

Abstract
Chronic exposure to carcinogens represents the major risk factor for head and neck squamous cell carcinoma (HNSCC). Beverages derived from broccoli sprout extracts (BSEs) that are rich in glucoraphanin and its bioactive metabolite sulforaphane promote detoxication of airborne pollutants in humans. Herein, we investigated the potential chemopreventive activity of sulforaphane using in vitro models of normal and malignant mucosal epithelial cells and an in vivo model of murine oral cancer resulting from the carcinogen 4-nitroquinoline-1-oxide (4NQO). Sulforaphane treatment of Het-1A, a normal mucosal epithelial cell line, and 4 HNSCC cell lines led to dose- and time-dependent induction of NRF2 and the NRF2 target genes NQO1 and GCLC, known mediators of carcinogen detoxication. Sulforaphane also promoted NRF2-independent dephosphorylation/inactivation of pSTAT3, a key oncogenic factor in HNSCC. Compared to vehicle, sulforaphane significantly reduced the incidence and size of 4NQO-induced tongue tumors in mice. A pilot clinical trial in 10 healthy volunteers evaluated the bioavailability and pharmacodynamic activity of three different BSE regimens, based upon urinary sulforaphane metabolites and NQO1 transcripts in buccal scrapings, respectively. Ingestion of sulforaphane-rich BSE demonstrated the greatest, most consistent bioavailability. Mucosal bioactivity, defined as 2-fold or greater upregulation of NQO1 mRNA, was observed in 6 of 9 evaluable participants ingesting glucoraphanin-rich BSE; 3 of 6 ingesting sulforaphane-rich BSE; and 3 of 9 after topical-only exposure to sulforaphane-rich BSE. Together, our findings demonstrate preclinical chemopreventive activity of sulforaphane against carcinogen-induced oral cancer, and support further mechanistic and clinical investigation of sulforaphane as a chemopreventive agent against tobacco-related HNSCC.
Introduction
Cancers of the upper aerodigestive tract, including head and neck squamous cell carcinoma (HNSCC), are strongly associated with chronic exposure to tobacco and alcohol. The concept of “condemned mucosa”, or epithelial field cancerization from environmental carcinogens, was introduced by Danely Slaughter in 1953 (1). Clinically, epithelial field cancerization manifests as an alarming rate of second primary tumor (SPT) formation in patients curatively treated for an initial primary HNSCC: 3–6%/year (2–5). Although smoking cessation reduces SPTs in HNSCC, moderation of risk is not observed for 5 years, and is insufficient to return risk to baseline (4, 6). Chemoprevention of SPTs by high-dose isotretinoin, a synthetic vitamin A analog, was demonstrated in a Phase III HNSCC chemoprevention study (7, 8). However, toxicities precluded chronic administration and risk reverted to baseline upon discontinuation; a subsequent study demonstrated that low-dose isotretinoin was tolerable but ineffective against SPTs (9). Molecular targeting of EGFR or COX-2 has shown preclinical chemopreventive efficacy against oral cancer (10–14), but clinical application has been hampered by poor efficacy and tolerability in humans (15–19). Thus, a tremendous unmet need remains for an effective and well-tolerated chemopreventive agent against HNSCC.
Epidemiologic studies indicate that diets rich in vegetables from the Brassica genus of the family Cruciferae (eg. broccoli, cabbage, cauliflower) are associated with reduced risk of HNSCC and SPTs (20–24). Broccoli extracts potently induce cytoprotective enzymes that promote detoxication of chemical carcinogens, including benzene, aldehydes, and polycyclic aromatic hydrocarbons found in tobacco smoke (25–28). The majority of inducer activity is driven by the phytochemical sulforaphane, a metabolite of glucoraphanin (28). Mechanistically, sulforaphane interacts with cysteine residues on Kelch-like ECH-associate protein 1 (KEAP1), a negative regulator of nuclear factor (erythroid-derived 2)-like 2 (NRF2) transcription factor (29). This interaction liberates NRF2 from proteasomal destruction and results in upregulation of NRF2 and NRF2 target genes (28, 30). Many known NRF2 target genes encode enzymes affecting carcinogen detoxication, including NAD(P)H quinone oxidoreductase 1 (NQO1), glutamate-cysteine ligase catalytic subunit (GCLC), glutathione S-transferases, and aldo-keto reductases. As glucoraphanin is 20–50 times more concentrated in broccoli seeds relative to mature plants (31, 32), various broccoli seed preparations are under development as chemopreventive agents against carcinogen-induced cancers.
Proof-of-concept clinical trials in healthy volunteers have shown that broccoli sprout extracts (BSEs) rich in glucoraphanin and/or sulforaphane are well-tolerated and promote rapid, sustained detoxication of the airborne pollutants acrolein and benzene (28, 33–36). In preclinical studies, sulforaphane has exhibited chemopreventive activity against carcinogen-induced stomach, skin and breast cancers (37–41). Studies in Nrf2−/− mice have shown that the chemopreventive effect of sulforaphane against benzo[a]pyrene-induced gastric cancer and 7,12-dimethylbenz(a)anthracene (DMBA)-induced skin cancer depends on the NRF2 signaling pathway (37, 38, 41). Neither sulforaphane nor BSEs have been investigated in oral cancer models. However, the relevance of NRF2 to oral cancer chemoprevention is highlighted by the enhanced susceptibility of Nrf2−/− mice to oral cancer induced by the carcinogen 4-nitroquinoline-1-oxide (4NQO), and the reduced susceptibility of Keap1−/− mice (42).
To investigate the chemopreventive activity of sulforaphane against HNSCC, we examined sulforaphane’s impact on NRF2 signaling in a normal mucosal epithelial cell line and four HNSCC cell lines. We also determined the effects of sulforaphane on apoptosis regulatory proteins, including STAT3. We investigated the in vivo chemopreventive activity of sulforaphane against carcinogen-induced oral cancer using the 4NQO murine model. Finally, we conducted a pilot clinical trial testing three BSE regimens in healthy volunteers, evaluating both bioavailability and effects on NRF2 signaling in oral epithelium: ingestion of glucoraphanin-rich BSE; ingestion of sulforaphane-rich BSE; and topical exposure to sulforaphane-rich BSE. Our findings provide the first preclinical demonstration that sulforaphane protects against carcinogen-induced oral cancer and support further mechanistic and clinical evaluation of broccoli-derived extracts and sulforaphane against tobacco-related HNSCC.
Materials and Methods
Reagents
R,S-sulforaphane was from LKT Laboratories, Inc., and cell culture reagents from Life Technologies. Annexin V/PI staining kits were from BD Pharmingen. All reagents for RNA purification and cDNA synthesis, including Trizol Reagent, RNAqueous-Micro Total RNA isolation kits, Superscript III First-Strand cDNA Synthesis kits, Platinum Taq DNA polymerase were purchased from Life Technologies. SYBR Green PCR Master Mix was from Applied Biosystems. UMSCC cell lines were from Thomas Carey (University of Michigan). Het-1A were obtained from ATCC (Manassas, VA). UPCI:SCC090 cells were provided by Susanne Gollin (University of Pittsburgh). Cell lines were authenticated genotypically using AmpFLSTR Profiler Plus Amplification Kit (A&B Applied Biosystem).
Immunoblotting
Cells were lysed and subjected to immunoblotting as previously described (43). Blots were probed with antibodies against NRF2, Mcl-1, Bik (Santa Cruz Biotechnology), phospho-STAT3, total STAT3, Bcl-XL, Bax, Bak (Cell Signaling), Bcl-2 (Dako), Bim (Stressgen Bioreagents), and β-actin (Sigma).
RNA purification and quantitative real-time PCR
Total RNA was purified from cells in culture using Trizol Reagent. RNA was purified from human buccal cells using RNAqueous-Micro Total RNA Isolation kits. cDNA was synthesized using Superscript III First-Strand cDNA Synthesis System. Quantitative real-time PCR (qPCR) was carried out using cDNA as template and the SYBR Green PCR Master Mix. Gene-specific primers for NQO1 were: 5′-GTCATTCTCTGGCCAATTCAGAGT-3′ (forward) and 5′-TTCCAGGATTTGAATTCGGG-3′ (reverse). Primers for GCLC were: 5′-GGCGATGAGGTGGAATACAT-3′ (forward) and 5′-GTCCTTTCCCCCTTCTCTTG-3′ (reverse). Primers for GAPDH were: 5′-GGACCTGACCTGCCGTCTAGAA-3′ (forward) and 5′-GGTGTCGCTGTTGAAGTGAGAG-3′ (reverse). GAPDH transcript levels were used as the internal control.
Treatment of mice
Female C57BL/6 mice (5–6 weeks; 18 mice/group) in both treatment groups were administered 4NQO (100 μg/ml) in ad libitum drinking water for 16 weeks. Stock 4NQO solutions were thawed, diluted to 12.5 mg/ml in propylene glycol, then 3.8 ml was added to each 450 ml bottle of sterilized drinking water. The 4NQO-containing water was changed weekly. In addition to 4NQO treatment, group 1 control mice were given PBS thrice weekly via oral gavage, and mice in group 2 were given 6 μmol of sulforaphane thrice weekly via oral gavage. All treatments were stopped after 16 weeks and mice were maintained for 8 additional weeks on normal tap water. After 24 total weeks, mice were sacrificed, tongue tissues harvested, and tongue tumors counted and measured.
Treatment of human subjects
The pilot clinical trial was approved by the IRB of the University of Pittsburgh and registered at clinicaltrials.gov (NCT02023931). All participants provided written, informed consent. Eligibility criteria, participant characteristics, beverage preparation, and toxicities are described in supplementary Materials and Methods.
