diabetes. 2009;58:1463–1467. doi: 10.2337/db08-1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Yaghootkar H, Stancáková A, Freathy RM, et al. Association analysis of 29,956 individuals confirms that a low frequency variant at CCND2 halves the risk of type 2 diabetes by enhancing insulin secretion. Diabetes. 2015;64:2279–2285. doi: 10.2337/db14-1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Steinthorsdottir V, Thorleifsson G, Sulem P, et al. Identification of low-frequency and rare sequence variants associated with elevated or reduced risk of type 2 diabetes. Nat Genet. 2014;46:294–298. doi: 10.1038/ng.2882. [DOI] [PubMed] [Google Scholar]
- 166.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012;44:981–990. doi: 10.1038/ng.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stamateris RE, Sharma RB, Kong Y, et al. Glucose induces mouse beta cell proliferation via IRS2, mTOR and cyclin D2 but not the insulin receptor. Diabetes. 2016 doi: 10.2337/db15-0529. doi:10.2337/d615-0529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Pal A, Potjer TP, Thomsen SK, et al. Loss-of-function mutations in the cell-cycle control gene CDKN2A impact on glucose homeostasis in humans. Diabetes. 2015;65:527–533. doi: 10.2337/db15-0602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Moreno-Asso A, Castaño C, Grilli A, Novials A, Servitja J-M. Glucose regulation of a cell cycle gene module is selectively lost in mouse pancreatic islets during ageing. Diabetologia. 2013;56:1761–1772. doi: 10.1007/s00125-013-2930-0. [DOI] [PubMed] [Google Scholar]
- 170.González-Navarro H, Vinué Á , Sanz MJ, et al. Increased dosage of Ink4/Arf protects against glucose intolerance and insulin resistance associated with ageing. Aging Cell. 2013;12:102–111. doi: 10.1111/acel.12023. [DOI] [PubMed] [Google Scholar]
- 171.Annicotte J-S, Blanchet E, Chavey C, et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol. 2009;11:1017–1023. doi: 10.1038/ncb1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Visel A, Zhu Y, May D, et al. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature. 2010;464:409–412. doi: 10.1038/nature08801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Bochenek G, Häsler R, El Mokhtari N-E, et al. The large non-coding RNA ANRIL, which is associated with atherosclerosis, periodontitis and several forms of cancer, regulates ADIPOR1, VAMP3 and C11ORF10. Hum Mol Genet. 2013;22:4516–4527. doi: 10.1093/hmg/ddt299. [DOI] [PubMed] [Google Scholar]
- 174.Svensson P-A, Wahlstrand B, Olsson M, et al. CDKN2B expression and subcutaneous adipose tissue expandability: possible influence of the 9p21 atherosclerosis locus. Biochem Biophys Res Commun. 2014;446:1126–1131. doi: 10.1016/j.bbrc.2014.03.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Horswell SD, Fryer LGD, Hutchison CE, et al. CDKN2B expression in adipose tissue of familial combined hyperlipidemia patients. J Lipid Res. 2013;54:3491–3505. doi: 10.1194/jlr.M041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Mettus RV, Rane SG. Characterization of the abnormal pancreatic development, reduced growth and infertility in Cdk4 mutant mice. Oncogene. 2003;22:8413–8421. doi: 10.1038/sj.onc.1206888. [DOI] [PubMed] [Google Scholar]
- 177.Fajas L. Re-thinking cell cycle regulators: the cross-talk with metabolism. Front Oncol. 2013;3:4. doi: 10.3389/fonc.2013.00004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Abella A, Dubus P, Malumbres M, et al. Cdk4 promotes adipogenesis through PPARγ activation. Cell Metab. 2005;2:239–249. doi: 10.1016/j.cmet.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 179.Lagarrigue S, Lopez-Mejia IC, Denechaud P-D, et al. CDK4 is an essential insulin effector in adipocytes. J Clin Invest. 2016;126:335–348. doi: 10.1172/JCI81480. