

Abstract
Intestinal cancer is a heterogeneous disease driven by genetic mutations and epigenetic changes. Approximately 80% of sporadic colorectal cancers are initiated by mutation and inactivation of the adenomatous polyposis coli (APC) gene, which results in unrestrained intestinal epithelial growth and formation of adenomas. Aberrant DNA methylation promotes cancer progression by the inactivation of tumor suppressor genes via promoter methylation. Additionally, global DNA hypomethylation is often seen before the formation of adenomas, suggesting it contributes to neoplastic transformation. Previous studies employed mice with a hypomorphic mutation in DNA methyltransferase 1 (Dnmt1), which exhibited constitutive global DNA hypomethylation and decreased tumorigenesis in the ApcMin/+ mouse model of intestinal cancer. However, the consequences of intestinal-epithelial specific acute hypomethylation during ApcMin/+ tumor initiation have not been reported.
Using temporally controlled intestinal epithelial-specific gene ablation, we show that total loss of Dnmt1 in the ApcMin/+ mouse model of intestinal cancer causes accelerated adenoma initiation. Deletion of Dnmt1 precipitates an acute response characterized by hypomethylation of repetitive elements and genomic instability, which, surprisingly is followed by remethylation with time. Two months post-Dnmt1 ablation, mice display increased macroadenoma load, consistent with a role for Dnmt1 and DNA methylation in maintaining genomic stability. These data suggest that DNA hypomethylation plays a previously unappreciated role in intestinal adenoma initiation.
Keywords: Colorectal cancer, intestinal epithelium, DNA methylation, Dnmt1
Introduction
Intestinal and colorectal cancers are multifactorial diseases with various inputs including diet, environment, genetic mutations, and epigenetic abnormalities. The disease first manifests as an over-proliferation defect in the form of polyps that, if not removed, can progress to precancerous adenomas. Further transition to invasive and metastatic cancer is due to the accumulation of multiple genetic mutations and epigenetic changes that alter gene expression patterns, driving neoplastic transformation and growth (1). However, the extent to which epigenetic modifications, and specifically DNA methylation, contribute to the initiation and progression of intestinal cancer is unclear.
Neoplastic tissues frequently display global DNA hypomethylation, which can occur passively through loss of the maintenance methyltransferase, DNMT1, or actively by TET enzyme-mediated oxidation of methyl-cytosine, followed by base excision repair (2–4). Alternatively, some tumors exhibit increased DNA methylation on specific tumor suppressor gene promoters, which requires the de novo DNA methyltransferases, DNMT3A and DNMT3B. Hypermethylation of tumor suppressor gene promoters causes decreased activity of the associated genes, and promotes cancer cell growth in vitro (5). Comparison of human colon adenomas to control tissue revealed that although proximal promoter CpG islands are often hypermethylated in tumors, DNA hypomethylation is found genome-wide, including neighboring CpG island shores and repetitive elements (6,7). Hypomethylated intergenic and intronic regions appear early in the transition from normal to neoplastic (8,9), suggesting a more important role for DNA hypomethylation in cancer initiation than previously appreciated.
Previously, the role of Dnmt1 in intestinal tumorigenesis was studied in the ApcMin/+ mouse model. This paradigm mimics the hereditary human colon cancer syndrome Familial Adenomatous Polyposis (FAP), which is caused by germline mutations of the APC gene (10). Loss of heterozygosity (LOH) at the APC locus causes β-catenin stabilization and unrestricted Wnt activity, resulting in the formation of macroscopic adenomas by 3 month of age (11–13). LOH is also a common triggering mechanism for sporadic intestinal and colorectal tumor development (1), rendering the ApcMin/+ mouse a useful model for the study of intestinal cancer initiation.
Multiple hypomorphic Dnmt1 paradigms show complete block of macroscopic tumor formation (14–18). The authors concluded that Dnmt1 and DNA methylation are required for adenoma development in the ApcMin/+ model. These studies employed hypomorphic Dnmt1 mice, which express constitutively reduced Dnmt1 levels from earliest development onward in all tissues (15), including non-epithelial cells, such as myofibroblasts. Mesenchymally expressed genes have been shown to be strong modifiers of the ApcMin/+ tumor load (19), and could thus contribute to the observed phenotype in the Dnmt1-hypomorphic ApcMin/+ intestine. Furthermore, continuous DNA hypomethylation may mask acute affects during the neoplastic transformation, which occurs prior to reported observations of macroadenomas at six months of age. Due to these limitations, the precise mechanism of how DNA methylation acts within the intestinal epithelium during adenoma initiation remains to be determined.
To address this important knowledge gap, we employed a temporally controlled, intestinal epithelial-specific gene ablation model to delete Dnmt1 and decrease maintenance DNA methylation. Ablation of Dnmt1 causes an acute phenotype characterized by global DNA hypomethylation, genome instability, and apoptosis. The severe effects of Dnmt1 ablation result in a dramatic increase in adenoma initiation by accelerated loss of heterozygosity at the Apc locus. These results strongly support a role for DNA hypomethylation in chromosomal instability and tumor initiation.
Materials and Methods
Mice
Dnmt1loxP/loxP mice were provided by Rudolf Jaenisch (20), Villin-Cre-ERT2 mice were received from Sylvia Robine (21), and ApcMin/+ mice from the Jackson Laboratories (10,12). For Dnmt1 deletion experiments, Cre-recombination activity was induced by three daily intraperitoneal injections of 1.6 mg tamoxifen (Sigma) in an ethanol/sunflower oil mixture. In all experiments, littermate controls without the VillinCreERT2 transgene were also tamoxifen treated. All procedures involving mice were conducted in accordance with approved Institutional Animal Care and Use Committee protocols.
Histology
Tissues were isolated and fixed using 4% paraformaldehyde in PBS and then embedded in paraffin. Antigen retrieval was performed using the 2100 Antigen-Retriever in Buffer A (Electron Microscopy Sciences) and standard immunostaining procedures were performed for Dnmt1 (Santa Cruz), E-Cadherin (BD Transduction Lab), Sox9 (Millipore), Ki67 (BD Pharmingen), β-catenin (BD Transduction Lab), CycD1 (Biocare Medical), Epcam (Abcam), and γH2AX (Cell Signaling). TUNEL staining was performed using TUNEL Label and Enzyme (Roche) and AlexaFluor 555-aha-dUTP (Life Technologies). Percentages of TUNEL+ nuclei were determined by counting the number of nuclei positive for TUNEL staining, and dividing by the total number of nuclei in the image. The same method was performed to quantify the percentage of γH2AX+ cells. All microscopy was performed on a Nikon Eclipse 80i.
Laser Capture Microdissection
Tumor and non-tumor epithelial cell DNA was collected using a Leica LMD7000 Laser Microdissection microscope and the Arcturus PicoPure DNA isolation kit (Applied Biosystems).
Targeted Bisulfite Sequencing
100 ng of mouse genomic DNA was bisulfite converted using the Epitect bisulfite kit (Qiagen). Template DNA was amplified using KAPA HIFI Uracel+ (KAPA) with primers directed to the LINE1 elements (22), the H19 imprinting control region, Olfm4 and Hes1 (23), Vdr, Hic1, Sfrp5, Mgmt, Dusp6, c-Fos, Msln, and S100a4. Primer sets and regions subsequently sequenced can be found in Supplemental Table 1. Sequencing libraries were prepared and analyzed using the BiSPCR2 strategy (LINE1, H19) or the standard protocol (Vdr, Hic1, Sfrp5, Mgmt, Dusp6, c-Fos, Msln, S100a4), described previously (23,24).
Tumor Evaluation
Macroscopic tumors were counted immediately after tissue isolation using a stereoscope. Adenoma size was calculated by measuring the width of neoplastic lesions on H&E stained small intestine sections using ImageJ. Histological pathology was performed by the Comparative Pathology Core at the University of Pennsylvania School of Veterinary Medicine following the guidelines as described previously (25). LOH was determined as described previously (26).
Phosphorylated H3 (pH3) cell cycle quantification
ApcMin/+;Dnmt1loxP/loxP;Villin-Cre-ERT2 mutants and sibling ApcMin/+;Dnmt1loxP/loxP controls were injected with tamoxifen at 1 month of age. Small intestines were collected 1 week, 1 month, and 2 months following tamoxifen administration, in separate cohorts of mice. Following fixation, tissue sections were co-stained for phosphorylated histone H3 (PH3) to identify cells in M-phase, and β-catenin to identify regions of hyper-proliferative neoplastic epithelium. We counted the number of PH3+ nuclei, and divided it by the total number of nuclei in the respective region. This calculation was also performed for neoplastic intestinal epithelium at both 1 month and 2 months post-tamoxifen time-points, for controls and mutants.
q-RTPCR
Intestines isolated 1 week post-tamoxifen were gently scraped to remove villi, and treated with EDTA to isolate crypt cells. Macroscopic tumors and adjacent normal tissue, from intestines isolated 2 months post-tamoxifen, were visualized and dissected using a stereoscope. RNA was extracted using the Trizol RNA isolation protocol (Invitrogen), followed by RNA cleanup using the RNeasy Mini Kit (Qiagen). mRNA expression was measured using quantitative RT-PCR, as described previously (27). The SYBR green qPCR master mix (Agilent) was used in all qPCR reactions, and the fold change was calculated relative to the geometric mean of Tbp and β-Actin, using the ΔCT method. The method of normalizing to the geometric mean of a set of reference genes has been described previously (28). Primer sequences can be found in Supplemental Table 1.