Ten participants underwent three 5-day interventions. On Days 1, 3, 4 and 5 of each intervention, buccal cells were collected by scraping the inner cheek with a curette, transferred immediately into RNAlater (Life Technologies), then frozen at −80°C until analysis. Overnight urine was self-collected from 5pm on Day 1 through the first morning void on Day 2, and repeated on Day 4–5. Urine volume was recorded, and two 15-mL aliquots were frozen at −20°C until analysis. On Days 2–4 of each intervention, participants self-administered the designated BSE beverage once daily (Regimen 1, 600 μmol of glucoraphanin-rich BSE/day; Regimen 2, 150 μmol of sulforaphane-rich BSE/day; Regimen 3, 150 μmol of sulforaphane-rich BSE was swished, gargled and expectorated for 6 minutes daily). A minimum 3-day washout was required between regimens. Cruciferous vegetables were avoided for 48 hours prior to and during each intervention.
The study was designed as a single-arm crossover trial in which all participants were exposed to the same sequence and doses of BSEs. As the goals of this pilot study were limited to feasibility, bioavailability, and obtaining preliminary pharmacodynamic estimates of NRF2 pathway modulation in oral mucosa for each BSE intervention, no formal hypothesis testing was planned. For purposes of descriptive reporting, mucosal bioactivity was defined as at least 2-fold upregulation of NQO1 buccal mRNA by qPCR, as compared to regimen baseline. A participant was considered evaluable for a regimen if mRNA yield was ≥ 18 ng/μl at baseline and at least one subsequent time point.
Analysis of urine specimens
Measurement of sulforaphane and sulforaphane-N-acetylcysteine in urine was performed by isotope dilution mass spectrometry, as previously reported (34, 36).
Statistical methods
ANOVAs and t-tests were employed for comparisons between groups. The assumptions of equal variances and Gaussian residuals were checked visually. Welch’s adjustment for unequal variances was applied if required. Count endpoints were analyzed using Poisson ANOVA. A linear mixed effects ANOVA was used to analyze qPCR data from the trial. All statistical tests were two-sided, and p<0.05 was required for significance.
Results
Sulforaphane activates NRF2 signaling in normal mucosal epithelial cells and HNSCC cell lines
The impact of sulforaphane on cellular expression levels of NRF2 was examined in a normal mucosal epithelial cell line, Het-1A, and the HNSCC cell lines UMSCC-22A, UMSCC-1, Cal33, and UPCI:SCC090. Het-1A cells are derived from normal squamous esophageal cells following transfection with SV40 large T antigen (44). Untreated or vehicle-treated (DMSO) Het-1A cells or HNSCC cells expressed only low levels of NRF2 protein (Fig. 1A and Supplementary Fig.1), commonly detected as a doublet in the 95–110 kDa range. Treatment with 10 μM sulforaphane led to marked induction of NRF2 in Het-1A cells as well as both HPV-negative (UMSCC-22A, UMSCC-1, Cal33) and HPV-positive (UPCI:SCC090) HNSCC cell lines (Fig. 1A and Supplementary Fig. 1). As cellular levels of NRF2 are known to be regulated by the proteasome, we also examined the effects of the proteasome inhibitors bortezomib, carfilzomib, and oprozomib (Supplementary Fig. 2). As expected, proteasome inhibition led to NRF2 upregulation.
Figure 1.
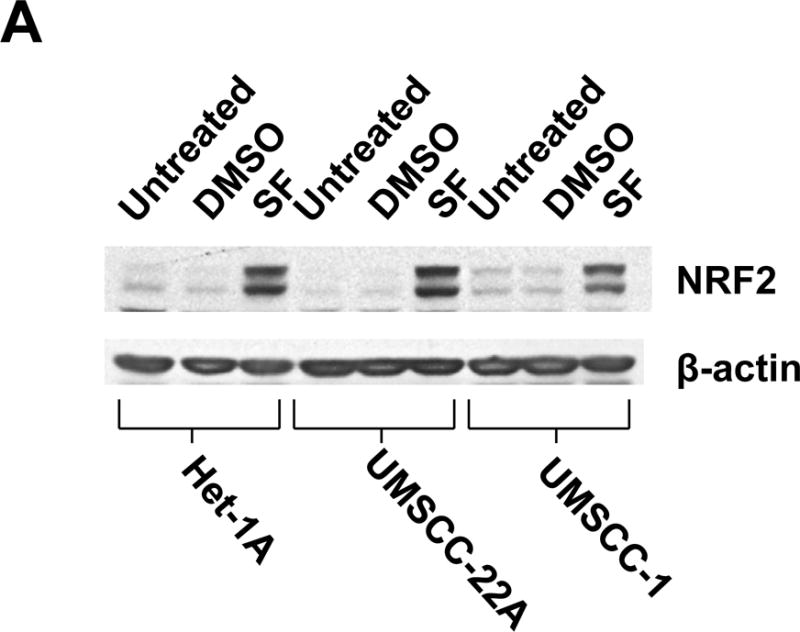
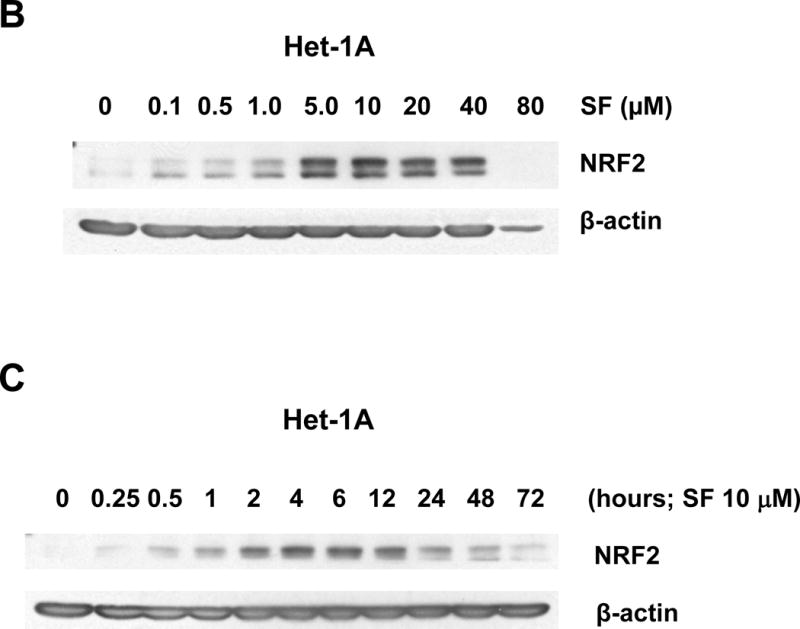
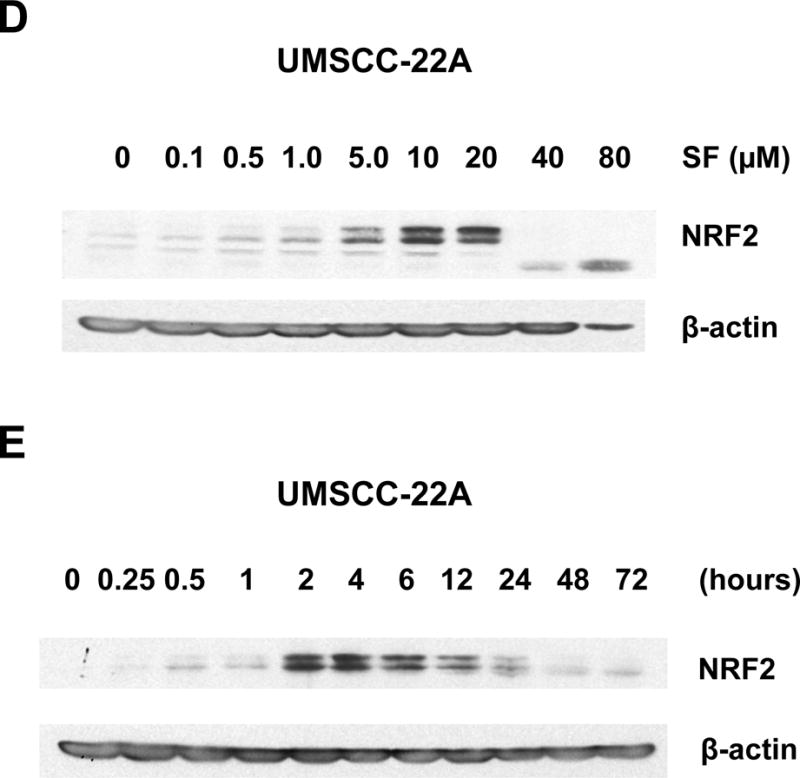
Sulforaphane elevates dose- and time-dependent expression of NRF2 in Het-1A cells and HNSCC cell lines. A, Het-1A, a normal mucosal epithelial cell line, or the HNSCC cell lines UMSCC-22A and UMSCC-1 were left untreated, or were treated for 6 hours with vehicle (0.1% DMSO) or 10 μM sulforaphane (SF). Whole cell lysates were subjected to anti-NRF2 immunoblotting. Blots were re-probed with anti-β-actin to demonstrate equal protein loading. B, Het-1A were treated for 24 hours with varying concentrations of SF, followed by immunoblotting for NRF2 or β-actin. C, Het-1A were treated with 10 μM SF for the indicated number of hours, then subjected to immunoblotting. D and E, UMSCC-22A were treated and analyzed as in Panels B and C, respectively. All experiments were performed a minimum of three times, with similar results.
Dose-response experiments revealed maximal elevation of NRF2 levels with 5.0 μM and 10.0 μM sulforaphane in Het-1A and UMSCC-22A, respectively (Figs. 1B and 1D). At high concentrations of sulforaphane (80 μM in Het-1A and 40 μM in UMSCC-22A), expression of intact NRF2 was lost, perhaps due to induction of apoptosis signaling. Time course analyses using a fixed dose of sulforaphane (10 μM) demonstrated elevation of NRF2 levels at 15 minutes in Het-1A and 30 minutes in UMSCC-22A (Figs. 1C and 1E), with peak elevation occurring at approximately 4 hours in both models. Following 12 hours of treatment, NRF2 levels declined, returning to near baseline levels by 48–72 hours.