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 180.Fuentes L, Wouters K, Hannou SA, et al. Downregulation of the tumour suppressor p16INK4A contributes to the polarisation of human macrophages toward an adipose tissue macrophage (ATM)-like phenotype. Diabetologia. 2011;54:3150–3156. doi: 10.1007/s00125-011-2324-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Wouters K, Cudejko C, Gijbels MJJ, et al. Bone marrow p16INK4A-deficiency does not modulate obesity, glucose homeostasis or atherosclerosis development. PLoS One. 2012;7:e32440. doi: 10.1371/journal.pone.0032440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Bantubungi K, Hannou S-A, Caron-Houde S, et al. Cdkn2a/p16Ink4a regulates fasting-induced hepatic gluconeogenesis through the PKA-CREB-PGC1α pathway. Diabetes. 2014;63:3199–3209. doi: 10.2337/db13-1921. [DOI] [PubMed] [Google Scholar]
- 183.Lee Y, Dominy JE, Choi YJ, et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature. 2014;510:547–551. doi: 10.1038/nature13267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Blanchet E, Annicotte J-S, Lagarrigue S, et al. E2F transcription factor-1 regulates oxidative metabolism. Nat Cell Biol. 2011;13:1146–1152. doi: 10.1038/ncb2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Morris AP. Fine mapping of type 2 diabetes susceptibility loci. Curr Diab Rep. 2014;14:549. doi: 10.1007/s11892-014-0549-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cotsapas C, Prokunina-Olsson L, Welch C, et al. Expression analysis of loci associated with type 2 diabetes in human tissues. Diabetologia. 2010;53:2334–2339. doi: 10.1007/s00125-010-1861-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fadista J, Vikman P, Laakso EO, et al. Global genomic and transcriptomic analysis of human pancreatic islets reveals novel genes influencing glucose metabolism. Proc Natl Acad Sci U S A. 2014;111:13924–13929. doi: 10.1073/pnas.1402665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Jarinova O, Stewart AFR, Roberts R, et al. Functional analysis of the chromosome 9p21.3 coronary artery disease risk locus. Arterioscler Thromb Vasc Biol. 2009;29:1671–1677. doi: 10.1161/ATVBAHA.109.189522. [DOI] [PubMed] [Google Scholar]
- 189.Cunnington MS, Santibanez Koref M, Mayosi BM, Burn J, Keavney B. Chromosome 9p21 SNPs associated with multiple disease phenotypes correlate with ANRIL expression. PLoS Genet. 2010;6:e1000899. doi: 10.1371/journal.pgen.1000899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Liu Y, Sanoff HK, Cho H, et al. INK4/ARF transcript expression is associated with chromosome 9p21 variants linked to atherosclerosis. PLoS One. 2009;4:e5027. doi: 10.1371/journal.pone.0005027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Holdt LM, Hoffmann S, Sass K, et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013;9:e1003588. doi: 10.1371/journal.pgen.1003588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Harismendy O, Notani D, Song X, et al. 9p21 DNA variants associated with coronary artery disease impair interferon-γ signalling response. Nature. 2011;470:264–268. doi: 10.1038/nature09753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Parker SCJ, Stitzel ML, Taylor DL, et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci U S A. 2013;110:17921–17926. doi: 10.1073/pnas.1317023110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Pasquali L, Gaulton KJ, Rodríguez-Seguí SA, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46:136–143. doi: 10.1038/ng.2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Wang X, Li W, Ma L, et al. Association study of the miRNA-binding site polymorphisms of CDKN2A/B genes with gestational diabetes mellitus susceptibility. Acta Diabetol. 2015;52:951–958. doi: 10.1007/s00592-015-0768-2. [DOI] [PubMed] [Google Scholar]
- 196.Gonzalez S, Klatt P, Delgado S, et al. Oncogenic activity of Cdc6 through repression of the INK4/ARF locus. Nature. 2006;440:702–706. doi: 10.1038/nature04585. [DOI] [PubMed] [Google Scholar]