Results and Discussion
Deletion of Dnmt1 in the intestinal epithelium of adult ApcMin/+ mice induces acute global hypomethylation
To determine the role of Dnmt1 in intestinal adenoma initiation, we employed ApcMin/+;Dnmt1loxP/loxP;Villin-Cre-ERT2 mice to inducibly delete Dnmt1 throughout the intestinal epithelia. ApcMin/+;Dnmt1loxP/loxP;Villin-Cre-ERT2 mice and their ApcMin/+;Dnmt1loxP/loxP siblings (referred to as “mutants” and “controls,” respectively) were injected with tamoxifen at four weeks of age to induce Dnmt1 deletion (Supplemental Figure 1). Based on our previous analysis of Dnmt1 ablation in the intestinal epithelium, we isolated the small intestine one week after tamoxifen treatment (23). To address the role of Dnmt1 in adenoma development, we analyzed a second cohort of mice in which we harvested the intestine and colon two months post-tamoxifen treatment, at three months of age. By three months of age, ApcMin/+ mice display macroscopic lesions (11,12). Thus, two-months post tamoxifen allowed us to contrast adenoma development in Dnmt1 mutants with ApcMin/+;Dnmt1loxP/loxP controls. At one week and two months following tamoxifen treatment, Dnmt1 deletion was effective throughout the mutant epithelia of small intestine (Figure 1A–D,G). Dnmt1 deletion was also maintained in mutant adenomas (Figure 1E–F,H). Since ApcMin/+ mice exhibit tumors predominantly in the small intestine (12,29), we focused our studies on adenoma development in the small intestinal epithelium. First, we analyzed the effects of Dnmt1 deletion in the non-neoplastic epithelium of ApcMin/+ mice.
Figure 1. Conditional ablation of Dnmt1 in the ApcMin/+ intestinal epithelium.

(A–F) Dnmt1 deletion is maintained one week (B) and two months (D) after tamoxifen treatment in the adult mouse small intestinal epithelium. Immunohistochemical staining of Dnmt1 protein is evident in the crypt epithelia located just above the submucosa (outlined in black) of control mice at both time points (A, C), but is absent in mutant mice (B, D). Neoplastic epithelia (outlined in red) display high levels of Dnmt1 protein in control animals (E). Deletion of Dnmt1 protein is maintained in the neoplastic epithelium (outlined in red) in mutant animals two months after tamoxifen treatment (F).
(G–H) q-RTPCR analysis comparing the relative gene expression levels of Dnmt1 in controls and mutants one-week (G) and two months (H) after tamoxifen treatment (n=3–5 per genotypes). Compared to controls, Dnmt1 mutants express significantly lower levels of Dnmt1 and deletion is maintained in macroscopic tumors isolated two-months post-tamoxifen. Gene expression is calculated relative to the geometric mean of TBP and β-actin.
All scale bars are 50 µm. *P<0.05, **P<0.01, Student’s t-test.
To estimate global DNA methylation levels, we performed targeted bisulfite sequencing of the H19 imprinting control region (ICR) and the Long Interspersed Nucleotide Element 1 (LINE1). The process of imprinting is dependent on DNA methylation, and refers to the methylation of alleles in a parental-specific manner in order to restrict gene expression to one allele (30). The H19 imprinting control region is methylated on the paternally inherited allele, thereby ensuring maternal allele expression (31). LINE1 retrotransposon loci comprise approximately 20% of the mouse and human genome, are maintained in a highly methylated state to inhibit transposase activity, and are a representative of genome-wide methylation levels (32–34). We employed laser capture microdissection (LCM) to collect crypts from mutant and control intestines one week following tamoxifen treatment. H19 methylation was maintained despite loss of Dnmt1 (Figure 2A), implicating a role for Dnmt3a and Dnmt3b in maintaining genomic imprints, as suggested previously (35,36). Strikingly, within one week following Dnmt1 deletion, we observed a 35% reduction in DNA methylation at the LINE1 loci in mutant crypts compared to the control crypt epithelium (Figure 2B). Thus, Dnmt1 is required acutely to maintain DNA methylation levels on LINE1 elements, but is not required for maintenance of imprinted loci in the rapidly proliferating intestinal epithelium.
Figure 2. DNA methylation dynamics following Dnmt1 deletion.

Targeted bisulfite sequencing of the H19 imprinting control region and the LINE1 repetitive elements was performed to estimate maintenance methylation of Dnmt1-deficient intestinal crypt cells. Crypt epithelium was isolated by laser capture microdissection from controls and mutants at one week (n=3–4 per group), one month (n=2 for controls, n=4 for mutants), or two months (n=4–7 per group) following tamoxifen treatment.
(A,C,E) Targeted bisulfite sequencing of six CpGs in the H19 imprinting control region (Chr7: 149,766,621-149,766,690). At one week (A), one month (C) and two months (E) following tamoxifen administration, H19 methylation levels are comparable between Dnmt1 mutants and controls.
(B,D,F) Targeted bisulfite sequencing of nine CpGs in the LINE1 repetitive elements (NCBI Accession #D84391: 976-1,072). LINE1 methylation levels one week after tamoxifen injection are significantly reduced in crypts of mutant mice compared to controls (B). One month (D) and two months (F) after tamoxifen-induced ablation of Dnmt1, global DNA methylation levels have been restored to baseline.
For all graphs, data are presented as average±SEM. **P<0.01 by two-tailed Student’s t-test.
To determine if global hypomethylation of repetitive elements was sustained over time, we isolated crypt epithelium from mutants and controls one month and two months following tamoxifen treatment. Surprisingly, we found no differences in methylation levels in all conditions tested, demonstrating that DNA methylation had been fully restored after temporary loss following Dnmt1 ablation (Figure 2C–F). These results were unexpected since Dnmt1 deletion was maintained at two months after tamoxifen administration (Figure 1C–F). Recovery of DNA methylation was likely driven by the de novo DNA methyltransferases Dnmt3a and 3b, which are also expressed in the intestinal epithelium (23).
Dnmt1 mutants display increased tumor initiation
One month and two months following tamoxifen injection, the small intestine was examined for neoplastic transformation. Surprisingly, hematoxylin and eosin staining revealed a dramatic increase in the number of neoplastic lesions throughout the mutant small intestine at two months, but not one month, post-tamoxifen (Figure 3A–D). Indeed, mutants displayed >6-fold more macroscopic adenomas compared to controls at two months following tamoxifen injection (Figure 3E; n=17–20 per group). In addition, mutant small intestinal tumors were on average twice as large as those of controls at two months post-tamoxifen injection (Figure 3F; n=5–8 per group). Furthermore, we observed 58% incidence of neoplastic transformation in colons of mutant mice (n=12) compared to 14% in controls (n=7) (Supplemental Figure 2).
Figure 3. Deletion of Dnmt1 in adult ApcMin/+ mice accelerates adenoma initiation.