To determine whether the NRF2 protein increased by sulforaphane was functionally active, we performed qPCR to examine expression levels of the NRF2 target genes NQO1 and GCLC (Fig. 2). Treatment with sulforaphane (10 μM) for 4 hours resulted in nearly 2-fold induction of NQO1 mRNA in Het-1A, UMSCC-22A, and UMSCC-1 cells. GCLC mRNA upregulation by sulforaphane was approximately 5-fold in Het-1A and approximately 2.5-fold in both HNSCC cell lines. Similar results were seen in HPV-negative and HPV-positive HNSCC cell lines (Supplementary Figs. 3 and 4). These results demonstrate that sulforaphane activates functional NRF2 signaling in normal mucosal epithelial cells and HNSCC cells.
Figure 2.
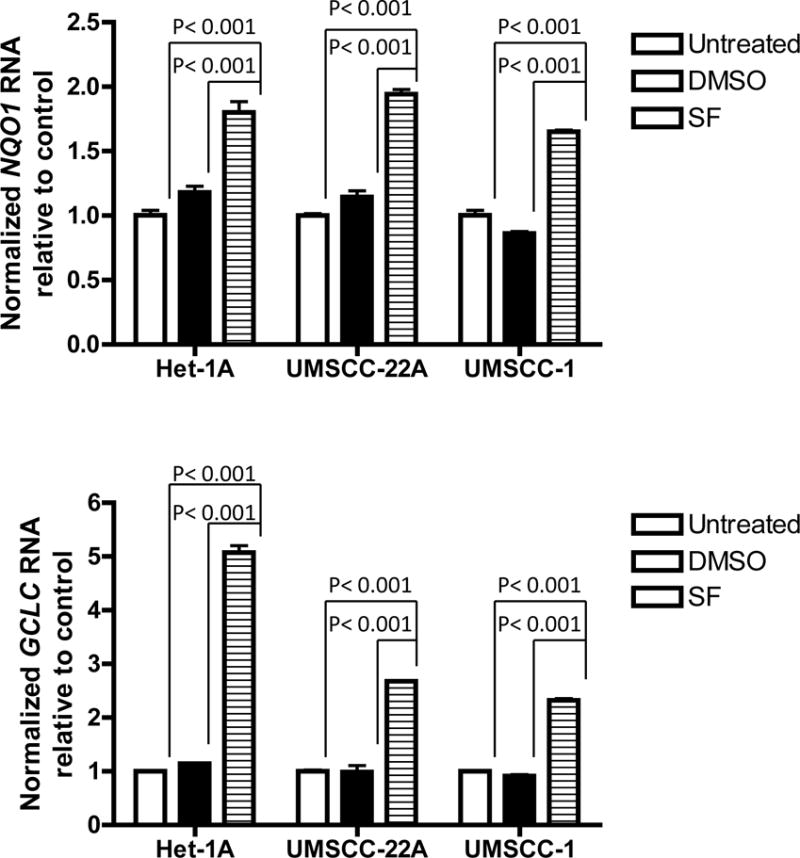
Induction of NRF2 target genes by sulforaphane. Het-1A, UMSCC-22A, and UMSCC-1 were left untreated, or treated for 4 hours with vehicle or 10 μM SF. Following treatment, RNA was purified and subjected to qPCR for NQO1 or GCLC, or GAPDH as internal control. Columns represent means and error bars standard deviations. Analysis was performed by one-way ANOVA with Tukey’s adjustment for multiple comparisons.
Sulforaphane suppresses STAT3 phosphorylation independently of NRF2
The impact of sulforaphane on STAT3 activation was assessed using an antibody directed against phospho-Tyr705 STAT3 (pSTAT3), the phosphorylated/activated form. Treatment of Het-1A or UMSCC-22A with increasing concentrations of sulforaphane led to loss of pSTAT3 expression at doses 10 μM or higher (Figs. 3A and 3C). By contrast, sulforaphane concentrations up to 40 μM in Het-1A and UMSCC-22A did not markedly alter the expression levels of total STAT3 protein, indicating the effects of sulforaphane on pSTAT3 were due to dephosphorylation. Time course analyses revealed rapid loss of pSTAT3, within 30 minutes, following treatment of Het-1A or UMSCC-22A with 10 μM sulforaphane (Figs. 3B and 3D). The levels of pSTAT3 remained low for 12–24 hours post-treatment and then returned to baseline. In contrast to effects on NRF2 and pSTAT3 levels, sulforaphane (10 μM, 24 hours) demonstrated little effect on the expression of anti-apoptotic or pro-apoptotic members of the Bcl-2 protein family (Fig. 3E), including anti-apoptotic Bcl-XL, a known pro-survival in HNSCC (45).
Figure 3.
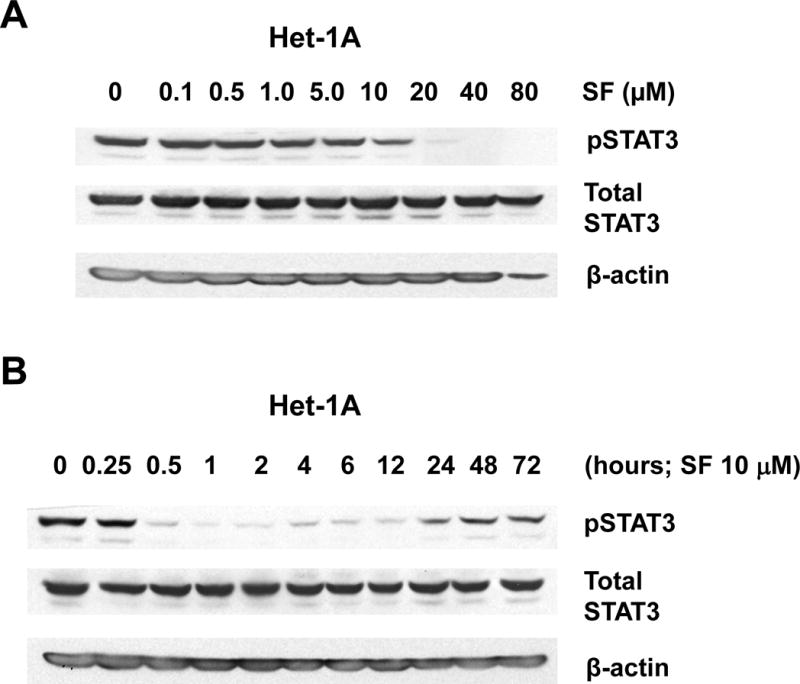
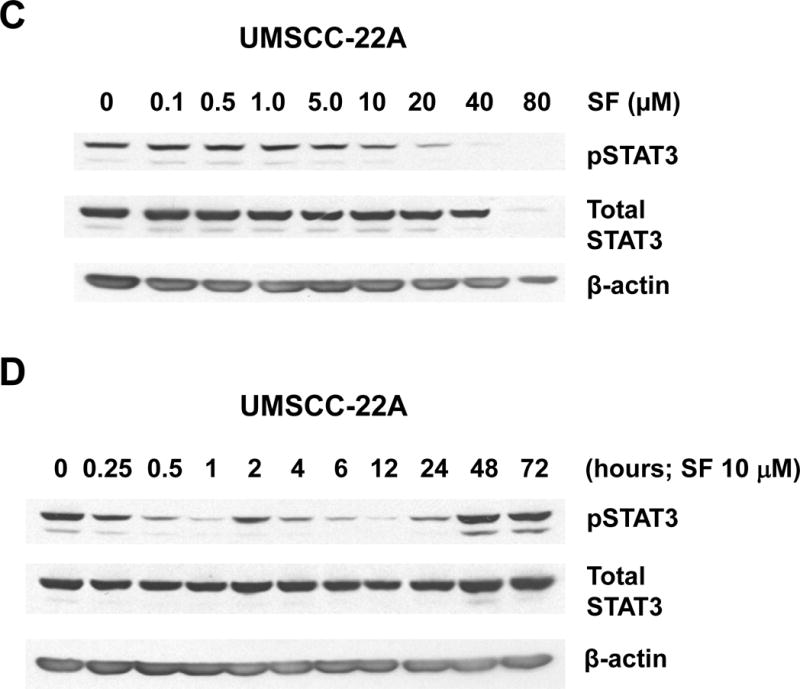
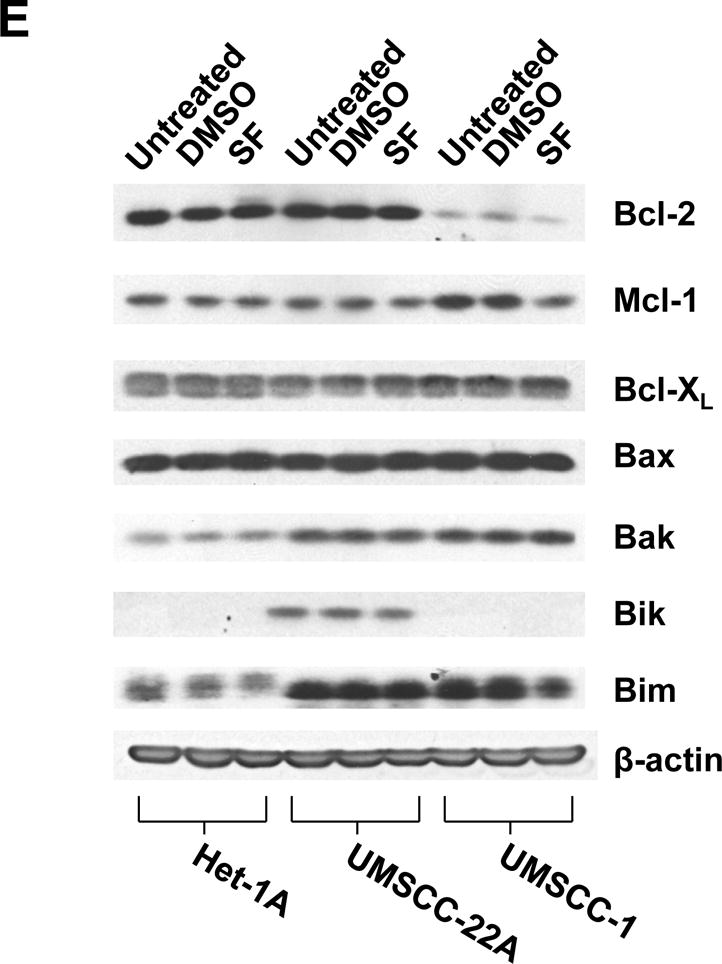
Sulforaphane induces rapid dephosphorylation of STAT3. A and B, Het-1A were treated for 12 hours with the indicated concentrations of SF (A), or were treated with 10 μM SF for the indicated times (B), followed by immunoblotting for phospho-Tyr705 STAT3 (pSTAT3), total STAT3, or β-actin. C and D, UMSCC-22A were treated and analyzed as in Panels A and B. E, Het-1A, UMSCC-22A, and UMSCC-1 were left untreated, or treated for 24 hours with DMSO control or 10 μM SF, followed by immunoblotting for anti- and pro-apoptotic Bcl-2 family members. Experiments were performed three times with similar results.