- 197.Agherbi H, Gaussmann-Wenger A, Verthuy C, Chasson L, Serrano M, Djabali M. Polycomb mediated epigenetic silencing and replication timing at the INK4A/ARF locus during senescence. PLoS One. 2009;4:e5622. doi: 10.1371/journal.pone.0005622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Poi MJ, Drosdeck J, Frankel WL, Muscarella P, Li J. Deletions of RDINK4/ARF enhancer in gastrinomas and nonfunctioning pancreatic neuroendocrine tumors. Pancreas. 2014;43:1009–1013. doi: 10.1097/MPA.0000000000000165. [DOI] [PubMed] [Google Scholar]
- 199.Dayeh TA, Olsson AH, Volkov P, Almgren P, Rönn T, Ling C. Identification of CpG-SNPs associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56:1036–1046. doi: 10.1007/s00125-012-2815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Popov N, Gil J. Epigenetic regulation of the INK4B-ARF-INK4A locus: in sickness and in health. Epigenetics. 2010;5:685–690. doi: 10.4161/epi.5.8.12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Jacobsen SC, Brøns C, Bork-Jensen J, et al. Effects of short-term high-fat overfeeding on genome-wide DNA methylation in the skeletal muscle of healthy young men. Diabetologia. 2012;55:3341–3349. doi: 10.1007/s00125-012-2717-8. [DOI] [PubMed] [Google Scholar]
- 202.Daniel M, Tollefsbol TO. Epigenetic linkage of ageing, cancer and nutrition. J Exp Biol. 2015;218:59–70. doi: 10.1242/jeb.107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Perry JRB, Frayling TM. New gene variants alter type 2 diabetes risk predominantly through reduced beta-cell function. Curr Opin Clin Nutr Metab Care. 2008;11:371–377. doi: 10.1097/MCO.0b013e32830349a1. [DOI] [PubMed] [Google Scholar]
- 204.Peng F, Hu D, Gu C, et al. The relationship between five widely-evaluated variants in CDKN2A/B and CDKAL1 genes and the risk of type 2 diabetes: a meta-analysis. Gene. 2013;531:435–443. doi: 10.1016/j.gene.2013.08.075. [DOI] [PubMed] [Google Scholar]
- 205.Grarup N, Rose CS, Andersson EA, et al. Studies of association of variants near the HHEX, CDKN2A/B, and IGF2BP2 genes with type 2 diabetes and impaired insulin release in 10,705 Danish subjects: validation and extension of genome-wide association studies. Diabetes. 2007;56:3105–3111. doi: 10.2337/db07-0856. [DOI] [PubMed] [Google Scholar]
- 206.Hribal ML, Presta I, Procopio T, et al. Glucose tolerance, insulin sensitivity and insulin release in European non-diabetic carriers of a polymorphism upstream of CDKN2A and CDKN2B. Diabetologia. 2011;54:795–802. doi: 10.1007/s00125-010-2038-8. [DOI] [PubMed] [Google Scholar]
- 207.`t Hart LM, Simonis-Bik AM, Nijpels G, et al. Combined risk allele score of eight type 2 diabetes genes is associated with reduced first-phase glucose-stimulated insulin secretion during hyperglycemic clamps. Diabetes. 2010;59:287–292. doi: 10.2337/db09-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Dimas AS, Lagou V, Barker A, et al. Impact of type 2 diabetes susceptibility variants on quantitative glycemic traits reveals mechanistic heterogeneity. Diabetes. 2014;63:2158–2171. doi: 10.2337/db13-0949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jonsson A, Ladenvall C, Ahluwalia TS, et al. Effects of common genetic variants associated with type 2 diabetes and glycemic traits on α- and β-cell function and insulin action in humans. Diabetes. 2013;62:2978–2983. doi: 10.2337/db12-1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Kirchhoff K, Machicao F, Haupt A, et al. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetologia. 2008;51:597–601. doi: 10.1007/s00125-008-0926-y. [DOI] [PubMed] [Google Scholar]
- 211.Heni M, Ketterer C, Thamer C, et al. Glycemia determines the effect of type 2 diabetes risk genes on insulin secretion. Diabetes. 2010;59:3247–3252. doi: 10.2337/db10-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Pascoe L, Tura A, Patel SK, et al. Common variants of the novel type 2 diabetes genes CDKAL1 and HHEX/IDE are associated with decreased pancreatic β-cell function. Diabetes. 2007;56:3101–3104. doi: 10.2337/db07-0634. [DOI] [PubMed] [Google Scholar]