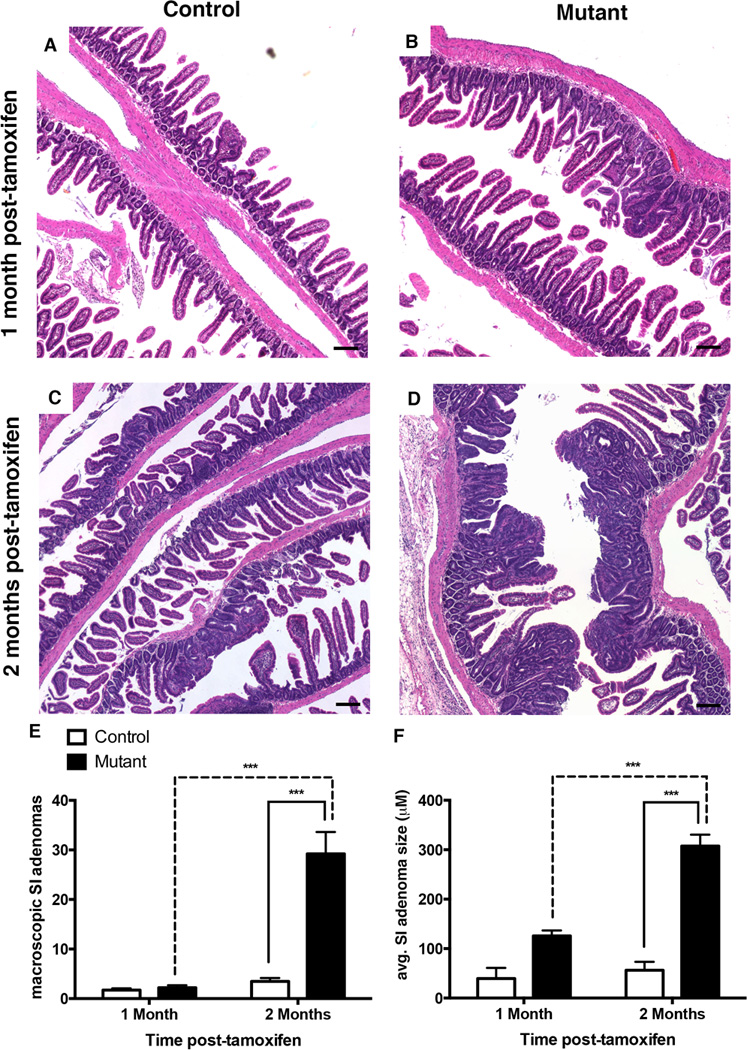
(A–D) Hematoxylin and Eosin (H&E) staining of control and mutant intestinal epithelium one month (A–B) and two months (C–D) after tamoxifen injection. H&E staining shows disruption of normal intestinal epithelial morphology in the small intestine of tamoxifen-treated mutants compared to control mice (B versus A, D versus C). Scale bars are 50µm.
(C) Intestinal epithelial-specific Dnmt1 deletion causes increased incidence of macroscopic tumors. Total number of macroscopic tumors was counted throughout the entire small intestine of mutant and control mice, one month (n=5–7 per group) and two months (n=17–20 per group) following tamoxifen-treatment.
(E) Dnmt1 deletion results in increased size of neoplastic lesions in the small intestine two months after tamoxifen treatment. Neoplastic lesions were measured in control and mutant mice at one month (n=2 for controls, n=3 for mutants) and two months (n=5 for controls, n=8 for mutants) following tamoxifen injection.
For all graphs, data are presented as average±SEM; ***P<0.001 by one-way ANOVA
Previously, it had been reported that partial loss of Dnmt1 produces a block in the progression of adenomas in the ApcMin/+ mouse paradigm (14–18), which is in sharp contrast to our findings of an increased number of macroscopic lesions in the ApcMin/+;Dnmt1loxP/loxP;VillinCreERT2 deletion model. To confirm that these lesions are bona fide adenomas, we consulted with a pathologist to perform histopathologic assessment on the intestines of mutant and control mice, one month and two months following tamoxifen treatment at four weeks of age. Using criteria described by Biovin et al. (25), we found 15-fold more lesions that had progressed to adenomas in mutant mice compared to ApcMin/+ controls (Supplemental Figure 3B) at two months post-tamoxifen injection. At one-month post-tamoxifen, all lesions observed in both controls and Dnmt1-mutants displayed similar histopathology, and were characterized as gastrointestinal intraepithelial neoplasias (Supplemental Figure 3A).
DNA hypomethylation is observed at specific genes in tumors
DNA methylation is important for maintaining appropriate intestinal stem cell gene expression, and loss of Dnmt1 in the intestinal epithelium results in increased crypt cell proliferation (23). Previously, we observed that one week following deletion of Dnmt1, DNA methylation was decreased at enhancers associated with genes critical for intestinal stem cell proliferation, Hes1 and Olfm4 (23). Thus, altered DNA methylation could be responsible for the dramatic increase in lesions observed in our mutant animals. We employed LCM to collect crypt epithelia and neoplastic tumors from mutant and control intestines two months following tamoxifen treatment. We employed targeted bisulfite sequencing of the H19 ICR and LINE1 loci to determine if global methylation levels were changed in tumors compared to epithelia in mice two months following tamoxifen injection. Interestingly, DNA methylation of the H19 ICR and LINE1 loci were unchanged in all conditions (Figure 4A–B). Control mice displayed increased methylation at Hes1 and Olfm4 in tumors compared to adjacent epithelia (Figure 4C). Mutant mice exhibited little change in methylation when comparing control and mutant epithelia (Figure 4C). However, mutant tumors are significantly hypomethylated at the Hes1 enhancer compared to control tumors (Figure 4C). Furthermore, mutant tumors are significantly hypomethylated at both Hes1 and Olfm4 loci compared to adjacent non-neoplastic epithelia (4C, dashed lines). These data are intriguing because Hes1 expression is increased in stem-cell like cells in colon cancer (37), and our results suggest that DNA methylation may play a role in activating Hes1 expression in tumors.
Figure 4. Dnmt1 mutant tumors show increased hypomethylation at potential oncogenes.

Targeted bisulfite sequencing was performed to estimate maintenance methylation of Dnmt1-deficient tumors, relative to controls tumors and to adjacent non-neoplastic crypt epithelium. Tumors and crypt epithelium were isolated by laser capture microdissection from controls and mutants two months following tamoxifen treatment.
(A–B) Analysis of nine CpGs in the LINE1 repetitive elements (NCBI Accession #D84391: 976-1,072) revealed no alterations in methylation of the repetitive transposable elements (A). Analysis of six CpGs in the H19 imprinting control region (Chr7: 149,766,621-149,766,690) showed that H19 methylation levels are comparable between Dnmt1 mutants and controls, in both tumor and non-tumor epithelium.
(C) Potential enhancer regions of genes involved in intestinal stem cell identity and proliferation, Olfm4 and Hes1 were analyzed for methylation changes in all conditions. Regional averages of seven CpGs in the Olfm4 enhancer (chr14:80399983-80400271) and six CpGs in the Hes1 enhancer (chr16:30055572-30055738) are shown. The Hes1 enhancer shows increased methylation in control tumors compared to adjacent tissue, which is abolished by Dnmt1 deletion.
(D) Genes with cancer-specific promoter hypermethylation, Vdr, Hic1, Sfrp5, Mgmt, were analyzed for methylation changes in all conditions. Regional averages of eight CpGs in the Vdr promoter (chr15:97731208-97731590), sixteen CpGs in Hic1 promoter (chr11:74983743-74983918), sixteen CpGs in the Sfrp5 promoter (chr19:42276328-42276453), and nineteen CpGs in Mgmt promoter (chr7:144086076-144086345) are shown. Only Vdr and Mgmt displayed significant hypermethylation in control tumors compared to adjacent tissue. Loss of Dnmt1 caused significant loss of hypermethylation at both loci suggesting hypermethylation of these genes is not required for tumor progression.
(E) Genes with cancer-specific promoter hypomethylation, Dusp6, c-Fos, Msln, S100a4, were analyzed for methylation changes in all conditions. Regional averages of eight CpGs in the Dusp6 promoter (chr10:98728738-98729028), fourteen CpGs in the c-Fos promoter (chr12:86817243-86817533), six CpGs in the Msln promoter (chr17:25891502-25891643), and three CpGs in the S100a4 promoter (chr3:90407319-90407489) are shown. Only Dusp6 and c-Fos displayed significant hypomethyation in control tumors compared to adjacent tissue. Dnmt1 deletion caused even more drastic loss of methylation at both promoters suggesting that loss of Dnmt1 may be driving activation of potential oncogenes during tumor growth.
Aberrant DNA methylation drives intestinal cancer cell growth in multiple paradigms (14–18) and could be responsible for the dramatic increase in lesion growth observed in our mutant animals. Hypermethylation of tumor-suppressor genes allows unrestrained growth and is a hallmark of the colorectal cancer CpG island methylator phenotype (CIMP) (38). We tested promoter methylation of four genes that have been previously shown to be hypermethylated in adenomas, Vdr, Hic1, Sfrp5 and Mgmt (39–43). To our surprise, only Vdr and Mgmt showed increased methylation in adenomas, relative to non-tumor epithelia (Figure 4D). Dnmt1 deficiency reduced methylation in mutant tumors compared to control tumors in an inconsistent manner (Figure 4D), suggesting that hypermethylation at these loci is not required for tumor growth. DNA hypomethylation has also been reported at several potential oncogenes including Dusp6, c-Fos, Msln and S100a4 (43–46). Although none of these gene promoters show significant loss of methylation in tumors compared to the respective normal epithelia, we observed significantly decreased methylation at Dusp6 and c-Fos in Dnmt1-mutant relative to control adenomas (Figure 4E). c-Fos is a potent driver of colon cancer cell growth (47), and its expression is regulated by DNA hyomethylation in several cancer types (48,49). These data suggest that hypomethylation at oncogenes may be driving tumor growth in Dnmt1-deficient ApcMin/+ mutant animals.