To determine whether dephosphorylation of STAT3 by sulforaphane treatment was dependent on NRF2, we utilized siRNA to suppress NRF2 expression. As expected, Het-1A and UMSCC-22A cells transfected with non-specific siRNA exhibited upregulation of NRF2 and loss of pSTAT3 following treatment with 10 μM sulforaphane (Fig. 4A). Transfection with siRNA directed against NRF2 mRNA resulted in nearly complete abrogation of NRF2 upregulation. Inhibition of NRF2 expression did not interfere with loss of pSTAT3 in sulforaphane-treated cells. Thus, inactivation of STAT3 by sulforaphane occurs independently of NRF2.
Figure 4.
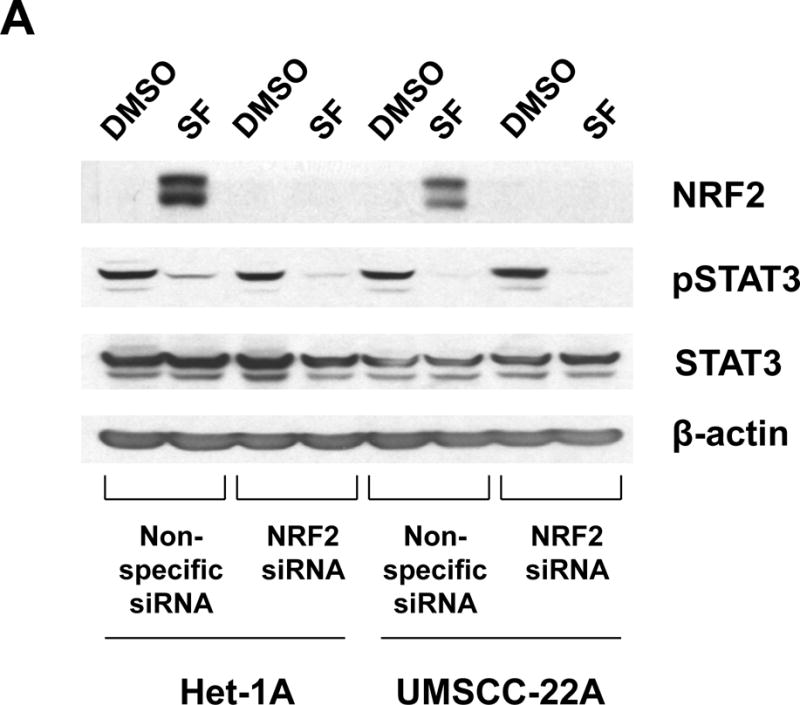
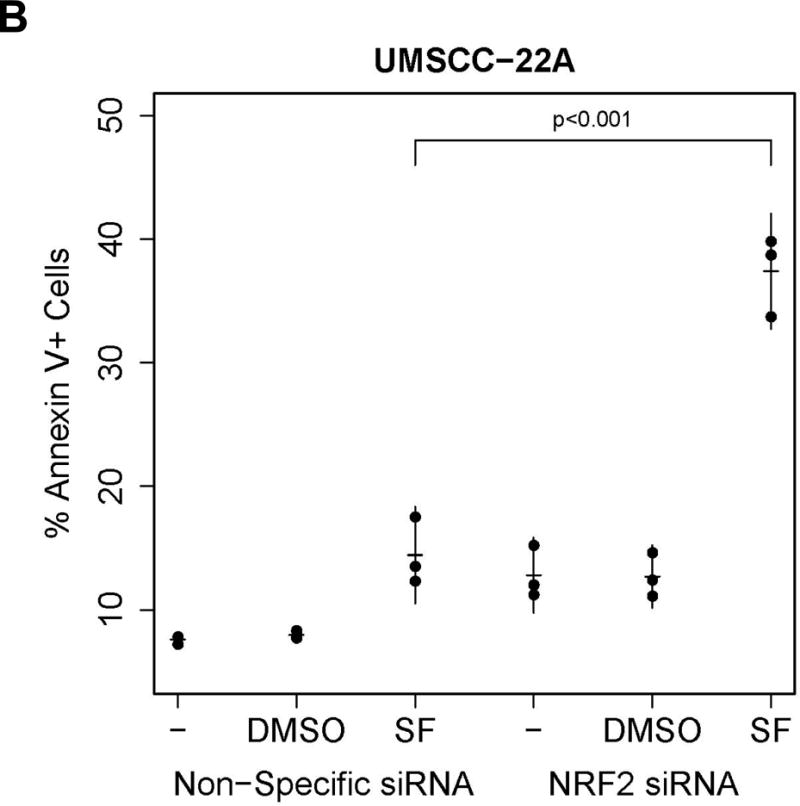
Role of NRF2 in sulforaphane-induced STAT3 inactivation and induction of apoptosis. A, Het-1A and UMSCC-22A were transfected for 6 hours with non-specific siRNA or NRF2 siRNA. Following transfection, cells were allowed to recover overnight before treatment for 12 hours with 0.1% DMSO or 10 μM SF. Cells were then subjected to immunoblotting for NRF2, pSTAT3, total STAT3, or β-actin. Similar results were seen in three independent experiments. B, UMSCC-22A transfected with non-specific siRNA or NRF2 siRNA as in Panel A were left untreated, or were treated for 24 hours with 0.1% DMSO or 10 μM SF, then analyzed by flow cytometry for Annexin V/PI staining. Numbers indicate the percentage of Annexin V-positive cells (p=0.001 by 2-way ANOVA when comparing SF-treated NRF2 siRNA versus SF-treated non-specific siRNA).
The inactivation of pro-survival pSTAT3 by sulforaphane suggested that sulforaphane may promote cell death in Het-1A and HNSCC cell lines. Indeed, while sulforaphane concentrations of 5–10 μM were effective at inducing NRF2 and NRF2-dependent detoxication enzymes, higher concentrations promoted cell death in Het-1A and five different HNSCC cell lines (Figs. 1&2 and Supplementary Fig. 5). Het-1A and UMSCC-22A exhibited sulforaphane IC50s of 21.3 μM and 21.1 μM, respectively, in 48-hour assays (Supplementary Fig. 5). To determine the role of NRF2 in regulating sulforaphane-induced cell death we again used siRNA to inhibit NRF2 expression. Suppression of NRF2 expression in UMSCC-22A significantly enhanced apoptosis by sulforaphane (10 μM, 24 hours), as determined by Annexin V staining (p=0.001; Fig. 4B). Collectively, these results indicate that NRF2 protects against apoptosis induced by high concentrations of sulforaphane, and does so without modulating sulforaphane-induced dephosphorylation of pSTAT3.
Sulforaphane prevents 4NQO-induced oral tumors
To investigate the chemopreventive activity of sulforaphane against carcinogen-induced oral cancer in vivo, two groups of C57BL/6J mice (n=18 mice/group) were treated with 4NQO in the presence of vehicle control vs. sulforaphane, as described in Materials and Methods. During the course of the 24-week experiment one mouse died in each group, leaving 17 evaluable animals per group.
The number of tongue tumors in mice treated with 4NQO plus sulforaphane was significantly lower than the number detected in mice treated with 4NQO plus vehicle (p=0.012; Fig. 5A), upon gross examination. Subsequent examination of hematoxylin/eosin-stained sections of the tongues confirmed a lower incidence of invasive SCC in mice treated with 4NQO plus sulforaphane versus 4NQO plus vehicle (Fig. 5B). Immunohistochemical staining of the invasive SCC specimens for NRF2 failed to reveal a statistically significant difference (p=0.46) between the two treatment groups (not shown), although this was not surprising since treatment with 4NQO/sulforaphane or 4NQO/vehicle was stopped 8 weeks prior to harvest of the tongues. The tongue tumors in sulforaphane-treated mice were also found to be significantly smaller than those observed in vehicle-treated mice (p=0.005; Fig. 5C. The median tumor volume was 0.90 mm3/mouse in the sulforaphane group, and 9.55 mm3/mouse in the control group. Together, these findings demonstrate that sulforaphane reduced the incidence and size of tongue tumors in 4NQO-treated mice.
Figure 5.