- 213.Groenewoud MJ, Dekker JM, Fritsche A, et al. Variants of CDKAL1 and IGF2BP2 affect first-phase insulin secretion during hyperglycaemic clamps. Diabetologia. 2008;51:1659–1663. doi: 10.1007/s00125-008-1083-z. [DOI] [PubMed] [Google Scholar]
- 214.Haupt A, Guthoff M, Schäfer SA, et al. The inhibitory effect of recent type 2 diabetes risk loci on insulin secretion is modulated by insulin sensitivity. J Clin Endocrinol Metab. 2009;94:1775–1780. doi: 10.1210/jc.2008-1876. [DOI] [PubMed] [Google Scholar]
- 215.Moore AF, Jablonski KA, McAteer JB, et al. Extension of type 2 diabetes genome-wide association scan results in the Diabetes Prevention Program. Diabetes. 2008;57:2503–2510. doi: 10.2337/db08-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ren Q, Han X, Tang Y, et al. Search for genetic determinants of sulfonylurea efficacy in type 2 diabetic patients from China. Diabetologia. 2014;57:746–753. doi: 10.1007/s00125-013-3146-z. [DOI] [PubMed] [Google Scholar]
- 217.Brito EC, Lyssenko V, Renström F, et al. Previously associated type 2 diabetes variants may interact with physical activity to modify the risk of impaired glucose regulation and type 2 diabetes: a study of 16,003 Swedish adults. Diabetes. 2009;58:1411–1418. doi: 10.2337/db08-1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ruchat S-M, Rankinen T, Weisnagel SJ, et al. Improvements in glucose homeostasis in response to regular exercise are influenced by the PPARG Pro12Ala variant: results from the HERITAGE Family Study. Diabetologia. 2010;53:679–689. doi: 10.1007/s00125-009-1630-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Hotta K, Kitamoto A, Kitamoto T, et al. Association between type 2 diabetes genetic susceptibility loci and visceral and subcutaneous fat area as determined by computed tomography. J Hum Genet. 2012;57:305–310. doi: 10.1038/jhg.2012.21. [DOI] [PubMed] [Google Scholar]
- 220.Gupta V, Vinay DG, Rafiq S, et al. Association analysis of 31 common polymorphisms with type 2 diabetes and its related traits in Indian sib pairs. Diabetologia. 2012;55:349–357. doi: 10.1007/s00125-011-2355-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Pulizzi N, Lyssenko V, Jonsson A, et al. Interaction between prenatal growth and high-risk genotypes in the development of type 2 diabetes. Diabetologia. 2009;52:825–829. doi: 10.1007/s00125-009-1291-1. [DOI] [PubMed] [Google Scholar]
- 222.van Hoek M, Dehghan A, Witteman JCM, et al. Predicting type 2 diabetes based on polymorphisms from genome-wide association studies: a population-based study. Diabetes. 2008;57:3122–3128. doi: 10.2337/db08-0425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Tam CHT, Ho JSK, Wang Y, et al. Use of net reclassification improvement (NRI) method confirms the utility of combined genetic risk score to predict type 2 diabetes. PLoS One. 2013;8:e83093. doi: 10.1371/journal.pone.0083093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Majithia AR, Florez JC. Clinical translation of genetic predictors for type 2 diabetes. Curr Opin Endocrinol Diabetes Obes. 2009;16:100–106. doi: 10.1097/med.0b013e3283292354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Congrains A, Kamide K, Hirose N, et al. Disease-associated polymorphisms in 9p21 are not associated with extreme longevity. Geriatr Gerontol Int. 2015;15:797–803. doi: 10.1111/ggi.12346. [DOI] [PubMed] [Google Scholar]
- 226.Landman GWD, van Vliet-Ostaptchouk JV, Kleefstra N, et al. Association between 9p21 genetic variants and mortality risk in a prospective cohort of patients with type 2 diabetes (ZODIAC-15) Cardiovasc Diabetol. 2012;11:138. doi: 10.1186/1475-2840-11-138. [DOI] [PMC free article] [PubMed] [Google Scholar]