Dnmt1-null tumors are initiated by loss of heterozygosity and Wnt activation
We further investigated tumor initiation to determine whether the observed changes in DNA methylation were a causative factor of neoplastic transformation. Adenoma formation in the ApcMin/+ mouse model is driven by loss of heterozygosity (LOH) at the Apc locus, which causes nuclear accumulation of β-catenin and activation of Wnt signaling (13). LOH is observed in virtually all ApcMin/+ intestinal tumors (26,50). To characterize adenoma initiation by LOH in detail, we employed laser capture microdissection to isolate neoplastic and normal intestinal epithelial cells from mutant mice and their control siblings, two months following tamoxifen treatment. Since the number of macroscopic lesions found in controls at one-month post-tamoxifen is very small (Figure 3E, Supplementary Figure 3), we focused on the two-month time-point for LOH analysis. DNA was isolated from these samples (approximately 1,000 cells per sample) and the status of the Apc alleles was examined as described previously (26). Mutant tumors displayed LOH at a rate similar to that of control tumors (Figure 5A), demonstrating that Dnmt1-mutant ApcMin/+ mice initiate tumors by the same mechanism.
Figure 5. Dnmt1-deficient ApcMin/+ intestinal tumors display increased Wnt signaling.
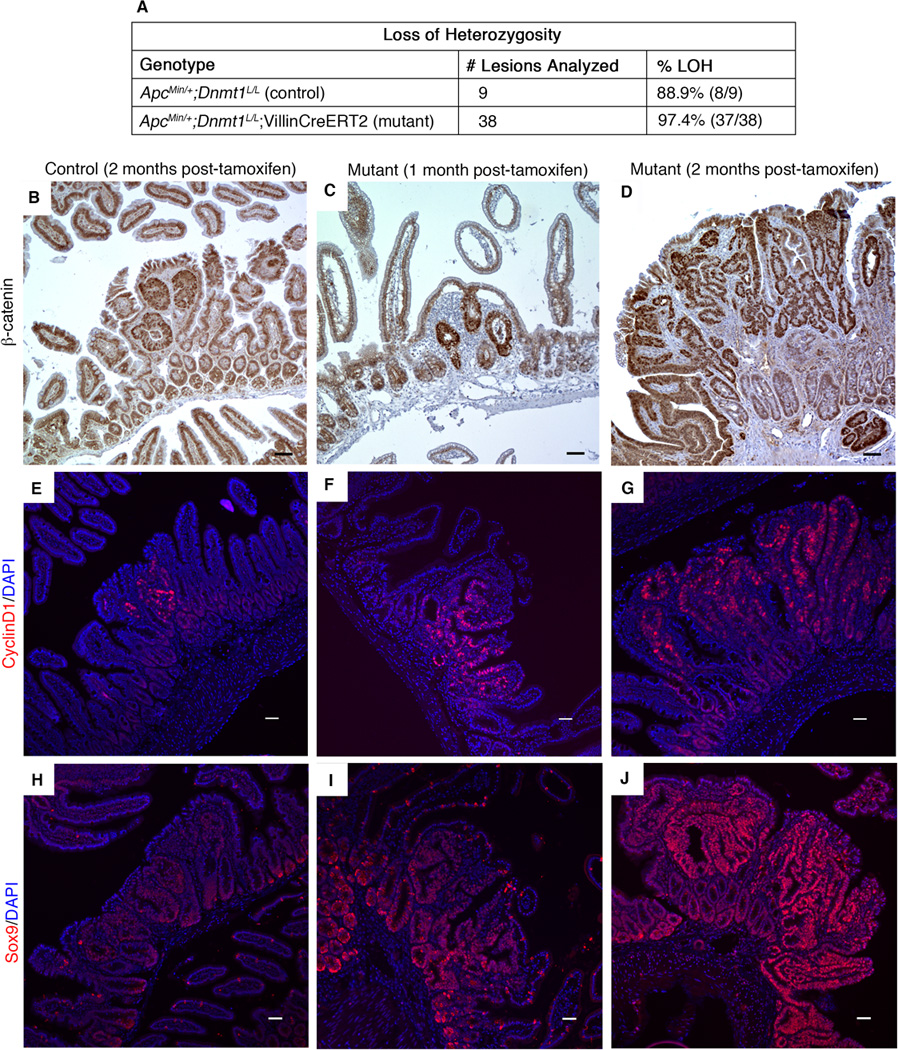
(A) Control and mutant mice were tamoxifen treated at 4 weeks of age, and intestines were harvested two months later. Intestinal tumors from control and mutant mice were isolated by laser capture microdissection for PCR testing of loss of heterozygosity (LOH) at the Apc locus. Results of PCR testing demonstrate that mutant tumors display LOH at a rate similar to that of controls.
(B–J) Control and mutant mice were tamoxifen treated at 4 weeks of age, and intestines were harvested one month or two months later for immunostaining analysis of Wnt signaling targets.
(B–D) Nuclear β-catenin, as visualized by immunohistochemical staining, is increased in Dnmt1-mutant intestine 2 months after tamoxifen treatment (D), compared to tamoxifen-treated, age-matched control small intestine (B). One month following Dnmt1 ablation, mutant small intestine displays nuclear β-catenin levels similar to that observed in the 2-month controls (C compared to B, respectively).
(E–G) Expression of the Wnt signaling target CyclinD1 (CycD1) is increased in Dnmt1-deficient tumors (G) relative to age-matched control tumors (E). At one-month post-Dnmt1 ablation, CycD1 staining appears slightly increased compared to the 2-month controls (F versus E, respectively). CycD1 (red), DAPI (blue).
(H–J) Sox9 immunofluorescent staining shows increased Sox9 levels in Dnmt1-deficient tumors (J) compared to control tumors (H) two months following tamoxifen treatment. At one month after Dnmt1 ablation, increased Sox9 expression is visible by immunostaining (I compared to control, H). (Sox9 (red), DAPI (blue). All scale bars are 50µm.
Several lines of evidence confirm that the numerous adenomas that develop in the Dnmt1-deficient ApcMin/+ mice are the result of Wnt pathway activation. Mutant adenomas at both one month and two months following tamoxifen treatment exhibited increased nuclear β-catenin protein (Figure 5B–D) relative to two-month controls. Multiple Wnt signaling targets, such as CyclinD1 (Figure 5E–G) and Sox9 (Figure 5H–J) were also strongly activated. These data demonstrate that total loss of Dnmt1 in the mature small intestinal epithelium promotes neoplastic progression, most likely through a mechanism that enhances LOH at the ApcMin/+ locus.
Dnmt1-mutant mice display acute increases in proliferation and apoptosis
Due to the dramatic difference in lesion size and hypomethylation at potential oncogenes observed in the inducible Dnmt1-mutant intestine, we investigated whether Dnmt1-deficiency had a direct impact on cell proliferation in ApcMin/+ adenomas or the non-neoplastic crypt epithelium. We determined percentages of control and mutant epithelial cells in M-phase by staining for phosphorylated-H3 (pH3), at one week, one month, and two months post-tamoxifen treatment. One week post-tamoxifen (Figure 6A–C), the percentage of pH3+ nuclei was significantly increased in the Dnmt1-deficient epithelium relative to control (n=4 per group). Interestingly, at one month (Figure 6D–F) and two months (Figure 6G–I) following tamoxifen injection, the proliferation rate was equivalent in Dnmt1-positive or -negative tumors, and in the adjacent crypt epithelium.
Figure 6. Loss of Dnmt1 in the ApcMin/+ intestine causes an acute increase in crypt cell proliferation.