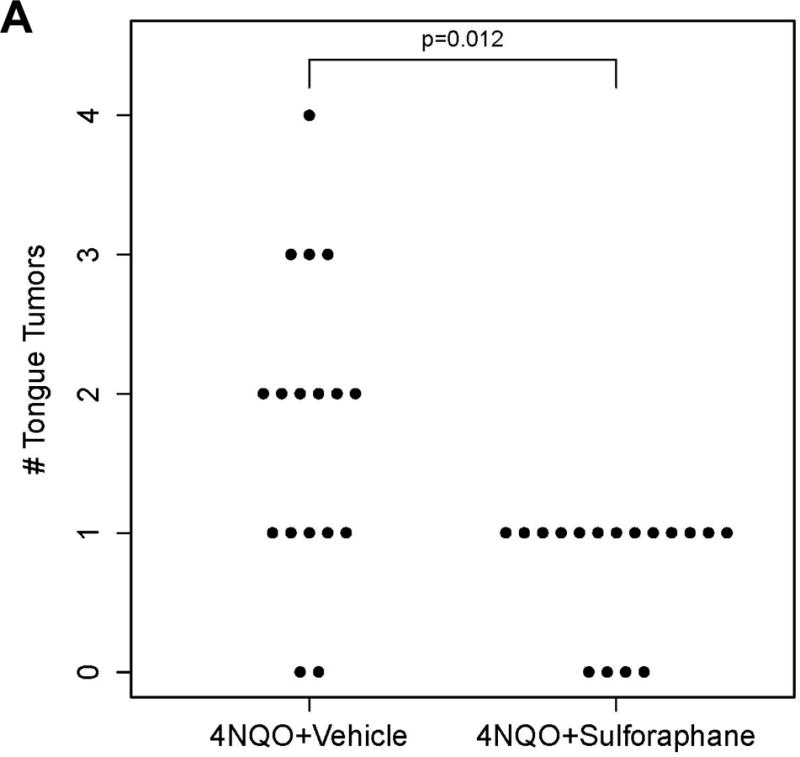

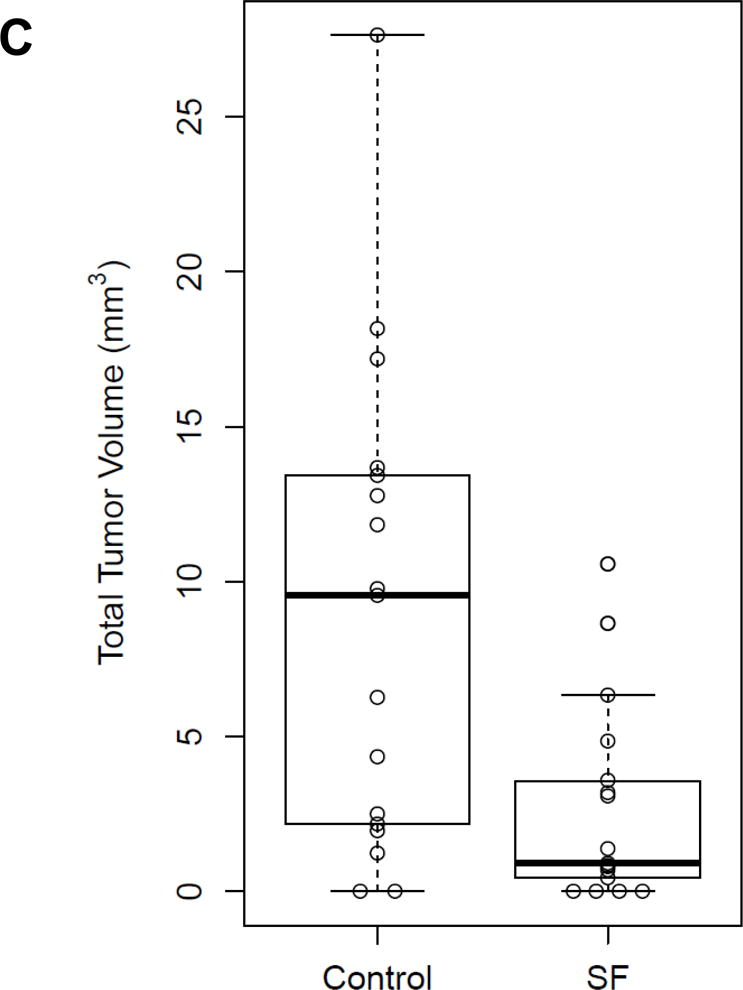
Sulforaphane reduces the incidence and size of tongue tumors in 4NQO-treated mice. A, Wild-type C57BL/6 mice (n=17/group) were treated for 16 weeks with 4NQO (100 μM in drinking water) plus vehicle (thrice weekly via oral gavage), or with 4NQO plus SF (6 μmole thrice weekly via oral gavage). After the 16-weeks, treatments were discontinued and mice given regular tap water for an additional 8 weeks. The number of tongue tumors in each mouse was counted, with significantly fewer in sulforaphane-treated mice (p=0.012 by Poisson ANOVA). B, Tongues from mice exhibiting tumors were fixed, embedded in paraffin, sectioned, and stained with hematoxylin/eosin. The stained sections were evaluated by a pathologist blinded to the treatment groups. The number of mice exhibiting normal tongue tissue only, squamous hyperplasia, dysplasia, or invasive SCC were scored. C, The total tumor volume per mouse (open circles) was significantly lower in sulforaphane-treated mice (p=0.005 by Welch’s two-sample t-test). Bold bars represent the median tumor volume (9.55 vs. 0.90 mm3), and boxes represent the 25th and 75th percentiles.
Evaluation of three BSE regimens in healthy volunteers
To evaluate the bioavailability and pharmacodynamic activity of oral and topical administration of BSE beverages, a pilot study was conducted in 10 healthy human volunteers. As described in Materials and Methods, participants were administered the same sequence of three BSE regimens: Regimen 1, ingestion of glucoraphanin-rich BSE; Regimen 2, ingestion of sulforaphane-rich BSE; and Regimen 3, topical exposure to sulforaphane-rich BSE. To assess bioavailability, overnight urine was collected at the beginning and end of each regimen and analyzed by isotope dilution mass spectrometry for free sulforaphane and its primary metabolite, the glutathione-derived conjugate sulforaphane-N-acetylcysteine. Total μmole of excreted sulforaphane plus its mercapturic acid metabolites were normalized to urinary volume and creatinine (Fig. 6A and Supplementary Table 1). Bioavailability was significantly greater for sulforaphane-rich BSE (p=0.0013) than with the other regimens. Urinary levels of sulforaphane and mercapturic acids were more variable when glucoraphanin-rich BSE was administered. Topical sulforaphane-rich BSE demonstrated negligible bioavailability, indicating compliance with non-ingestion and trivial systemic absorption through oral mucosa.
Figure 6.
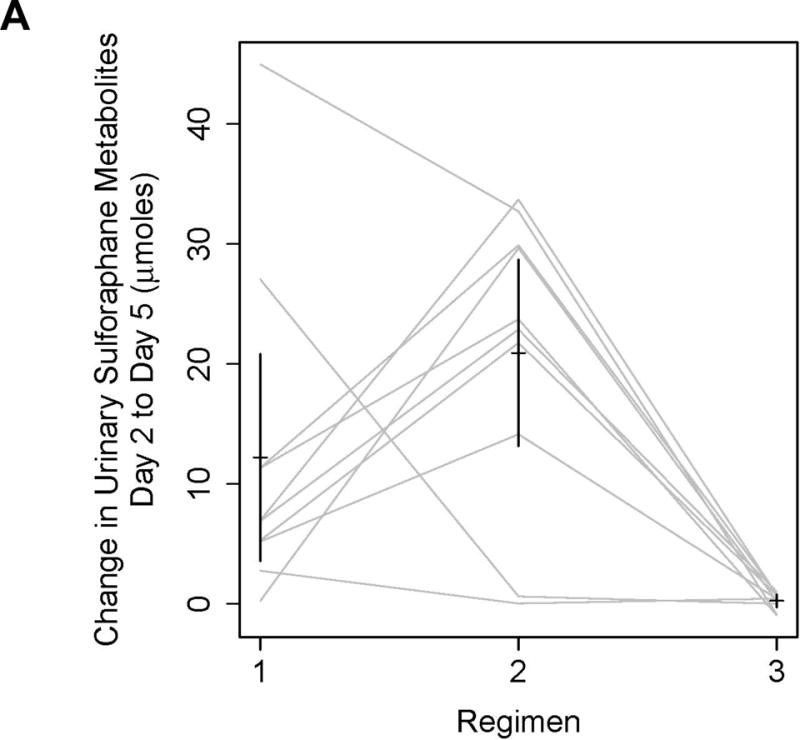
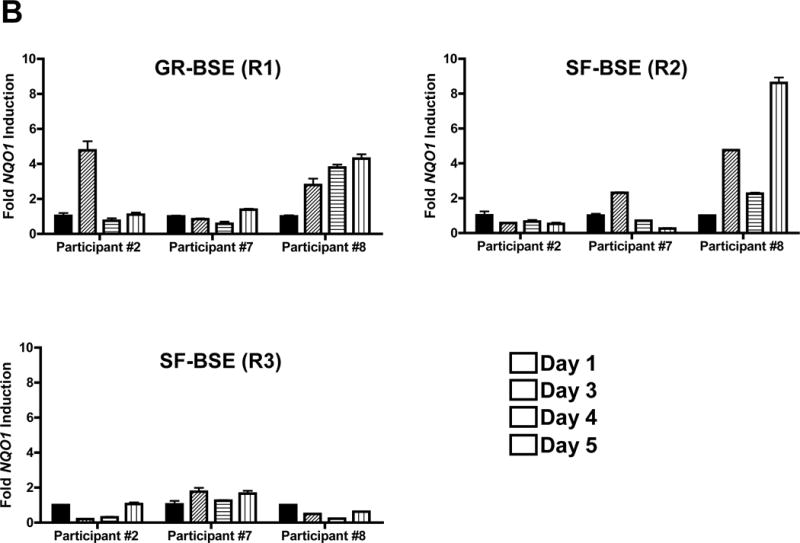
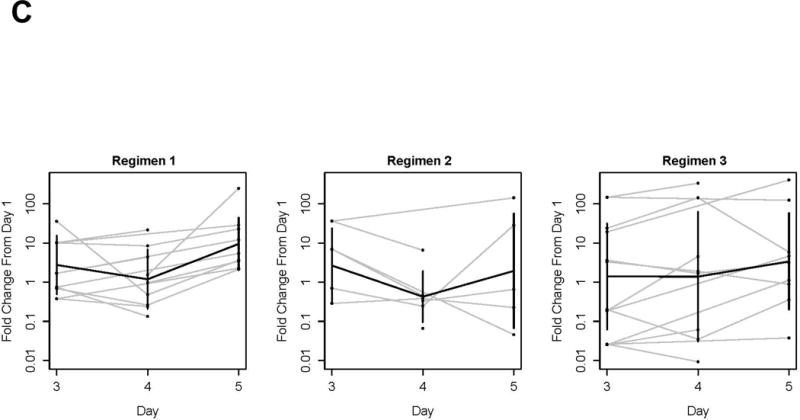
Broccoli sprout extracts are bioavailable and bioactive in the oral mucosa of healthy volunteers. A, Overnight urine was collected at baseline, and following the final BSE dose in each regimen. Regimen 1, 600 μmol of glucoraphanin-rich BSE/day; Regimen 2, 150 μmol of sulforaphane-rich BSE/day; Regimen 3, 150 μmol of sulforaphane-rich BSE was swished, gargled and expectorated for 6 minutes daily. Sulforaphane and sulforaphane-N-acetylcysteine were quantified by isotope dilution mass spectrometry, and normalized to urine creatinine. Sulforaphane-rich BSE was significantly more bioavailable than either glucoraphanin-rich BSE or topical sulforaphane-rich BSE (p=0.0013 by mixed-effects ANOVA). Bars represent 90% confidence intervals. B, Buccal cells were collected at baseline, and days 3–5 of each BSE regimen. mRNA transcripts for NQO1 were measured by qPCR. Three participants had adequate mRNA at every regimen time point and are displayed. The protocol definition of mucosal bioactivity, ≥2-fold upregulation of NQO1 transcripts, is shown for glucoraphanin-rich BSE in participants #2 and #8, for sulforaphane-rich BSE in participants #7 and #8, and for topical sulforaphane-rich BSE in none. C, A mixed-effects ANOVA was conducted to explore the effect of regimen and day on NQO1 qPCR. Fold change in ΔCT is presented by participant (gray lines), regimen, and day. Bold lines represent the means estimated from the mixed-effects ANOVA.