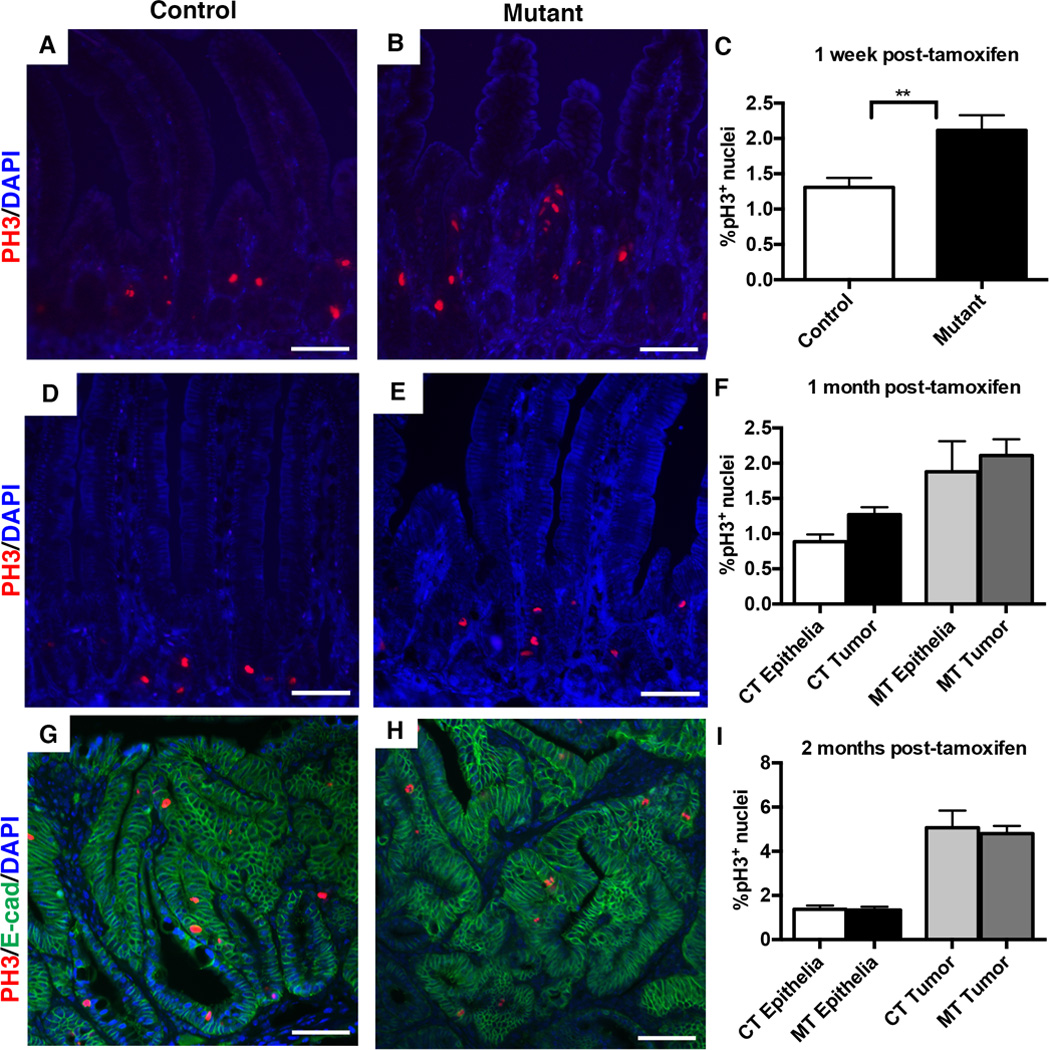
Percentage of cells in mitosis was determined by immunostaining and counting of phospho-H3 positive (pH3+) nuclei in the small intestine one week, one month, and two months following tamoxifen treatment. The percentage of pH3+ cells was calculated as the number of pH3+ nuclei, divided by the total number of epithelial nuclei.
(A–C) One week following tamoxifen injection, mutants displayed a significantly increased percentage of pH3+ cells (C, n=4 per genotype). Representative images of pH3 immunostaining are shown for controls (A) and Dnmt1 mutants (B).
(D–F) One month after tamoxifen treatment, mutants (E) and controls (D) displayed comparable percentages of pH3+ cells, in both tumors and non-cancerous epithelia (F). 24 tumors across 5 mice were counted for Dnmt1-mutants; 4 tumors across 4 mice were counted for controls. ~1,500 non-cancerous cells were counted from the same biological replicates to calculate pH3 frequency in non-tumor tissue.
(G–I) Two months after tamoxifen treatment, mutants (H) and controls (G) displayed comparable percentages of pH3+ cells, in both tumors and non-cancerous epithelia (I). 24 tumors across 7 mice were counted for Dnmt1-mutants; 8 tumors across 3 mice were counted for controls. Non-tumor epithelia quantified as in (D–F). E-cadherin (green) outlines the intestinal epithelium.
All scale bars are 50µm. For all graphs, data are presented as average±SEM; **P<0.01 by Students’s t-test (C). One-way ANOVA was performed for (F, I), and the results were not significant.
We also examined the small intestine at all three time-points for signs of cell death using terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL), which detects DNA fragmentation in apoptotic cells. We quantified the percent of epithelial cells that were TUNEL+, similar to the pH3 staining quantification. Mutants one week after tamoxifen treatment displayed a significantly higher proportion of TUNEL+ cells compared to controls (Figure 7A–C). Both control and mutant adenomas exhibited only rare epithelial cells positive for TUNEL, and we found no differences in cell death between mutant and control neoplastic and non-neoplastic epithelia, at one month (Figure 7D–F) or two months (Figure 7G–I) following tamoxifen administration. However, many subepithelial cells were TUNEL positive (Figure 7D–E, 7G–H). These data demonstrate that loss of Dnmt1 in the adult intestinal epithelium in vivo has acute temporary effects on intestinal proliferation and apoptosis, but no effect on ApcMin/+ adenoma proliferation or cell death. Overall, our results suggest that the increase in adenoma number and size found in the Dnmt1-mutant mice is driven by accelerated tumor initiation through loss of heterozygosity (LOH).
Figure 7. Loss of Dnmt1 results in a short-term increase in intestinal epithelial apoptosis, but is not altered one month or two months following Dnmt1 deletion.
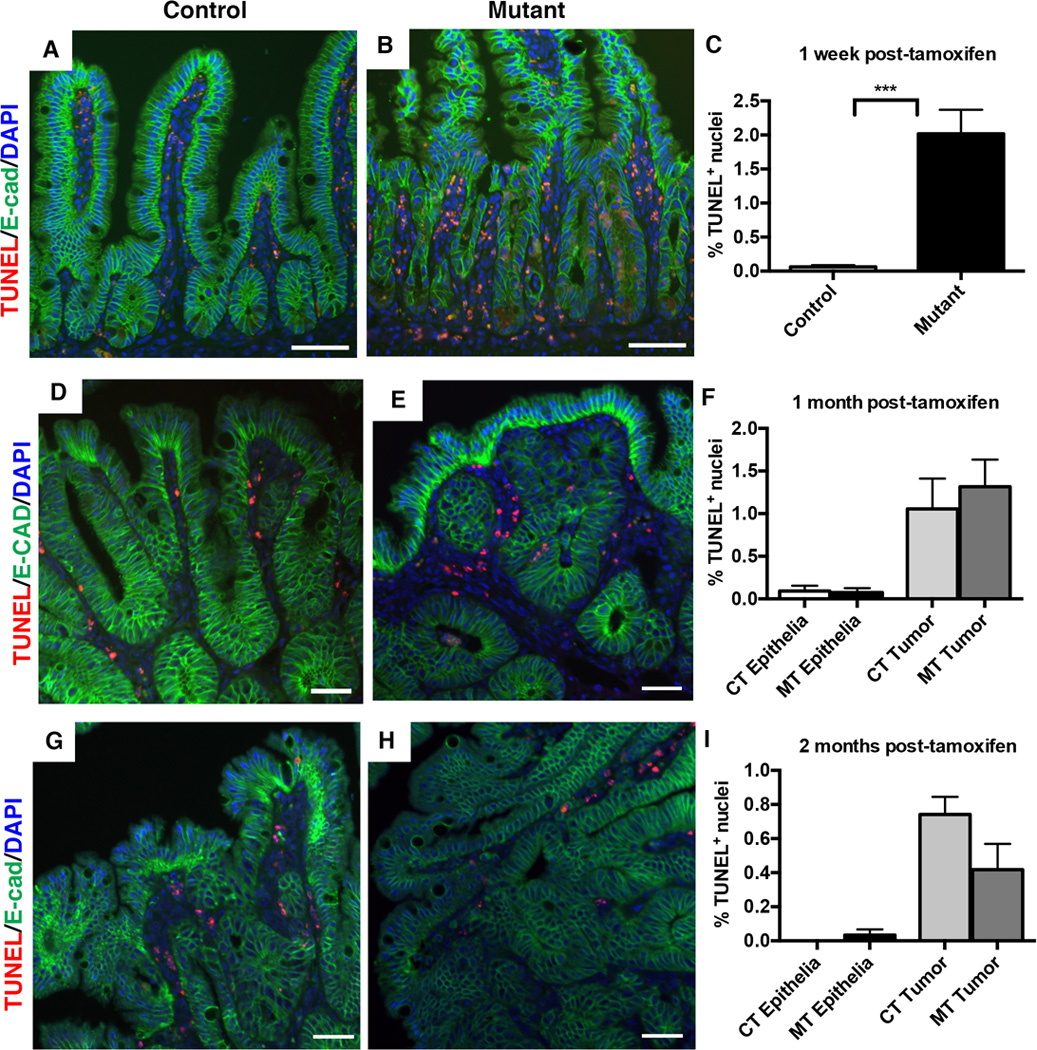
TUNEL staining (red) was performed to detect apoptotic cells, with E-cadherin (green) to outline the epithelium. The percentage of apoptotic cells was determined by counting of the TUNEL-positive (TUNEL+) nuclei in the small intestine one week, one month, and two months following tamoxifen treatment. The percentage of TUNEL+ cells was calculated as the number of TUNEL+ nuclei, divided by the total number of epithelial nuclei.
(A–C) One week following tamoxifen treatment, Dnmt1 mutants have significantly higher rates of apoptosis compared to controls (B versus A; quantitation in C). N=4 per group. ***P<0.001 by Students’s t-test.
(D–F) One month after tamoxifen injection, both tumor and non-tumor epithelia contain TUNEL-positive cells, which is unchanged in mutant mice (D versus E, quantitation in F). N=3 biological replicates per group, with 8 tumors counted from controls and 14 tumors counted from mutants.
(G–I) Two months after tamoxifen injection, both tumor and non-tumor epithelia contain TUNEL-positive cells at low frequency in control mice, which is unchanged in mutant mice (H versus G, quantitation in I). N=3–5 biological replicates per group; two tumors were counted from controls, and 10 tumors were counted from mutants.
All scale bars are 50µm. For all graphs, data are presented as average±SEM. One-way ANOVA test was performed in (F, I), and results were not significant.
Dnmt1-deficient ApcMin/+ intestinal epithelia exhibit increased genomic instability
Next, we considered the molecular mechanisms behind the accelerated LOH we observed in the Dnmt1-deficient intestinal epithelium. We hypothesized that Dnmt1’s established role in the preservation of genomic stability might contribute to the phenotype. DNA methylation has been linked to mismatch repair (MMR) deficiency and genomic instability in multiple contexts, in both cell lines and in disease. In the HCT116 colorectal cancer cell line, ablation of catalytically active DNMT1 causes cell cycle arrest and apoptosis due to increased chromosomal instability (51,52). In mouse ES cells, loss of Dnmt1 also causes global hypomethylation and increased mutation rates (53).
To determine levels of DNA damage in mutant and control mice, we performed γH2AX staining, which visualizes DNA double-strand breaks as a marker of chromosomal instability. One week following tamoxifen administration, we discovered a dramatic increase in the fraction of γH2AX+ epithelial cells in Dnmt1-mutant versus controls, which contained no γH2AX+ epithelial cells (Figure 8A–C). One month post tamoxifen injection, mutants and controls displayed a similar number of γH2AX+ nuclei (Figure 8D–F). There was a significant difference between neoplastic and non-neoplastic epithelium in control mice, and a similar statistically relevant change was observed in mutants (Figure 8F). Two months following tamoxifen treatment, non-tumor epithelia in both the control and mutant mice showed very little γH2AX (Figure 8G–I). Although we found many more epithelial cells within Dnmt1-deficient tumors to be γH2AX+ compared to control tumors (Figure 8I), these data were not statistically significant. These results demonstrate that increased genomic instability occurs as a result of Dnmt1 deletion in the ApcMin/+ model, and that genomic instability occurs prior to tumor development. We propose that this temporary decrease in genomic stability, combined with elevated proliferation and apoptosis levels, contributes to accelerated loss of heterozygosity and enhanced tumorigenesis in the inducible ApcMin/+;Dnmt1loxP/loxP;Villin-Cre-ERT2 mice.
Figure 8. The Dnmt1-deficient ApcMin/+ intestine displays increased chromosomal instability.
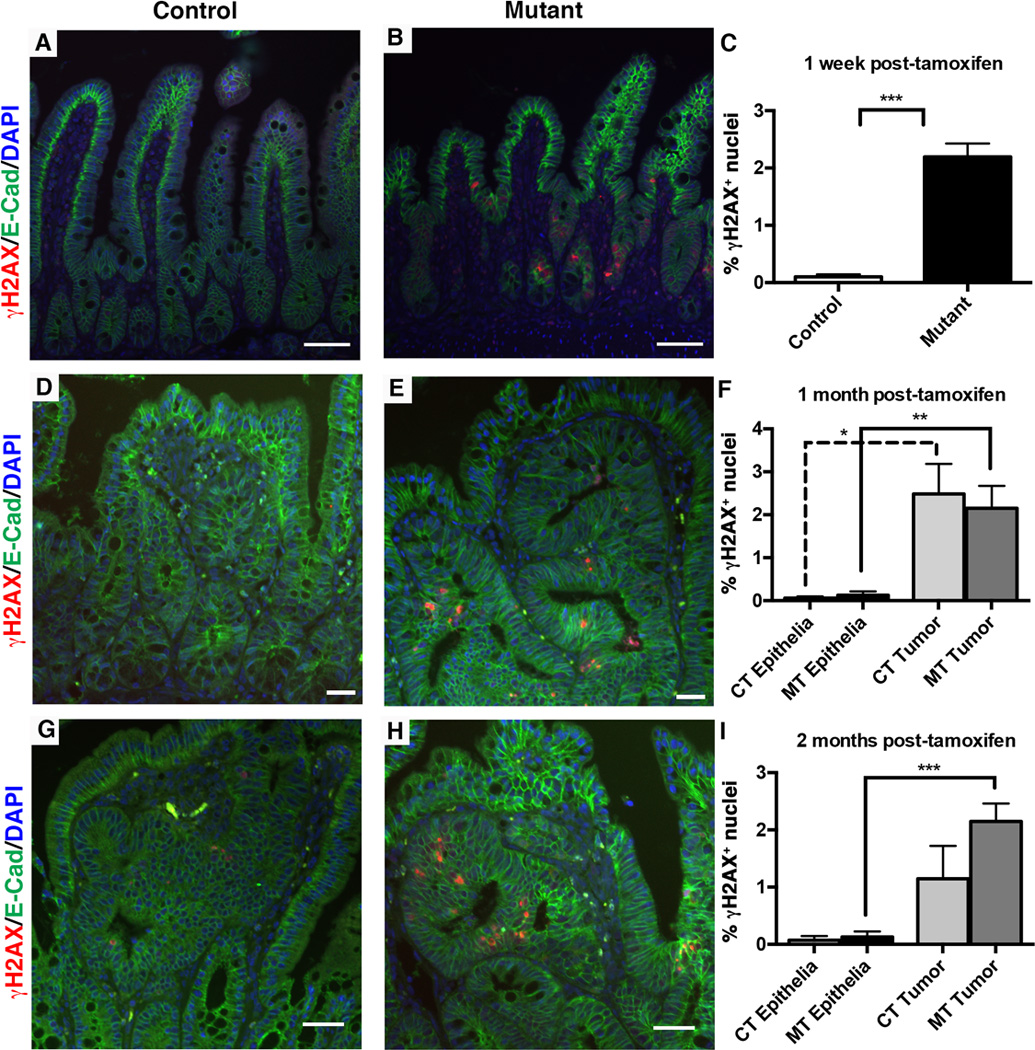
Mice were tamoxifen treated at four weeks of age, and intestines were harvested one week, one month, or two months later for immunofluorescent staining analysis. γH2AX (red) was used to visualize DNA double strand breaks as a marker of genome instability and E-cadherin (green) was used to outline the epithelium. The percentage of γH2AX positive (γH2AX+) cells was calculated as the number of γH2AX+ nuclei, divided by the total number of epithelial nuclei.
(A–C) One week following tamoxifen treatment, the number of γH2AX+ nuclei is increased in mutant compared to control small intestines (B compared to A, quantification in C). N=4 biological replicates per group. ***P<0.001 by Students’s t-test.
(D–F) One month post-tamoxifen injection, both mutant and control non-neoplastic epithelia display low levels of DNA damage, which is significantly increased in tumors of both groups (F). N=3 biological replicates per group; 2 tumors were counted for controls, 8 tumors were counted for mutants.
(G–I) Two months following tamoxifen treatment, increased levels of γH2AX+ foci continue to be seen in mutant tumors compared to mutant non-neoplastic epithelium (I). There is no significant difference between control and mutant γH2AX+ levels (G versus H, quantification in I). N=3–4 biological replicates per group; 3 tumors were counted from controls, and 12 tumors were counted from mutants.
All scale bars are 50µm. For (F, I), *P<0.05, **P<0.01, ***P<0.001, one-way ANOVA.
Discussion
The results presented above have major implications for the fields of DNA methylation and intestinal cancer biology. We show that loss of the DNA methyltransferase Dnmt1 in the ApcMin/+ cancer model results in acute hypomethylation and DNA damage, but does not affect tumor cell proliferation or apoptosis. We posit that decreased genomic stability accelerates loss of heterozygosity (LOH) at the Apc locus, resulting in increased tumor initiation, and larger tumors over time. Our work adds to the body of evidence that implicates a crucial role for Dnmt1 and DNA methylation in maintaining genome stability.