To evaluate whether BSEs modulate NRF2 signaling in oral epithelium, as measured by NQO1 qPCR, buccal cells were collected at baseline and on Days 3–5 of each regimen. Median total RNA yields were: Regimen 1, 461 ng (range 326–2,000); Regimen 2, 455 ng (range 234–1,324); Regimen 3, 528 ng (range 302–1,316). Total mRNA yield was < 18 ng/μl at 20 of 120 time points; these time points failed to meet quality control criteria and were excluded from the primary descriptive analysis. Following pre-specified definitions for evaluability and mucosal bioactivity, at least 2-fold upregulation of NQO1 buccal mRNA was observed with variable kinetics in 6 of 9 evaluable participants ingesting glucoraphanin-rich BSE; 3 of 6 ingesting sulforaphane-rich BSE; and 3 of 9 after topical exposure to sulforaphane-rich BSE. Three participants (#2, #7, and #8) provided sufficient buccal cell RNA at all regimen time points for quantitative analysis. qPCR of RNA from these participants demonstrated greater than 2-fold upregulation of NQO1 transcript in Participants #2 and #8 during Regimen 1, and greater than 2-fold upregulation in Participants #7 and #8 during Regimen 2 (Fig. 6B). Upregulation of NQO1 was not detected in any of the three participants during Regimen 3. Fold change in ΔCT, compared to day 1, is shown by regimen for all evaluable participants (Fig 6C). Although underpowered, a mixed-effects ANOVA was conducted to explore the effect of regimen and day on NQO1 qPCR. All ΔCT values derived from time points meeting quality criteria (mRNA > 18 ng) were included. NQO1 mRNA changed significantly by regimen (p=0.0014) and day (p=0.029), however no significant interaction between day and regimen was detected (p=0.66). As compared to baseline, NQO1 mRNA was significantly upregulated on day 5 in Regimen 1 (p<0.0001; Westfall’s correction for multiple testing). To address the low RNA yield from buccal cell collection by curette, a feasibility barrier for future studies, we subsequently collected buccal cells by cytobrush from 10 volunteers; this technique achieved substantially higher yields (median 1,490 ng of RNA; range 285–4,668 ng).
Discussion
Despite preclinical successes, including the prevention of murine oral cancers by retinoids or COX inhibitors in the DMBA model and by an EGFR inhibitor in the 4NQO model, no tolerable and effective agent to prevent HNSCC has been successfully translated to the clinic. The developmental failure of isolated micronutrient and molecular targeting interventions has invigorated interest in “green chemoprevention,” cost-conscious and tolerable interventions based upon whole plants or their simple extracts (46). In this study, we utilize preclinical in vitro and in vivo models as well as a pilot clinical trial to investigate the potential chemopreventive activity of sulforaphane, a phytochemical found in cruciferous vegetables, against oral environmental carcinogenesis. We observe induction of functional NRF2 signaling by sulforaphane in both a normal mucosal and several HNSCC cell lines. Notably, we provide first-time demonstration that concurrent oral administration of sulforaphane significantly protects against carcinogen-induced oral cancer in mice. We further demonstrate that the systemic administration of BSEs, rich in sulforaphane or its precursor glucoraphanin, is well-tolerated and shows preliminary evidence of NRF2 pathway activation in the oral mucosa of healthy human volunteers.
The impact of sulforaphane on normal epithelial cells and HNSCC cells is likely to be complex and dependent on dose. Sulforaphane induction of NRF2 occurred at concentrations as low at 0.1–0.5 μM, whereas induction of apoptosis typically required concentrations 10 μM or higher. The elevation of NRF2 protein and consequent induction of NRF2 target genes at low concentrations of sulforaphane promotes detoxication of environmental carcinogens and cellular protection from oxidative damage (30). These effects may prevent transformation of normal or condemned mucosal epithelial cells in at-risk patients, particularly where exposure to carcinogens continues to be high (eg. smokers). However, NRF2-mediated detoxication may lessen the potency of chemotherapy or radiation, as previously reported (47), suggesting that sulforaphane-based chemoprevention against second primary tumors would ideally start after completion of curative-intent treatment for HNSCC. At higher concentrations of sulforaphane (≥10 μM) Het-1A and HNSCC cells underwent apoptosis, consistent with reports of sulforaphane-induced apoptosis in other cancer models (48, 49). Interestingly, NRF2 acted to inhibit sulforaphane-induced apoptosis, as suppression of NRF2 expression led to elevated levels of cell death in UMSCC-22A cells. Higher concentrations of sulforaphane also led to rapid dephosphorylation of STAT3 via an NRF2-independent pathway. Similar dephosphorylation of STAT3 has also been reported in prostate cancer cells (50). Since phosphorylated/activated STAT3 plays a key role in driving the proliferation and survival of HNSCC, it will be important to determine whether inactivation of STAT3 represents a primary mechanism of the pro-apoptotic activity of sulforaphane in this disease.
The chemopreventive potential of sulforaphane against carcinogen-induced oral cancer had not previously been investigated. Our findings demonstrate that co-treatment with sulforaphane markedly reduced both the incidence and size of tongue tumors in mice exposed to 4NQO. Our pilot study in mice did not answer whether sulforaphane prevents tumor initiation, promotion, or both. Determining where and how sulforaphane acts in the continuum of oral carcinogenesis may be important to the design of chemoprevention studies. NRF2 has been described as a “double-edged sword” in environmental carcinogenesis (51). NRF2 is a critical mammalian pathway for maintaining homeostatic reduction potential in the face of chronic oxidative stress. Paradoxically, activating Nrf2 or inactivating Keap1 mutations were found in 14% and 5% of HPV-negative HNSCC tumors sequenced by the Cancer Genome Atlas (52), respectively, raising the possibility that constitutive NRF2 pathway activation promotes HNSCC development. However, in vivo, sulforaphane did not potentiate 4NQO-induced tongue tumors. Rather, both incidence and size of established tumors were markedly reduced, the latter suggesting that NRF2 activation did not provide a proliferation or survival advantage to established neoplasias. Future experiments testing sequential 4NQO followed by sulforaphane will determine whether sulforaphane can prevent post-initiation tumor formation, potentially analogous to preventing SPTs in smokers. Experiments with Nrf2−/− mice will clarify whether the preventive effects of sulforaphane are due to NRF2-mediated detoxication, prevention of tumor initiation and/or progression, or induction of apoptosis by sulforaphane in developing tumors. In either case, our in vivo results support the evaluation of sulforaphane, or more realistically, sulforaphane-rich plant extracts, as a chemopreventive agent in patients exposed to high levels of environmental carcinogens.