Importantly, our study demonstrates increased intestinal adenoma formation after deletion of Dnmt1. Our experimental paradigm results in intestine-specific, temporal loss of Dnmt1 and acute hypomethylation, followed by recovery of global genome DNA methylation, in contrast to prior work, which had employed germline hypomorphic Dnmt1 alleles (14–18). These previous studies thus differed both in the timing of Dnmt1 deletion and in the lack of tissue specificity, and described a constitutive hypomethylation phenotype which may be a contributing factor to our disparate results. Our Dnmt1-deficient tumors two months after tamoxifen administration exhibit significant hypomethylation of several potential oncogenes, yet display minimal DNA methylation changes genome-wide. We cannot rule out that the genome remethylation occurring from one week to one month post-tamoxifen contributes to accelerated tumorigenesis in our conditional Dnmt1 ablation intestinal cancer model. However, the significantly enhanced genomic instability, proliferation, and apoptosis observed immediately following Dnmt1 ablation, combined with the increased adenoma formation at one and two months post-tamoxifen, strongly suggest a role for Dnmt1 in maintaining genomic stability and preventing intestinal cancer initiation.
Interestingly, our model results in acute global hypomethylation at LINE1 repetitive elements. DNA methylation is associated with the silencing of LINE1 transcription (54), and LINE1 hypomethylation is highly variable in colorectal carcinomas (55). It has been hypothesized that LINE1 retrotransposition events can interrupt critical genes, such as APC, and drive tumorigenesis (54). In particular contexts, such as APC heterozygosity, changes in genome stability are critical drivers of cancer initiation. Our data support the general theory that DNA hypomethylation causes increased DNA damage, and LINE1 hypomethylation may also be contributing to our observed phenotype.
The DNA demethylating agents azacytidine and decitabine have been tested as anti-cancer therapeutics in colorectal cancer, to abrogate DNA hypermethylation and the silencing of tumor suppressor genes (56,57). Decitabine and azacytidine are most commonly used to treat acute myeloid leukemia (AML) and myelodysplastic syndromes, with variable success rates (58–60). Our results indicate that DNA demethylation actually contributes to cancer formation, and warrant caution in treating GI cancer patients with a potential tumor-enhancing drug.
In conclusion, we show that deletion of Dnmt1 in the adult intestinal epithelium of ApcMin/+ mice causes accelerated formation of adenomas. Loss of Dnmt1 results in acute hypomethylation and genomic instability, accompanied by increased proliferation and apoptosis. Although Dnmt1-deficient adenomas eventually recover global DNA methylation, they continue to display hypomethylation at several potential oncogenes, highlighting an unappreciated role for DNA hypomethylation in intestinal tumor development. These results support a fundamental role for DNA methylation in preserving genomic integrity during intestinal tumor formation.
Supplementary Material
Acknowledgments
Grant Support: This work was supported by National Institutes of Health grant R37-DK053839 to K.H.K.
We thank Haleigh Zillges, Joe Grubb, Jonathan Schug and Elizabeth Buza for technical assistance. A special thank you to services provided by the University of Pennsylvania Functional Genomics Core (P30-DK19525). We thank the University of Pennsylvania Department of Dermatology, especially Dr. John Seykora and Dr. Stephen Prouty, for Leica LCM training and equipment access. We acknowledge the support of Dr. Adam Bedenbaugh and the Morphology Core of the Penn Center for the Study of Digestive and Liver Diseases (P30-DK050306).
Footnotes
Conflicts of interest: The authors disclose no conflicts.
Author Contributions: K.L.S., E.N.E., and K.H.K planned experiments and prepared the manuscript. K.L.S. and E.N.E. performed experiments. All authors have read, commented on, and approved the manuscript.
References
- 1.Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507. doi: 10.1146/annurev-pathol-011110-130235. [DOI] [PubMed] [Google Scholar]
- 2.Smith ZD, Meissner A. DNA methylation: roles in mammalian development. Nat Rev Genet. 2013;14(3):204–220. doi: 10.1038/nrg3354. [DOI] [PubMed] [Google Scholar]
- 3.Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324(5929):930–935. doi: 10.1126/science.1170116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A, et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146(1):67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, et al. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature. 2002;416(6880):552–556. doi: 10.1038/416552a. [DOI] [PubMed] [Google Scholar]
- 6.Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y, et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44(1):40–46. doi: 10.1038/ng.969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S, Carter KT, Morris SM, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014;147(2):418–429. e8. doi: 10.1053/j.gastro.2014.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goelz SE, Vogelstein B, Hamilton SR, Feinberg AP. Hypomethylation of DNA from benign and malignant human colon neoplasms. Science. 1985;228(4696):187–190. doi: 10.1126/science.2579435. [DOI] [PubMed] [Google Scholar]
- 9.Feinberg AP, Gehrke CW, Kuo KC, Ehrlich M. Reduced genomic 5-methylcytosine content in human colonic neoplasia. Cancer Res. 1988;48(5):1159–1161. [PubMed] [Google Scholar]
- 10.Su LK, Kinzler KW, Vogelstein B, Preisinger AC, Moser AR, Luongo C, et al. Multiple intestinal neoplasia caused by a mutation in the murine homolog of the APC gene. Science. 1992;256(5057):668–670. doi: 10.1126/science.1350108. [DOI] [PubMed] [Google Scholar]
- 11.Cheung AF, Carter AM, Kostova KK, Woodruff JF, Crowley D, Bronson RT, et al. Complete deletion of Apc results in severe polyposis in mice. Oncogene. 2010;29(12):1857–1864. doi: 10.1038/onc.2009.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moser AR, Pitot HC, Dove WF. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science. 1990;247(4940):322–324. doi: 10.1126/science.2296722. [DOI] [PubMed] [Google Scholar]
- 13.Sansom OJ, Reed KR, Hayes AJ, Ireland H, Brinkmann H, Newton IP, et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes Dev. 2004;18(12):1385–1390. doi: 10.1101/gad.287404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamada Y, Jackson-Grusby L, Linhart H, Meissner A, Eden A, Lin H, et al. Opposing effects of DNA hypomethylation on intestinal and liver carcinogenesis. Proc Natl Acad Sci U S A. 2005;102(38):13580–13585. doi: 10.1073/pnas.0506612102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung WE, Li E, et al. Suppression of intestinal neoplasia by DNA hypomethylation. Cell. 1995;81(2):197–205. doi: 10.1016/0092-8674(95)90329-1. [DOI] [PubMed] [Google Scholar]
- 16.Eads CA, Nickel AE, Laird PW. Complete genetic suppression of polyp formation and reduction of CpG-island hypermethylation in Apc(Min/+) Dnmt1-hypomorphic Mice. Cancer Res. 2002;62(5):1296–1299. [PubMed] [Google Scholar]
- 17.Cormier RT, Dove WF. Dnmt1N/+ reduces the net growth rate and multiplicity of intestinal adenomas in C57BL/6-multiple intestinal neoplasia (Min)/+ mice independently of p53 but demonstrates strong synergy with the modifier of Min 1(AKR) resistance allele. Cancer Res. 2000;60(14):3965–3970. [PubMed] [Google Scholar]
- 18.Trasler J, Deng L, Melnyk S, Pogribny I, Hiou-Tim F, Sibani S, et al. Impact of Dnmt1 deficiency, with and without low folate diets, on tumor numbers and DNA methylation in Min mice. Carcinogenesis. 2003;24(1):39–45. doi: 10.1093/carcin/24.1.39. [DOI] [PubMed] [Google Scholar]
- 19.Perreault N, Sackett SD, Katz JP, Furth EE, Kaestner KH. Foxl1 is a mesenchymal Modifier of Min in carcinogenesis of stomach and colon. Genes Dev. 2005;19(3):311–315. doi: 10.1101/gad.1260605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson-Grusby L, Beard C, Possemato R, Tudor M, Fambrough D, Csankovszki G, et al. Loss of genomic methylation causes p53-dependent apoptosis and epigenetic deregulation. Nat Genet. 2001;27(1):31–39. doi: 10.1038/83730. [DOI] [PubMed] [Google Scholar]
- 21.el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, et al. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39(3):186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- 22.Lane N, Dean W, Erhardt S, Hajkova P, Surani A, Walter J, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35(2):88–93. doi: 10.1002/gene.10168. [DOI] [PubMed] [Google Scholar]
- 23.Sheaffer KL, Kim R, Aoki R, Elliott EN, Schug J, Burger L, et al. DNA methylation is required for the control of stem cell differentiation in the small intestine. Genes Dev. 2014;28(6):652–664. doi: 10.1101/gad.230318.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bernstein DL, Kameswaran V, Le Lay JE, Sheaffer KL, Kaestner KH. The BisPCR(2) method for targeted bisulfite sequencing. Epigenetics Chromatin. 2015;8:27. doi: 10.1186/s13072-015-0020-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boivin GP, Washington K, Yang K, Ward JM, Pretlow TP, Russell R, et al. Pathology of mouse models of intestinal cancer: consensus report and recommendations. Gastroenterology. 2003;124(3):762–777. doi: 10.1053/gast.2003.50094. [DOI] [PubMed] [Google Scholar]
- 26.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of Apc+ in intestinal adenomas from Min mice. Cancer Res. 1994;54(22):5947–5952. [PubMed] [Google Scholar]
- 27.Gupta RK, Gao N, Gorski RK, White P, Hardy OT, Rafiq K, et al. Expansion of adult beta-cell mass in response to increased metabolic demand is dependent on HNF-4alpha. Genes Dev. 2007;21(7):756–769. doi: 10.1101/gad.1535507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3(7) doi: 10.1186/gb-2002-3-7-research0034. RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moser AR, Dove WF, Roth KA, Gordon JI. The Min (multiple intestinal neoplasia) mutation: its effect on gut epithelial cell differentiation and interaction with a modifier system. J Cell Biol. 1992;116(6):1517–1526. doi: 10.1083/jcb.116.6.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141(9):1805–1813. doi: 10.1242/dev.101428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tremblay KD, Saam JR, Ingram RS, Tilghman SM, Bartolomei MS. A paternal-specific methylation imprint marks the alleles of the mouse H19 gene. Nat Genet. 1995;9(4):407–413. doi: 10.1038/ng0495-407. [DOI] [PubMed] [Google Scholar]
- 32.Yang AS, Estécio MR, Doshi K, Kondo Y, Tajara EH, Issa JP. A simple method for estimating global DNA methylation using bisulfite PCR of repetitive DNA elements. Nucleic Acids Res. 2004;32(3):e38. doi: 10.1093/nar/gnh032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuramochi-Miyagawa S, Watanabe T, Gotoh K, Totoki Y, Toyoda A, Ikawa M, et al. DNA methylation of retrotransposon genes is regulated by Piwi family members MILI and MIWI2 in murine fetal testes. Genes Dev. 2008;22(7):908–917. doi: 10.1101/gad.1640708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gregory TR. Synergy between sequence and size in large-scale genomics. Nat Rev Genet. 2005;6(9):699–708. doi: 10.1038/nrg1674. [DOI] [PubMed] [Google Scholar]
- 35.Riggs AD, Xiong Z. Methylation and epigenetic fidelity. Proc Natl Acad Sci U S A. 2004;101(1):4–5. doi: 10.1073/pnas.0307781100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen T, Ueda Y, Dodge JE, Wang Z, Li E. Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol Cell Biol. 2003;23(16):5594–5605. doi: 10.1128/MCB.23.16.5594-5605.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao F, Zhang Y, Wang S, Liu Y, Zheng L, Yang J, et al. Hes1 is involved in the self-renewal and tumourigenicity of stem-like cancer cells in colon cancer. Sci Rep. 2014;4:3963. doi: 10.1038/srep03963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Issa JP. CpG island methylator phenotype in cancer. Nat Rev Cancer. 2004;4(12):988–993. doi: 10.1038/nrc1507. [DOI] [PubMed] [Google Scholar]
- 39.Marik R, Fackler M, Gabrielson E, Zeiger MA, Sukumar S, Stearns V, et al. DNA methylation-related vitamin D receptor insensitivity in breast cancer. Cancer Biol Ther. 2010;10(1):44–53. doi: 10.4161/cbt.10.1.11994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Abouzeid HE, Kassem AM, Abdel Wahab AH, El-mezayen HA, Sharad H, Abdel Rahman S. Promoter hypermethylation of RASSF1A, MGMT, and HIC-1 genes in benign and malignant colorectal tumors. Tumour Biol. 2011;32(5):845–852. doi: 10.1007/s13277-011-0156-7. [DOI] [PubMed] [Google Scholar]
- 41.Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–422. doi: 10.1038/ng1330. [DOI] [PubMed] [Google Scholar]
- 42.Qi J, Zhu YQ, Luo J, Tao WH. Hypermethylation and expression regulation of secreted frizzled-related protein genes in colorectal tumor. World J Gastroenterol. 2006;12(44):7113–7117. doi: 10.3748/wjg.v12.i44.7113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grimm C, Chavez L, Vilardell M, Farrall AL, Tierling S, Böhm JW, et al. DNA-methylome analysis of mouse intestinal adenoma identifies a tumour-specific signature that is partly conserved in human colon cancer. PLoS Genet. 2013;9(2):e1003250. doi: 10.1371/journal.pgen.1003250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rosty C, Ueki T, Argani P, Jansen M, Yeo CJ, Cameron JL, et al. Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am J Pathol. 2002;160(1):45–50. doi: 10.1016/S0002-9440(10)64347-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi EK, Uyeno S, Nishida N, Okumoto T, Fujimura S, Aoki Y, et al. Alterations of c-fos gene methylation in the processes of aging and tumorigenesis in human liver. Mutat Res. 1996;354(1):123–128. doi: 10.1016/0027-5107(96)00056-5. [DOI] [PubMed] [Google Scholar]
- 46.Sato N, Maitra A, Fukushima N, van Heek NT, Matsubayashi H, Iacobuzio-Donahue CA, et al. Frequent hypomethylation of multiple genes overexpressed in pancreatic ductal adenocarcinoma. Cancer Res. 2003;63(14):4158–4166. [PubMed] [Google Scholar]
- 47.Pandey MK, Liu G, Cooper TK, Mulder KM. Knockdown of c-Fos suppresses the growth of human colon carcinoma cells in athymic mice. Int J Cancer. 2012;130(1):213–222. doi: 10.1002/ijc.25997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shukla V, Coumoul X, Lahusen T, Wang RH, Xu X, Vassilopoulos A, et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res. 2010;20(11):1201–1215. doi: 10.1038/cr.2010.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dizik M, Christman JK, Wainfan E. Alterations in expression and methylation of specific genes in livers of rats fed a cancer promoting methyl-deficient diet. Carcinogenesis. 1991;12(7):1307–1312. doi: 10.1093/carcin/12.7.1307. [DOI] [PubMed] [Google Scholar]
- 50.Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54(22):5953–5958. [PubMed] [Google Scholar]
- 51.Chen T, Hevi S, Gay F, Tsujimoto N, He T, Zhang B, et al. Complete inactivation of DNMT1 leads to mitotic catastrophe in human cancer cells. Nat Genet. 2007;39(3):391–396. doi: 10.1038/ng1982. [DOI] [PubMed] [Google Scholar]
- 52.Spada F, Haemmer A, Kuch D, Rothbauer U, Schermelleh L, Kremmer E, et al. DNMT1 but not its interaction with the replication machinery is required for maintenance of DNA methylation in human cells. J Cell Biol. 2007;176(5):565–571. doi: 10.1083/jcb.200610062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen RZ, Pettersson U, Beard C, Jackson-Grusby L, Jaenisch R. DNA hypomethylation leads to elevated mutation rates. Nature. 1998;395(6697):89–93. doi: 10.1038/25779. [DOI] [PubMed] [Google Scholar]
- 54.Miousse IR, Koturbash I. The Fine LINE: Methylation Drawing the Cancer Landscape. Biomed Res Int. 2015;2015:131547. doi: 10.1155/2015/131547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Estécio MR, Gharibyan V, Shen L, Ibrahim AE, Doshi K, He R, et al. LINE-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS One. 2007;2(5):e399. doi: 10.1371/journal.pone.0000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Braiteh F, Soriano AO, Garcia-Manero G, Hong D, Johnson MM, Silva LeP, et al. Phase I study of epigenetic modulation with 5-azacytidine and valproic acid in patients with advanced cancers. Clin Cancer Res. 2008;14(19):6296–6301. doi: 10.1158/1078-0432.CCR-08-1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Goel A, Boland CR. Epigenetics of colorectal cancer. Gastroenterology. 2012;143(6):1442–1460. e1. doi: 10.1053/j.gastro.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kadia TM, Jabbour E, Kantarjian H. Failure of hypomethylating agent-based therapy in myelodysplastic syndromes. Semin Oncol. 2011;38(5):682–692. doi: 10.1053/j.seminoncol.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nebbioso A, Carafa V, Benedetti R, Altucci L. Trials with 'epigenetic' drugs: an update. Mol Oncol. 2012;6(6):657–682. doi: 10.1016/j.molonc.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–232. doi: 10.1016/S1470-2045(09)70003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.