Towards the development of a “green chemoprevention” regimen for patients at risk for HNSCC, we performed a pilot study in healthy human volunteers to evaluate the bioavailability and pharmacodynamic activity of three BSE regimens. The safety, tolerability and pharmacokinetics of various broccoli seed preparations have been well characterized in healthy volunteers, and standard preparations with defined concentrations of sulforaphane or glucoraphanin have been developed (28, 33–36, 53). Notably, consumption of a BSE beverage containing 70 μmol sulforaphane-equivalent by residents of a polluted region of China promoted significant and sustained detoxication of the air pollutants acrolein and benzene, carcinogens also found in tobacco smoke (36). Moreover, bioactivity was observed throughout a 12-week exposure and healthy volunteers demonstrated excellent compliance, raising the promise of sustainable chronic administration. In our pilot study, both tolerability and bioavailability of BSE beverages were consistent with observations from prior trials. Compliance was excellent for ingestion of glucoraphanin-rich or sulforaphane-rich BSE. Bioavailability was superior for ingested sulforaphane versus glucoraphanin, consistent with prior studies and thought to reflect individual variability of the gut microbiome and hence, intestinal conversion of glucoraphanin to sulforaphane (28, 34). Importantly, ingestion of either beverage demonstrated preliminary evidence of NRF2 pathway activation in oral mucosa. Because the majority of tobacco-related HNSCC occurs in the oral cavity and oropharynx, oral rinses are an attractive and plausible method to concentrate drug delivery to at-risk tissue. However, we observed low bioavailability of the topical sulforaphane-rich beverage in Regimen 3. Although the median topical exposure time was 6 minutes, as recommended, 6 participants also described Grade 1 gum, buccal or pharyngeal irritation, attributed to the acidic juice medium. Therefore, we do not recommend development of this preparation as an oral rinse. Future studies are planned with encapsulated broccoli seed powder to enhance ease of dispensing and acceptability.
In summary, our studies illustrate the chemopreventive potential of sulforaphane-rich broccoli seed preparations against oral environmental carcinogenesis, and justify prospective clinical investigation in patients at risk for tobacco-related HNSCC and SPTs. Further pharmacodynamic and dose-response studies are planned in patients who have completed curative treatment for a first tobacco-related HNSCC. Ultimately, interventions based upon whole plants or their simple extracts, as compared to pharmaceutical agents, may ease the production, distribution, and sustainability of chemoprevention against HNSCC – particularly in resource-poor settings. The identification of a cost-effective, green chemopreventive agent would have a major global impact on mortality and quality of life in patients at risk, as the burden of HNSCC disproportionately affects the developing world.
Supplementary Material
Acknowledgments
Grant Support
This work was supported by NIH grant P50CA097190 (J.E. Bauman, D.P. Normolle, J.R. Grandis, D.E. Johnson) and R01CA190610 (T.W. Kensler). This project also received support from the Lewis B. and Dorothy Cullman Foundation (J.W. Fahey), and also used UPCI shared resources supported in part by NIH grant P30 CA47904.
Footnotes
Disclosure of Potential Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Slaughter DP, Southwick HW, Smejkal W. Field cancerization in oral stratified squamous epithelium; clinical implications of multicentric origin. Cancer. 1953 Sep;6(5):963–8. doi: 10.1002/1097-0142(195309)6:5<963::aid-cncr2820060515>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 2.Leon X, Quer M, Diez S, Orus C, Lopez-Pousa A, Burgues J. Second neoplasm in patients with head and neck cancer. Head Neck. 1999 May;21(3):204–10. doi: 10.1002/(sici)1097-0347(199905)21:3<204::aid-hed4>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 3.Lee DH, Roh JL, Baek S, Jung JH, Choi SH, Nam SY, et al. Second cancer incidence, risk factor, and specific mortality in head and neck squamous cell carcinoma. Otolaryngology–head and neck surgery: official journal of American Academy of Otolaryngology-Head and Neck Surgery. 2013 Oct;149(4):579–86. doi: 10.1177/0194599813496373. [DOI] [PubMed] [Google Scholar]
- 4.Day GL, Blot WJ, Shore RE, McLaughlin JK, Austin DF, Greenberg RS, et al. Second cancers following oral and pharyngeal cancers: role of tobacco and alcohol. J Natl Cancer Inst. 1994 Jan 19;86(2):131–7. doi: 10.1093/jnci/86.2.131. [DOI] [PubMed] [Google Scholar]
- 5.Lippman SM, Hong WK. Second malignant tumors in head and neck squamous cell carcinoma: the overshadowing threat for patients with early-stage disease. Int J Radiat Oncol Biol Phys. 1989 Sep;17(3):691–4. doi: 10.1016/0360-3016(89)90126-0. [DOI] [PubMed] [Google Scholar]
- 6.Khuri FR, Kim ES, Lee JJ, Winn RJ, Benner SE, Lippman SM, et al. The impact of smoking status, disease stage, and index tumor site on second primary tumor incidence and tumor recurrence in the head and neck retinoid chemoprevention trial. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 2001 Aug;10(8):823–9. [PubMed] [Google Scholar]
- 7.Hong WK, Endicott J, Itri LM, Doos W, Batsakis JG, Bell R, et al. 13-cis-retinoic acid in the treatment of oral leukoplakia. N Engl J Med. 1986 Dec 11;315(24):1501–5. doi: 10.1056/NEJM198612113152401. [DOI] [PubMed] [Google Scholar]
- 8.Hong WK, Lippman SM, Itri LM, Karp DD, Lee JS, Byers RM, et al. Prevention of second primary tumors with isotretinoin in squamous-cell carcinoma of the head and neck. N Engl J Med. 1990 Sep 20;323(12):795–801. doi: 10.1056/NEJM199009203231205. [DOI] [PubMed] [Google Scholar]
- 9.Khuri FR, Lee JJ, Lippman SM, Kim ES, Cooper JS, Benner SE, et al. Randomized phase III trial of low-dose isotretinoin for prevention of second primary tumors in stage I and II head and neck cancer patients. J Natl Cancer Inst. 2006 Apr 5;98(7):441–50. doi: 10.1093/jnci/djj091. [DOI] [PubMed] [Google Scholar]
- 10.Leeman-Neill RJ, Seethala RR, Singh SV, Freilino ML, Bednash JS, Thomas SM, et al. Inhibition of EGFR-STAT3 signaling with erlotinib prevents carcinogenesis in a chemically-induced mouse model of oral squamous cell carcinoma. Cancer prevention research. 2011 Feb;4(2):230–7. doi: 10.1158/1940-6207.CAPR-10-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perkins TM, Shklar G. Delay in hamster buccal pouch carcinogenesis by aspirin and indomethacin. Oral surgery, oral medicine, and oral pathology. 1982 Feb;53(2):170–8. doi: 10.1016/0030-4220(82)90283-3. [DOI] [PubMed] [Google Scholar]
- 12.Cornwall H, Odukoya O, Shklar G. Oral mucosal tumor inhibition by ibuprofen. Journal of oral and maxillofacial surgery: official journal of the American Association of Oral and Maxillofacial Surgeons. 1983 Dec;41(12):795–800. doi: 10.1016/s0278-2391(83)80046-9. [DOI] [PubMed] [Google Scholar]
- 13.Tanaka T, Nishikawa A, Mori Y, Morishita Y, Mori H. Inhibitory effects of non-steroidal anti-inflammatory drugs, piroxicam and indomethacin on 4-nitroquinoline 1-oxide-induced tongue carcinogenesis in male ACI/N rats. Cancer Lett. 1989 Dec;48(3):177–82. doi: 10.1016/0304-3835(89)90115-8. [DOI] [PubMed] [Google Scholar]
- 14.Shiotani H, Denda A, Yamamoto K, Kitayama W, Endoh T, Sasaki Y, et al. Increased expression of cyclooxygenase-2 protein in 4-nitroquinoline-1-oxide-induced rat tongue carcinomas and chemopreventive efficacy of a specific inhibitor, nimesulide. Cancer Res. 2001 Feb 15;61(4):1451–6. [PubMed] [Google Scholar]
- 15.Laine L, Bombardier C, Hawkey CJ, Davis B, Shapiro D, Brett C, et al. Stratifying the risk of NSAID-related upper gastrointestinal clinical events: results of a double-blind outcomes study in patients with rheumatoid arthritis. Gastroenterology. 2002 Oct;123(4):1006–12. doi: 10.1053/gast.2002.36013. [DOI] [PubMed] [Google Scholar]
- 16.Mulshine JL, Atkinson JC, Greer RO, Papadimitrakopoulou VA, Van Waes C, Rudy S, et al. Randomized, double-blind, placebo-controlled phase IIb trial of the cyclooxygenase inhibitor ketorolac as an oral rinse in oropharyngeal leukoplakia. Clin Cancer Res. 2004 Mar 1;10(5):1565–73. doi: 10.1158/1078-0432.ccr-1020-3. [DOI] [PubMed] [Google Scholar]
- 17.Bertagnolli MM. Chemoprevention of colorectal cancer with cyclooxygenase-2 inhibitors: two steps forward, one step back. Lancet Oncol. 2007 May;8(5):439–43. doi: 10.1016/S1470-2045(07)70139-0. [DOI] [PubMed] [Google Scholar]
- 18.Papadimitrakopoulou VA, William WN, Jr, Dannenberg AJ, Lippman SM, Lee JJ, Ondrey FG, et al. Pilot randomized phase II study of celecoxib in oral premalignant lesions. Clin Cancer Res. 2008 Apr 1;14(7):2095–101. doi: 10.1158/1078-0432.CCR-07-4024. [DOI] [PubMed] [Google Scholar]
- 19.Saba NF, Hurwitz SJ, Kono SA, Yang CS, Zhao Y, Chen Z, et al. Chemoprevention of head and neck cancer with celecoxib and erlotinib: results of a phase ib and pharmacokinetic study. Cancer prevention research. 2014 Mar;7(3):283–91. doi: 10.1158/1940-6207.CAPR-13-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Day GL, Shore RE, Blot WJ, McLaughlin JK, Austin DF, Greenberg RS, et al. Dietary factors and second primary cancers: a follow-up of oral and pharyngeal cancer patients. Nutr Cancer. 1994;21(3):223–32. doi: 10.1080/01635589409514321. [DOI] [PubMed] [Google Scholar]
- 21.Chainani-Wu N. Diet and oral, pharyngeal, and esophageal cancer. Nutr Cancer. 2002;44(2):104–26. doi: 10.1207/S15327914NC4402_01. [DOI] [PubMed] [Google Scholar]
- 22.Pavia M, Pileggi C, Nobile CG, Angelillo IF. Association between fruit and vegetable consumption and oral cancer: a meta-analysis of observational studies. Am J Clin Nutr. 2006 May;83(5):1126–34. doi: 10.1093/ajcn/83.5.1126. [DOI] [PubMed] [Google Scholar]
- 23.Fowke JH. Head and neck cancer: a case for inhibition by isothiocyanates and indoles from cruciferous vegetables. European journal of cancer prevention: the official journal of the European Cancer Prevention Organisation. 2007 Aug;16(4):348–56. doi: 10.1097/01.cej.0000236258.80522.fb. [DOI] [PubMed] [Google Scholar]
- 24.Bravi F, Bosetti C, Filomeno M, Levi F, Garavello W, Galimberti S, et al. Foods, nutrients and the risk of oral and pharyngeal cancer. Br J Cancer. 2013 Nov 26;109(11):2904–10. doi: 10.1038/bjc.2013.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Prochaska HJ, Santamaria AB, Talalay P. Rapid detection of inducers of enzymes that protect against carcinogens. Proc Natl Acad Sci U S A. 1992 Mar 15;89(6):2394–8. doi: 10.1073/pnas.89.6.2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, et al. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun. 1997 Jul 18;236(2):313–22. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 27.Kensler TW. Chemoprevention by inducers of carcinogen detoxication enzymes. Environmental health perspectives. 1997 Jun;105(Suppl 4):965–70. doi: 10.1289/ehp.97105s4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kensler TW, Egner PA, Agyeman AS, Visvanathan K, Groopman JD, Chen JG, et al. Keap1-nrf2 signaling: a target for cancer prevention by sulforaphane. Topics in current chemistry. 2013;329:163–77. doi: 10.1007/128_2012_339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong F, Freeman ML, Liebler DC. Identification of sensor cysteines in human Keap1 modified by the cancer chemopreventive agent sulforaphane. Chemical research in toxicology. 2005 Dec;18(12):1917–26. doi: 10.1021/tx0502138. [DOI] [PubMed] [Google Scholar]
- 30.Kensler TW, Wakabayashi N. Nrf2: friend or foe for chemoprevention? Carcinogenesis. 2010 Jan;31(1):90–9. doi: 10.1093/carcin/bgp231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fahey JW, Zhang Y, Talalay P. Broccoli sprouts: an exceptionally rich source of inducers of enzymes that protect against chemical carcinogens. Proc Natl Acad Sci U S A. 1997 Sep 16;94(19):10367–72. doi: 10.1073/pnas.94.19.10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Tang L. Discovery and development of sulforaphane as a cancer chemopreventive phytochemical. Acta pharmacologica Sinica. 2007 Sep;28(9):1343–54. doi: 10.1111/j.1745-7254.2007.00679.x. [DOI] [PubMed] [Google Scholar]
- 33.Kensler TW, Chen JG, Egner PA, Fahey JW, Jacobson LP, Stephenson KK, et al. Effects of glucosinolate-rich broccoli sprouts on urinary levels of aflatoxin-DNA adducts and phenanthrene tetraols in a randomized clinical trial in He Zuo township, Qidong, People’s Republic of China. Cancer Epidemiol Biomarkers Prev. 2005 Nov;14(11 Pt 1):2605–13. doi: 10.1158/1055-9965.EPI-05-0368. [DOI] [PubMed] [Google Scholar]
- 34.Egner PA, Chen JG, Wang JB, Wu Y, Sun Y, Lu JH, et al. Bioavailability of Sulforaphane from two broccoli sprout beverages: results of a short-term, cross-over clinical trial in Qidong, China. Cancer prevention research. 2011 Mar;4(3):384–95. doi: 10.1158/1940-6207.CAPR-10-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kensler TW, Ng D, Carmella SG, Chen M, Jacobson LP, Munoz A, et al. Modulation of the metabolism of airborne pollutants by glucoraphanin-rich and sulforaphane-rich broccoli sprout beverages in Qidong, China. Carcinogenesis. 2012 Jan;33(1):101–7. doi: 10.1093/carcin/bgr229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Egner PA, Chen JG, Zarth AT, Ng DK, Wang JB, Kensler KH, et al. Rapid and sustainable detoxication of airborne pollutants by broccoli sprout beverage: results of a randomized clinical trial in China. Cancer prevention research. 2014 Aug;7(8):813–23. doi: 10.1158/1940-6207.CAPR-14-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fahey JW, Haristoy X, Dolan PM, Kensler TW, Scholtus I, Stephenson KK, et al. Sulforaphane inhibits extracellular, intracellular, and antibiotic-resistant strains of Helicobacter pylori and prevents benzo[a]pyrene-induced stomach tumors. Proc Natl Acad Sci U S A. 2002 May 28;99(11):7610–5. doi: 10.1073/pnas.112203099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu C, Huang MT, Shen G, Yuan X, Lin W, Khor TO, et al. Inhibition of 7,12-dimethylbenz(a)anthracene-induced skin tumorigenesis in C57BL/6 mice by sulforaphane is mediated by nuclear factor E2-related factor 2. Cancer Res. 2006 Aug 15;66(16):8293–6. doi: 10.1158/0008-5472.CAN-06-0300. [DOI] [PubMed] [Google Scholar]
- 39.Cornblatt BS, Ye L, Dinkova-Kostova AT, Erb M, Fahey JW, Singh NK, et al. Preclinical and clinical evaluation of sulforaphane for chemoprevention in the breast. Carcinogenesis. 2007 Jul;28(7):1485–90. doi: 10.1093/carcin/bgm049. [DOI] [PubMed] [Google Scholar]
- 40.Zhang Y, Kensler TW, Cho CG, Posner GH, Talalay P. Anticarcinogenic activities of sulforaphane and structurally related synthetic norbornyl isothiocyanates. Proceedings of the National Academy of Sciences of the United States of America. 1994 Apr 12;91(8):3147–50. doi: 10.1073/pnas.91.8.3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ramos-Gomez M, Kwak MK, Dolan PM, Itoh K, Yamamoto M, Talalay P, et al. Sensitivity to carcinogenesis is increased and chemoprotective efficacy of enzyme inducers is lost in nrf2 transcription factor-deficient mice. Proc Natl Acad Sci U S A. 2001 Mar 13;98(6):3410–5. doi: 10.1073/pnas.051618798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohkoshi A, Suzuki T, Ono M, Kobayashi T, Yamamoto M. Roles of Keap1-Nrf2 system in upper aerodigestive tract carcinogenesis. Cancer prevention research. 2013 Feb;6(2):149–59. doi: 10.1158/1940-6207.CAPR-12-0401-T. [DOI] [PubMed] [Google Scholar]
- 43.Zang Y, Kirk CJ, Johnson DE. Carfilzomib and oprozomib synergize with histone deacetylase inhibitors in head and neck squamous cell carcinoma models of acquired resistance to proteasome inhibitors. Cancer Biol Ther. 2014 Sep;15(9):1142–52. doi: 10.4161/cbt.29452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stoner GD, Kaighn ME, Reddel RR, Resau JH, Bowman D, Naito Z, et al. Establishment and characterization of SV40 T-antigen immortalized human esophageal epithelial cells. Cancer Res. 1991 Jan 1;51(1):365–71. [PubMed] [Google Scholar]
- 45.Trask DK, Wolf GT, Bradford CR, Fisher SG, Devaney K, Johnson M, et al. Expression of Bcl-2 family proteins in advanced laryngeal squamous cell carcinoma: correlation with response to chemotherapy and organ preservation. Laryngoscope. 2002 Apr;112(4):638–44. doi: 10.1097/00005537-200204000-00009. [DOI] [PubMed] [Google Scholar]
- 46.Fahey JW, Talalay P, Kensler TW. Notes from the field: “green” chemoprevention as frugal medicine. Cancer prevention research. 2012 Feb;5(2):179–88. doi: 10.1158/1940-6207.CAPR-11-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rushworth SA, Zaitseva L, Murray MY, Shah NM, Bowles KM, MacEwan DJ. The high Nrf2 expression in human acute myeloid leukemia is driven by NF-kappaB and underlies its chemo-resistance. Blood. 2012 Dec 20;120(26):5188–98. doi: 10.1182/blood-2012-04-422121. [DOI] [PubMed] [Google Scholar]
- 48.Singh AV, Xiao D, Lew KL, Dhir R, Singh SV. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis. 2004 Jan;25(1):83–90. doi: 10.1093/carcin/bgg178. [DOI] [PubMed] [Google Scholar]
- 49.Pham NA, Jacobberger JW, Schimmer AD, Cao P, Gronda M, Hedley DW. The dietary isothiocyanate sulforaphane targets pathways of apoptosis, cell cycle arrest, and oxidative stress in human pancreatic cancer cells and inhibits tumor growth in severe combined immunodeficient mice. Mol Cancer Ther. 2004 Oct;3(10):1239–48. [PubMed] [Google Scholar]
- 50.Hahm ER, Singh SV. Sulforaphane inhibits constitutive and interleukin-6-induced activation of signal transducer and activator of transcription 3 in prostate cancer cells. Cancer prevention research. 2010 Apr;3(4):484–94. doi: 10.1158/1940-6207.CAPR-09-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hayes JD, McMahon M. The double-edged sword of Nrf2: subversion of redox homeostasis during the evolution of cancer. Mol Cell. 2006 Mar 17;21(6):732–4. doi: 10.1016/j.molcel.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 52.Cancer Genome Atlas N. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015 Jan 29;517(7536):576–82. doi: 10.1038/nature14129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cipolla BG, Mandron E, Lefort JM, Coadou Y, Della Negra E, Corbel L, et al. Effect of Sulforaphane in Men with Biochemical Recurrence after Radical Prostatectomy. Cancer prevention research. 2015 May 12; doi: 10.1158/1940-6207.CAPR-14-0459. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.